

Abstract
Because both the host and pathogen require iron, the innate immune response carefully orchestrates control over iron metabolism to limit its availability during times of infection. Nutritional iron deficiency can impair host immunity, while iron overload can cause oxidative stress to propagate harmful viral mutations. An emerging enigma is that many viruses use the primary gatekeeper of iron metabolism, the transferrin receptor, as a means to enter cells. Why and how this iron gate is a viral target for infection are the focus of this review.
Keywords: iron, infection, innate immunity, receptor-mediated endocytosis, mTOR
1. INTRODUCTION
Iron is essential for oxygen transport, ATP (adenosine triphosphate) production, mitochondrial function, DNA replication, and other vital metabolic processes. Since both hosts and pathogens require iron, our innate immune response includes carefully orchestrated control over iron metabolism to limit its availability during times of infection. However, too much sequestered iron can be toxic due to oxidative stress and the generation of free radicals that damage lipids, DNA, and other cellular constituents. Recent reviews have focused on how mammalian cells balance their needs for iron with strategies to limit overload (4, 19, 60, 75) and how iron status is lowered during the innate immune response to fight infection by invading microorganisms (43, 162, 164, 165). While significant advances have been made in our understanding of iron metabolism and homeostasis, an emerging enigma is that many viruses use the primary gatekeeper of iron metabolism, the transferrin receptor, as a means to enter cells. Why and how this iron gate is a target for viral infection are the focus of this review.
2. IRON DELIVERY
Although the human body contains a total of 2–4 g of iron, only 1–2 mg of iron is absorbed from the diet each day (60). This small but necessary amount is sufficient to maintain losses caused by the turnover of gastrointestinal cells and skin epithelia, along with unregulated body excretions, such as sweat and tears. The balance of iron in the body is maintained by a high degree of conservation in its use rather than a reliance on dietary intake. While there is constant demand for iron to produce hemoglobin for new red blood cells, most of the supply is met by clearing damaged and senescent cells to liberate used iron for recycling. It has been estimated that about 25–30 mg of iron is turned over each day by the macrophages of the reticuloendothelial system. The distribution of iron between these different pools is carried out by transferrin (Tf) and its receptor, which mediate delivery and uptake by peripheral tissues (53, 68).
2.1. Transferrin
Tf is an ~80-kDa glycoprotein that binds up to two atoms of iron with very high affinity [Ka (association constant) ~ 1020 M–1]. It is a single chain polypeptide with two lobes (N- and C-terminal) that can each bind a single ferric cation (Fe3+), with carbonate as a counteranion (104). While Tf has two asparagine-linked glycosylation sites in the C-lobe, the oligosaccharides do not appear to influence Tf binding to its receptor (104). Its half-life in circulation is 8–10 days (15), although desialylation may enhance the protein’s clearance by asialoglycoprotein receptors (104). The well-defined pattern of Tf glycosylation is commonly used to screen individuals for congenital disorders of glycosylation (54).
Tf is produced primarily in the liver, with much lower transcript levels detected in some extrahepatic tissues (thymus, adrenal glands, uterus, and ovaries) (173). Its synthesis is modulated by iron deficiency, inflammation, and hypoxia (68). Hypotransferrinemia may be associated with alcoholism, hemodialysis, depression, and urinary protein loss (9). Tf is one of the most abundant plasma proteins (~3 mg/mL), but in healthy individuals only 20–50% of the available Tf binding sites have iron bound. Tf iron-binding capacity (TIBC) and serum iron are routine clinical laboratory measurements from which the iron saturation of Tf can be derived; values less than 15% denote iron deficiency, and those greater than 45% indicate iron overload (128). Circulating Tf concentrations are subject to diurnal variation (34), but in general, because TIBC is typically in excess, large and immediate increases in iron binding can be accommodated. These free Tf iron-binding sites are needed to help distribute large fluxes of iron to peripheral tissues after dietary iron absorption by the intestine, iron recycling after phagocytosis of red blood cells by macrophages of the reticuloendothelial system, and iron release from hepatic stores at times of demand, such as during blood loss, pregnancy, and hypoxia. The reversible binding of iron to Tf and its excess buffering capacity also significantly limit potentially damaging free nontransferrin- bound iron (NTBI) and help to maintain the metal in its ferric form under physiological pH and oxygen tension, conditions that would otherwise promote ferric iron oxidation and generate redox activity to produce free radical damage. Interestingly, individuals with hypotransferrinemia survive and their condition can be corrected by supplementation with exogenous protein (15). Also, hypotransferrinemic (Hpx) mice survive with the help of transferrin injections (9). Although iron assimilation into hemoglobin is impaired by hypotransferrinemia, tissue iron loading occurs due to the presence of high levels of NTBI (154).
2.2. Transferrin Receptor-1
The major receptor for Tf is transferrin receptor-1 (TfR1), a homodimeric type II membrane glycoprotein (~95 kDa) that binds to and facilitates entry of its ligand into cells for the delivery of iron (Figure 1) (75). Also known as CD71, human TfR1 has a short cytoplasmic N-terminal tail (residues 1–67) with an endosomal uptake signal motif (YTRF) (29). This intracellular domain is also lipidated (29, 141). A single transmembrane-spanning domain connects to an extracellular stalk region that houses the two disulfide bonds (Cys89 and Cys98), covalently linking the large homodimeric ectodomains (113, 142). Extracellular O-linked glycosylation atThr104 influences cleavage of the stalk at Arg100 (137). Cleavage at this site releases a circulating form of TfR1 (soluble Tf receptor) that is frequently used as a biomarker to assess iron status since it reflects turnover of TfR1, particularly in erythropoietic cells (128).
Figure 1.
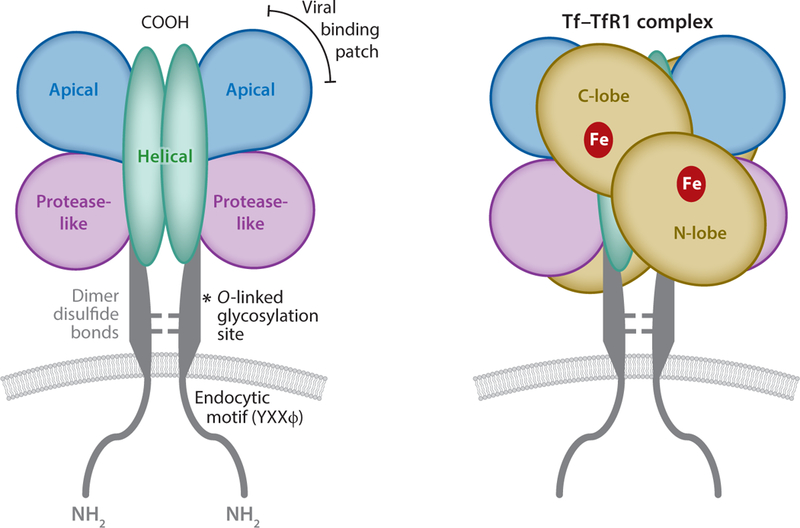
Structural representation of the transferrin–transferrin receptor-1 (Tf–TfR1) complex. The receptor is a type II membrane protein with apical, helical, and protease-like domains. Two cysteine residues in the so-called stalk region form disulfide links between monomers. The viral binding patch is located in the apical domain. The Tf–TfR1 complex includes Tf binding to each dimer. The ligand has N- and C-lobes that each contain an iron binding site.
The receptor’s ectodomain binds Tf and is characterized by an apical domain, a helical domain, and a protease-like domain (97). The latter adopts its name due to its structural resemblance to carboxypeptidase II and other proteases. The N-lobe of Tf contacts this protease-like domain, while the C-lobe interacts with the receptor’s helical domain (65). The individual subunits of the receptor dimer contain one Tf binding site, yielding a 2:2 stoichiometry for the overall complex. Each receptor subunit contains three N-linked glycosylation sites that influence TfR1 trafficking to the cell surface as well as Tf binding (172).
TfR1 binds iron-bound holo-Tf with greater affinity than iron-free apo-Tf (68, 142), and Tf binding to the receptor is pH dependent (104, 147). In addition, metal binding by Tf is also pH dependent (93). These pH-sensitive interactions enable iron delivery through the membrane trafficking pathway of the receptor, which carries Tf into acidic intracellular compartments where iron can be discharged, reduced to Fe2+, and translocated into the cytosol for different metabolic purposes (see Section 3). Interestingly, apo-Tf binds TfR1 with greater affinity at low pH, enabling it to be trafficked back to the cell surface, with release accommodated at the extracellular neutral pH (36). TfR1 constitutively traffics with Tf from the cell surface to intracellular endosomal compartments and back, such that at any given time, only ~20% of the total cellular receptor pool appears at the cell surface (105).
TfR1 is ubiquitously expressed, with protein levels reflecting both iron utilization and availability. For example, erythroid cells require a large amount of iron to manufacture hemoglobin and have up to 106 Tf receptors (78). Most actively proliferating cells in culture have approximately 104 to 105 cell surface receptors to sustain their metabolic needs. In general, the number of cellular receptors reflects the metabolic demand for iron, such that levels increase with deficiency and decrease with sufficiency (68). At the cellular level, TfR1 levels respond to fluctuating iron levels due to the activity of iron response element (IRE) binding proteins (IRPs) (75). IRPs bind to IREs in the 3’ untranslated region of TfR1 messenger RNA when iron is low. Activation of RNA binding activity under these conditions prevents transcript degradation and subsequently raises the level of TfR1 expression. When iron levels increase, IRP-IRE interactions are reduced, and, consequently, receptor expression is lowered.
Given that Tf-deficient mice survive and even accumulate tissue iron, it is perhaps surprising that global deletion of the TfR1 gene in mice causes embryonic lethality (100). Tissue-specific deletion of the receptor either in heart (170) or skeletal muscle (8) also significantly impairs iron metabolism. One explanation for the contrasting phenotypes of Tf- and TfR1-deficient mice is that in the absence of Tf, NTBI is available to tissues, while in TfR1-deficient mice, the presence of Tf makes iron much less available (154). Alternatively, TfR1 may have functions unrelated to iron metabolism that could explain these differences. Mouse genetic studies have implicated iron- independent roles for TfR1 in the early development of the nervous system (100), maintenance of intestinal homeostasis (23), craniofacial development (98), and lymphocyte development (120). At the cellular level, TfR1 is proposed to function in T cell signaling (10, 139), NF-κB (nuclear factor κB) activation (90), and JNK regulation (141). A human mutation in TfR1 has been identified in patients with combined immunodeficiency, revealing the receptor’s indirect role in adaptive immunity (81).
Although classically studied as a receptor for Tf, human (but not mouse) TfR1 also has been characterized to bind and internalize ferritin (101). In this pathway, ferritin is released intracellularly and targeted for lysosomal degradation to ultimately deliver iron to cells. This function may play a significant part in brain iron metabolism since Tf levels are low while ferritin levels are high in neuronal interstitial fluid. Another reported ligand for TfR1 is polymeric A1 isotype immunoglobulin (pIgA1) (117). Bound pIgA1 does not compete with holo-Tf and potentially may be involved in signaling functions important to nephropathies and celiac disease (116). The unique functions of these alternative ligands and TfR1 suggest that there may be additional undiscovered roles for the receptor.
2.3. Transferrin Receptor-2
In 1999, Kawabata et al. (89) characterized a second Tf receptor (TfR2) that has a close structural relationship to TfR1 but a more restricted pattern of expression, mainly in the liver and erythropoietic cells (168). Like TfR1, TfR2 binds holo-Tf, but with 25-fold lower affinity; when Tf is bound, turnover of this receptor decreases, resulting in greater TfR2 levels (166). Unlike for TfR1, N-linked glycosylation of the TfR2 ectodomain does not influence Tf binding and trafficking to the cell surface, but this modification is required for the stabilization of TfR2 by the ligand (178). Because of its properties, TfR2 can be thought of as an iron sensor, such that higher iron levels and the corresponding increase in Tf saturation are reflected by a greater number of TfR2s at the cell surface (84, 134). Although in vitro studies show that TfR2 can mediate the uptake of iron from Tf (89, 166), its reduced affinity for the ligand, altered intracellular trafficking with Tf, and the limited pattern of receptor expression suggest that its true physiological function is quite different from TfR1 (133, 134). Both expression analyses (177) and functional studies (27) indicate that TfR2 levels predominate over TfR1 in the liver and some other tissues, but that the larger flux of Tf-bound iron taken up by hepatocytes most likely proceeds through TfR1. Heterodimers between TfR1 and TfR2 have been reported, but the functional consequences of such complexes are unclear (155). Conceptually, TfR1 can be thought of as the major player in the endocytic delivery of iron, while TfR2 is more analogous to a coincidence detector that signals the increasing iron saturation of Tf. The idea that TfR2 helps to generate a homeostatic signal to reflect TIBC levels is compatible with the observations that liver-specific deletion of TfR2 in mice promotes tissue iron overload (156) and that human TfR2 mutations are associated with the iron overload disease type 3 hereditary hemochromatosis (20, 51, 168). Consistent with TfR2’s role as an iron sensor, hemochromatosis patients with defective TfR2 have impaired responses to acute iron challenge (66).
2.4. Transferrin and Transferrin Receptor Function in Iron Homeostasis
As noted above, Tf binds iron that enters circulation via three major pathways: (a) dietary absorption by the intestine, (b) erythrophagocytosis and iron recycling of red blood cells, and (c) release from liver stores (Figure 2). In each circumstance, iron export is carried out by the membrane transporter ferroportin and is regulated by the liver-derived iron hormone hepcidin (60). Hepcidin binds to ferroportin, causing its internalization and degradation (122). Increased hepcidin reduces the amount of iron available to bind Tf, and, conversely, decreased hepcidin is associated with increased Tf saturation. Thus, the amount of iron bound to Tf and the amount of iron entering circulation can be coordinated through hepcidin.
Figure 2.
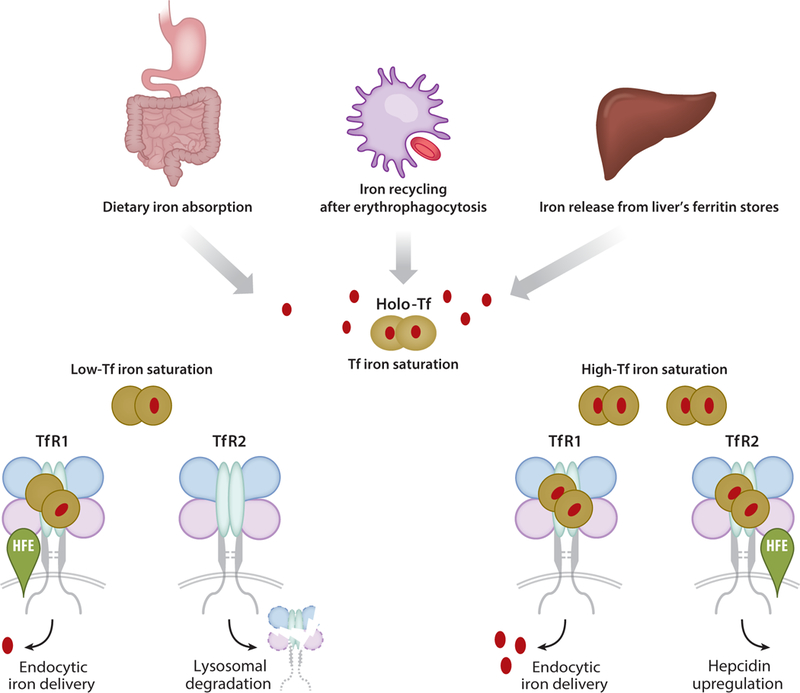
Transferrin (Tf) and transferrin receptor (TfR) function in iron homeostasis. Tf binds iron entering circulation via dietary absorption, iron recycling of red blood cells, or release from liver stores. These export pathways are regulated by the hormone hepcidin, which responds to iron saturation sensed by Tf–TfR through the hereditary hemochromatosis protein HFE. If there is iron surplus, Tf binding competes with HFE for association with TfR1, enabling it to bind TfR2 and participate in signaling hepcidin upregulation. Under these conditions, TfR1 mediates iron delivery, ultimately targeting the nutrient for metabolism or storage. When Tf saturation becomes reduced under low iron conditions, TfR2 is destabilized, and endocytic delivery is adjusted to meet functional demands.
A major element ensuring appropriate levels of hepcidin is the hereditary hemochromatosis protein (HFE) (61). Mutations in HFE are associated with type 1 hereditary hemochromatosis (49). HFE is an MHC (major histocompatibility complex) class I–like molecule that associates with both TfR1 and TfR2; however, its interactions with each receptor involve different structural domains. While the sites for Tf and HFE binding to TfR1 overlap (64, 65), Tf and HFE bind to independent sites on TfR2 (25). When there is iron surplus, increased Tf saturation promotes holo-Tf binding to TfR1 to displace HFE. Consequently, HFE–TfR2 interactions are enabled, especially because increased concentrations of holo-Tf stabilize TfR2, and HFE does not interfere with Tf binding to this receptor. Ultimately, increasing iron saturation of Tf confers a signal to upregulate hepcidin synthesis (70). It is likely that this signal involves hemojuvelin and other factors in the BMP/SMAD signaling pathway (7, 33). The idea of a Tf iron-sensing regulatory complex is well supported by the known consequences of genetic alterations in HFE, hemojuvelin, and TfR2, which, respectively, give rise to type 1,2, and 3 hereditary hemochromatosis because of impaired hepcidin regulation (168). This particular role is concordant with high TfR2 levels in the liver, the major site of hepcidin synthesis (180); other studies suggest that high levels of TfR2 in erythroid progenitors help to signal adaptive responses in red blood cell production to adjust systemic iron levels (119). Ultimately, high metabolic demand for iron by peripheral tissues also must be matched by endocytic delivery of Tf, which can be enhanced due to IRP-controlled levels of TfR1. The net result of the feedback loop is to reduce iron saturation and, consequently, diminish hepcidin production. In this manner, hepcidin and HFE help Tf to coordinate the distinct functions of both Tf receptors to maintain iron homeostasis (Figure 2).
3. THE INTRACELLULAR PATHWAY FOR IRON DELIVERY
The mechanism used for intracellular membrane trafficking by the Tf–TfR1 complex is integral to the process of iron delivery (68). Given the high-affinity receptor binding for Tf (nanomolar) coupled with high circulating levels of the serum protein (50 μM), it is conceivable that all TfR1 receptors are fully occupied most of the time with bound ligand. While the internalization of most growth factor receptors is induced by ligand binding, the view has emerged that TfR1 is constitutively internalized with or without bound Tf (2, 160). However, there is limited evidence that Tf binding triggers the internalization of the receptor (21, 67). The receptor’s cytoplasmic domain can be phosphorylated by Src tyrosine kinase (82), and it has been suggested that binding of Tf is associated with the activation of Src and recruitment of the endocytic machinery to TfR1 to stimulate constitutive endocytosis (21). Such signaling functions of TfR1 remain a key area to be explored.
A crucial feature of Tf–TfR1 membrane trafficking is the acidification of the endosomal recycling pathway. First, the lower pH reduces the binding affinity of Tf for iron. Second, the deepening pH gradient induces conformational changes in TfR1 itself that promote the release of iron and increased affinity for apo-Tf (44). Third, the acidic milieu enhances the solubility of the ferric form of iron. Once released, iron can be reduced by the ferrireductase STEAP1 and transported across the endosomal membrane by the pH-dependent divalent metal transporter-1 (DMT1) or other transport proteins (118). In erythroid cells, the demand for iron to maintain heme biosynthesis may be met by directed delivery of Tf iron from endocytic vesicles into the mitochondria (143). Figure 3a summarizes features of Tf–TfR1 membrane trafficking.
Figure 3.
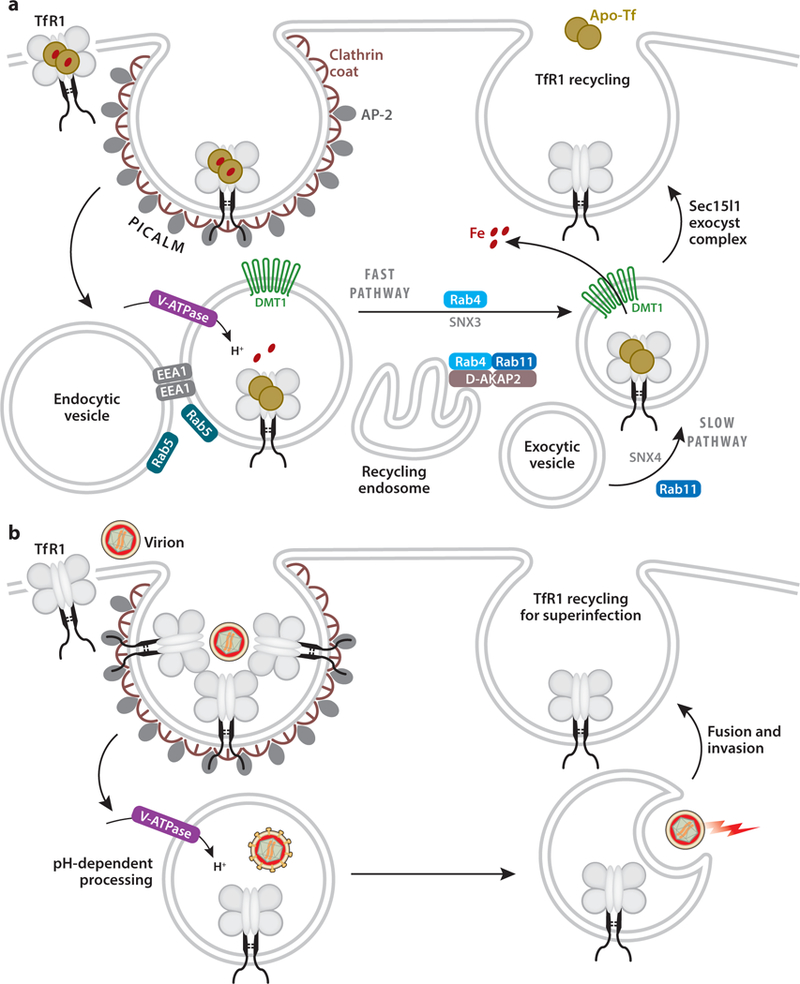
(a) The intracellular pathway for iron delivery. Transferrin receptor-1 (TfR1) clusters into clathrin-coated pits through sorting interactions with AP-2 (clathrin adaptor protein-2) and PICALM (phosphatidylinositol binding clathrin assembly lymphoid myeloid leukemia). Endocytic vesicles rapidly bud, EEA1 (early endosome associated antigen-1) is recruited, and endosomes undergo homotypic fusion mediated by Rab5. Endosomes acidify due to V-ATPase (vacuolar H+-ATPase) activity, and low pH promotes the discharge of iron, while apo-Tf remains bound to TfR1. Divalent metal transporter-1 (DMT1) mediates the release of iron to the cytoplasm. Recycling involves slow (Rab4/Rab11) and fast (Rab4) pathways; D-AKAP2 (dual-specific A kinase anchoring protein-2) regulates trafficking through the endocytic recycling compartment into these pathways. Sorting nexins (SNX3 and SNX4) help direct the Tf–TfR1 complex to return to the cell surface where the exocyst complex mediates fusion. Sec15l1, a component of the exocyst, helps direct TfR1 recycling. (b) Viral entry follows the TfR1 membrane trafficking pathway. Invasion begins with virions interacting with surface receptors, capturing the capsid for entry into the clathrin-mediated pathway. Uptake may involve the clustering of a few receptors, which deliver the virion to low-pH endocytic compartments. Here, pH-dependent processing can occur, and fusion of the virion with the endosomal membrane and invasion into the cytoplasm takes place. The recycling of TfR1 may allow for superinfection to occur.
3.1. Endocytosis
TfR1 is the textbook example of the canonical clathrin-mediated endocytosis of a plasma transmembrane receptor (112). Its cytoplasmic domain contains a well-recognized tyrosine-based motif (YXXϕ) that directs endocytic membrane traffic. The recognition of this signal motif and the clustering of receptors at the surface of the plasma membrane into clathrin-coated pits (46) is aided by sorting interactions with clathrin adaptor protein-2 (AP-2) (30, 115). Erythroid cells, which have high levels of TfR1, utilize an additional clathrin adaptor called PICALM (phosphatidylino- sitol binding clathrin assembly lymphoid myeloid leukemia) for Tf endocytosis (80). Both global knockout and conditional knockdown of PICALM in mice result in anemia. Genome-wide association studies have linked polymorphisms in PICALM to Alzheimer’s disease (73), but associations with human iron deficiency or overload diseases have yet to be reported. In a mouse model of polycythemia vera, red blood cell production was suppressed by Picalm deletion, suggesting new strategies to control Tf endocytosis that could block iron uptake when necessary (80).
Both AP-2 and PICALM bind phosphatidylinositol-4,5-bisphosphate (PIP2) (39, 80, 112). These endocytic factors cooperate to assemble clathrin at the membrane surface, sorting cargo into invaginating clathrin-coated membrane domains. Live cell imaging shows that fluorescently labeled Tf colocalizes with newly formed clathrin-coated areas within 15 seconds of their formation (46). Clathrin-coated pits of the plasma membrane eventually bud off vesicles with the help of dynamin, a GTPase (guanosine triphosphatase) that mediates the scission event to form the intracellular vesicle (30). These vesicles shed their clathrin coats rapidly and fuse with early endosomes (30, 92). The appearance of Tf in this compartment occurs with a half-life of 2 minutes, as TfR1 rapidly moves into the intracellular endolysosomal pathway (110).
Early endosomal vesicles are identified by the presence of Rab5, a low-molecular-weight GTPase that mediates homotypic membrane fusion. Rab GTPases, such as Rab5, coordinate vectorial membrane traffic between intracellular compartments, and they interact with numerous effectors of lipid metabolism to enable targeted membrane fusion coupled to appropriate protein sorting (157). For example, lipid remodeling by the phosphatidylinositol-3-kinase (PI3K)–Vps34–Vps15 complex is activated by Rab5, increasing membrane levels of phosphatidylinositol-3-phosphate (PI3P), which, in turn, helps to recruit early endosome associated antigen-1 (EEA1). EEA1 couples vesicle docking with membrane fusion machinery to direct homotypic membrane fusion between early endocytic vesicles and endosomes (26, 157).
Early endosomes are acidified by vacuolar H+-ATPase (V-ATPase), which is an electrogenic proton pump. The lower pH of these intracellular organelles promotes the dissociation of ligands—such as low-density lipoprotein- or mannose-6-phosphate-containing hydrolases—from their receptors and supports the recycling of unoccupied receptors to the cell surface (111). In contrast, cargo iron is released from Tf at low pH, with apo-Tf remaining bound to its receptor for return to the cell surface (44). Because of the importance of endosomal acidification in iron delivery, inhibitors of V-ATPase, such as bafilomycin A1, disrupt this process (176).
3.2. Recycling
TfR1 constitutively recycles between the cell surface and intracellular compartments in contrast to growth factor receptors that become downregulated by trafficking to late endosomal and lysosomal compartments. Geometrical sorting within endosomes helps to return the Tf–TfR1 complex to the plasma membrane (110). There are at least two different pathways for TfR1 recycling: a rapid pathway directed by Rab4, and a Rab4/Rab11-mediated slow pathway that involves further trafficking through the endosomal recycling compartment. Colocalization and trafficking experiments have shown that DMT1 also moves through similar intracellular compartments, suggesting that iron release from Tf at low pH is functionally coupled to its transport into and out of sorting and recycling endosomes (152).
The recycling of both TfR1 (108, 146) and DMT1 (152) is sensitive to wortmannin, a PI3 kinase inhibitor. These observations indicate that lipid-mediated sorting events must occur to return the iron transport machinery to the cell surface. The recycling of membrane proteins from intracellular compartments to the cell surface involves a multimolecular complex called retromer (83). Retromer is thought to recognize returning cargo molecules with the help of sorting nexins (SNXs) (31). A conceptual framework for these events is that SNXs interact with PI3P to help identify receptors and couple their retrieval by the retromer complex. Structural studies confirm that the DMT1 recycling signal is situated at the interface between sorting nexin-3 (SNX3) and subunits of the retromer complex (103). In addition to DMT1 recycling, SNX3 also retrieves TfR1 (24). The suppression of SNX3 impairs iron delivery, with Tf accumulation in early endosomes. SNX3 is highly expressed in developing red blood cells, suggesting a critical role in erythroid development and hematopoiesis (24). It also has been shown that sorting nexin-4 (SNX4) is important for endosomal sorting and retrieval of TfR1 (153). Traer et al. (153) postulated that SNX4 is involved in sorting interactions that deliver TfR1 targeted by the slower route to recycling endosomes, from which the second sorting event must occur to deliver the receptor to the plasma membrane. While the fast recycling route may be regulated by the Rab4 GTPase, the slow or indirect pathway through the endosomal recycling compartment appears to involve both Rab4 and Rab11 (110, 157). These two GTPases have been shown to interact with D-AKAP2 (dual-specific A kinase anchoring protein-2) in a manner that regulates TfR1 trafficking through the recycling compartment and perhaps directs the receptor to the most appropriate pathway (slow or fast) for return to the cell surface (45).
Whether originating from rapid or slow recycling compartments, vesicles that contain TfR1 must tether to and fuse with the plasma membrane, a process that involves another complex of proteins called the exocyst (151). A mutation in one member of the exocyst complex, Sec15l1, promotes hypochromic anemia in Hbd mice (102, 167). The Hbd mutation disrupts the rapid trafficking pathway of TfR1 recycling to the cell surface, but does not impair the delivery of iron to cells (62). Thus, iron must be released at some point earlier in Tf–TfR1’s journey through the endocytic pathway. The final arrival of the apo-Tf–TfR1 complex at the plasma membrane completes this process in which the ligand–receptor complex can dissociate due to the neutral pH of the extracellular milieu (36) or competitive displacement by holo-Tf (99), or both.
3.3. Cellular Destinations for Iron
Once iron enters into the cell, it can be transferred to mitochondria for heme or Fe–S cluster synthesis, used as cofactor for many essential redox-active enzymes, or stored into ferritin as a supply for future metabolic demands (75, 118, 132). The amount of free iron composing the labile iron pool in most cells is in the micromolar range. The labile iron pool is limited to reduce the risk of iron toxicity, and it appears to be controlled by the chaperones poly-C binding protein-1 and −2 (PCBP1 and PCBP2), which have been reported to transfer iron from DMT1 (171) and deliver it to ferritin (144). Other chaperone-like factors include glutaredoxins, which are involved in Fe–S cluster assembly and that help to deliver iron to mitochondria and nonheme enzymes in the cytosol (129). PCBP2 activity also has been implicated in the export of iron from cells by ferroportin (171).
Since iron in excess of metabolic needs remains stored in ferritin, an immediate supply can be liberated under times of duress (for example, during nutritional deprivation, hemolytic anemia, or hypoxia induced by high altitude). Recent evidence indicates that stress-induced microautophagy of ferritin enables the release of iron within the acidic endolysosomal system (6). Ferritinophagy is mediated by the autophagic cargo receptor nuclear coactivator-4 (NCOA4), which binds to ferritin and targets its uptake by autophagosomes that then fuse with lysosomes for degradation and the release of iron (14, 41, 106, 138). This nutrient recycling mechanism may allow apo-Tf to recapture iron within the intracellular trafficking pathway for release to systemic circulation. Such reverse transport could be particularly important in the liver where ~ 10gof iron is stored in ferritin and where Tf synthesis occurs. In the absence of Tf, there is some evidence that constitutively recycling receptors are more susceptible to higher rates of proteolysis. The degradation of TfR1 occurs in the lysosome after receptor ubiquitinylation (55) and in an iron-dependent manner that contributes to maintaining receptor levels in response to iron status (150).
4. THE TRANSFERRIN RECEPTOR-1 ENDOCYTIC PATHWAY AND VIRAL ENTRY
Although individual viruses have different mechanisms of invasion, a primary initiating event involves interactions with a receptor or set of plasma membrane surface markers that enable the pathogen to identify its targeted host. TfR1 is one of the more highly expressed plasma membrane components, making it an attractive target for the virus to initiate host cell infection. Subsequent events in the infection process lead to the fusion of the virion with the cell membrane in order to unleash its replication machinery. Sometimes this process occurs at the cell surface, but more often the reorganization of viral capsid proteins must be triggered by uptake into the endolysosomal pathway, where pH-induced structural rearrangements can be accommodated. For these reasons, TfR1 also represents an ideal portal for viruses to enter cells. An emerging body of evidence indicates that it is exploited by a number of different pathogens as summarized below and depicted in Figure 3b.
4.1. Mechanism of Viral Invasion
It has been documented that multiple viruses recognize and bind TfR1 to target the endosomal compartment. Most cells have a metabolic pool of receptors available and ready to bind capsid factors to initiate viral invasion. The efficient process of clathrin-dependent coat assembly is promoted by TfR1’s cytoplasmic endocytic motif (32). Factors such as PICALMmay even further expedite this process (80). By co-opting the TfR1 endocytic pathway, the viral particle can be brought into the cell as cargo to enable pH-dependent membrane fusion. Furthermore, this receptor constitutively recycles back to the cell surface, unlike many other receptors, which become downregulated or degraded through internalization, or both. Indeed, TfR1 interacts with factors such as SNX3, SNX4, and Sec15l1 that enable its selective and targeted recycling. Because Tf receptors are redirected back to the cell surface, multiple rounds of infection may be possible, and superinfection has been shown to occur with some viruses that use TfR1 as an entry factor (63).
4.2. Species Selectivity
Entry factors contribute to the species specificity of host–pathogen interactions. All vertebrates appear to have TfR1 orthologs (96), but human TfR1 displays species selectivity in ligand interactions (88). While combined interactions between the protease-like domain and the helical domain provide a ligand binding pocket for Tf and enable pH-dependent iron release (65), the role of the receptor’s apical domain in iron metabolism is less clear. In studies that have mapped the sites of interaction between TfR1 and certain viruses, the apical domain represents a unique target for viral recognition that does not interfere with interactions with Tf or its iron delivery. For example, specific residues in the apical domain of feline and canine TfR1 have been shown to direct species selectivity, and receptor glycosylation can contribute to these interactions (69, 125). This so-called viral binding patch can be targeted by pathogens, such as parvoviruses, in a selective manner (69). In the absence of a clear functional role, the apical domain represents an attractive landscape to be mined by viral invaders and is a substrate for the coevolution of host and pathogen.
4.3. The Iron Gate
Several reinforcing concepts emerge from these functional and structural considerations that support an iron-gate paradigm for viral invasion: (a) The ubiquity of TfR1 expression across various cell types provides a common entry target; (b) structural aspects of TfR1 could contribute to host cell restriction as well as zoogenesis; and (c) the apical domain permits viral binding without interfering with iron delivery. Most importantly, viral invaders seek the exact same route as iron delivery: internalization into intracellular acidic compartments, pH-dependent cellular entry across endosomal domains, and entry into the cytoplasm (Figure 3). The key functional importance of TfR1 in iron delivery from Tf has led to our understanding of an incredibly efficient recycling mechanism for membrane traffic. However, relatively little attention has been paid to how pathogens might usurp this pathway to invade host cells.
5. VIRUSES ENTERING THE IRON GATE
In 2001, Parker et al. (127) first reported that canine parvovirus (CPV) and feline panleukopenia virus (FPV) infected cells through the TfR1 trafficking pathway. Taking advantage of Chinese hamster ovary cells that lack TfR1 (TRVb cells) (114), these investigators showed that viral entry and infection depend on receptor expression. Another parvovirus, mink enteritis virus (MEV), was subsequently shown to infect these cells via the feline TfR1 (126). More recently, TfR1 has been associated with infections caused by a number of New World hemorrhagic fever arenaviruses (140). Viruses that interact with TfR1 and other factors in the iron uptake pathway are summarized in Table 1 and discussed below.
Table 1.
Viruses that interact with transferrin receptor-1 (TfR1) and other factors in the iron uptake pathway
| Virus (abbreviation) | Description |
|---|---|
| New World arenaviruses | Emerging pathogens into humans via zoonotic transmission; cause hemorrhagic fever with poor prognosis; natural hosts include rodents in the Muridae family; they are a bioterrorism threat due to aerosolized transmission; bind to TfR1 apical domains |
| Machupo (MACV) | Found in Bolivia; infects human and cat TfR1; the large vesper mouse is the endogenous host; mouse and rat models have not been efficiently infected |
| Junin (JUNV) | Responsible for Argentine hemorrhagic fever; the drylands vesper mouse is the principal host; mouse and rat models have not been efficiently infected |
| Ocozocoautla de Espinosa (OCEV) | Isolated in Mexican deer mice near site of 1967 hemorrhagic fever epidemic; does not infect human TfR1, but replacement of three amino acids with bat ortholog made an efficient target of the human receptor; related to Tacaribe virus |
| Tacaribe (TCRV) | Found in bats; nonfatal human laboratory-acquired human infections have been observed |
| Guanarito (GTOV) | Cane mouse is the principal host; unlike MACV and JUNV does not infect vesper mouse TfR1 ortholog; mouse and rat models have not been efficiently infected; identified in Venezuela |
| Sabia (SABV) | Host is not yet identified; found in Brazil; only small number of infections known |
| Chapare (CHAV) | Found in Bolivia; suspected cause of 1960s hemorrhagic fever outbreak in Chiapas, Mexico |
| Mouse mammary tumor virus (MMTV) | Infects Mus musculus; invasion is incompatible with human TfR1; likely to have had a larger host range of rodent-related species in the past; milk-borne pathogen |
| Human and simian immunodeficiency viruses (HIV, SIV) |
HIV Nef protein downregulates HFE, a regulator of iron metabolism and a TfR1 binding partner; SIV Nef induces downregulation of TfR1 |
| Parvoviruses | First class of pathogens recognized to invade via TfR1; infect actively dividing cells of the lymph system and crypt cells of the intestine; bind to TfR1 apical domain |
| Canine parvovirus (CPV) | Originated in early carnivores; reemerged with acquisition of species-specific glycosylation site in canine TfR1 |
| Feline panleukopenia virus (FPV) | Causes distemper in cats; infects feline and mink cells, but not canine cells; binds to determinants in TfR1 apical domain |
| Mink enteritis virus (MEV) | Important economic pathogen in mink industry; downregulates TfR1 expression after infection |
| Hepatitis C virus (HCV) | TfR1 is among other entry factors, but it is likely required for endocytic uptake; virus attacks host iron metabolism; iron depletion is beneficial to infected individuals |
| Human adenovirus | Gene therapy has engineered adenovirus 5 to recognize TfR1 for enhanced efficiency and to cross the blood–brain barrier (e.g., Tf-AdLac) |
| Alphaviruses | Sindbis virus uses DMT1 as entry receptor into endocytic pathway; first identified in Egypt; causes Sindbis fever in humans; transmitted by mosquitoes |
5.1. New World Hemorrhagic Fever Arenaviruses
Infections with New World hemorrhagic fever arenaviruses can cause up to 30% mortality and are considered potential bioterrorism threats because of their aerosol transmission (140). The development of antivirals is a particularly critical concern, and because TfR1 is a cellular receptor for pathogenic strains, potential interactions enabling these emerging pathogens to cross the iron gate are a focus of keen attention. In 2007, Radoshitzky et al. (130) first demonstrated a specific high-affinity association between TfR1 and the entry glycoprotein of Machupo virus, a New World arenavirus. The expression of human TfR1, but not human TfR2, enhanced the infection of viruses pseudotyped with the infective glycoprotein G1 of Machupo, Guanarito, and Junin viruses, but not with those of Old World arenaviruses (Lassa fever or lymphocytic choriomeningitis viruses). An anti-TfR1 antibody efficiently inhibited the entry of Machupo, Guanarito, Junin, Sabia, and Chapare viruses, but not Lassa fever virus (74, 130). Iron depletion of culture medium enhanced, while iron supplementation decreased, the efficiency of infection by Junin and Machupo viruses, suggesting that iron-dependent levels of TfR1 correlate with viral entry. Based on these multiple lines of evidence, it is clear that TfR1 is a key cellular receptor for these emerging New World hemorrhagic fever arenaviruses.
Crystallographic studies of the Machupo G1 glycoprotein bound to TfR1 revealed that the virus binds to the apical domain of the receptor without overlap with the protease-like and helical domains (1). Consistent with this structural information, the presence of Tf does not interfere with viral invasion (130), and an antibody to an apical domain epitope blocks the binding of the envelope G1 of several New World arenaviruses, including Junin, Guanarito, Sabia, Chapare, and Machupo (74). Because the structure of G1 appears to be the same whether or not it is bound to TfR1 (1, 16), it is unlikely that the receptor binding induces changes in the capsid to mediate viral invasion. Instead, the receptor’s membrane trafficking pathway appears to accommodate viral fusion with the early endosomal membrane due to entry into the low pH of these compartments (22, 123). TfR1 does not appear to be downregulated by Junin virus infection, suggesting that receptors are capable of recycling and enabling superinfection to occur (63).
Zoonotic transmission of New World arenaviruses is thought to originate through their use of orthologous TfR1 in rodent hosts (131). There is evidence to support selective pressure by viruses onTfR1 in host species (37,123,174,179). For example, it has been shown that a virus related to the Whitewater Arroyo virus was able to infect cells through human TfR1, possibly pointing to the source of three human fatalities reported in the US Southwest (179). Conversely, by targeting the apical domain of TfR1, receptor mutations that might either block viral entry or facilitate infection do not necessarily interfere with iron uptake. These coevolutionary interactions balance the opposing selective pressures of host-virus interactions (37).
Although the precise cellular targets for arenavirus infection remain unknown, it is thought that macrophages and dendritic cells are infected in the lung by airborne exposures (140). In a similar fashion, milk-borne infection by mouse mammary tumor virus (MMTV) also targets dendritic cells of the intestinal mucosa, with TfR1 serving as a portal for infection (see Section 5.2). An attenuated strain of Junin virus is used in Argentina as a protective vaccine against hemorrhagic fever, but more advanced antiviral therapies remain to be developed, with the apical domain of TfR1 being a candidate drug target for disrupting infection (123, 140). The apical-domain-targeting anti-TfR1 antibody, for example, could offer an important therapeutic approach (74). Alternatively, antibodies that bind to the viral G1 protein and that mimic TfR1 interactions are a potential immunological solution (175).
5.2. Mouse Mammary Tumor Virus
MMTV is a well-characterized retrovirus that is associated with the pathogenesis of mammary adenocarcinomas and T cell lymphomas in rodents. The seminal discovery by Varmus and Bishop that MMTV activated proto-oncogenes led to their Nobel prize in 1989. Results from low-level polymerase chain reaction fueled controversy about whether this retrovirus could possibly be linked to human breast cancer (109). However, biochemical studies showed that MMTV uses only mouse TfR1 for infection (136). Although the mouse virus might be able to interact with human receptors, an interesting finding is that only mouse TfR1 is capable of mediating viral entry into low-pH compartments (159). Because MMTV membrane fusion requires pH 5 or lower, late endosomal domains are thought to be necessary for invasion of the mouse virus. It has been suggested that a complex with mouse TfR1 can be diverted to more-acidic late-endosomal compartments and that viral binding sites (separate from Tf binding sites) alter the receptor’s intracellular trafficking pathway, inducing receptor-specific signals or association with coreceptors to promote fusion of the viral envelope (158). Like arenaviruses, interactions with the MMTV retrovirus do not interfere with the housekeeping functions of TfR1 in iron transport. An evolutionary analysis of these interactions suggests positive selective pressure for apical domain- binding sites (37). Thus, the host range of MMTV-like viruses may have once been much larger, but resistant mutations in TfR1 appear to have won the host–virus arms race.
5.3. Human and Simian Immunodeficiency Viruses
The lentiviral gene product Nef captured attention when early patients identified as being infected with Nef-deficient HIV-1 did not appear to progress to AIDS. Because Nef appeared to be a necessary factor for full virulence, its function in pathogenesis became a focus of great attention (52). Nef activity was associated with the downregulation of surface molecules through its interactions with the clathrin adaptor protein AP-2 (145). Among these targets, HIV-1 Nef was reported to downregulate the hereditary hemochromatosis protein HFE in macrophages that were infected ex vivo (42). As a consequence of this action, these cells became iron loaded, suggesting that this virus influences iron metabolism. More recently, it has become clear that simian immunodeficiency virus (SIV) Nef inhibits Tf endocytosis (94). Researchers have found that simian Nef reduces TfR1 internalization by interacting with an AP-2 binding motif that does not normally function in HIV- and SIV-induced downregulation of other surface receptors. This proposed function may limit viral replication and represents a host-virus adaption in monkeys (94). Ultimately, when it was learned that patients infected with Nef-deficient HIV-1 suffered disease progression, interest in Nef’s function in endocytosis, viral entry, and iron delivery waned (52). Fortunately for these and other patients, antiviral therapies have now helped to significantly lower the burden of disease and reduce spread of the AIDS epidemic. Nonetheless, it is important to discern why lentivirus interactions may induce iron loading or iron deficiency, depending on the host and cellular target.
5.4. Canine Parvovirus
As mentioned above, CPV was the first pathogen recognized to invade cells through the TfR1 iron gate (127). CPV infections can cause severe gastroenteritis, especially in puppies, and are a serious concern among dog breeders. The parvoviruses are closely related to FPV and MEV, but only CPV invades canine cells (127). The process of CPV invasion involves clathrin-mediated endocytosis, with subsequent invasion into the cell from endocytic compartments. Anti-TfR1 antibodies were found to block CPV infection (127), and later studies found that soluble TfR1 could block infection and decrease mortality in CPV-infected dogs (163). Similar to MMTV and the New World arenaviruses, the apical domains of the receptor appear to be involved in parvovirus recognition and binding (69).
The mechanism of CPV entry through TfR1 interactions also has been characterized by using fluorescence live cell imaging (32). This study suggests that the relatively low affinity of CPV viral capsids for the receptor allows TfR1 to cluster at the cell surface into clathrin-coated structures. Those virus particles that do not rapidly encounter a clathrin domain dissociate from the cell surface. It has been reported that CPVbinds relatively fewTfR1s (five to seven receptor complexes) (32, 72), so that this lateral diffusion mechanism improves the efficiency of virus internalization via clathrin-coated membrane invaginations. Interestingly, the spread of CPV infection had been previously associated with the acquisition of the parvovirus’s specific recognition of canine TfR1 (77), but more recent evolutionary studies suggest that the virus may have readapted to recognize a resistant ortholog of TfR1 previously used in a former carnivore host (86). Once again, host–virus coevolution is likely supported by viral binding to the TfR1 apical domain, which is not required for iron delivery.
5.5. Feline Panleukopenia Virus
FPV causes distemper in cats, affecting rapidly dividing cells of the intestine and bone marrow. Parker et al. (127) initially reported that FPV and CPV bind to human TfR1 using heterologous expression in TRVb cells. The use of blocking antibodies prevented viral invasion, confirming the role of this receptor. Moreover, expression of feline TfR1 conferred susceptibility to infection. Although both FPV and CPV recognize TfR1, the host range appears to be restricted. Subsequent investigation showed that CPV selectively interacts with the canine TfR1 (77). Using a series of chimeric FPV receptors, and by studying specific point mutations represented in the canine receptor (69, 125), researchers later determined that CPV infection is mediated by a canine-specific TfR1 glycosylation site (Asn383), while adding this site to the feline receptor reduced FPV binding and infection. Instead, a critical residue in the apical domain (Leu221) was identified as influencing parvovirus uptake of feline TfR1 (69). In human TfR1, this site is adjacent to a residue found to be critical for arenavirus binding; interestingly, feline, but not canine, TfR1 orthologs bind to arenaviruses (1), suggesting there are subtle structural alterations and that the additional glycosylation site in canine TfR1 orthologs must affect interactions between these viruses and the receptor’s apical domain. Such evidence supports the idea that pathogen infections of new hosts may be determined by simple amino acid substitutions in TfR1 (28).
5.6. Mink Enteritis Virus
MEV is a variant of FPV and also displays homology with CPV. MEV invades mesenteric lymph nodes and intestinal epithelium to cause enteritis, and it is an economically challenging pathogen in the mink industry. Similar to the other parvoviruses discussed above, TfR1 has been implicated as a viral receptor. In a screen of small noncoding microRNAs, miR-320a and mi-R140 were identified as becoming upregulated upon MEV infection to inhibit its entry. These microRNAs target the 3′ untranslated region of TfR1 to downregulate receptor expression; thus, it appears that MEV selectively represses the receptor to block further rounds of infection (149). This work not only supports the role of TfR1 in viral infection, it further suggests that some invading pathogens may manipulate the expression of the receptor to limit superinfection. It also points to potential therapeutic avenues for limiting infection through the use of small interfering RNAs (siRNAs) to alter TfR1 expression.
5.7. Hepatitis C Virus
About 3–5 million people in the United States are infected with hepatitis C virus (HCV), which leads to chronic liver disease and hepatocellular carcinoma. Fortunately, treatment with direct-acting antivirals now results in a greater than 90% cure rate (3). However, large numbers of HCV patients load excessive liver iron, and iron metabolism appears to be a viral target, including the downregulation of TfR1 (87,107). Infection by HCV is complex and involves multiple host surface molecules as cofactors for attachment. Using a small molecule that targets TfR1 but not TfR2 (18, 76), Martin & Uprichard (107) determined that the receptor is an additional HCV entry factor. Their findings suggest that TfR1 most likely acts later than other surface factors that bind the virus, but it is necessary to mediate internalization to endosomal compartments. The use of a blocking antibody suggests that virus recognition by the receptor involves its apical domain, which is similar to New World arenaviruses (130). After infection, the invading virus appears to have strong influence on host cell cellular iron metabolism, most likely to avoid inhibition of replication due to iron-induced inhibition of the HCV viral polymerase NS5B (50). This host–pathogen interaction would explain the apparent downregulation of TfR1 in infected hepatocytes (50, 87, 130).
5.8. Human Adenoviruses for Gene Therapy
More than 400 human gene therapy trials have focused on human adenovirus (Ad) vectors. Because many disorders involve metabolic gene defects in the central nervous system, a specific challenge of viral-based therapies is to cross the blood–brain barrier. Xia et al. (169) addressed this issue by engineering recombinant Ad5 to recognize and specifically bind to human TfR1. Their study determined several peptide sequences that allowed gene transfer into human brain microcapillary endothelial cells when introduced into the HI loop of Ad5 fiber (Ad5GFP-B6HI and AdGFP- B8HI). Likewise, Joung et al. (85) engineered Ad5 virus particles to contain a peptide mimetic for Tf (THRPPMWSPVWP) and showed effective delivery of Tf-AdLac into neuroglia through TfR1. Importantly, the ability of the latter construct to infect cells was not altered by the presence of the ligand Tf, raising the possibility of in vivo infection without significant perturbations to cellular iron metabolism. These investigations demonstrate the feasibility of exploiting the iron gate to effectively target gene delivery.
5.9. Alphaviruses
Alphaviruses infect both insect and vertebrate cells and are spread by mosquitos. Emerging pathogens of this class include the Chikungunya virus, which causes fever and joint pain, and there have been recent outbreaks of infection in the United States, Latin America, and the Caribbean. More well-characterized laboratory strains include the Sindbis and Semliki Forest viruses (148). Host factors required for infection by alphaviruses remained poorly understood until a recent genome-wide RNA interference screen identified the product of the malvolio (Mvl) gene as the cellular receptor for the Sindbis virus (135). This Drosophila homolog of Nramp2 or DMT1 participates in the delivery of iron across endosomal membranes (see Section 3). These findings are consistent with the known entry of alphaviruses by clathrin-mediated endocytosis (91) and provide yet another example of the co-opted use of the TfR1 iron-delivery pathway for cellular invasion by viruses.
6. VIRAL INFECTIONS AND IRON STATUS
An often overlooked virulence factor is the host’s nutritional status (13). The recent emergence of infectious pathogens such as New World arenaviruses has demonstrated our vulnerability to viral attacks. Our coevolution with infectious pathogens raises the risk of outbreaks due to acquired sensitivity or resistance, or both, in host factors such as TfR1. As described earlier, TfR1 is highly regulated by iron status and plays a central part in the host’s iron homeostasis by participating in the signaling pathway controlling hepcidin levels. How could host iron metabolism influence out-breaks of emerging disease? Perhaps the best evidence supporting the role of the host’s nutritional status on emerging pathogens comes from studies of selenium status and its role in coxsackie, polio, influenza, and other viral infections (12, 13). The effects of host micronutrient status on inducing and propagating viral mutations have been described, and they are thought to originate from oxidative stress due to imbalanced levels of critical vitamins and minerals, as well as the altered immunity of the host under these conditions. Among the plausible molecular mechanisms in host–pathogen interactions that contribute to evolutionary pressures are redox regulation, leading to faster viral replication, and direct oxidative damage, leading to genomic alterations. Below are some brief examples of how both iron overload and iron deficiency might alter the course of human infection and the emergence of pathogenicity. For a more comprehensive perspective on iron metabolism and inflammation, the reader is referred to recent reviews (43, 162, 164, 165)
6.1. Iron Overload
The role of excess iron in promoting viral infections has been discussed for many years (161). During infections, hepcidin levels become elevated, and numerous studies have demonstrated a hypoferremic response to viral infections (60). Higher hepcidin levels are associated with lower ferroportin levels, but the latter effect serves to elevate the intracellular iron stores of macrophages and hepatocytes. Thus, cells of the reticuloendothelial system and the liver accumulate excess iron during inflammation (60). In the case of HCV infection, such hepatic iron loading contributes to the progression of disease (57). The infection also produces systemic iron loading with increased Tf saturation, serum ferritin, and hepatic iron due to the dysregulation of hepcidin synthesis by the liver (38, 56). In vitro infection studies of hepatocytes indicate that the virus may directly manipulate the host’s iron status to promote its replication; since the liver is the site of hepcidin synthesis, such changes appear to actively subvert the host’s iron homeostasis (50). Iron depletion strategies have proven to be beneficial to HCV patients by reducing liver overload (58). These and other observations led Drakesmith & Prentice (43) to hypothesize that oxidative stress due to liver iron loading could modify the evolution of HCV viral quasispecies, which are associated with poor outcomes (48).
6.2. Iron Deficiency
The host’s immune response can be significantly compromised in individuals with nutritional iron deficiency because activities of important iron-requiring enzymes that combat infection become impaired (e.g., myeloperoxidase, inducible nitric oxide synthase) (reviewed in Reference 164). Iron is also a necessary cofactor for ribonucleotide reductase, a key enzyme in nucleic acid synthesis that is critical for the proliferation of lymphocytes in order to mount a defense against invading pathogens. However, whether iron intake should be supplemented when the body undergoes iron restriction during infection is an open question. In the case of HIV-1 infection, viremia has been associated with decreased plasma iron and increased hepcidin (5). Iron is required for HIV-1 replication (43), and iron supplementation of HIV-positive individuals may worsen the progression of the disease (121). However, some studies have shown that many HIV-positive individuals are iron deficient (95), with increased risks of mortality and disease progression (79). These findings reinforce the idea that low iron status increases susceptibility and reduces the host’s capacity to limit infection, particularly when the virus attacks immune cells (47, 124).
7. THE IRON GATE PARADIGM
Information from studies of the pathogenicity of the viruses listed in Table 1 and described in Section 5 not only sheds light on their invasion and intracellular trafficking strategies but also supports the idea that TfR1 and host iron metabolism are particularly critical to combating emerging infectious diseases. Given the significant influence of iron status on viral infections, obvious strategies include careful monitoring of the nutritional status of world populations at higher risk for emerging epidemics. However, decisions about whether to provide supplementation to iron-deficient individuals during infection need to be informed by pathogen-specific interactions; for example, does the virus invade hepatocytes and macrophages that become iron rich during the anemia of inflammation induced by infection, or are cells of the immune system the target of infection, such that the host response under iron-deficiency conditions becomes further compromised (43)? Targeted therapeutic approaches that have been discussed for pathogens that use TfR1 as an entry receptor include the use of anti-TfR1 antibodies (74) and antiviral peptide mimetics that neutralize the sites of interaction in the TfR1 apical domain (175). Novel strategies to reduce the risk of infection by lowering receptor levels using siRNA could be explored (149).
Many lines of evidence support why TfR1 represents such a vulnerable target for viral infection: (a) It is an abundantly expressed, cell-surface membrane protein; (b) it is endocytosed and recycled in a constitutive manner; (c) pH-dependent viral invasion can be supported by the intracellular trafficking pathway of the receptor; (d) specialized cellular machinery that accommodates efficient iron delivery via this vesicular route can be co-opted for efficient infection; (e) orthologous Tf receptors exist across mammalian hosts; (f the receptor’s molecular structure includes a large apical domain that permits capsid binding; and (g) viral interactions do not interfere with iron delivery and its regulation by the receptor because these function are supported by other domains of TfR1.
A remaining enigma to be resolved is how TfR1 participates in the process of infection. It has been largely assumed that TfR1 is simply a passive target for infection. Could the receptor play a more active role in signaling the metabolic fitness of host? For example, an iron-poor host with nutritional deficiency or the anemia of inflammation, or both, would significantly impair the survival of the pathogen. Although iron deficiency might increase TfR1 levels due to IRP control, these receptors would be unoccupied by ligands since systemic iron deficiency lowers Tf saturation. Virus interactions with TfR1 do not necessarily interfere with Tf binding, but whether the presence of a ligand is necessary for viral recognition and invasion has not been rigorously addressed. It is conceivable that the Tf–TfR1 complex is the more precise viral recognition target since the presence of a ligand would signal the host’s iron sufficiency status to invading pathogens (Figure 4).
Figure 4.
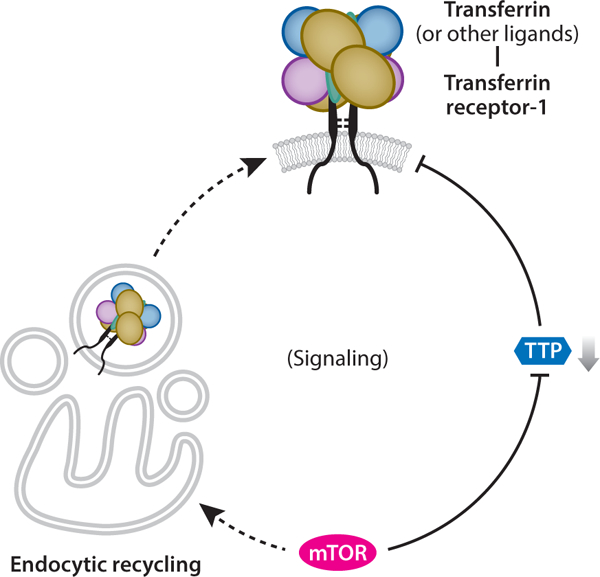
The active roles of transferrin receptor-1 (TfR1) during infection may signal the host’s metabolic fitness to pathogens. Holo-transferrin (Tf ) binding to TfR1 reflects systemic iron sufficiency, such that the ligand–receptor complex itself might be the entry target and not simply TfR1 alone. Alternatively, other ligands (e.g., ferritin, polymeric A1 isotype immunoglobulin, unknown moieties) could participate in viral invasion as coreceptors. TfR1 also has iron-independent functions, including the activation of signaling pathways that might be involved with infections to suppress the cellular stress response. Mammalian target of rapamycin (mTOR), a master regulator of energy metabolism, appears to positively regulate TfR1 by maintaining the endocytic recycling pathway to guard against receptor degradation and to suppress tristetraprolin (TTP) degradation of TfR1 messenger RNA. Thus, cell surface TfR1 reports cellular energy status to the virus, reflecting mTOR control.
Other active roles might be imparted by alternative TfR1 ligands (e.g., ferritin, pIGA1, or other unknown factors). Alternatively, direct effects of iron-independent TfR1 signaling (e.g., NF-κB activation, JNK regulation, or other implicated pathways) could be involved (90, 141). Although iron-independent functions of TfR1 have yet to be fully elucidated, these activities could modify viral interactions and support invasion.
TfR1 levels and iron flux are also correlated with the activity of mTOR (mammalian target of rapamycin), a master regulator of energy metabolism. mTOR is a component of mTORC1 (mTOR complex 1), an essential complex that also includes Raptor and mLST8 (40). mTOR is active when energy and nutrients are in surplus, but under stress conditions, it is inhibited to promote cell survival by conserving these resources. In fact, many viruses have evolved complex mechanisms to overcome mTOR inhibition caused by stress responses to infection (17). Early siRNA screening studies indicated that the mTOR signaling pathway was a positive regulator of Tf uptake (59). More recent studies identified at least one downstream target of mTOR that is involved in TfR1 regulation, tristetraprolin (TTP). TTP is an anti-inflammatory response protein and a mammalian homolog of the yeast Cth1 and Cth2 proteins, which are known to regulate iron levels under deficiency conditions to promote survival (71). In contrast to IRP control, TTP binds to TfR1 messenger RNA and enhances its degradation (11). In its active state, mTORC1 blocks this effect, while rapamycin inhibition induces TfR1 degradation. Thus, TTP appears to counterbalance IRP control, which is iron responsive but not energy sensitive. An additional layer to the regulation of iron flux by this signaling pathway is that mTORC1 maintains the recycling pathway necessary for the constitutive membrane trafficking of TfR1 (35). TfR1 levels must be coordinated with iron delivery within endocytic compartments, and the process of membrane sorting and trafficking appears to be tightly controlled by mTOR. Under stress conditions, TfR1 is redirected to late endosomal and lysosomal compartments, where it is degraded (55, 150). A deeper understanding of the relationship between iron status and metabolic fitness would not only increase our understanding of the mTOR/TTP/TfR1 signaling pathway (Figure 4), but also help to illuminate the precise role of TfR1 in host–pathogen interactions.
8. CONCLUSIONS
Our innate immune response carefully orchestrates control over iron metabolism to limit its availability during times of infection. An emerging enigma is that many viruses use the primary gatekeeper of iron metabolism, TfR1, as a means to enter cells. TfR1 is a vulnerable target as it is an abundantly expressed cell-surface membrane protein that undergoes constitutive endocytosis and recycling that would bring virus particles to acidic compartments for pH-dependent invasion. Moreover, orthologous receptors exist across mammalian hosts, and within these structures there is a large apical domain that permits capsid binding. Finally, viral interactions do not interfere with iron delivery and its regulation by the receptor, thereby supporting the survival of the host cell. Although the iron gate paradigm explains why the transferrin receptor is targeted for viral infection, how it may participate is unknown. Although the receptor is thought to have a passive role in pathogen invasion, this review has considered whether it may be a more active participant in viral infections by signaling the metabolic fitness of the host via interactions that reflect the iron saturation of Tf, by interactions with other ligands or Tf-independent functions, or through regulation of the receptor by the master energy regulator mTOR.
ACKNOWLEDGMENTS
The author is supported by National Institutes of Health grants DK064750 and ES014638.
Glossary
- Tf
transferrin
- TIBC
transferrin iron-binding capacity
- NTBI
nontransferrin-bound iron
- TfR
transferrin (Tf) receptor
- IRE
iron response element
- IRP
iron response element (IRE) binding protein
- DMT1
divalent metal transporter-1
- AP-2
clathrin adaptor protein-2
- PICALM
phosphatidylinositol binding clathrin assembly lymphoid myeloid leukemia
- PI3P
phosphatidylinositol-3-phosphate
- V-ATPase
vacuolar H+-ATPase
- SNX
sorting nexin
- D-AKAP2
dual-specific A-kinase anchoring protein-2
- mTOR
mammalian target of rapamycin
Footnotes
DISCLOSURE STATEMENT
The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Abraham J, Corbett KD, Farzan M, Choe H, Harrison SC 2010. Structural basis for receptor recognition by New World hemorrhagic fever arenaviruses. Nat. Struct. Mol. Biol. 17:438–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ajioka RS, Kaplan J. 1986. Intracellular pools of transferrin receptors result from constitutive internalization of unoccupied receptors. PNAS 83:6445–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alkhouri N, Lawitz E, Poordad F. 2017. Novel treatments for chronic hepatitis C: closing the remaining gaps. Curr. Opin. Pharmacol. 37:107–11 [DOI] [PubMed] [Google Scholar]
- 4.Andrews NC. 2008. Forging a field: the golden age of iron biology. Blood 112:219–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armitage AE, Stacey AR, Giannoulatou E, Marshall E, Sturges P, et al. 2014. Distinct patterns of hepcidin and iron regulation during HIV-1, HBV, and HCV infections. PNAS 111:12187–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asano T, Komatsu M, Yamaguchi-Iwai Y, Ishikawa F, Mizushima N, Iwai K. 2011. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol. Cell. Biol. 31:2040–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, et al. 2006. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 38:531–39 [DOI] [PubMed] [Google Scholar]
- 8.Barrientos T, Laothamatas I, Koves TR, Soderblom EJ, Bryan M, et al. 2015. Metabolic catastrophe in mice lacking transferrin receptor in muscle. EBioMedicine 2:1705–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartnikas TB. 2012. Known and potential roles of transferrin in iron biology. BioMetals 25:677–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batista A, Millan J, Mittelbrunn M, Sanchez-Madrid F, Alonso MA. 2004. Recruitment of transferrin receptor to immunological synapse in response to TCR engagement. J. Immunol. 172:6709–14 [DOI] [PubMed] [Google Scholar]
- 11.Bayeva M, Khechaduri A, Puig S, Chang HC, Patial S, et al. 2012. mTOR regulates cellular iron homeostasis through tristetraprolin. Cell Metab. 16:645–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beck MA. 2007. Selenium and vitamin E status: impact on viral pathogenicity. J. Nutr. 137:1338–40 [DOI] [PubMed] [Google Scholar]
- 13.Beck MA, Handy J, Levander OA. 2004. Host nutritional status: the neglected virulence factor. Trends Microbiol. 12:417–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellelli R, Federico G, Matte A, Colecchia D,Iolascon A, et al. 2016. NCOA4 deficiency impairs systemic iron homeostasis. Cell Rep. 14:411–21 [DOI] [PubMed] [Google Scholar]
- 15.Boshuizen M, van der Ploeg K, von Bonsdorff L, Biemond BJ, Zeerleder SS, et al. 2017. Therapeutic use of transferrin to modulate anemia and conditions of iron toxicity. Blood Rev. 31:400–5 [DOI] [PubMed] [Google Scholar]
- 16.Bowden TA, Crispin M, Graham SC, Harvey DJ, Grimes JM, et al. 2009. Unusual molecular architecture of the Machupo virus attachment glycoprotein. J. Virol. 83:8259–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. 2008. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol. 6:266–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byrne SL, Buckett PD, Kim J, Luo F, Sanford J, et al. 2013. Ferristatin II promotes degradation of transferrin receptor-1 in vitro and in vivo. PLOS ONE 8:e70199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Camaschella C 2015. Iron-deficiency anemia. N. Engl. J. Med. 373:485–86 [DOI] [PubMed] [Google Scholar]
- 20.Camaschella C, Roetto A, Cali A, De Gobbi M Garozzo G, et al. 2000. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet. 25:14–15 [DOI] [PubMed] [Google Scholar]
- 21.Cao H, Chen J, Krueger EW, McNiven MA. 2010. Src-mediated phosphorylation of dynamin and cortactin regulates the “constitutive” endocytosis of transferrin. Mol. Cell. Biol. 30:781–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castilla V, Mersich SE. 1996. Low-pH-induced fusion of Vero cells infected with Junin virus. Arch. Virol. 141:1307–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen AC, Donovan A, Ned-Sykes R, Andrews NC. 2015. Noncanonical role of transferrin receptor 1 is essential for intestinal homeostasis. PNAS 112:11714–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen C, Garcia-Santos D, Ishikawa Y, Seguin A, Li L, et al. 2013. Snx3 regulates recycling of the transferrin receptor and iron assimilation. CellMetab. 17:343–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Chloupkova M, Gao J, Chapman-Arvedson TL, Enns CA 2007. HFE modulates transferrin receptor 2 levels in hepatoma cells via interactions that differ from transferrin receptor 1-HFE interactions. J. Biol. Chem. 282:36862–70 [DOI] [PubMed] [Google Scholar]
- 26.Christoforidis S, McBride HM, Burgoyne RD, Zerial M. 1999. The Rab5 effector EEA1 is a core component of endosome docking. Nature 397:621–25 [DOI] [PubMed] [Google Scholar]
- 27.Chua AC, Herbison CE, Drake SF, Graham RM, Olynyk JK, Trinder D 2008. The role of Hfe in transferrin-bound iron uptake by hepatocytes. Hepatology 47:1737–44 [DOI] [PubMed] [Google Scholar]
- 28.Coffin JM. 2013. Virions at the gates: receptors and the host-virus arms race. PLOS Biol. 11:e1001574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Collawn JF, Lai A, Domingo D, Fitch M, Hatton S, Trowbridge IS. 1993. YTRF is the conserved internalization signal of the transferrin receptor, and a second YTRF signal at position 31–34 enhances endocytosis. J. Biol. Chem. 268:21686–92 [PubMed] [Google Scholar]
- 30.Conner SD, Schmid SL. 2003. Regulated portals of entry into the cell. Nature 422:37–44 [DOI] [PubMed] [Google Scholar]
- 31.Cullen PJ. 2008. Endosomal sorting and signalling: an emerging role for sorting nexins. Nat. Rev. Mol. Cell Biol. 9:574–82 [DOI] [PubMed] [Google Scholar]
- 32.Cureton DK, Harbison CE, Cocucci E, Parrish CR, Kirchhausen T. 2012. Limited transferrin receptor clustering allows rapid diffusion of canine parvovirus into clathrin endocytic structures. J. Virol. 86:5330–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Alessio F, Hentze MW, Muckenthaler MU. 2012. The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation. J. Hepatol. 57:1052–60 [DOI] [PubMed] [Google Scholar]
- 34.Dale JC, Burritt MF, Zinsmeister AR. 2002. Diurnal variation of serum iron, iron-binding capacity, transferrin saturation, and ferritin levels. Am. J. Clin. Pathol. 117:802–8 [DOI] [PubMed] [Google Scholar]
- 35.Dauner K, Eid W, Raghupathy R, Presley JF, Zha X. 2017. mTOR complex 1 activity is required to maintain the canonical endocytic recycling pathway against lysosomal delivery. J. Biol. Chem. 292:5737–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dautry-Varsat A, Ciechanover A, Lodish HF. 1983. pH and the recycling of transferrin during receptor- mediated endocytosis. PNAS 80:2258–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Demogines A, Abraham J, Choe H, Farzan M, Sawyer SL. 2013. Dual host-virus arms races shape an essential housekeeping protein. PLOS Biol. 11:e1001571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Di Bisceglie AM, Axiotis CA, Hoofnagle JH, Bacon BR. 1992. Measurements of iron status in patients with chronic hepatitis. Gastroenterology 102:2108–13 [DOI] [PubMed] [Google Scholar]
- 39.Di Paolo G, De Camilli P. 2006. Phosphoinositides in cell regulation and membrane dynamics. Nature 443:651–57 [DOI] [PubMed] [Google Scholar]
- 40.Dibble CC, Manning BD. 2013. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 15:555–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, et al. 2014. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 16:1069–79 [DOI] [PubMed] [Google Scholar]
- 42.Drakesmith H, Chen N, Ledermann H, Screaton G, Townsend A, Xu XN. 2005. HIV-1 Nef down-regulates the hemochromatosis protein HFE, manipulating cellular iron homeostasis. PNAS 102:11017–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drakesmith H, Prentice AM. 2012. Hepcidin and the iron-infection axis. Science 338:768–72 [DOI] [PubMed] [Google Scholar]
- 44.Eckenroth BE, Steere AN, Chasteen ND, Everse SJ, Mason AB. 2011. How the binding of human transferrin primes the transferrin receptor potentiating iron release at endosomal pH. PNAS 108:13089–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eggers CT, Schafer JC, Goldenring JR, Taylor SS. 2009. D-AKAP2 interacts with Rab4 and Rab11 through its RGS domains and regulates transferrin receptor recycling. J. Biol. Chem. 284:32869–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ehrlich M, Boll W, Van Oijen A, Hariharan R, Chandran K, et al. 2004. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell 118:591–605 [DOI] [PubMed] [Google Scholar]
- 47.Ekiz C, Agaoglu L, Karakas Z, Gurel N, Yalcin I. 2005. The effect of iron deficiency anemia on the function of the immune system. Hematol. J. 5:579–83 [DOI] [PubMed] [Google Scholar]
- 48.Farci P, Shimoda A, Coiana A, Diaz G, Peddis G, et al. 2000. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science 288:339–44 [DOI] [PubMed] [Google Scholar]
- 49.Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, et al. 1996. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat. Genet. 13:399–408 [DOI] [PubMed] [Google Scholar]
- 50.Fillebeen C, Pantopoulos K. 2013. Hepatitis C virus infection causes iron deficiency in Huh7.5.1 cells. PLOS ONE 8:e83307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fleming RE, Ahmann JR, Migas MC, Waheed A, Koeffler HP, et al. 2002. Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. PNAS 99:10653–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foster JL, Garcia JV. 2008. HIV-1 Nef: at the crossroads. Retrovirology 5:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frazer DM, Anderson GJ. 2014. The regulation of iron transport. BioFactors 40:206–14 [DOI] [PubMed] [Google Scholar]
- 54.Freeze HH. 2013. Understanding human glycosylation disorders: Biochemistry leads the charge. J. Biol. Chem. 288:6936–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fujita H, Iwabu Y, Tokunaga K, Tanaka Y. 2013. Membrane-associated RING-CH (MARCH) 8 mediates the ubiquitination and lysosomal degradation of the transferrin receptor. J. Cell Sci. 126:2798–809 [DOI] [PubMed] [Google Scholar]
- 56.Fujita N, Sugimoto R, Takeo M, Urawa N, Mifuji R, et al. 2007. Hepcidin expression in the liver: relatively low level in patients with chronic hepatitis C. Mol. Med. 13:97–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fujita N, Sugimoto R, Urawa N, Araki J, Mifuji R, et al. 2007. Hepatic iron accumulation is associated with disease progression and resistance to interferon/ribavirin combination therapy in chronic hepatitis C. J. Gastroenterol. Hepatol. 22:1886–93 [DOI] [PubMed] [Google Scholar]
- 58.Fujita N, Takei Y. 2007. Iron, hepatitis C virus, and hepatocellular carcinoma: Iron reduction preaches the gospel for chronic hepatitis C. J. Gastroenterol. 42:923–26 [DOI] [PubMed] [Google Scholar]
- 59.Galvez T, Teruel MN, Heo WD, Jones JT, Kim ML, et al. 2007. siRNA screen of the human signaling proteome identifies the PtdIns(3,4,5)P3-mTOR signaling pathway as a primary regulator of transferrin uptake. Genome Biol. 8:R142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ganz T 2013. Systemic iron homeostasis. Physiol Rev. 93:1721–41 [DOI] [PubMed] [Google Scholar]
- 61.Gao J, Chen J, Kramer M, Tsukamoto H, Zhang AS, Enns CA. 2009. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 9:217–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garrick MD, Garrick LM. 2007. Loss of rapid transferrin receptor recycling due to a mutation in Sec15l1 in hbd mice. Biochim. Biophys. Acta 1773:105–8 [DOI] [PubMed] [Google Scholar]
- 63.Gaudin R, Kirchhausen T. 2015. Superinfection exclusion is absent during acute Junin virus infection of Vero and A549 cells. Sci. Rep. 5:15990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Giannetti AM, Bjorkman PJ. 2004. HFE and transferrin directly compete for transferrin receptor in solution and at the cell surface. J. Biol. Chem. 279:25866–75 [DOI] [PubMed] [Google Scholar]
- 65.Giannetti AM, Snow PM, Zak O, Bjorkman PJ. 2003. Mechanism for multiple ligand recognition by the human transferrin receptor. PLOS Biol. 1:e51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Girelli D, Trombini P, Busti F, Campostrini N, Sandri M,et al. 2011A time course of hepcidin response to iron challenge in patients with HFE and TFR2 hemochromatosis. Haematologica 96:500–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Girones N, Davis RJ. 1989. Comparison of the kinetics of cycling of the transferrin receptor in the presence or absence of bound diferric transferrin. Biochem. J. 264:35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gkouvatsos K, Papanikolaou G, Pantopoulos K. 2012. Regulation of iron transport and the role of transferrin. Biochim. Biophys. Acta 1820:188–202 [DOI] [PubMed] [Google Scholar]
- 69.Goodman LB, Lyi SM, Johnson NC, CifUente JO, Hafenstein SL, Parrish CR. 2010. Binding site on the transferrin receptor for the parvovirus capsid and effects of altered affinity on cell uptake and infection. J. Virol. 84:4969–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goswami T, Andrews NC. 2006. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J. Biol. Chem. 281:28494–98 [DOI] [PubMed] [Google Scholar]
- 71.Guan P, Wang N. 2014. Mammalian target of rapamycin coordinates iron metabolism with iron-sulfur cluster assembly enzyme and tristetraprolin. Nutrition 30:968–74 [DOI] [PubMed] [Google Scholar]
- 72.Hafenstein S, Palermo LM, Kostyuchenko VA, Xiao C, Morais MC, et al. 2007. Asymmetric binding of transferrin receptor to parvovirus capsids. PNAS 104:6585–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, et al. 2009. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 41:1088–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Helguera G, Jemielity S, Abraham J, Cordo SM, Martinez MG, et al. 2012. An antibody recognizing the apical domain of human transferrin receptor 1 efficiently inhibits the entry of all New World hemorrhagic fever arenaviruses. J. Virol. 86:4024–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hentze MW, Muckenthaler MU, Andrews NC. 2004. Balancing acts: molecular control of mammalian iron metabolism. Cell 117:285–97 [DOI] [PubMed] [Google Scholar]
- 76.Horonchik L, Wessling-Resnick M. 2008. The small-molecule iron transport inhibitor ferristatin/NSC306711 promotes degradation of the transferrin receptor. Chem. Biol. 15:647–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hueffer K, Parker JS, Weichert WS, Geisel RE, Sgro JY, Parrish CR. 2003. The natural host range shift and subsequent evolution of canine parvovirus resulted from virus-specific binding to the canine transferrin receptor. J. Virol. 77:1718–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Iacopetta BJ, Morgan EH, Yeoh GC. 1982. Transferrin receptors and iron uptake during erythroid cell development. Biochim. Biophys. Acta 687:204–10 [DOI] [PubMed] [Google Scholar]
- 79.Isanaka S, Mugusi F, Urassa W, Willett WC, Bosch RJ, et al. 2012. Iron deficiency and anemia predict mortality in patients with tuberculosis. J. Nutr. 142:350–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ishikawa Y, Maeda M, Pasham M, Aguet F, Tacheva-Grigorova SK, et al. 2015. Role of the clathrin adaptor PICALM in normal hematopoiesis and polycythemia vera pathophysiology. Haematologica 100:439–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jabara HH, Boyden SE, Chou J, Ramesh N, Massaad MJ, et al. 2016. A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nat. Genet. 48:74–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jain A, Chasoo G, Singh SK, Saxena AK,Jain SK. 2011. Transferrin-appended PEGylated nanoparticles for temozolomide delivery to brain: in vitro characterisation. J. Microencapsul. 28:21–28 [DOI] [PubMed] [Google Scholar]
- 83.Johannes L, Wunder C. 2011. Retrograde transport: Two (or more) roads diverged in an endosomal tree? Traffic 12:956–62 [DOI] [PubMed] [Google Scholar]
- 84.Johnson MB, Enns CA. 2004. Diferric transferrin regulates transferrin receptor 2 protein stability. Blood 104:4287–93 [DOI] [PubMed] [Google Scholar]
- 85.Joung I, Harber G, Gerecke KM, Carroll SL, Collawn JF, Engler JA. 2005. Improved gene delivery into neuroglial cells using a fiber-modified adenovirus vector. Biochem. Biophys. Res. Commun. 328:1182–87 [DOI] [PubMed] [Google Scholar]
- 86.Kaelber JT, Demogines A, Harbison CE, Allison AB, Goodman LB, et al. 2012. Evolutionary reconstructions of the transferrin receptor of caniforms supports canine parvovirus being a re-emerged and not a novel pathogen in dogs. PLOS Pathog. 8:e1002666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kaito M 2007. Molecular mechanism of iron metabolism and overload in chronic hepatitis C. J. Gastroenterol. 42:96–99 [DOI] [PubMed] [Google Scholar]
- 88.Kawabata H, Tong X, Kawanami T, Wano Y, Hirose Y, et al. 2004. Analyses for binding of the transferrin family of proteins to the transferrin receptor 2. Br. J. Haematol. 127:464–73 [DOI] [PubMed] [Google Scholar]
- 89.Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, et al. 1999. Molecular cloning of transferrin receptor 2: a new member of the transferrin receptor-like family. J. Biol. Chem. 274:20826–32 [DOI] [PubMed] [Google Scholar]
- 90.Kenneth NS, Mudie S, Naron S, Rocha S. 2013. TfR1 interacts with the IKK complex and is involved in IKK–NF-κB signalling. Biochem.J. 449:275–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kielian M, Chanel-Vos C, Liao M. 2010. Alphavirus entry and membrane fusion. Viruses 2:796–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kirchhausen T, Owen D, Harrison SC. 2014. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb. Perspect. Biol. 6:a016725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Klausner RD, Ashwell G, van Renswoude J, Harford JB, Bridges KR. 1983. Binding of apotransferrin to K562 cells: explanation of the transferrin cycle. PNAS 80:2263–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koppensteiner H, Hohne K, Gondim MV, Gobert FX, Widder M, et al. 2014. Lentiviral Nef suppresses iron uptake in a strain specific manner through inhibition of Transferrin endocytosis. Retrovirology 11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kupka R, Msamanga GI, Mugusi F, Petraro P, Hunter DJ, Fawzi WW. 2007. Iron status is an important cause of anemia in HIV-infected Tanzanian women but is not related to accelerated HIV disease progression. J. Nutr. 137:2317–23 [DOI] [PubMed] [Google Scholar]
- 96.Lambert LA. 2012. Molecular evolution of the transferrin family and associated receptors. Biochim. Biophys. Acta 1820:244–55 [DOI] [PubMed] [Google Scholar]
- 97.Lawrence CM, Ray S, Babyonyshev M, Galluser R, Borhani DW, Harrison SC 1999. Crystal structure of the ectodomain of human transferrin receptor. Science 286:779–82 [DOI] [PubMed] [Google Scholar]
- 98.Lei R, Zhang K, Liu K, Shao X, Ding Z, et al. 2016. Transferrin receptor facilitates TGF-β and BMP signaling activation to control craniofacial morphogenesis. Cell Death Dis. 7:e2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Leverence R, Mason AB, Kaltashov IA. 2010. Noncanonical interactions between serum transferrin and transferrin receptor evaluated with electrospray ionization mass spectrometry. PNAS 107:8123–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC. 1999. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat. Genet. 21:396–99 [DOI] [PubMed] [Google Scholar]
- 101.Li L, Fang CJ, Ryan JC, Niemi EC, Lebron JA, et al. 2010. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. PNAS 107:3505–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lim JE, Jin O, Bennett C, Morgan K, Wang F, et al. 2005. A mutation in Sec15l1 causes anemia in hemoglobin deficit (hbd) mice. Nat. Genet. 37:1270–73 [DOI] [PubMed] [Google Scholar]
- 103.Lucas M, Gershlick DC, Vidaurrazaga A, Rojas AL, Bonifacino JS, Hierro A. 2016. Structural mechanism for cargo recognition by the retromer complex. Cell 167:1623–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Luck AN, Mason AB. 2012. Transferrin-mediated cellular iron delivery. Curr. Top. Membr. 69:3–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Luck AN, Mason AB. 2013. Structure and dynamics of drug carriers and their interaction with cellular receptors: focus on serum transferrin. Adv. Drug Deliv. Rev. 65:1012–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. 2014. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509:105–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Martin DN, Uprichard SL. 2013. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. PNAS 110:10777–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Martys JL, Wjasow C, Gangi DM, Kielian MC, McGraw TE, Backer JM. 1996. Wortmannin-sensitive trafficking pathways in Chinese hamster ovary cells: differential effects on endocytosis and lysosomal sorting. J. Biol. Chem. 271:10953–62 [DOI] [PubMed] [Google Scholar]
- 109.Mason AL, Gilady SY, Mackey JR. 2011. Mouse mammary tumor virus in human breast cancer: red herring or smoking gun? Am. J. Pathol. 179:1588–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Maxfield FR, McGraw TE. 2004. Endocytic recycling. Nat. Rev. Mol. Cell Biol. 5:121–32 [DOI] [PubMed] [Google Scholar]
- 111.Maxson ME, Grinstein S. 2014. The vacuolar-type H+-ATPase at a glance—more than a proton pump. J. Cell Sci. 127:4987–93 [DOI] [PubMed] [Google Scholar]
- 112.Mayle KM, Le AM, Kamei DT. 2012. The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta 1820:264–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McClelland A, Kuhn LC, Ruddle FH. 1984. The human transferrin receptor gene: genomic organization, and the complete primary structure of the receptor deduced from a cDNA sequence. Cell 39:267–74 [DOI] [PubMed] [Google Scholar]
- 114.McGraw TE, Greenfield L, Maxfield FR. 1987. Functional expression of the human transferrin receptor cDNA in Chinese hamster ovary cells deficient in endogenous transferrin receptor. J. Cell Biol. 105:207–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Motley A, Bright NA, Seaman MN, Robinson MS. 2003. Clathrin-mediated endocytosis in AP-2-depleted cells. J. Cell Biol. 162:909–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Moura IC, Arcos-Fajardo M, Sadaka C, Leroy V, Benhamou M, et al. 2004. Glycosylation and size of IgA1 are essential for interaction with mesangial transferrin receptor in IgA nephropathy. J. Am. Soc. Nephrol. 15:622–34 [DOI] [PubMed] [Google Scholar]
- 117.Moura IC, Centelles MN, Arcos-Fajardo M, Malheiros DM, Collawn JF, et al. 2001. Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J. Exp. Med. 194:417–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Muckenthaler MU, Rivella S,Hentze MW, Galy B 2017. Ared carpet for iron metabolism. Cell 168:344–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nai A, Lidonnici MR, Rausa M, Mandelli G, Pagani A, et al. 2015. The second transferrin receptor regulates red blood cell production in mice. Blood 125:1170–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ned RM, Swat W, Andrews NC. 2003. Transferrin receptor 1 is differentially required in lymphocyte development. Blood 102:3711–18 [DOI] [PubMed] [Google Scholar]
- 121.Nekhai S, Kumari N, Dhawan S. 2013. Role of cellular iron and oxygen in the regulation of HIV-1 infection. Future Virol. 8:301–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, et al. 2004. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306:2090–93 [DOI] [PubMed] [Google Scholar]
- 123.Nunberg JH, York J. 2012. The curious case of arenavirus entry, and its inhibition. Viruses 4:83–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Oppenheimer SJ. 2001. Iron and its relation to immunity and infectious disease. J. Nutr. 131:616S–33S [DOI] [PubMed] [Google Scholar]
- 125.Palermo LM, Hueffer K, Parrish CR. 2003. Residues in the apical domain of the feline and canine transferrin receptors control host-specific binding and cell infection of canine and feline parvoviruses. J. Virol. 77:8915–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Park GS, Best SM, Bloom ME. 2005. Two mink parvoviruses use different cellular receptors for entry into CRFK cells. Virology 340:1–9 [DOI] [PubMed] [Google Scholar]
- 127.Parker JS, Murphy WJ, Wang D, O’Brien SJ, Parrish CR. 2001. Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J. Virol. 75:3896–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Peng YY, Uprichard J. 2017. Ferritin and iron studies in anaemia and chronic disease. Ann. Clin. Biochem. 54:43–48 [DOI] [PubMed] [Google Scholar]
- 129.Philpott CC, Ryu MS. 2014. Special delivery: distributing iron in the cytosol of mammalian cells. Front. Pharmacol. 5:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Radoshitzky SR, Abraham J, Spiropoulou CF, Kuhn JH, Nguyen D, et al. 2007. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 446:92–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Radoshitzky SR, Kuhn JH, Spiropoulou CF, Albarino CG, Nguyen DP, et al. 2008. Receptor determinants of zoonotic transmission of New World hemorrhagic fever arenaviruses. PNAS 105:2664–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Recalcati S, Gammella E, Buratti P, Cairo G. 2017. Molecular regulation of cellular iron balance. IUBMB Life 69:389–98 [DOI] [PubMed] [Google Scholar]
- 133.Robb AD, Ericsson M, Wessling-Resnick M 2004. Transferrin receptor 2 mediates a biphasic pattern of transferrin uptake associated with ligand delivery to multivesicular bodies. Am. J. Physiol. Cell Physiol. 287:C1769–75 [DOI] [PubMed] [Google Scholar]
- 134.Robb AD, Wessling-Resnick M. 2004. Regulation of transferrin receptor 2 protein levels by transferrin. Blood 104:4294–99 [DOI] [PubMed] [Google Scholar]
- 135.Rose PP, Hanna SL, Spiridigliozzi A, Wannissorn N, Beiting DP, et al. 2011. Natural resistance-associated macrophage protein is a cellular receptor for Sindbis virus in both insect and mammalian hosts. Cell Host Microbe 10:97–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ross SR, Schofield JJ, Farr CJ, Bucan M. 2002. Mouse transferrin receptor 1 is the cell entry receptor for mouse mammary tumor virus. PNAS 99:12386–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rutledge EA, Enns CA. 1996. Cleavage of the transferrin receptor is influenced by the composition of the O-linked carbohydrate at position 104. J. Cell. Physiol. 168:284–93 [DOI] [PubMed] [Google Scholar]
- 138.Ryu MS, Zhang D, Protchenko O, Shakoury-Elizeh M, Philpott CC 2017PCBP1 and NCOA4regulate erythroid iron storage and heme biosynthesis. J. Clin. Investig. 127:1786–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Salmeron A, Borroto A, Fresno M, Crumpton MJ, Ley SC, Alarcon B 1995. Transferrin receptor induces tyrosine phosphorylation in T cells and is physically associated with the TCR zeta-chain. J. Immunol. 154:1675–83 [PubMed] [Google Scholar]
- 140.Sarute N, Ross SR. 2017. New World arenavirus biology. Annu. Rev. Virol. 4:141–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Senyilmaz D, Virtue S, Xu X, Tan CY, Griffin JL, et al. 2015. Regulation of mitochondrial morphology and function by stearoylation ofTFR1. Nature 525:124–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sheftel AD, Mason AB, Ponka P. 2012. The long history of iron in the Universe and in health and disease. Biochim. Biophys. Acta 1820:161–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sheftel AD, Zhang AS, Brown C, Shirihai OS, Ponka P. 2007. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 110:125–32 [DOI] [PubMed] [Google Scholar]
- 144.Shi H, Bencze KZ, Stemmler TL, Philpott CC. 2008. A cytosolic iron chaperone that delivers iron to ferritin. Science 320:1207–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Shu ST, Emert-Sedlak LA, Smithgall TE 2017. Cell-based fluorescence complementation reveals a role for HIV-1 Nef protein dimerization in AP-2 adaptor recruitment and CD4 co-receptor down-regulation. J. Biol. Chem. 292:2670–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Spiro DJ, Boll W, Kirchhausen T, Wessling-Resnick M. 1996. Wortmannin alters the transferrin receptor endocytic pathway in vivo and in vitro. Mol. Biol. Cell 7:355–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Steere AN, Chasteen ND, Miller BF, Smith VC, MacGillivray RT, Mason AB. 2012. Structure-based mutagenesis reveals critical residues in the transferrin receptor participating in the mechanism of pH- induced release of iron from human serum transferrin. Biochemistry 51:2113–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stiles KM, Kielian M. 2011. Alphavirus entry: NRAMP leads the way. Cell Host Microbe 10:92–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sun JZ, Wang J, Wang S, Yuan D, Li Z, et al. 2014. MicroRNA miR-320a and miR-140 inhibit mink enteritis virus infection by repression of its receptor, feline transferrin receptor. Virol. J. 11:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tachiyama R, Ishikawa D, Matsumoto M, Nakayama KI, Yoshimori T, et al. 2011. Proteome of ubiquitin/MVB pathway: possible involvement of iron-induced ubiquitylation of transferrin receptor in lysosomal degradation. Genes Cells 16:448–66 [DOI] [PubMed] [Google Scholar]
- 151.TerBush DR, Maurice T, Roth D, Novick P. 1996. The exocyst is a multiprotein complex required for exocytosis in Saccharomyces cerevisiae. EMBOJ. 15:6483–94 [PMC free article] [PubMed] [Google Scholar]
- 152.Touret N, Furuya W, Forbes J, Gros P, Grinstein S. 2003. Dynamic traffic through the recycling compartment couples the metal transporter Nramp2 (DMT1) with the transferrin receptor. J. Biol. Chem. 278:25548–57 [DOI] [PubMed] [Google Scholar]
- 153.Traer CJ, Rutherford AC, Palmer KJ, Wassmer T, Oakley J, et al. 2007. SNX4 coordinates endosomal sorting of TfnR with dynein-mediated transport into the endocytic recycling compartment. Nat. Cell Biol. 9:1370–80 [DOI] [PubMed] [Google Scholar]
- 154.Trenor CC 3rd, Campagna DR, Sellers VM, Andrews NC, Fleming MD. 2000. The molecular defect in hypotransferrinemic mice. Blood 96:1113–18 [PubMed] [Google Scholar]
- 155.Vogt TM, Blackwell AD, Giannetti AM, Bjorkman PJ, Enns CA. 2003. Heterotypic interactions between transferrin receptor and transferrin receptor 2. Blood 101:2008–14 [DOI] [PubMed] [Google Scholar]
- 156.Wallace DF, Summerville L, Subramaniam VN. 2007. Targeted disruption of the hepatic transferrin receptor 2 gene in mice leads to iron overload. Gastroenterology 132:301–10 [DOI] [PubMed] [Google Scholar]
- 157.Wandinger-Ness A, Zerial M. 2014. Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb. Perspect. Biol. 6:a022616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wang E, Albritton L, Ross SR. 2006. Identification of the segments of the mouse transferrin receptor 1 required for mouse mammary tumor virus infection. J. Biol. Chem. 281:10243–49 [DOI] [PubMed] [Google Scholar]
- 159.Wang E, Obeng-Adjei N, Ying Q, Meertens L, Dragic T, et al. 2008. Mouse mammary tumor virus uses mouse but not human transferrin receptor 1 to reach a low pH compartment and infect cells. Virology 381:230–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Watts C 1985. Rapid endocytosis of the transferrin receptor in the absence of bound transferrin. J. Cell Biol. 100:633–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Weinberg ED. 1996. Iron withholding: a defense against viral infections. BioMetals 9:393–99 [DOI] [PubMed] [Google Scholar]
- 162.Weiss G, Carver PL. 2017. Role of divalent metals in infectious disease susceptibility and outcome. Clin. Microbiol. Infect. 24:16–23 [DOI] [PubMed] [Google Scholar]
- 163.Wen J, Pan S, Liang S, Zhong Z, He Y, et al. 2013. Soluble form of canine transferrin receptor inhibits canine parvovirus infection in vitro and in vivo. BioMed Res. Int. 2013:172479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wessling-Resnick M 2010. Iron homeostasis and the inflammatory response. Annu. Rev. Nutr. 30:105–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wessling-Resnick M 2015. Nramp1 and other transporters involved in metal withholding during infection. J. Biol. Chem. 290:18984–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.West AP Jr., Bennett MJ, Sellers VM, Andrews NC, Enns CA, Bjorkman PJ 2000. Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J. Biol. Chem. 275:38135–38 [DOI] [PubMed] [Google Scholar]
- 167.White RA, Boydston LA, Brookshier TR, McNulty SG, Nsumu NN, et al. 2005. Iron metabolism mutant hbd mice have a deletion in Sec15l1, which has homology to a yeast gene for vesicle docking. Genomics 86:668–73 [DOI] [PubMed] [Google Scholar]
- 168.Worthen CA, Enns CA. 2014. The role of hepatic transferrin receptor 2 in the regulation of iron homeostasis in the body. Front. Pharmacol. 5:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Xia H, Anderson B, Mao Q, Davidson BL. 2000. Recombinant human adenovirus: Targeting to the human transferrin receptor improves gene transfer to brain microcapillary endothelium. J. Virol. 74:11359–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Xu W, Barrientos T, Mao L, Rockman HA, Sauve AA, Andrews NC. 2015. Lethal cardiomyopathy in mice lacking transferrin receptor in the heart. CellRep. 13:533–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yanatori I, Yasui Y, Tabuchi M, Kishi F. 2014. Chaperone protein involved in transmembrane transport of iron. Biochem. J. 462:25–37 [DOI] [PubMed] [Google Scholar]
- 172.Yang B, Hoe MH, Black P, Hunt RC. 1993. Role of oligosaccharides in the processing and function of human transferrin receptors: effect of the loss of the three N-glycosyl oligosaccharides individually or together. J. Biol. Chem. 268:7435–41 [PubMed] [Google Scholar]
- 173.Yang FM, Friedrichs WE, Buchanan JM, Herbert DC, Weaker FJ, et al. 1990. Tissue specific expression of mouse transferrin during development and aging. Mech. Ageing Dev. 56:187–97 [DOI] [PubMed] [Google Scholar]
- 174.Zapata JC, Salvato MS 2013. Arenavirus variations due to host-specific adaptation. Viruses 5:241–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zeltina A, Krumm SA, Sahin M, Struwe WB, Harlos K,et al. 2017. Convergent immunological solutions to Argentine hemorrhagic fever virus neutralization. PNAS 114:7031–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhang AS, Sheftel AD, Ponka P. 2005. Intracellular kinetics of iron in reticulocytes: evidence for endo- some involvement in iron targeting to mitochondria. Blood 105:368–75 [DOI] [PubMed] [Google Scholar]
- 177.Zhang AS, Xiong S, Tsukamoto H, Enns CA. 2004. Localization of iron metabolism—related mRNAs in rat liver indicate that HFE is expressed predominantly in hepatocytes. Blood 103:1509–14 [DOI] [PubMed] [Google Scholar]
- 178.Zhao N, Enns CA. 2013. N-linked glycosylation is required for transferrin-induced stabilization of transferrin receptor 2, but not for transferrin binding or trafficking to the cell surface. Biochemistry 52:3310–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zong M, Fofana I, Choe H. 2014. Human and host species transferrin receptor 1 use by North American arenaviruses. J. Virol. 88:9418–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zumerle S, Mathieu JR, Delga S, Heinis M, Viatte L,et al. 2014. Targeted disruption ofhepcidin in the liver recapitulates the hemochromatotic phenotype. Blood 123:3646–50 [DOI] [PubMed] [Google Scholar]