

SUMMARY
The lymphatic vasculature requires intraluminal valves to maintain forward lymph flow. Lymphatic valves form and are constantly maintained by oscillatory fluid flow throughout life, yet the earliest steps of how lymphatic endothelial cells are able to respond to fluid shear stress remain unknown. Here, we show that the adherens junction protein VE-cadherin is required for the upregulation of valve-specific transcription factors. Conditional deletion of VE-cadherin in vivo prevented valve formation in the embryo and caused postnatal regression of nearly all lymphatic valves in multiple tissues. Since VE-cadherin is known to signal through β-catenin and the VEGFR/AKT pathway, each pathway was probed. Expression of a constitutively active β-catenin mutant or direct pharmacologic activation of AKT in vivo significantly rescued valve regression in the VE-cadherin-deficient lymphatic vessels. In conclusion, VE-cad-herin-dependent signaling is required for lymphatic valve formation and maintenance and therapies to augment downstream pathways hold potential to treat lymphedema in patients.
Graphical Abstract
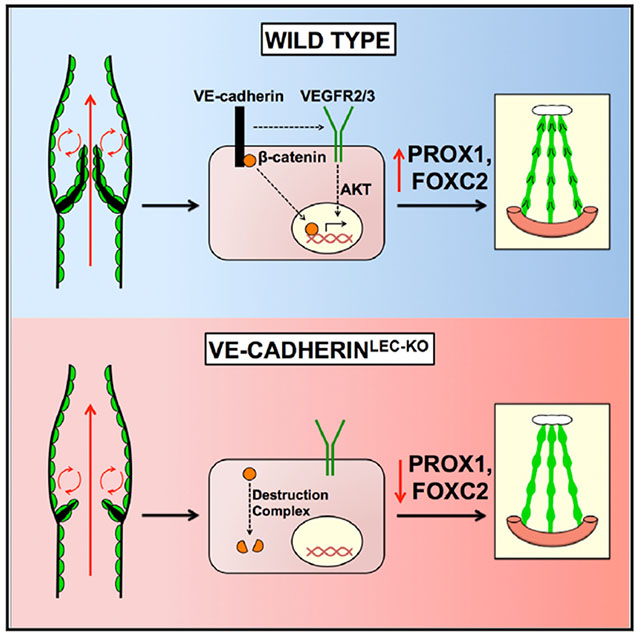
In Brief
Oscillatory fluid flow increases the expression of nuclear transcription factors that orchestrate lymphatic valve morphogenesis. Yang et al. investigate how signaling events mediated by VE-cadherin at the cell membrane regulate valve development. They find that VE-cadherin is required for β-catenin and AKT signaling that regulate nuclear Prox1 and Foxc2 expression.
INTRODUCTION
In contrast to the blood vasculature, the lymphatic vasculature experiences oscillatory fluid flow. Oscillatory fluid flow activates nuclear transcription factors that orchestrate the formation of intraluminal bileaflet valves that are dependent on fluid shear for their lifelong maintenance (Sabine et al., 2012). Thus, in the absence of flow, lymphatic valves undergo regression (Sweet et al., 2015). Since lymphatic vessels transport fluid from the tissues to the bloodstream against an increasing hydrostatic pressure, lymphatic valves serve to prevent retrograde lymph flow and edema.
Mutations in lymphatic valve genes have been identified in many human syndromes (Brouillard et al., 2014) and lead to lymphedema, severe tissue swelling characterized by fibrosis and infections. Lymphedema can also be acquired, most commonly after lymph node surgery in breast cancer patients (Petrek et al., 2001; Stanton et al., 2006). Valve defects likely instigate lymphedema because the most common gene mutations in human lymphedema occur in transcription factors regulating valve formation and because retrograde lymph flow is observed in patients (Rasmussen et al., 2009). However, it remains unclear how fluid shear stress exerted on the cell membrane activates transcription factors located in the cell nucleus. The lack of a defined signaling pathway has precluded the development of new clinical therapies for lymphedema.
Endothelial cells sense fluid shear stress through the mechanotransduction pathway. In this pathway, PECAM1 senses shear stress and initiates SRC phosphorylation of VEGFR2 and VEGFR3 to activate PI3K/AKT signaling. These proteins are held in a complex by VE-cadherin (Baeyens and Schwartz, 2016; Tzima et al., 2005). A role for PECAM1 in embryonic lymphatic valve formation was shown recently, supporting its role as a mechanosensor in the lymphatic vasculature (Wang et al., 2016). Whether VE-cadherin regulates mechanotransduction in the lymphatic vasculature remains unknown. While lymphatic-specific deletion of VE-cadherin causes lymphatic valve loss (Hägerling et al., 2018), it is unknown whether this is due to defective mechanotransduction signaling or due to vessel instability, as vascular structures have been reported to disintegrate upon loss of VE-cadherin (Carmeliet et al., 1999; Gory-Fauré et al., 1999). Here, we demonstrate that VE-cadherin regulates cell alignment with flow and its deletion inhibits lymphatic valve formation in the embryo and leads to severe valve regression in postnatal mice, prior to the onset of vessel disintegration and loss of integrity.
RESULTS
Generation and Validation of a Conditional VE-Cadherin Allele
Oscillatory shear stress created by pulsatile lymph flow has been shown to be involved in the early steps of lymphatic valve formation (Sabine et al., 2012, 2015; Sweet et al., 2015), indicating that a mechanotransduction signaling complex may be essential for this process. To test whether VE-cadherin was required for lymphatic valve development, we generated a conditional floxed Cdh5 allele through homologous recombination (Figure S1A). Previous studies have demonstrated that global loss of VE-cadherin is embryonic lethal at embryonic day (E) ~10.5 (Carmeliet et al., 1999; Gory-Fauré et al., 1999). To test that our floxed allele disrupts Cdh5 gene expression, we crossed it with the constitutive pan-endothelial Tie2Cre allele to obtain Tie2Cre;Cdh5flox/flox embryos (Figures S1B, S1C, and S1D). As reported for global Cdh5−/− embryos (Carmeliet et al., 1999; Gory-Fauré et al., 1999), Tie2Cre;Cdh5-flox/flox embryos at E10.5 exhibited stunted growth, a dilated pericardial sac, and underdeveloped vascular structures that were prone to hemorrhaging (Figure S1D). Additionally, the yolk sac vasculature was malformed and appeared as a collection of blood islands (Figure S1C). When pups were genotyped at birth from Tie2Cre;Cdh5+/fl x Cdh5fl/fl matings, the expected ratios of Cdh5+/fl, Cdh5fl/fl, and Tie2Cre;Cdh5+/fl were born (30%, 24%, and 22%, respectively; n = 150 pups), but no conditional null Tie2Cre;Cdh5fl/fl pups were obtained. To test whether pups could be obtained from matings with a constitutive lymphatic-specific Cre strain, we mated Lyve1Cre;Cdh5+/fl with Cdh5fl/fl and genotyped the pups at birth. The expected ratios of Lyve1Cre;Cdh5+/fl, Cdh5+/fl, and Cdh5fl/ fl mice were obtained (29%, 27%, and 19%, respectively; n = 64 pups), while no conditional null Lyve1Cre;Cdh5fl/fl pups were produced. To determine whether we could obtain lymphatic-specific VE-cadherin null embryos at a later stage than pan-endothelial null embryos, we searched for LyvelCre;Cdh5fl/fl embryos at E11.5, but found that they displayed stunted growth and appeared to be undergoing resorption (data not shown). Therefore, both Lyve1Cre;Cdh5fl/fl and Tie2-Cre;Cdh5fl/fl mice are embryonic lethal at ~E10.5, presumably due to the expression of Lyve1 in the embryonic arterial and venous endothelium as well as the yolk sac vasculature (Gordon et al., 2008).
Embryonic Lymphatic-Specific Deletion of VE-Cadherin Results in Lymphatic Valve Defects
To study the role of VE-cadherin in the lymphatic vasculature during embryonic valve development, we resorted to using a tamoxifen-inducible lymphatic-specific CreERT2 strain with a high recombination efficiency (Bazigou et al., 2011). The Cdh5flox/flox and Prox1CreERT2 strains were interbred to generate Cdh5flox/flox controls and Prox1CreERT2;Cdh5flox/flox knockout mice (hereafter referred to as VE-cadherinLEC-KO). To visualize the lymphatic vasculature in freshly dissected tissues, the Prox1-GFP reporter strain (Choi et al., 2011) was intercrossed with these strains. Inactivation of genes that are involved in lymphatic valve formation typically result in valve loss when they are quantified in the mouse mesentery (Bazigou et al., 2009; Kazenwadel et al., 2015; Sabine et al., 2012, 2015; Sweet et al., 2015). We induced recombination of the VE-cadherin gene at E14.5, which is approximately 1 day after lymphatic endothelial cell (LEC) progenitors begin to form a mesenteric lymphatic plexus (Stanczuk et al., 2015) (Figures 1A and 1B). To determine whether loss of VE-cadherin resulted in embryonic lymphatic valve defects, embryos were collected at E18.5 and mesenteries were imaged immediately at low and high magnification. As shown in Figure 1C, control mesenteries had several GFP+ lymphatic vessels that each contained numerous GFPhi bright spots, which were confirmed to be lymphatic valves at higher magnification (Figure 1D). In contrast, several dilated GFP+ lymphatic vessels were found in the VE-cadherinLEC-KO mesenteries and no GFPhi spots were ever visualized (Figures 1E and 1F), indicating a complete absence of lymphatic valves. At high magnification, the lymphatic structures in the VE-cadherinLEC-KO mesenteries were reminiscent of a lymphatic plexus that had failed to remodel into defined collecting vessels (Figure 1F), similar to that reported for Clec2−/− mice that lacked lymph flow (Sweet et al., 2015).
Figure 1. Embryonic Deletion of VE-Cadherin Causes Loss of Lymphatic Valves.

(A) Alleles used to delete VE-cadherin from lymphatic endothelium while simultaneously enabling the visualization of Prox1-expressing cells.
(B) Schematic of the tamoxifen schedule used to delete VE-cadherin from the embryonic mesentery. TM, tamoxifen.
(C–F) Direct fluorescence imaging of GFP (green) of freshly dissected mesenteries from E18.5 Cdh5flox/flox (C and D) and Prox1CreERT2;Cdh5flox/flox (E and F) embryos.
(D and F) High-magnification images of the areas outlined by the white dashed boxes in (C) and (E), respectively. White arrows indicate lymphatic valves. Images are representative of n = 3 or more mesenteries per genotype. (See also Figure S2 for loss of lymphatic valves in embryonic back skin and axillary regions.)
(G) Tamoxifen schedule used for early embryonic deletion of VE-cadherin. TM, tamoxifen.
(H and I) E14.5 control (H) and VE-cadherinLEC-KO (I) embryos. White arrowheads denote severe edema.
(J and K) E16.5 control (J) and VE-cadherinLEC-KO (K) embryos. White arrowheads denote edema.
(L and M) Immunostaining of frontal sections of E14.5 control (L) and VE-cadherinLEC-KO (M) embryos for PROX1 (green) and PECAM1 (red). LS, lymph sac; SCV, subclavian vein; IJV, internal jugular vein.
(N and O) Immunostaining of frontal sections of E16.5 control (N) and VE-cadherinLEC-KO (O) embryos for VEGFR3 (green), GFP (red), and PECAM1 (blue). White arrows indicate lymphovenous valves while yellow arrows indicate venous valves only.
(P and Q) Immunostaining of frontal sections of E16.5 control (P) and VE-cadherinLEC-KO (Q) embryos as in (N) and (O), but omitting the green channel to show Prox1-GFP expression (red channel).
Scale bars are 200 μm in (C)–(F); 2 μm in (H)–(K); and 100 μm for (L)–(Q).
Demonstrating that the lymphatic valve defects were not confined to the mesentery alone, we also assessed valve morphology in other tissues outside the abdomen (Figure S2). In the developing axillary lymphatic vessels that connect the inguinal and axillary lymph nodes at E18.5, the control lymphatic vessels were highly branched with numerous areas of valve formation that were completely absent in the VE-cadherinLEC-KO (Figures S2A and S2B). Similarly, in embryonic back skin at E18.5, the developing lymphatic plexus contains several valve-forming areas denoted by GFPhi spots that are completely absent from the VE-cadherinLEC-KO back skin (Figures S2C–S2F). Further, we found that the lymphatic vessels appeared to migrate to the midline faster in the VE-cadherinLEC-KO back skin and appeared to be disintegrating (Figures S2C and S2D). Together, these data indicate that VE-cadherin is required for lymphatic valve development in the embryo and for the maturation of the early lymphatic capillary plexus. As these two processes have previously been shown to require lymph flow in vivo (Sweet et al., 2015), and oscillatory shear stress upregulates valve specification genes in vitro (Cha et al., 2016; Kazenwadel et al., 2015; Sabine et al., 2012; Sweet et al., 2015), we hypothesized that a mechanotransduction complex similar to that identified in blood vessels regulates lymphatic valve formation.
VE-Cadherin Deletion Impairs Valve Maturation
Lymphatic valve formation occurs in the embryonic mesentery from E16.5 to E18.5 (Geng et al., 2017; Norrmén et al., 2009; Sabine et al., 2012; Tatin et al., 2013). Since VE-cadherin-deficient lymphatic vessels completely lacked valves at E18.5, we checked the earliest stages of lymphatic valve formation at E16.5 and E17.5. Single PROX1+ cells first appear in the mouse mesentery at E13.5, which later assemble into vessel structures that then initiate valve development (Stanczuk et al., 2015). The initiation of valve development first appears as PROX1hi cell clusters at E16.5 in mouse mesentery (Geng et al., 2017; Stanczuk et al., 2015). To assess the role of VE-cadherin in lymphatic valve formation, inactivation of the VE-cadherin gene was induced by injection of tamoxifen at E13.5 and E14.5, followed by harvesting mesenteries at E16.5 (Figure S3A). Control and VE-cadherinLEC-KO mesenteries with the Prox1-GFP reporter possessed many GFPhi spots that indicated the normal initiation of valve formation (Figures S3B–S3E). Both the morphology and number of GFPhi areas appeared similar in the controls compared to the VE-cadherinLEC-KO lymphatic vessels. To directly confirm the efficient deletion of VE-cadherin, mesenteries were immunostained for both VE-cadherin and PROX1 (Figures S3F–S3I). Again, control and VE-cadherinLEC-KO lymphatic vessels both contained PROX1hi cell clusters that indicated lymphatic valve formation was initiated normally and all PROX1+ vessels lacked VE-cadherin expression in the VE-cadherinLEC-KO tissues. Together, these results demonstrate that VE-cadherin is dispensable for the specification of lymphatic valve cell clusters.
Next, we checked whether the lymphatic valve cell clusters that appeared at E16.5 entered into the second and third steps of valve formation, condensation and leaflet elongation, at E17.5 (Sabine et al., 2012). VE-cadherin was deleted as before prior to harvesting mesenteries at E17.5 (Figure S4A). To check valve morphology, we performed immunostaining for PROX1 and Integrin-α9, a receptor for fibronectin EIIIA that is upregulated on lymphatic valve endothelium (Bazigou et al., 2009). In control E17.5 mesenteries, the valve-forming territories had upregulated PROX1 expression that was accompanied by increased expression of Integrin-α9 compared to the surrounding LECs (Figures S4B and S4C). The VE-cadherinLEC-KO valve territories still expressed high levels of PROX1, similar to E16.5, and these areas were associated with higher levels of Integrin-α9 (Figures S4D and S4E). When their morphologies were compared, there appeared to be no defects in valve elongation as defined by Sabine et al. (2012). The fourth and final step of valve formation is valve maturation that occurs between E17.5 and postnatal day (P) 7. No mature valve leaflets were observed at E18.5, precluding further analysis, and demonstrated that VE-cadherin is required for the valve maturation step of valve formation.
VE-Cadherin Is Required for Lymphovenous Valve Development in the Embryo
Recently, it was proposed that the embryonic edema in mutant mouse models of lymphatic disease is indicative of defects in the formation of the lymphovenous valves (LVVs), which form at the junction between the lymphatic and blood vasculatures to prevent blood from entering the lymphatic vessels (Geng et al., 2016). The first valve structures that form in the developing embryo are the leaflets of the LVVs, which begin protruding from the lymph sac at E12.5 into the future subclavian vein (Srinivasan and Oliver, 2011). Two layers of endothelial cells contribute to the LVV leaflets: lymphatic endothelium from the lymph sac and venous endothelium from the subclavian vein. By E16.5, well-defined valve leaflets extend from the lymph sacs into the subclavian vein (Geng et al., 2016). To determine whether loss of VE-cadherin affected the formation of the LVV, recombination was induced at E10.5, 1 day after the specification of LECs had begun but prior to the formation of the lymph sac and primitive lymphatic plexus (Srinivasan et al., 2007; Yang et al., 2012) (Figure 1G). Embryos were obtained at E14.5 and E16.5 and were sectioned frontally to visualize the LVV. Gross examination of the VE-cadherinLEC-KO embryos at E14.5 and E16.5 revealed profound edema, indicating the presence of lymphatic vascular defects (Figures 1H–1K). At E14.5, the LVV leaflets appeared normal and seemed to make continuous connections with the subclavian vein in both the controls and knockouts (Figures 1L and 1M). In contrast, the lymph sacs of the E16.5 VE-cadherinLEC-KO embryos failed to upregulate PROX1, lacked valve leaflets, and remained completely disconnected from the subclavian vein (Figures 1N–1Q). This revealed that although VE-cadherin expression was dispensable for the early development of LVV valves at E14.5, the complete formation of the LVV leaflets required VE-cadherin.
LEC Nuclei Do Not Align with Flow in the Absence of VE-Cadherin
A hallmark of endothelial mechanotransduction signaling is the ability of cells to align parallel to the direction of flow (Tzima et al., 2005). Loss of VE-cadherin from blood vessel endothelium prevents cell alignment in the direction of flow (Tzima et al., 2005), while loss of Pecam1 or Sdc4 from the lymphatic vasculature has been shown to prevent alignment of LECs with flow and rounder cells (Wang et al., 2016). To assess LEC alignment with flow, we measured whether PROX1+ nuclei were aligned in the direction of lymph flow in straight, unbranched vessels and measured the nuclear length-to-width ratio to assess roundness (Figure 2). PROX1+ LEC nuclei in control lymphatic vessels at E18.5 appeared elongated along the vessel axis, compared with VE-cadherin-deficient LECs that appeared round and randomly oriented (Figures 2A and 2B). When this was quantified, with 0° set parallel to the vessel length, control nuclei were within approximately 3° of the flow axis, while VE-cadherinLEC-KO nuclei were on average oriented ~40° away from the flow axis with higher variability (Figure 2C; 2.9 ± 0.6 versus 38.5 ± 7.4, n = 3, p < 0.05). Similarly, the length-to-width ratio was higher in the control nuclei, indicating elongation, while this was significantly reduced in the VE-cadherinLEC-KO nuclei (Figure 2D; 3.7 ± 0.7 versus 1.6 ± 0.1, n = 3, p < 0.05). Thus, VE-cadherin-deficient LECs do not align with flow. These data strongly implicate VE-cadherin in mechanotransduction signaling in the developing lymphatic vasculature.
Figure 2. VE-Cadherin Is Required for Lymphatic Endothelial Cell Alignment with Flow in the Embryo.

(A and B) E18.5 mesenteries from control (A) and VE-cadherinLE-KO (B) embryos immunostained for PROX1.
(C) Nucleus orientation was measured using NIH FIJI software. The flow axis was defined as 0° and the absolute values of the angles were reported.
(D) Nucleus roundness was measured as the length-to-width ratio.
All values are means ± SEM of n = 3 independent experiments. *p < 0.05. Scale bars are 50 μm.
VE-Cadherin Regulates Lymphatic Vessel Integrity and Valve Morphology Postnatally
Oscillatory shear stress upregulates Prox1 and Foxc2 in LECs, with Foxc2 regulating the constant expression of connexins and calcineurin signaling to maintain the valve structures once they have formed (Sabine et al., 2012, 2015). Thus, persistent oscillatory flow induces constant cellular signaling pathways that prevent the regression of lymphatic valves during postnatal and adult life. This essential role for flow in lymphatic valve development was recently confirmed in vivo (Sweet et al., 2015). If VE-cadherin is necessary for mechanotransduction signaling, then we would expect that its continued expression would be required for valve maintenance postnatally. Controls and VE-cadherinLEC-KO pups were administered tamoxifen at P1 and P3 to completely delete VE-cadherin at a time when many lymphatic valves had already formed (Figure 3A). Pups were then examined for gross lymphatic vascular defects at P8 and P14. At P14, but not at P8, we observed the accumulation of chylous ascites in the VE-cadherinLEC-KO pups (Figure 3C) but not in controls (Figure 3B). When the mesenteric lymphatic vessels were examined directly under brightfield microscopy, we visualized leakage of milky fluid into the peritoneum from lymphatic collecting vessels, which appeared as a white haze around the vessels (Figure 3E). This was expected due to the fact that VE-cadherin is also a crucial adherens junction protein that controls blood vessel endothelial integrity (Carmeliet et al., 1999; Dejana et al., 2008). Due to the absorption of dietary lipids, the lymphatic collecting vessels appeared white in both groups, which enabled the visualization of the valves. Normal V-shaped valves were apparent in the controls (Figure 3F), but were not observed in any of the VE-cadherinLEC-KO lymphatic collecting vessels (Figure 3G). Instead, many lymphatic vessels were devoid of valves and the valve structures that were occasionally present were constricted ring structures that lacked leaflets (Figure 3G).
Figure 3. Postnatal Loss of VE-Cadherin Leads to Chylous Ascites and Loss of Valves.

(A) Tamoxifen schedule used for postnatal deletion of VE-cadherin. TM, tamoxifen.
(B and C) Chylous ascites fluid of VE-cadherinLEC-KO mice (C, black arrowhead) compared to control (B).
(D and E) Lymph leakage from mesenteric lymphatic collecting vessels of VE-cadherinLEC-KO mesentery (E) compared to control (D).
(F and G) Valve structures in the lymphatic collecting vessels of control (F, white arrow) and VE-cadherinLEC-KO (G, yellow arrows) mesenteries. Insets are zoomed-in views of the valves indicated by the arrows.
(H and I) In situ fluorescence imaging of the mesenteric lymphatic vasculature at P8. Images are of control (H) and VE-cadherinLEC-KO (I) tissues expressing the Prox1-GFP reporter (green) at low magnification.
(J and K) High-magnification images of P8 lymphatic valves of control (J) and VE-cadherinLEC-KO (K) mesenteries.
(L) The total number of lymphatic valves in P8 control and VE-cadherinLEC-KO mesenteries.
(M and N) In situ fluorescence imaging of the mesenteric lymphatic vasculature at P14 expressing the Prox1-GFP at low magnification for control (M) and VE-cadherinLEC-KO (N).
(O and P) High-magnification images of P14 lymphatic valves of control (O) and VE-cadherinLEC-KO (P) mesenteries.
(Q) The total number of lymphatic valves in the P14 mouse mesentery of control and VE-cadherinLEC-KO animals. Unpaired Student’s t tests were performed to compare the total valve number between control and VE-cadherinLEC-KO tissues at P8 and P14 (n = 3 per genotype per stage).
All values are means ± SEM. *p < 0.05. Scale bars are 5 mm in (B) and (C); 500 μm in (D) and (E); 250 μm in (F) and (G); and 200 μm in (H)–(P).
Quantification of the Total Number of Lymphatic Valves Reveals a Valve Maintenance Defect
To enable quantification of the total number of valves in the control and VE-cadherinLEC-KO mesenteries at P8 and P14, valves were visualized in situ with fluorescence microscopy (Figures 3H–3P).
The valves of control mesenteries at both stages exhibited the typical V shape and were composed of PROX1hi LECs (Figures 3J and 3O), whereas the lymphatic vessels in the VE-cadherinLEC-KO mesenteries were either constricted (Figure 3K) or possessed ring structures composed of PROX1hi LECs (Figure 3P). Long, perfectly formed valve leaflets were rare in the VE-cadherinLEC-KO mesenteries. Small collateral branches at the defective valve sites were common (Figure 3P). In P8 control mesenteries, numerous regularly spaced valves were counted, whereas the VE-cadherinLEC-KO mesenteries had 75% fewer valves (Figures 3H, 3I, and 3L) (743 ± 14 versus 173 ± 4, n = 3, p < 0.05). Approximately 1 week later, the P14 VE-cadherinLEC-KO mesenteries had 90% fewer valves than the controls (1,083 ± 48 versus 115 ± 8, n = 3, p < 0.05), indicating that over time the valves were regressing (Figures 3M, 3N, and 3Q). As the loss of lymphatic valves in the VE-cadherinLEC-KO animals could also be explained by a loss of lymph flow instead of a direct loss of VE-cadherin signaling, we fed postnatal pups 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-hexadecanoic acid (BODIPY FL C16), a fluorescent lipid that is selectively absorbed by the lymphatic collecting vessels draining the intestine. Others have shown that without forward flow, mesenteric lymphatic collecting vessels will not fill with BODIPY FL C16 and, thus, will not exhibit any fluorescence (Sweet et al., 2015). Mesenteric lymphatic collecting vessels from control and VE-cadherinLEC-KO pups at P8 and P14 were filled with the fluorescent lipid 3 h after feeding (Figure S6), indicating that these vessels had patent lumens and forward flow. Another possibility is that loss of VE-cadherin might decrease the expression of other junction proteins prior to the loss of valves at P8, and the loss of several junction proteins would collectively lead to an enhanced permeability and reduction in lymph flow to indirectly cause valve regression. However, genetic deletion of VE-cadherin from adult blood vessel endothelium (Frye et al., 2015) or adult lymphatic endothelium (Hägerling et al., 2018) was recently shown to be without effect on the expression of other junction proteins or the ultrastructure of interendothelial junctions by electron microscopy. To determine whether the loss of other junction proteins from the postnatal lymphatic vasculature could explain the valve regression in P8 VE-cadherinLEC-KO mice, we examined the tight junction proteins, Claudin-5 and ZO-1 (Figure S5). Similar to the adult lymphatic vasculature, we show that postnatal deletion of VE-cadherin at an early time point does not affect the expression of other junction proteins. Together, these data demonstrate that VE-cadherin signaling is required for postnatal valve maintenance, and postnatal valve regression occurs in the presence of lymph flow and prior to the onset of chylous ascites or altered junction protein expression.
VE-Cadherin Regulates PROX1 and FOXC2 Expression In Vivo and Inhibits Cell Death
Interestingly, the postnatal valve phenotypes of our VE-cadherinLEC-KO mice appeared similar to another mouse model that has demonstrated postnatal valve regression, which is a conditional, lymphatic-specific deletion of Foxc2, a major transcription factor required for valve formation and maintenance (Sabine et al., 2015). Thus, we hypothesized that VE-cadherin-mediated signaling regulated the expression of Foxc2 through mechanotransduction signaling. Immunostaining was performed on E18.5 and P8 mesenteries to determine whether PROX1 and/or FOXC2 were downregulated upon loss of VE-cadherin (Figure 4). At E18.5, collecting lymphatic vessels from control mesenteries exhibited well-defined valves that had higher PROX1 expression than the surrounding lymphangion cells and expressed VEGFR3 and VE-cadherin (Figures 4A and 4B). In contrast, the lymphatic collecting vessels from VE-cadherinLEC-KO mesenteries expressed PROX1 uniformly in every LEC (Figure 4C). We also verified our deletion efficiency by staining for VE-cadherin, which was deleted from >95% of LECs (Figure 4D). Similarly, control lymphatic collecting vessels demonstrated widespread expression of FOXC2 in every cell, as this is before the developmental stage when FOXC2 becomes restricted to the valve areas, but FOXC2 was highly upregulated in the valves (Figures 4E and 4F). Upon loss of VE-cadherin, the expression of FOXC2 was uniform throughout the lymphatic vasculature at a level similar to non-valve lymphangion cells (Figures 4G and 4H). Postnatal loss of Foxc2 has been shown to cause valve regression through apoptosis in a caspase-3-dependent manner (Sabine et al., 2015). Embryonic loss of VE-cadherin similarly led to clusters of apoptotic cells around branched regions in the VE-cadherinLEC-KO lymphatic collecting vessels (Figures 4K and 4L), while no staining was observed in the controls (Figures 4I and 4J). To investigate how the postnatal loss of VE-cadherin affected the expression of PROX1 and FOXC2, VE-cadherin deletion was induced at P1/P3 and immunostaining was performed on P8 mesenteries. In control mesenteries, PROX1hi cells were observed in the valve regions (Figures 4M and 4N), whereas in VE-cadherinLEC-KO lymphatic collecting vessels, LECs expressed PROX1 uniformly (Figures 4O and 4P). At P8, FOXC2 expression appeared more restricted to the valve regions in the controls (Figures 4Q and 4R), but these FOXC2hi cells were mostly absent in the VE-cadherinLEC-KO lymphatic vessels, and every LEC expressed uniform levels of FOXC2 (Figures 4S and 4T). In later stages (> P14), we found that FOXC2 staining in control mesenteries only labels the valve LEC, whereas in the VE-cadherinLEC-KO mesenteries, FOXC2 could not be detected by immunostaining (data not shown). Together, these data indicate that VE-cadherin signaling enables the proper regulation of PROX1 and FOXC2 expression during valve development and sustains high FOXC2 expression in the valve cells throughout postnatal life. VE-cadherin deletion was recently shown to affect YAP/TAZ protein expression (Hägerling et al., 2018), and YAP/TAZ signaling regulates PROX1 expression and lymphatic valves (Cho et al., 2019). In control valves TAZ was highly expressed in the LEC nuclei, but in VE-cadherinLEC-KO valves TAZ was downregulated (Figures 4U–4X).
Figure 4. VE-Cadherin-Deficient Lymphatic Valve Endothelial Cells Fail to Upregulate Prox1 and Foxc2 and Undergo Apoptosis.

(A and C) Whole mount immunostaining of mesenteries collected from E18.5 control (A) and VE-cadherinLEC-KO (C) embryos for VEGFR3 (green), PROX1 (red), and VE-cadherin (blue).
(B and D) The same images as in (A) and (C) showing only PROX1 (red) in control (B) and VE-cadherinLEC-KO (D).
(E-H) Whole mount immunostaining of mesenteries collected from E18.5 control (E) and VE-cadherinLEC-KO (G) embryos expressing the Prox1-GFP reporter for GFP (green), FOXC2 (red), and 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI; blue). Images of only FOXC2 (red) are shown for control (F) and VE-cadherinLEC-KO (H).
(I–L) Whole mount immunostaining of mesenteries collected from E18.5 control (I) and VE-cadherinLEC-KO (K) embryos for VEGFR3 (green), Casp3 (red), and PECAM1 (blue). Images without VEGFR3 are shown for control (J) and VE-cadherinLEC-KO (L).
(M and O) Whole mount immunostaining of mesenteries collected from P8 control (M) and VE-cadherinLEC-KO (O) pups for VEGFR3 (green), PROX1 (red), and VE-cadherin (blue).
(N and P) The same images as in (M) and (O) showing only PROX1 (red) for control (N) and VE-cadherinLEC-KO (P).
(Q–T) Whole mount immunostaining of mesenteries collected from P8 control (Q) and VE-cadherinLEC-KO (S) pups for PECAM1 (green), FOXC2 (red), and DAPI (blue). Images of only FOXC2 (red) are shown for control (R) and VE-cadherinLEC-KO (T).
(U–X) Whole mount immunostaining of mesenteries collected from P8 control (U) and VE-cadherinLEC-KO (W) pups for PROX1 (green), TAZ (red), and VE-cadherin (blue). Images of only TAZ (red) are shown for control (V) and VE-cadherinLEC-KO (X). Insets at the lower right of some panels are the expanded views of the areas outlined by white boxes.
Scale bars are 100 μm.
VE-Cadherin Is Required for Lymphatic Endothelial Shear Stress Responses In Vitro
Our data indicate that VE-cadherin is essential for the formation and maintenance of lymphatic valves, at least in part by modulating PROX1 and FOXC2 expression. However, whether VE-cadherin is able to regulate these and other transcription factors involved in valve development in response to oscillatory shear stress (OSS) is unknown. To address this issue, we cultured human dermal LECs in the absence or presence of OSS and transfected LECs in both conditions with a scrambled small hairpin RNA (shRNA) or with a targeted shRNA against CDH5. qRT-PCR was then performed to determine the changes in expression of several genes known to be involved in lymphatic valve development or responses to endothelial shear stress (Figure 5). Since β-catenin directly binds VE-cadherin (Taddei et al., 2008) and regulates embryonic lymphatic valve development by binding the promoter regions of Prox1 and Foxc2 (Cha et al., 2016), we also inhibited the degradation of β-catenin with the chemical inhibitor 6-bromoindirubin-3’-oxime (BIO), a GSK3β antagonist.
Figure 5. VE-Cadherin-Dependent Signaling Is Required for Transcription Factor Upregulation in Response to Oscillatory Flow In Vitro.

(A) Western blot of FOXC2 and CDH5 in human dermal lymphatic endothelial cells (hdLECs) cultured for 48 h in the absence or presence of oscillatory flow with or without a targeted shRNA against CDH5. Cells were treated with vehicle or with a GSK3-β antagonist (BIO).
(B) qRT-PCR was performed for the indicated genes using hdLECs cultured under no flow (black bars) or oscillatory flow (red bars), transfected with a control scramble or shRNA against CDH5 (as indicated), and treated with vehicle or BIO (gray and blue bars). All values are means ± SEM of n = 3 experiments. One-way ANOVA was performed with Sidak’s post hoc test for multiple comparisons (*p < 0.05).
(C) hdLECs cultured under no-flow (static) conditions and immunostained for DAPI (blue), VE-cadherin (violet), PROX1 (red), and β-catenin (green) after infection with a scramble or shRNA targeting CDH5 for 48 h.
(D) Western blot for β-catenin, pAKT, and total AKT in hdLECs under static or OSS conditions (for 5 min) treated with scramble or shRNA against CDH5.
(E) Immunostaining for Prox1-GFP (green), VE-cadherin (red), and β-catenin (violet) in control and VE-cadherinLEC-KO lymphatic vessels.
Scale bars are 50 μm in (C) and 100 μm in (E).
The total amount of CDH5 protein was reduced by ~95% by the shRNA construct compared to the scrambled construct (Figure 5A). Additionally, treatment of the LECs with BIO increased FOXC2 protein expression in both static (no flow) and oscillatory flow conditions (Figure 5A). At the message level, the major transcription factors controlling valve development, GATA2 and FOXC2, were upregulated significantly in response to OSS in the scramble control transfected LECs, but their expression did not change in response to OSS in the CDH5-knockdown LECs (Figure 5B). BIO treatment of scramble control LECs exposed to OSS increased the expression of GATA2 and FOXC2 further than OSS alone, while BIO treatment of CDH5 knockdown LECs exposed to OSS significantly increased the expression of GATA2 and FOXC2 to near control levels. Similarly, the earliest shear-responsive transcription factor (Dekker et al., 2002; Hamik et al., 2007; Sangwung et al., 2017), KLF4, was upregulated significantly in response to OSS in scramble control LECs, but was downregulated in the CDH5-knockdown LECs and did not change in response to OSS. Another gene known to be upregulated in the lymphatic valves is Connexin 37 (GJA4), whose expression is controlled by PROX1 and FOXC2 (Sabine et al., 2012). Our data show that CX37 was upregulated in response to OSS only in the scramble control LECs, whereas in the CDH5 knockdown LECs CX37 expression did not change in response to OSS, and its overall expression was lower. However, BIO treatment did not significantly increase the expression of either KLF4 or CX37 in scramble control or CDH5 knockdown LECs, but instead reduced KLF4, but not CX37, expression. Since AXIN2 is a known direct target of nuclear β-catenin signaling (Jho et al., 2002), we determined whether it responded to OSS. AXIN2 expression was upregulated in response to OSS in the scramble control LECs, indicating β-catenin nuclear activity in response to OSS, but did not change in response to OSS in the CDH5 knockdown LECs. As expected, the expression of AXIN2 was significantly increased by BIO treatment regardless of transfection with scramble control or CDH5 knockdown shRNA. While PROX1 is normally highly expressed in the valve LECs in vivo, previous reports have established that it is not upregulated in vitro in response to OSS (Cha et al., 2016; Sweet et al., 2015). For a negative control, we found that the expression of both PROX1 and VEGFR3 did not change in response to OSS in either the scramble control or CDH5 knockdown LECs, as expected. Confirming a previous study (Cha et al., 2016), PROX1 expression was increased by BIO treatment in both static and OSS conditions.
Since these data indicated that β-catenin signaling was inhibited in the absence of VE-cadherin, we performed immunostaining (Figure 5C) and western blot (Figure 5D) for β-catenin. Indeed, when VE-cadherin expression was silenced, β-catenin expression was nearly abolished. This finding indicated that β-catenin expression depends on sequestration at the membrane by binding VE-cadherin and that β-catenin nuclear activity is responsible for changes in gene expression in response to OSS. To test this in vivo, we performed immunostaining for β-catenin in control and VE-cadherinLEC-KO collecting lymphatic vessels and valves (Figure 5E). We found that in vivo, loss of VE-cadherin led to a near complete loss of β-catenin.
VE-Cadherin/β-Catenin Signaling In Vivo Links Mechanotransduction to Valve Maintenance
Because our in vitro data showed that stimulation of β-catenin signaling was able to enhance the expression of some valve-specific transcription factors in LECs lacking VE-cadherin, we next attempted to confirm whether constitutively active β-catenin signaling in vivo could rescue the lymphatic valve maintenance defect in our VE-cadherinLEC-KO animals. Conditional deletion of exon 3 from the β-catenin gene (Ctnnb1) prevents the targeting of β-catenin for degradation by the destruction complex (Harada et al., 1999). Therefore, we crossed the previously published Ctnnb1lox(ex3> allele with our control and VE-cadherinLEC-KO mice expressing the Prox1-GFP reporter. Confirming our previous data, loss of VE-cadherin resulted in a significant loss of 90% of lymphatic valves compared to controls at P16 (Figures 6A–6D and 6G) (1,081 ± 42 versus 143 ± 4, n = 3, p < 0.05). However, the simultaneous overactivation of nuclear β-catenin signaling significantly rescued the maintenance defect and increased the number of remaining valves to 43% of control (Figures 6E–6G) (460 ± 33 versus 143 ± 4, n = 3, p < 0.05). To examine whether the valve morphology was also rescued, we performed high-magnification confocal imaging (Figures 6H–6M). The control vessels had mature bileaflet valves that were positive for Integrin-α9 and VE-cadherin (Figures 6H and 6I). As expected, the VE-cadherinLEC-KO lymphatic vessels were negative for VE-cadherin and the valve areas appeared to be regressing with faint Integrin-α9 expression (Figures 6J and 6K). In contrast, the VE-cadherinLEC-KO valves that expressed a constitutive active β-catenin appeared similar to the control valve leaflets and expressed high levels of Integrin-α9 despite the absence of VE-cadherin (Figures 6L and 6M). We performed further characterization of this rescue experiment by examining PROX1, FOXC2, GATA2, and KLF4 expression in vivo (Figure S7). All of these transcription factors had lower expression in regressing valves that was restored by constitutive activation of β-catenin. Thus, these data are consistent with mechanotransduction signaling through VE-cadherin at the cell membrane enabling the nuclear translocation of β-catenin and subsequent upregulation of the flow-sensitive genes involved in valve maintenance.
Figure 6. Constitutively Active β-Catenin Signaling Partially Rescues Valve Regression in the Absence of VE-Cadherin.

(A–F) Direct fluorescence imaging of Prox1-GFP (green) of freshly dissected mesenteries from P16 Cdh5fl/fl, VE-cadherinLEC-KO, and VE-cadherinLEC-KO;Ctnnb1+/lox(ex3) pups at low (A, C, and E) and high (B, D, and F) magnification.
(G) Quantification of the total number of valves in the mesenteries from each genotype. All values are means ± SEM of n = 3 littermates per genotype.
(H–M) Confocal imaging of lymphatic valves stained for Integrin-α9, GFP, and VE-cadherin from control (H and I), VE-cadherinLEC-KO (J and K), or rescue conditions(L and M) to show valve morphology.
One-way ANOVA with Tukey’s post hoc test was performed for multiple comparisons (*p < 0.05). Scale bars are 500 μm in (A), (C), and (E); 200 μm in (B), (D), and(F); and 50 μm in (H)–(M).
Direct Activation of AKT Partially Rescues Postnatal Valve Regression Caused by Loss of VE-Cadherin
We found that cultured LECs exposed to OSS had increased expression of phosphorylated AKT, but this response was inhibited by knockdown of VE-cadherin (Figure 5D). Because shear stress activates VEGFR/PI3K/AKT signaling downstream of VE-cadherin in vascular endothelium, we investigated this pathway further (Coon et al., 2015). Postnatal pups were administered a small molecule that directly activates AKT (SC79) daily from P2-P7 (Jo et al., 2012). To confirm that SC79 activated AKT signaling in LEC, we performed immunostaining for AKT target genes (Figures 7A–7D). Previous studies demonstrated that FOXC2 is a downstream target of VEGFR3/PI3K/AKT signaling (Petrova et al., 2004; Tammela et al., 2011). As expected, vehicle-treated lymphatic vessels expressed high levels of FOXC2 in the valve leaflets, but very low expression in the neighboring lymphangion LECs (Figures 7A and 7B). In contrast, lymphatic vessels from mice treated with SC79 expressed FOXC2 not only in the valves, but also in many LECs outside of the valve areas in a mosaic pattern (Figures 7C and 7D). Therefore, SC79 appeared to be activating the AKT pathway to stimulate ectopic FOXC2 expression in the non-valve LECs. To determine how AKT activation affected lymphatic valve development, VE-cadherin deletion was induced at P1/P3 as before and SC79 or vehicle was administered to postnatal pups (Figures 7E–7M). Control mesenteries treated with SC79 contained ~30% more lymphatic valves than control mesenteries treated with the vehicle alone (Figures 7E–7H and 7M) (790 ± 15 versus 1,010 ± 12, n = 4–5, p < 0.05). Inactivation of VE-cadherin resulted in a 76% loss of lymphatic valves, as before, and treatment with SC79 partially inhibited this lymphatic valve regression (Figures 7I–7L and 7M) (190 ± 21 versus 349 ± 13, n = 3, p < 0.05), restoring the total number of lymphatic valves to 45% of control. High-magnification imaging revealed that SC79 treatment of control Cdh5fl/fl mice did not affect valve morphology (Figures 7F and 7H). However, SC79 treatment of the VE-cadherinLEC-KO lymphatics stimulated the growth of morphologically mature valves (Figures 7J and 7L). These data show that AKT activation stimulates valve formation in healthy lymphatic vessels and that AKT signaling regulates valve maintenance downstream of VE-cadherin.
Figure 7. Direct Activation of Akt Partially Restores Valve Maintenance in the Absence of VE-Cadherin.

(A-D) P8 wild-type lymphatic vessels from mice treated with vehicle (DMSO; A and B) or the small molecule AKT activator SC79 (C and D) and immunostained for FOXC2 (red) and VE-cadherin (green).
(E-L) Direct fluorescence imaging of Prox1-GFP (green) of freshly dissected mesenteries from P8 pups treated as indicated at low(E, G, I, and K) and high (F, H, J, and L) magnification.
(M) Quantification of the total number of valves in the mesenteries from each genotype. All values are means ± SEM of n = 3 experiments per genotype.
One-way ANOVA with Tukey’s post hoc test was performed for multiple comparisons (*p < 0.05). Scale bars are 100 μm for(A)-(D); 500 μm for (E), (G), (I), and (K); and 100 μm for (F), (H), (J), and (L).
DISCUSSION
Oscillatory fluid flow is a major inducer of lymphatic valve development. Several studies have demonstrated that OSS induces the upregulation of lymphatic valve-specific transcription factors in cultured LECs (Cha et al., 2016; Kazenwadel et al., 2015; Sabine et al., 2012; Sweet et al., 2015) and in vivo (Sweet et al., 2015). However, the mechanism by which LECs convert mechanical force exerted at the cell membrane into cell signals to regulate nuclear gene expression has remained elusive. Here, we identify VE-cadherin, a protein located at the cell membrane in adherens junctions, as a regulator of the formation and maintenance of lymphatic valves. We found that VE-cadherin was required for cell alignment to flow, and for the upregulation of lymphatic valve transcription factors in vivo during valve development and in vitro in response to OSS.
Control of Transcription Factors by VE-Cadherin
The present study elucidated how membrane signals are transmitted to the nucleus to regulate gene expression. VE-cadherin forms protein complexes at the cell membrane and two of its binding partners are known to regulate lymphatic gene expression. VE-cadherin binds β-catenin, which was recently shown to be necessary for lymphatic valve formation in the embryo (Cha et al., 2016). Here, we show that VE-cadherin prevents the cytosolic degradation of β-catenin. VE-cadherin also binds VEGFR2 and VEGFR3 through its transmembrane domain to elicit AKT signaling (Coon et al., 2015). Since the ratio of VEGFR3 to VEGFR2 is known to dictate the cell sensitivity to shear stress, it stands to reason that LECs are highly sensitive to low levels of shear due to a high expression level of VEGFR3. Additionally, AKT signaling has been shown to regulate lymphatic valve development (Zhou et al., 2010), potentially by modulating Foxc2 expression (Petrova et al., 2004; Tammela et al., 2011). We show that AKT signaling is impaired in the absence of VE-cadherin. Thus, we propose that these two signaling pathways operate in parallel to regulate lymphatic valve maturation and maintenance. Supporting the involvement of these two signaling pathways, our data demonstrate that constitutive activation of β-catenin rescued the upregulation of GATA2 and FOXC2 in vitro in response to shear and significantly restored the total number of lymphatic valves in vivo to approximately half of controls. We also showed that knockdown of VE-cadherin in vitro or knockout of VE-cadherin in vivo resulted in a dramatic loss of β-catenin protein at the cell membrane. In aggregate, these findings suggest that the junctional pool of β-catenin is responsible for nuclear signaling. Our results agree with a recent study showing that β-catenin directly binds to the promoters of Prox1 and Foxc2, and that β-catenin signaling can regulate GATA2 protein expression (Cha et al., 2016). To directly stimulate AKT signaling, we used a well-characterized pharmacologic activator of AKT and found that this was able to significantly rescue the number of lymphatic valves to half of control levels. Mechanistically, we show that stimulation of the AKT pathway leads to ectopic expression of FOXC2 outside of valve leaflet LECs and this likely leads to new valve growth. Thus, our data show that AKT and β-catenin signaling work together to control valve formation, and both are necessary to prevent valve regression.
Role for VE-Cadherin in Valve Maturation
With respect to the earliest events of valve formation in the embryo, we show that VE-cadherin expression does not regulate the initiation (or specification) step of valve formation or the condensation or elongation steps of valve formation. Instead, it appears that VE-cadherin is particularly required for the valve maturation step that takes place between E17.5 and E18.5 because we did not observe any valve structures at E18.5 in the absence of VE-cadherin. Similarly, we found that the LVV leaflets began to form at E14.5, but were completely absent at E16.5. However, a previous study demonstrated that lymph flow is required for lymphatic valve formation at E16.5 in the mesentery, the earliest time that PROX1hi cell clusters can be observed in this tissue (Sweet et al., 2015). Together with our results, it seems to suggest that separate shear stress signaling pathways are likely responsible for discrete steps of valve formation and that while VE-cadherin regulates some aspects of shear stress signaling, it is likely not responsible for all of mechanotransduction signaling in the lymphatic valves. Consistent with this idea, the mechano-sensitive ion channel PIEZO1 was recently shown to regulate only the condensation and elongation steps of valve formation (Fotiou et al., 2015; Nonomura et al., 2018). Another explanation is that some residual β-catenin protein could remain in the cytoplasm in the VE-cadherin-deficient LEC to respond to Wnt ligands to initiate valve specification (Cha et al., 2018).
VE-Cadherin in Lymphatic Mechanotransduction Signaling
The present study demonstrates that a member of the blood endothelial cell (BEC) mechanotransduction pathway is required for lymphatic valve development. A separate study demonstrated that PECAM1, which acts as a mechanosensor upstream of VE-cadherin in BECs (Tzima et al., 2005), regulates lymphatic valve formation in the embryo along with SDC4 (Wang et al., 2016). While the double-knockout mice exhibited severe lymphatic valve defects and reduced survival, the Pecam1−/− embryos in that study had a relatively subtle defect in valve development and survived well into adulthood, arguing against an essential role in valve maintenance. In contrast, we show that the loss of VE-cadherin leads to defective cell alignment, severe regression of lymphatic valves, loss of PROX1hi and FOXC2hi cell clusters, abnormal branches at the postnatal regressing valves, and LEC apoptosis at the branch points of the embryonic lymphatic plexus. These phenotypes are reminiscent of the postnatal deletion of Foxc2, after which TAZ signaling becomes upregulated, leading to unchecked proliferation and apoptosis (Sabine et al., 2015). Another report showed that lymphatic-specific deletion of VE-cadherin also leads to increased expression of TAZ (Hägerling et al., 2018). In contrast, we found that VE-cadherin deletion led to a near complete loss of TAZ expression in the collecting lymphatic vessels and valve LECs. The reason for the difference in TAZ expression is likely because Hägerling et al. (2018) examined the expression of TAZ in hyperproliferative LEC sheets that grew in adult mesenteries after loss of VE-cadherin, whereas the present study is focused on the postnatal expression of TAZ in the valves and lymphangions of collecting lymphatic vessels that are not hyperproliferative. Our result is consistent with the finding that TAZ expression is decreased in the intestinal lacteal LECs reported by Hägerling et al. (2018). Our data are also consistent with another study that found that genetic deletion of both YAP and TAZ results in a near-complete loss of valves (Cho et al., 2019). Future studies should investigate whether lymphatic valves in this model can be rescued by targeting YAP/TAZ signaling.
Regulation of Lymphatic Integrity
While VE-cadherin regulates vascular mechanotransduction, it is also a well-established regulator of endothelial permeability (Dejana and Vestweber, 2013; Giannotta et al., 2013) and its differential expression in the lymphatic vasculature helps establish discontinuous ‘‘button’’ junctions in the lymphatic capillaries and continuous ‘‘zipper’’ junctions in the larger lymphatic collecting vessels (Baluk et al., 2007). As expected, we show that the early postnatal deletion of VE-cadherin results in chylous ascites and severe lymph leakage. Since it is possible that increased lymphatic vessel permeability or vessel disintegration may lead to a reduction in lymph flow, the loss of valves may be interpreted as an indirect effect due to vessel disintegration and not due to mechanotransduction signaling events. However, our data strongly argue against these possibilities. First, we did not observe chylous ascites or leaky, hazy lymphatic vessels at P8, but they were consistently present at P14. Additionally, we did not observe lymphatic vessel disintegration at P8, when valve number was already reduced by 75%, or at E18.5, when valves are completely absent. Thus, the loss of lymphatic valves in our mouse model precedes lymphatic vessel leakage. Second, we demonstrated that VE-cadherin-deficient mice had lymph flow at the same time point as littermate controls using a fluorescent lipid at both P8 and P14. Finally, loss of lymph flow in the mesenteric lymphatic vessels causes the lymphatics to no longer appear visibly white (Zhang et al., 2018). We found that VE-cadherin-deficient lymphatic vessels were consistently filled with white chyle at P8 and P14.
Conclusion
In conclusion, our data explain how fluid forces at the LEC membrane regulate nuclear transcription factors to control valve formation and maintenance. We found that augmenting AKT signaling is capable of stimulating valve formation in otherwise wild-type mice. Future studies are needed to investigate the AKT signaling pathway to identify therapeutic targets to safely enhance valve formation in lymphedema patients.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Request for reagents must be directed to the Lead Contact, Joshua Scallan (jscallan@health.usf.edu). Upon completion of a Material Transfer Agreement (MTA), we will share any reagents.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
Generation of the Cdh5flox allele is described in Figure S1. The Tie2Cre, LyvelCre, Prox1CreERT2, Prox1-GFP, β-cateninex3(loxP> strains were described previously (Kisanuki et al., 2001; Pham et al., 2010; Bazigou et al., 2011; Choi et al., 2011; Harada et al., 1999). Prox1CreERT2;Cdh5fl/fl;Prox1-GFP and Pmx1CreERT2;Cdh5ml;β-catenin+/ex3(loxP);Prox1-GFP mice were generated by crossbreeding. All mice were maintained on a mixed genetic background (NMRI x C57BL/6J x SV129) and both sexes were used. Embryonic deletion of VE-cadherin was induced by intraperitoneal injections of 5 mg tamoxifen each into pregnant dams at the indicated developmental stages. Pups were injected twice with 100 μg tamoxifen in 5 μL sunflower oil subcutaneously on postnatal days P1 and P3. To directly activate AKT, the small molecule SC79 (Jo et al., 2012)(Millipore Sigma) or vehicle (DMSO) was injected into pups (40 mg/kg, i.p.) once daily from P2 to P7 and pups were sacrificed on P8. All experiments were performed in accordance with the University of South Florida guidelines and were approved by the institutional IACUC committee.
Cell Lines
Primary human dermal lymphatic endothelial cells (hdLEC, Lonza and PromoCell) were cultured on fibronectin-coated six-well plates and were maintained in EBM-2 media (Lonza and PromoCell). All experiments were performed using cells at passage 6-7 and verified to express PROX1, a marker of lymphatic identity.
METHOD DETAILS
Whole Mount Immunostaining Procedure
All procedures were carried out at 4°C on an orbital shaker (Belly Dancer, IBI Scientific) unless otherwise specified. Tissues were harvested and fixed overnight in 1% paraformaldehyde. The next day, tissues were washed with PBS three times 10 minutes each, permeabilized with PBS + 0.3% Triton X-100 (PBST) for one hour, and blocked with 3% donkey serum in PBST for two hours. Primary antibodies were mixed in PBST, added to the tissue, and incubated overnight. To remove primary antibodies, tissues were washed five times 15 minutes each with PBST. Secondary and conjugated antibodies were mixed in PBST, added to the tissues, and incubated at room temperature for two hours on an orbital shaker (Belly Button, IBI Scientific). Tissues were then washed five times 15 minutes each with PBST. To provide nuclear contrast, DAPI was dissolved in PBS, added to the tissues, and incubated at room temperature for 5 minutes, followed by a wash in PBS for 5 minutes. Tissues were then mounted on glass slides (Superfrost Plus Microscope slides, Fisherbrand) with ProLong Diamond Antifade Mountant containing DAPI (Invitrogen) and stored at 4°C overnight before imaging on an inverted fluorescence microscope (Axio Observer Z1, Zeiss) or a Leica SP8 confocal microscope. Images were acquired with Zen 2 Pro software and figures were created using Adobe Photoshop. Individual primary and secondary antibodies are listed in the Key Resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Prox1 | abcam | Cat# ab101851; RRID: AB_10712211 |
| Goat polyclonal anti-Prox1 | R&D | Cat# AF2727; RRID: AB_2170716 |
| Rabbit polyclonal anti-Prox1 | AngioBio | Cat# 11-002; RRID: AB_10013720 |
| Rat monoclonal anti-VE-cadherin | BD | Cat# 550548; RRID: AB_2244723 |
| Rabbit polyclonal anti-VE-cadherin | ThermoFisher | Cat# 36-1900; RRID: AB_2533243 |
| Rabbit polyclonal anti-VE-cadherin | Enzo | Cat# ENZ-ABS661-0100 |
| Sheep polyclonal anti-Foxc2 | R&D | Cat# AF6989; RRID: AB_10973139 |
| Mouse monoclonal anti-Foxc2 | Santa Cruz | Cat# SC-515234; |
| Goat polyclonal anti-Vegfr3 | R&D | Cat# AF743; RRID: AB_355563 |
| Rat monoclonal anti-Pecam1 | BD | Cat# 550274; RRID: AB_393571 |
| Rabbit polyclonal anti-GFP | Life Technologies | Cat# A11122; RRID: AB_221569 |
| Rabbit polyclonal anti-Casp3 | Cell Signaling | Cat# 9661; RRID: AB_2341188 |
| Rabbit polyclonal anti-Taz | Sigma-Aldrich | Cat# HPA007415; RRID: AB_1080602 |
| Rabbit polyclonal anti-Phospho-Akt (Ser473) | Cell Signaling | Cat# 9271; RRID: AB_329825 |
| Rabbit monoclonal anti-Akt (pan) (C67E7) | Cell Signaling | Cat# 4691; RRID: AB_915783 |
| Mouse monoclonal anti-β-Catenin (L54E2) | Cell Signaling | Cat# 2677; RRID: AB_1030943 |
| Rabbit polyclonal anti-β-Catenin | Millipore | Cat# 06-734; RRID: AB_310231 |
| Goat polyclonal anti-Klf4 | R&D | Cat# AF3158; RRID: AB_2130245 |
| Goat polyclonal anti-Integrin α9 | R&D | Cat# AF3827; RRID: AB_2128452 |
| Rabbit polyclonal anti-Gata2 | Novus Biologicals | Cat# NBP1-82581; RRID: AB_11026191 |
| Rabbit polyclonal anti-Claudin 5 | ThermoFisher | Cat# 34-1600; RRID: AB_2533157 |
| Rabbit polyclonal anti-ZO-1 | ThermoFisher | Cat# 61-7300; RRID: AB_2533938 |
| Mouse monoclonal anti-ACTB | Sigma-Aldrich | Cat# A5441; RRID: AB_47674 |
| Alexa Fluor 488 conjugated GFP | Life Technologies | Cat# A21311; RRID: AB_221477 |
| Alexa Fluor 488 Donkey Anti-Rabbit IgG (H+L) | Life Technologies | Cat# A21206; RRID: AB_141708 |
| Alexa Fluor 488 Donkey Anti-Goat IgG (H+L) | Life Technologies | Cat# A11055; RRID: AB_2534102 |
| Alexa Fluor 488 Donkey Anti-Sheep IgG (H+L) | Life Technologies | Cat# A11015; RRID: AB_141362 |
| Alexa Fluor 488 Donkey Anti-Rat IgG (H+L) | Life Technologies | Cat# A21208; RRID: AB_141709 |
| Alexa Fluor 594 Donkey Anti-Rabbit IgG (H+L) | Life Technologies | Cat# A21207 RRID: AB_141637 |
| Alexa Fluor 594 Donkey Anti-Goat IgG (H+L) | Life Technologies | Cat# A11058; RRID: AB_2534105 |
| Alexa Fluor 594 Donkey Anti-Sheep IgG (H+L) | Life Technologies | Cat# A11016; RRID: AB_2534083 |
| Alexa Fluor 594 Donkey Anti-Rat IgG (H+L) | Life Technologies | Cat# A21209; RRID: AB_2535795 |
| Alexa Fluor 647 Donkey Anti-Rabbit IgG (H+L) | Life Technologies | Cat# A-31573; RRID: AB_2536183 |
| Alexa Fluor 647 Donkey Anti-Goat IgG (H+L) | Life Technologies | Cat# A-21447; RRID: AB_2535864 |
| Alexa Fluor 647 Donkey Anti-Rat IgG (H+L) | Jackson ImmunoResearch | Cat# 712-605-153; RRID: AB_2340694 |
| Bacterial and Virus Strains | ||
| pLV[shRNA]-EGFP-control | This paper | N/A |
| pLV[shRNA]-EGFP-CDH5-shRNA#1 | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BIO | Sigma-Aldrich | Cat# B1686 |
| Human Fibronectin | Corning | Cat# 354008 |
| SC-79 | Millipore | Cat# 123871; CAS 305834-79-1 |
| BODIPY FL C16 | Invitrogen | Cat# D3821 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| Critical Commercial Assays | ||
| Trizol | Thermo Fisher Scientific | Cat# 15596026 |
| PowerUp SYBR Green Master Mix | Applied Biosystems | Cat# A25742 |
| iScript Advanced cDNA Synthesis Kit | Bio-Rad | Cat# 172-5038 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23227 |
| EGM-2 EC Growth Medium-2 Bullet Kit | Lonza | Cat# CC-4176 |
| Endothelial Cell Growth Medium MV 2 | PromoCell | Cat# C-22121 |
| SuperSignal West Femto Maximum Sensitivity Substrate Kit | Thermo Fisher Scientific | Cat# 34095 |
| Experimental Models: Cell Lines | ||
| Human neonatal dermal lymphatic endothelial cells (HLECs): HMVEC-dLyNeo-Der | Lonza | Cat# CC-2812 |
| Human dermal lymphatic endothelial cells (HDLEC), juvenile foreskin | PromoCell | Cat# C-12216 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Tg(Tek-Cre)1Ywa | Kisanuki et al., 2001 | N/A |
| Mouse: Lyve1tm1.1(EGFP/Cre)Cys | Pham et al., 2010 | N/A |
| Mouse: Cdh5flox | This paper | N/A |
| Mouse: Tg(Prox1-cre/ERT2)#aTmak | Bazigou et al., 2011 | N/A |
| Mouse: Tg(Prox1-EGFP)221Gsat/Mmcd | Choi et al., 2011 | N/A |
| Mouse: Ctnnb1tm1Mmt | Harada et al., 1999 | N/A |
| Oligonucleotides | ||
| Please see Table S1 | N/A | N/A |
| Recombinant DNA | ||
| pLV[shRNA]-EGFP-control | This paper | N/A |
| pLV[shRNA]-EGFP-CDH5-shRNA#1 | This paper | N/A |
| Software and Algorithms | ||
| Adobe Photoshop | Adobe system | https://www.adobe.com/products/photoshop.html |
| ImageJ | N/A | https://imagej.nih.gov/ij/ |
| GraphPad Prism 7 | GraphPad Software Inc | https://www.graphpad.com/scientific-software/prism/ |
| Zen 2 Pro | ZEISS | https://www.zeiss.com/microscopy/us/microscope-cameras.html |
Lymphangiography
Postnatal pups (at P8 or P14) were fed 1 μL of 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-hexadecanoic acid (BODIPY FL C16, ThermoFisher Scientific) that was dissolved in pure olive oil at a concentration of 10 μg/μL. Three hours after feeding, pups were euthanized and the mesenteric lymphatic vasculature was imaged immediately with a fluorescence stereo zoom microscope (Zeiss Axio Zoom V16).
Lymphatic Valve Quantification
Postnatal pups expressing the Prox1-GFP reporter strain were euthanized on postnatal day P8 or P14, and a loop of intestine was exteriorized through a midline incision and pinned onto a cushion made of Sylgard 184. Lymphatic valves, visualized as GFPhi spots, were counted individually in situ in each section of the mesentery with the aid of a hand tally counter beginning at the duodenum and terminating at the cecum. The number of valves for each section was then totaled. Control and knockout littermates were analyzed in pairs or triplets from at least 3 independent litters.
Cell Culture and Oscillatory Shear
Cells were infected with lentiviral particles expressing an shRNA targeting CDH5 ora control scramble construct expressing GFP for 48 hours. Subsequently, hdLEC were cultured in the presence of oscillatory shear stress (OSS) for 48 hours in the presence of DMSO or BIO (0.5 mM). RNA was then extracted and analyzed by quantitative real-time PCR (qRT-PCR) for selected genes, with RNA normalized to GAPDH. The OSS experiments were performed according to our previously published protocol (Cha et al., 2016). Specifically, hdLEC were cultured to 95% confluency in six-well plates and exposed to OSS using a test tube rocker (Thermolyne Speci-Mix aliquot mixer, model M71015, Barnstead Internat ional) with a preset frequency of 18 rpm. Cells in each well were covered with 1 μL of fluid. Based on previously described calculations, the cells were exposed to a total shear stress of 0.3 dynes/cm2. The entire experiment was performed inside a sterile humidified incubator containing 5% CO2.
RNA Isolation, qRT-PCR, and Western Blot Analyses
For quantitative real-time PCR, total RNA was isolated from human dermal lymphatic endothelial cells (hdLECs; Lonza and Promo-Cell) using TRIZOL reagent (Invitrogen) according to the manufacturer’s protocol. cDNA was synthesized from total RNA (0.1 −1.0 μg) with Superscript III First-Strand Synthesis System (Invitrogen). qRT-PCR was performed using SYBR Green PCR Master Mix reagent (Bio-Rad) in a CFX96 Real-Time System (Bio-Rad). PCR conditions were 95°C (5 min) and 40 cycles at 95°C (30 s) and 60°C (1 min). The threshold cycle (Ct) value for each gene was normalized to the Ct value for GAPDH. The primer sequences are listed in Table S1.
For western blot, hdLECs were lysed in lysis buffer [20mM Tris-HCl (pH 7.5), 150mM NaCl, 1.0% Triton X-100, 20mM NaF, 2mM EDTA, 2mM Na-orthovanadate, 1mM phenylmethylsulfonyl fluoride (PMSF), 5 mg/ml leupeptin A]. Lysates were centrifuged for 15 min at 15,000 g in a cold room and the supernatant was collected and used for immunoblotting. Equal amounts of protein were resolved on 10% SDS-PAGE and transferred to a PVDF membrane. After blocking with 5% nonfat skim milk in TBS with 0.1% Tween-20 for 1 hour, the membranes were incubated with the indicated antibodies overnight at 4°C. Bound antibodies were visualized by ECL (Thermo Fisher Scientific) using HRP-conjugated antibodies. The antibodies used for western blot are listed in the Key Resources Table.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are represented as means ± SEM. For datasets containing exactly two groups, an unpaired two-sided Student’s t test was used to determine significant differences (p < 0.05). For datasets with more than two groups, one-way ANOVA with Tukey’s posthoc analysis was performed to test for significant differences, where p < 0.05 was considered significant. Graphpad Prism software (version 6) was used for all statistical analyses and to plot quantitative data.
DATA AND CODE AVAILABILITY
This study did not generate/analyze datasets or code.
Supplementary Material
Highlights.
VE-cadherin regulates mechanotransduction signaling in the lymphatic vasculature
Loss of VE-cadherin impairs lymphatic valve formation and maintenance
VE-cadherin stabilizes β-catenin to enable regulation of PROX1 and FOXC2
AKT signaling regulates FOXC2 and is impaired in the absence of VE-cadherin
ACKNOWLEDGMENTS
We thank Drs. Taija Makinen, Young-Kwon Hong, and Makoto Taketo for kindly providing the Prox1CreERT2, Prox1-GFP, and Ctnnb1lox(ex3) strains, respectively. We thank Dr. Dietmar Vestweber for helpful discussion of our findings. We thank Dr. Jerome Breslin for the use of his cell culture facility and reagents. Funding for this work was supported by NIH NHLBI grants R01HL145397 (to Y.Y.), R00HL124142 and R01HL142905 (to J.P.S.), and R01HL131652 and R01HL133216 (to R.S.S.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.072.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Baeyens N, and Schwartz MA (2016). Biomechanics of vascular mechano-sensation and remodeling. Mol. Biol. Cell 27, 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, Vestweber D, Corada M, Molendini C, Dejana E, and McDonald DM (2007). Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 204, 2349–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazigou E, Xie S, Chen C, Weston A, Miura N, Sorokin L, Adams R, Muro AF, Sheppard D, and Makinen T (2009). Integrin-alpha9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Dev. Cell 17, 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazigou E, Lyons OT, Smith A, Venn GE, Cope C, Brown NA, and Makinen T (2011). Genes regulating lymphangiogenesis control venous valve formation and maintenance in mice. J. Clin. Invest. 121, 2984–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillard P, Boon L, and Vikkula M (2014). Genetics of lymphatic anomalies. J. Clin. Invest. 124, 898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, Balconi G, Spagnuolo R, Oosthuyse B, Dewerchin M, et al. (1999). Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 08, 147–157. [DOI] [PubMed] [Google Scholar]
- Cha B, Geng X, Mahamud MR, Fu J, Mukherjee A, Kim Y, Jho EH, Kim TH, Kahn ML, Xia L, et al. (2016). Mechanotransduction activates canonical Wnt/β-catenin signaling to promote lymphatic vascular patterning and the development of lymphatic and lymphovenous valves. Genes Dev. 80, 1454–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha B, Geng X, Mahamud MR, Zhang JY, Chen L, Kim W, Jho EH, Kim Y, Choi D, Dixon JB, et al. (2018). Complementary Wnt Sources Regulate Lymphatic Vascular Development via PROX1-Dependent Wnt/b-Catenin Signaling. Cell Rep. 25, 571–584. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Kim J, Ahn JH, Hong YK, Makinen T, Lim DS, and Koh GY (2019). YAP and TAZ Negatively Regulate Prox1 During Developmental and Pathologic Lymphangiogenesis. Circ. Res. 124, 225–242. [DOI] [PubMed] [Google Scholar]
- Choi I, Chung HK, Ramu S, Lee HN, Kim KE, Lee S, Yoo J, Choi D, Lee YS, Aguilar B, and Hong YK (2011). Visualization of lymphatic vessels by Prox1-promoterdirected GFP reporter in a bacterial artificial chromosome-based transgenic mouse. Blood 117, 362–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon BG, Baeyens N, Han J, Budatha M, Ross TD, Fang JS, Yun S, Thomas JL, and Schwartz MA (2015). Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechano-sensory complex. J. Cell Biol. 208, 975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E, and Vestweber D (2013). The role of VE-cadherin in vascular morphogenesis and permeability control. Prog. Mol. Biol. Transl. Sci. 116, 119–144. [DOI] [PubMed] [Google Scholar]
- Dejana E, Orsenigo F, and Lampugnani MG (2008). The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 121,2115–2122. [DOI] [PubMed] [Google Scholar]
- Dekker RJ, van Soest S, Fontijn RD, Salamanca S, de Groot PG, Van-Bavel E, Pannekoek H, and Horrevoets AJ (2002). Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kr€ppel-like factor (KLF2). Blood 100, 1689–1698. [DOI] [PubMed] [Google Scholar]
- Fotiou E, Martin-Almedina S, Simpson MA, Lin S, Gordon K, Brice G, Atton G, Jeffery I, Rees DC, Mignot C, et al. (2015). Novel mutations in PIEZO1 cause an autosomal recessive generalized lymphatic dysplasia with non-immune hydrops fetalis. Nat. Commun. 6, 8085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, Dierkes M, Küppers V, Vockel M, Tomm J, Zeuschner D, Rossaint J, Zarbock A, Koh GY, Peters K, et al. (2015). Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J. Exp. Med. 212, 2267–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng X, Cha B, Mahamud MR, Lim KC, Silasi-Mansat R, Uddin MKM, Miura N, Xia L, Simon AM, Engel JD, et al. (2016). Multiple mouse models of primary lymphedema exhibit distinct defects in lymphove-nous valve development. Dev. Biol. 409, 218–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng X, Cha B, Mahamud MR, and Srinivasan RS (2017). Intraluminal valves: development, function and disease. Dis. Model. Mech. 10,1273–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannotta M, Trani M, and Dejana E (2013). VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev. Cell 26, 441–454. [DOI] [PubMed] [Google Scholar]
- Gordon EJ, Gale NW, and Harvey NL (2008). Expression of the hyaluronan receptor LYVE-1 is not restricted to the lymphatic vasculature; LYVE-1 is also expressed on embryonic blood vessels. Dev. Dyn. 287, 1901–1909. [DOI] [PubMed] [Google Scholar]
- Gory-Fauré S, Prandini MH, Pointu H, Roullot V, Pignot-Paintrand I, Vernet M, and Huber P (1999). Role of vascular endothelial-cadherin in vascular morphogenesis. Development 126, 2093–2102. [DOI] [PubMed] [Google Scholar]
- Hägerling R, Hoppe E, Dierkes C, Stehling M, Makinen T, Butz S, Vestweber D, and Kiefer F (2018). Distinct roles of VE-cadherin for development and maintenance of specific lymph vessel beds. EMBO J. 87, e98271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamik A, Lin Z, Kumar A, Balcells M, Sinha S, Katz J, Feinberg MW, Gerzsten RE, Edelman ER, and Jain MK (2007). Kruppel-like factor 4 regulates endothelial inflammation. J. Biol. Chem. 282, 13769–13779. [DOI] [PubMed] [Google Scholar]
- Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, and Taketo MM (1999). Intestinal polyposisin mice with adominant stable mutation of the beta-catenin gene. EMBO J. 18, 5931–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, and Costantini F (2002). Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol. 22, 1172–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo H, Mondal S, Tan D, Nagata E, Takizawa S, Sharma AK, Hou Q, Shanmugasundaram K, Prasad A, Tung JK, et al. (2012). Small molecule-induced cytosolic activation of protein kinase Akt rescues ischemia-elicited neuronal death. Proc. Natl. Acad. Sci. USA 109, 10581–10586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazenwadel J, Betterman KL, Chong CE, Stokes PH, Lee YK, Secker GA, Agalarov Y, Demir CS, Lawrence DM, Sutton DL, et al. (2015). GATA2 is required for lymphatic vessel valve development and maintenance. J. Clin. Invest. 125, 2979–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, and Yanagisawa M (2001). Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 280, 230–242. [DOI] [PubMed] [Google Scholar]
- Nonomura K, Lukacs V, Sweet DT, Goddard LM, Kanie A, Whitwam T, Ranade SS, Fujimori T, Kahn ML, and Patapoutian A (2018). Mechanically activated ion channel PIEZO1 is required for lymphatic valve formation. Proc. Natl. Acad. Sci. USA 115, 12817–12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrmén C, Ivanov KI, Cheng J, Zangger N, Delorenzi M, Jaquet M, Miura N, Puolakkainen P, Horsley V, Hu J, et al. (2009). FOXC2 controls formation and maturation of lymphatic collecting vessels through cooperation with NFATc1. J. Cell Biol. 185, 439–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrek JA, Senie RT, Peters M, and Rosen PP (2001). Lymphedema in a cohort of breast carcinoma survivors 20 years after diagnosis. Cancer 92, 1368–1377. [DOI] [PubMed] [Google Scholar]
- Petrova TV, Karpanen T, Norrmen C, Mellor R, Tamakoshi T, Finegold D, Ferrell R, Kerjaschki D, Mortimer P, Yla-Herttuala S, et al. (2004). Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat. Med. 10, 974–981. [DOI] [PubMed] [Google Scholar]
- Pham TH, Baluk P, Xu Y, Grigorova I, Bankovich AJ, Pappu R, Coughlin SR, McDonald DM, Schwab SR, and Cyster JG (2010). Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J. Exp. Med. 207, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen JC, Tan IC, Marshall MV, Fife CE, and Sevick-Muraca D M. (2009). Lymphatic imaging in humans with near-infrared fluorescence. Curr. Opin. Biotechnol. 20, 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabine A, Agalarov Y, Maby-El Hajjami H, Jaquet M, Hägerling R, Pollmann C, Bebber D, Pfenniger A, Miura N, Dormond O, et al. (2012). Mechanotransduction, PROX1, and FOXC2 cooperate to control connexin37 and calcineurin during lymphatic-valve formation. Dev. Cell 22, 430–445. [DOI] [PubMed] [Google Scholar]
- Sabine A, Bovay E, Demir CS, Kimura W, Jaquet M, Agalarov Y, Zangger N, Scallan JP, Graber W, Gulpinar E, et al. (2015). FOXC2 and fluid shear stress stabilize postnatal lymphatic vasculature. J. Clin. Invest. 125, 3861–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangwung P, Zhou G, Nayak L, Chan ER, Kumar S, Kang DW, Zhang R, Liao X, Lu Y, Sugi K, et al. (2017). KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight 2, e91700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan RS, and Oliver G (2011). Prox1 dosage controls the number of lymphatic endothelial cell progenitors and the formation of the lymphovenous valves. Genes Dev. 25, 2187–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan RS, Dillard ME, Lagutin OV, Lin FJ, Tsai S, Tsai MJ, Samokhvalov IM, and Oliver G (2007). Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes Dev. 21, 2422–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanczuk L, Martinez-Corral I, Ulvmar MH, Zhang Y, Lavina B, Fruttiger M, Adams RH, Saur D, Betsholtz C, Ortega S, et al. (2015). cKit Lineage Hemogenic Endothelium-Derived Cells Contribute to Mesenteric Lymphatic Vessels. Cell Rep. 10, 1708–1721. [DOI] [PubMed] [Google Scholar]
- Stanton AW, Modi S, Mellor RH, Peters AM, Svensson WE, Levick JR, and Mortimer PS (2006). A quantitative lymphoscintigraphic evaluation of lymphatic function in the swollen hands of women with lymphoedema following breast cancer treatment. Clin. Sci. (Lond.) 110, 553–561. [DOI] [PubMed] [Google Scholar]
- Sweet DT, Jimenez JM, Chang J, Hess PR, Mericko-Ishizuka P, Fu J, Xia L, Davies PF, and Kahn ML (2015). Lymph flow regulates collecting lymphatic vessel maturation in vivo. J. Clin. Invest. 125, 2995–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, Pirazzoli V, Potente M, Daly C, Dimmeler S, and Dejana E (2008). Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 10, 923–934. [DOI] [PubMed] [Google Scholar]
- Tammela T, Zarkada G, Nurmi H, Jakobsson L, Heinolainen K, Tvorogov D, Zheng W, Franco CA, Murtomaki A, Aranda E, et al. (2011). VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat. Cell Biol. 13, 1202–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatin F, Taddei A, Weston A, Fuchs E, Devenport D, Tissir F, and Makinen T (2013). Planar cell polarity protein Celsr1 regulates endothelial adherens junctions and directed cell rearrangements during valve morphogenesis. Dev. Cell 26, 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, and Schwartz MA (2005). A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437, 426–431. [DOI] [PubMed] [Google Scholar]
- Wang Y, Baeyens N, Corti F, Tanaka K, Fang JS, Zhang J, Jin Y, Coon B, Hirschi KK, Schwartz MA, and Simons M (2016). Syndecan 4 controls lymphatic vasculature remodeling during mouse embryonic development. Development 143, 4441–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Garcia-Verdugo JM, Soriano-Navarro M, Srinivasan RS, Scallan JP, Singh MK, Epstein JA, and Oliver G (2012). Lymphatic endothelial progenitors bud from the cardinal vein and intersomitic vessels in mammalian embryos. Blood 120, 2340–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Zarkada G, Han J, Li J, Dubrac A, Ola R, Genet G, Boye K, Michon P, Kiinzel SE, et al. (2018). Lacteal junction zippering protects against diet-induced obesity. Science 361, 599–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Chang Z, Zhang L, Hong YK, Shen B, Wang B, Zhang F, Lu G, Tvorogov D, Alitalo K, et al. (2010). Akt/Protein kinase B is required for lymphatic network formation, remodeling, and valve development. Am. J. Pathol. 177, 2124–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets or code.