

Abstract
Hypersecretion and alterations in the biological activity of the incretin hormone, glucose-dependent insulinotropic polypeptide (GIP), have been postulated as contributing factors in the development of obesity-related diabetes. However, recent studies also point to weight-reducing effects of GIP receptor activation. Therefore, generating precise experimental tools, such as specific and effective GIP receptor (GIPR) antagonists, is of key significance to better understand GIP physiology. Thus, the primary aim of the current study was to uncover improved GIPR antagonists for use in rodent studies, using human and mouse GIP sequences with N- and C-terminal deletions. Initial in vitro studies revealed that the GIPR agonists, human (h) GIP(1-42), hGIP(1-30) and mouse (m) GIP(1-30), stimulated (P < 0.01 to P < 0.001) insulin secretion from rat BRIN-BD11 cells. Analysis of insulin secretory effects of the N- and C-terminally cleaved GIP peptides, including hGIP(3-30), mGIP(3-30), h(Pro3)GIP(3-30), hGIP(5-30), hGIP(3-42) and hGIP(5-42), revealed that these peptides did not modulate insulin secretion. More pertinently, only hGIP(3-30), mGIP(3-30) and h(Pro3)GIP(3-30) were able to significantly (P < 0.01 to P < 0.001) inhibit hGIP(1-42)-stimulated insulin secretion. The human-derived GIPR agonist sequences, hGIP(1-42) and hGIP(1-30), reduced (P < 0.05) glucose levels in mice following conjoint injection with glucose, but mGIP(1-30) was ineffective. None of the N- and C-terminally cleaved GIP peptides affected glucose homeostasis when injected alone with glucose. However, hGIP(5-30) and mGIP(3-30) significantly (P < 0.05 to P < 0.01) impaired the glucose-lowering action of hGIP(1-42). Further evaluation of these most effective sequences demonstrated that mGIP(3-30), but not hGIP(5-30), effectively prevented GIP-induced elevations of plasma insulin concentrations. These data highlight, for the first time, that mGIP(3-30) represents an effective molecule to inhibit GIPR activity in mice.
Keywords: Glucose-dependent insulinotropic polypeptide (GIP), insulin secretion, glucose homeostasis, species specificity
Introduction
The gut hormone glucose-dependent insulinotropic polypeptide (GIP), secreted postprandially from enteroendocrine K cells, has been linked to the aetiology of obesity-related type 2 diabetes mellitus (T2DM) through its actions as a key regulator of insulin secretion and lipid metabolism.1,2 Remarkably, in rodents both activation3,4 and inhibition5,6 of GIP receptors (GIPRs) have been associated with benefits in obesity. In addition, prolonged administration of a GIPR neutralizing antibody was shown to enhance the weight loss induced by glucagon-like peptide-1 (GLP-1), the sister incretin of GIP, in mice.7 In some contrast, a stable GIPR agonist was recently demonstrated to augment GLP-1-induced body weight reduction.8 Further to this, competitive inhibition of the GIPR by (Pro3)GIP had no additional impact on body weight when administered together with the GLP-1 receptor (GLP-1R) agonist, exendin-4, in mice.9 Taken together, it is clear from these rodent-based studies that there are still many unanswered questions as to the exact role of GIPR signalling in the pathophysiology of obesity.
Studies in humans have attempted to answer some of these uncertainties, but the picture is far from clear. Excess consumption of fat is known to be a potent stimulus of GIP secretion.10 As such, in obesity circulating GIP levels are elevated,11 which is believed to be partially linked to excessive deposition of visceral and subcutaneous fat. Thus, increased secretion and action of GIP can predispose individuals to obesity.12 In keeping with this, bariatric surgery, the only proven method to result in sustained weight loss in humans, points to decreased GIP secretion as a primary beneficial metabolic effect of the surgical process.13-15 On the contrary, GIPR activation was recently evidenced to enhance the weight-reducing and appetite suppressive effects of GLP-1 in T2DM patients.16,17 Yet in some contrast, GLP-1 infusion in obese men decreased energy intake, with simultaneous GIP infusion having no impact on this.18 This uncertainty in the physiological role, and potential therapeutic application, of GIP necessitates generation of useful tools to provide unambiguous answers, such as specific and potent GIPR antagonists.
Fortunately, there have already been some advancements in this area. Several peptide-derived prospective GIPR antagonists have been produced, using N-terminally truncated GIP peptides19-21 as well as amino acid substituted analogues.22-25 Originally, human (h) h(Pro3)GIP was indicated as the lead candidate GIPR antagonist.26 Indeed, numerous studies have described beneficial weight-reducing effects of sustained h(Pro3)GIP administration in obese-diabetic rodents, linked to impairment of GIPR signalling,27,28 with no weight-reducing effects in normal mice.29 However, more recent research has revealed h(Pro3)GIP to be a partial GIPR agonist,20 with species-specific activity also noted in different mammalian systems.30 In addition, (Pro3)GIP was also revealed to exert partial GIPR agonist activity in human embryonic kidney cells,31 suggesting possible additional tissue-specific actions. Nonetheless, further work has identified hGIP(3-30) as a credible GIPR antagonist in humans,32,33 but not rodents.34 Given that much knowledge on the physiology of GIP stems from work in rodents, such as rats and especially mice, it is vitally important that a potent and specific GIPR antagonist be developed for these species studies. Therefore, the present study aims to assess the GIPR antagonistic characteristics of various N- and C-terminally truncated GIP peptides, of both human (h) and mouse (m) lineage, in a rodent pancreatic beta cell line and mice. We focused predominantly on human sequences for possible translation of observations to the human setting. However, the current results suggest that mouse-derived GIP sequences are likely to provide more effective GIPR antagonists for rodent studies than human sequence counterparts.
Materials and Methods
Peptides
All peptides (Table 1) were purchased from Syn Peptide (Shanghai, China) at greater than 95% purity. In-house confirmation of peptide purity and molecular weight was carried out by Reversed-phase high-performance liquid chromatography (RP-HPLC) and matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI-ToF MS), as previously described.35
Table 1.
In-house characterisation of GIP peptides by HPLC and MALDI-ToF MS.
| Peptides | Name | Sequence | % HPLC purity | Theoretical Mass (Da) |
Experimental Mass (Da) |
|---|---|---|---|---|---|
| 1 | hGIP(1-42) | YAEGTFISDYSIAMDKIHQQDFVNWLLAQKGKKNDWKHNITQ | 98.2 | 4983.6 | 4982.8 |
| 2 | hGIP(1-30) | YAEGTFISDYSIAMDKIHQQDFVNWLLAQK-NH2 | 98.7 | 3531.9 | 3530.6 |
| 3 | mGIP(1-30) | YAEGTFISDYSIAMDKIRQQDFVNWLLAQR-NH2 | 97.5 | 3579.0 | 3578.4 |
| 4 | hGIP(3-30) | EGTFISDYSIAMDKIHQQDFVNWLLAQK-NH2 | 96.7 | 3297.7 | 3296.3 |
| 5 | mGIP(3-30) | EGTFISDYSIAMDKIRQQDFVNWLLAQR-NH2 | 96.9 | 3344.7 | 3342.5 |
| 6 | h(Pro3)GIP(3-30) | PGTFISDYSIAMDKIHQQDFVNWLLAQK-NH2 | 98.5 | 3265.7 | 3263.4 |
| 7 | hGIP(3-42) | EGTFISDYSIAMDKIHQQDFVNWLLAQKGKKNDWKHNITQ | 98.3 | 4749.3 | 4749.2 |
| 8 | hGIP(5-30) | TFISDYSIAMDKIHQQDFVNWLLAQK-NH2 | 99.0 | 3111.5 | 3110.2 |
| 9 | hGIP(5-42) | TFISDYSIAMDKIHQQDFVNWLLAQKGKKNDWKHNITQ | 97.4 | 4563.1 | 4562.3 |
Amino acid sequence of peptides using one letter amino acid nomenclature. Differences in the mouse sequence compared with the human GIP sequence are highlighted by underlined bold text. HPLC was performed on a Phenomenex Aeris Peptide 3.6 µ XB-C18 250*15 mm HPLC column equilibrated with 0.12% trifluoroacetic acid (TFA)/H2O at a rate of 6 mL/min using 0.1% TFA in 70% acetonitrile/H2O on a Surveyor Plus Liquid Chromatography/HPLC system (Thermo Finnigan, California, USA). Absorbance was measured at 214 nm and purity determined from peak areas. Peptide molecular mass was confirmed using MALDI-ToF on a Voyager-DE BioSpectrometry Workstation. Abbreviations: GIP, glucose-dependent insulinotropic polypeptide; HPLC, high-performance liquid chromatography; MALDI-ToF MS, matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry; hGIP, human glucose-dependent insulinotropic polypeptide; mGIP, mouse glucose-dependent insulinotropic polypeptide.
Acute effects of peptides on in vitro insulin secretion from BRIN-BD11 cells
The in vitro insulin secretory activity of test peptides was examined in BRIN-BD11 cells, cultured and maintained as previously described.36 For experimentation, BRIN-BD11 cells were seeded in 24-well plates at a cell density of 150 000 cells/well and allowed to attach overnight at 37°C. Following preincubation with Krebs-Ringer bicarbonate buffer (KRBB) (pH 7.4) supplemented with 0.5% (w/v) bovine serum albumin and 1.1 mM glucose (40 min; 37°C), cells were then incubated with hGIP(1-42), hGIP(1-30) and mGIP(1-30) (10−6 to 10−12 M) at 5.6 mM glucose for 20 minutes, to confirm GIPR agonist activity. Importantly, we have routinely shown that 5.6 mM glucose has stimulatory effects in BRIN-BD11 cells, which are significantly augmented by GIP.19,20,22,23,26 In a separate set of experiments, cells were seeded as before and incubated with N- and C-terminally truncated human and mouse GIP test peptides (10−12 to 10−6 M), namely hGIP(3-30), mGIP(3-30), h(Pro3)GIP(3-30), hGIP(5-30), hGIP(3-42) and hGIP(5-42), alone and in the presence of hGIP(1-42) (10−7 M) at 5.6 mM glucose for 20 minutes. Following 20-minute test incubations, aliquots of assay buffer (200 μL) were collected and stored at −20°C prior to assessment of insulin concentrations by an in-house radioimmunoassay (RIA).37
Animals
All animal studies were carried out using male NIH Swiss mice (12-14 weeks of age, Envigo Ltd, UK), housed individually in an air-conditioned room at 22 ± 2°C with a 12 -hour light:12 -hour dark cycle. Animals were maintained on a standard rodent chow diet (10% fat, 30% protein and 60% carbohydrate, Trouw Nutrition, UK), with ad libitum access to diet and water. All animal experiments were carried out in accordance with the UK Animal Scientific Procedures Act 1986 and approved by the University of Ulster Animal Welfare and Ethical Review Body (AWERB).
Acute effects on glucose tolerance in mice
Blood glucose and plasma insulin concentrations, where appropriate, were determined immediately prior to and 15, 30 and 60 minutes after intraperitoneal injection of glucose control (18 mmol/kg bw), as well as glucose together with N- and C-terminally truncated GIP peptides (50 nmol/kg bw) alone, or in combination with hGIP(1-42) (50 nmol/kg bw), in 4-hour fasted mice.
Biochemical analysis
An incision to the tail vain of conscious mice was used to obtain blood samples for biochemical analysis. Blood glucose was measured directly by an Ascencia Contour glucose metre (Bayer, Newbury, UK). For plasma insulin analyses, blood samples were collected into chilled fluoride/heparin glucose microcentrifuge tubes (Sarstedt, Numbrecht, Germany) and immediately centrifuged using a Beckman microcentrifuge (Beckman Instruments, Galway, Ireland) for 1 minute at 13 000 × g and stored at −20°C prior to insulin RIA.37
Statistical analysis
GraphPad PRISM (Version 5) was used for statistical analyses. Results are expressed as mean ± standard error of mean, and data compared by 1-way analysis of variance (ANOVA) followed by Student-Newman-Keuls post hoc test or 2-way ANOVA followed by Bonferroni posttests or unpaired Student t test, where appropriate. Incremental area under the curve (AUC) data were calculated using the trapezoidal rule with baseline subtraction. Significant difference was considered between data sets with P < 0.05.
Results
Effects of hGIP(1-42), hGIP(1-30) and mGIP(1-30) on insulin release from BRIN-BD11 cells
Figure 1 demonstrates the abilities of hGIP(1-42), as well as the C-terminally truncated human and mouse GIP forms, hGIP(3-30) and mGIP(3-30), to stimulate insulin secretion from BRIN-BD11 cells at 5.6 mM glucose. hGIP(1-42) and mGIP(1-30) significantly increased (P < 0.01 to P < 0.001) insulin secretion at concentrations of 10−8 M and above (Figure 1). hGIP(1-30) was less potent, with significant augmentation of insulin secretion above control levels only observed at 10−6 M peptide incubations (Figure 1). In addition, mGIP(1-30) was more efficacious (P < 0.05 to P < 0.01) than hGIP(1-30) and hGIP(1-42) at 10−8 and 10−6 M (Figure 1).
Figure 1.
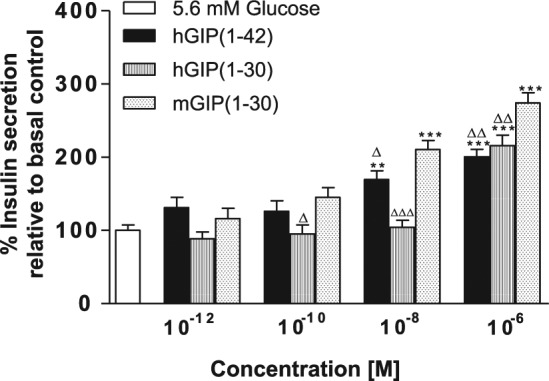
Effects of hGIP(1-42), hGIP(1-30) and mGIP(1-30) on insulin release from BRIN-BD11 cells.
BRIN-BD11 cells were incubated (20 min) with test peptides (10−12 to 10−6 M) in the presence of 5.6 mM glucose. Insulin was measured by radioimmunoassay. Values are mean ± standard error of mean (n = 8) for insulin release.
Abbreviations: hGIP indicates human glucose-dependent insulinotropic polypeptide; mGIP, mouse glucose-dependent insulinotropic polypeptide.
**P < 0.01, ***P < 0.001 compared with 5.6 mM glucose alone. ∆P < 0.05,∆∆P < 0.01, ∆∆∆P < 0.01 compared with mouse GIP(1-30).
Effects of N- and C-terminally truncated human and mouse GIP peptides on secretion from BRIN-BD11 cells
None of the N- and C-terminally truncated human and mouse GIP peptides, hGIP(3-30), mGIP(3-30), h(Pro3)GIP(3-30), hGIP(5-30), hGIP(3-42) and hGIP(5-42), stimulated insulin secretion from BRIN-BD11 cells at 5.6 mM glucose (Figure 2A to F). However, hGIP(3-30), mGIP(3-30) and h(Pro3)GIP(3-30) significantly (P < 0.01 to P < 0.001) inhibited hGIP(1-42)-stimulated elevations of insulin secretion under these conditions (Figure 2A to C). None of these peptides completely annulled the insulin secretory effects of hGIP(1-42) (Figure 2A to C). In contrast, hGIP(5-30), hGIP(3-42) and hGIP(5-42) were completely ineffective in terms of reducing 10−7 M hGIP(1-42) induced insulin release (Figure 2D to F).
Figure 2.

Effects of N- and C-terminally truncated GIP peptides alone or in combination with hGIP(1-42) (10−7 M) on insulin release from BRIN-BD11 cells.
BRIN-BD11 cells were incubated (20 min) with test peptides (10−12 to 10−6 M) alone or in combination with hGIP(1-42) (10−7 M) in the presence of 5.6 mM glucose. Insulin was measured by RIA. Values are mean ± standard error of mean (n = 8) for insulin release.
Abbreviations: GIP indicates glucose-dependent insulinotropic polypeptide; hGIP, human glucose-dependent insulinotropic polypeptide; mGIP, mouse glucose-dependent insulinotropic polypeptide.
***P < 0.001 compared with relevant glucose alone. ∆∆P < 0.01, ∆∆∆P < 0.001 compared with hGIP(1-42).
Effects of hGIP(1-42), hGIP(1-30) and mGIP(1-30) on glucose homeostasis in mice
As expected, hGIP(1-42) induced significant (P < 0.05 toP < 0.01) reductions in individual and 0 to 60-minute overall AUC glucose values when administered conjointly with glucose to mice (Figure 3A and B). hGIP(1-30) had similar beneficial (P < 0.05) glucose-lowering actions, whereas mGIP(1-30) was devoid of glucose homeostatic effects (Figure 3A and B). hGIP(1-42) appeared to be marginally the most effective glucose-lowering agent in mice (Figure 3A and B). Thus, the ability of the six N- and C-terminally truncated human and mouse GIP peptides to inhibit the glucose homeostatic actions of hGIP(1-42) was next assessed.
Figure 3.

Effects of hGIP(1-42), hGIP(1-30) and mGIP(1-30) on glucose tolerance in mice. Blood glucose concentrations (A) were measured before and 15, 30 and 60 minutes after intraperitoneal injection of glucose alone (18 mmol/kg bw), or in combination with test peptides (each at 50 nmol/kg bw) in fasted mice. (B) Blood glucose area under the curve (AUC) values for 0 to 60 minutes post injection.
Abbreviations: AUC indicates area under the curve; hGIP, human glucose-dependent insulinotropic polypeptide; mGIP, mouse glucose-dependent insulinotropic polypeptide.
Values represent means ± standard error of mean (n = 5-6). *P < 0.05, **P < 0.01 compared with glucose alone.
Effects of N- and C-terminally truncated human and mouse GIP peptides on GIP-induced glucose homeostasis and insulin secretion in mice
While administration of hGIP(3-30), h(Pro3)GIP(3-30), hGIP(3-42) or hGIP(5-42) in combination with hGIP(1-42) was associated with marginal inhibitory effects on hGIP(1-42)-mediated improvements of glucose homeostasis, this failed to reach significance in terms of individual or overall glucose levels (Figure 4A, C, E and F). Notably, only mGIP(3-30) and hGIP(5-30) were effective (P < 0.05 to P < 0.01) in terms of completely annulling 0 to 60-minute overall hGIP(1-42)-induced reductions in blood glucose levels (Figure 4B and D). As such, the ability of both peptides to counter GIP-induced elevations of insulin secretion was also examined (Table 2). mGIP(3-30) completely prevented any increase of insulin secretion by hGIP(1-42), whereas plasma insulin concentrations increased by 14% when hGIP(5-30) was administered in combination with hGIP(1-42) to mice (Table 2).
Figure 4.

Effects of N- and C-terminally truncated GIP peptides on glucose tolerance and GIP-mediated reductions of blood glucose in mice.
Blood glucose concentrations (A-F) were measured before and 15, 30, and 60 minutes after intraperitoneal injection of glucose alone (18 mmol/kg bw), or in combination with test peptides or test peptides combined with hGIP(1-42) (each peptide at 50 nmol/kg bw) in fasted mice. Blood glucose AUC values for 0 to 60 minutes post injection are shown in insets.
Abbreviations: AUC indicates area under the curve; GIP, glucose-dependent insulinotropic polypeptide; hGIP, human glucose-dependent insulinotropic polypeptide; mGIP, mouse glucose-dependent insulinotropic polypeptide.
Values represent means ± standard error of mean (n = 5-6). *P < 0.05, **P < 0.01 compared with glucose alone. ∆P < 0.05, ∆∆P < 0.01 compared with hGIP(1-42).
Table 2.
Effects of mGIP(3-30) and hGIP(3-30) on GIP-induced elevations of plasma insulin concentrations in mice.
| Peptide | 0-60-min AUC plasma insulin secretion (% of control) | Delta (Δ) |
|---|---|---|
| mGIP(3-30) | 100 ± 8 | −2 % |
| mGIP(3-30) + hGIP(1-42) | 98 ± 7 | |
| hGIP(5-30) | 100 ± 2 | 14 % |
| hGIP(5-30) + hGIP(1-42) | 114 ± 9 |
Plasma insulin concentrations were measured before and 15, 30 and 60 minutes after intraperitoneal injection of glucose in combination with mGIP(3-30) or hGIP(3-30) alone and in combination with hGIP(1-42) (each peptide at 50 nmol/kg bw) in fasted mice. Plasma insulin 0 to 60-minute AUC values are shown. Values represent means ± standard error of mean (n = 5-6). Abbreviations: AUC, area under the curve; GIP, glucose-dependent insulinotropic polypeptide; hGIP, human glucose-dependent insulinotropic polypeptide; mGIP, mouse glucose-dependent insulinotropic polypeptide.
Discussion
Recent work has revealed hGIP(3-30) as an efficacious GIPR antagonist in humans.33 This accords with the view that only amino acids from position 3-30 of GIP are necessary for GIPR binding.21 However, the same research group has also established that hGIP(3-30) is selective for primate, but not rodent, GIPRs.34 Given the plethora of potentially translatable information acquired from GIP studies conducted in rodents, and particularly mice, it is imperative to determine an effective GIPR antagonist for such rodent-based studies. Indeed, because GIP is regarded as the major physiological incretin hormone in man,38 as suggested over 15 years previously in rodents,26 the need is even more imperative.
In terms of GIPR agonism, the first two N-terminal amino acids of GIP are known to be essential for agonist properties.21 In keeping with this, and the idea that only amino acids 3-30 are required for GIPR interaction, several studies have established hGIP(1-30) as a full GIPR agonist at rat39-41 and human30 receptors. This is despite knowledge that the final 12 C-terminal amino acid residues of GIP are believed to enhance intrinsic receptor activity.19 However, GIP(1-30) is expressed in pancreatic α-cells and within some GIP-producing intestinal K cells,42 suggesting physiological importance. In keeping with this, hGIP(1-30) and mGIP(1-30) augmented insulin secretion from the rodent-derived BRIN-BD11 pancreatic beta cell line in the current study. However, in mice, mGIP(1-30) appeared to be ineffective in terms of regulating glucose homeostasis, whereas both hGIP(1-42) and hGIP(1-30) displayed good bioactivity. This was unexpected, especially given the reported species specificity of the GIPR signalling system.21,30 Moreover, mGIP(1-30) is naturally expressed in mice42 and long-acting forms display good bioactivity when administered subchronically to diabetic mice43,44; thus, our observations could relate to the strain of mouse or peptide dose employed. Nonetheless, together this suggests that omission of the 31-42 C-terminal sequence of GIP should have no detrimental effect on GIPR interaction of prospective antagonistic GIP-derived peptide sequences.
Consistent with this view, the current study demonstrated that GIP peptides retaining C-terminal residues 31-42, namely hGIP(3-42) and hGIP(5-42), were unable to counter GIP-induced insulin secretion in vitro, or GIP-mediated reductions in glucose levels in mice. Interestingly, GIP(3-42) represents the naturally occurring dipeptidyl peptidase-4 (DPP-4) degradation product of GIP(1-42) and is recognised to circulate at relatively high levels in the bloodstream.45 It was initially considered as a probable GIPR antagonist and postulated to physiologically moderate the insulin secreting and metabolic actions of GIP in vivo.46 However, the GIP inhibitory effects of hGIP(3-42) are weak, as described here and elsewhere,47 discounting it as a pharmacologically useful GIPR antagonistic. Given the above, it may be expected that GIP(3-30) should also be discoverable in the circulation, given the ubiquitous expression of DPP-4,48 but surprisingly this has yet to be confirmed. Nonetheless, mGIP(3-30) and hGIP(3-30), as well as h(Pro3)GIP(3-30), countered the insulinotropic action of GIP in vitro. Thus, although (Pro3)GIP(1-42) was shown to have agonist activity at hGIP receptors,30 it is considered a competitive antagonist at rat and mGIP receptors.20,30 In the same experimental setting, hGIP(5-30) was less effective, despite suggestion that it functions as a high-affinity competitive GIPR antagonist.21 However, such differences could be related to the reported species- or tissue-specific actions of GIP peptides.30,31 As such, substitution of histidine in human GIP, for arginine in mouse and rat GIP at position 18, could have some importance in terms of modulation of the GIPR in our experimental systems. In addition, assessment of receptor binding affinities of the peptides may also help to further understand any differences in GIPR inhibitory activity.
When administered conjointly with GIP to mice, only mGIP(3-30) and hGIP(5-30) fully countered the glucose-lowering action of GIP. Interestingly, hGIP(3-30) and hGIP(5-30) have previously been identified as competitive antagonists of the human GIPR.21 Given the primary physiological action of GIP is glucose-dependent insulin secretion,49 we further investigated the ability of mGIP(3-30) and hGIP(5-30) to modulate this response in mice. In keeping with species specificity of GIP-mediated biological actions,30 only mGIP(3-30) effectively prevented GIP-induced elevations of insulin secretion in mice. Thus, hGIP(5-30) is perhaps less useful as a GIPR antagonist in murine systems. Indeed, rat GIP(3-30) is recognised as a high affinity competitive antagonist at the level of the rat, but not human, GIPR.32 It may also have been interesting to consider the effect of mGIP(3-30) and hGIP(5-30) on GIP-induced glucagon secretion,50 and further studies in this regard would be required.
As well as being useful tools to enable better understanding of GIP physiology, there is important possible therapeutic application of GIPR antagonists for obesity.12 For instance, studies employing GIPR blockade in rodents through genetic deletion of the GIPR,51,52 active or passive immunisation,7,9,53 small molecular weight receptor antagonists54,55 or peptide-based GIP inhibitors19,20,23,56 all provide clear evidence for anti-obesity effects of attenuation of GIPR signalling. In humans, GIP induces cytokine expression, lipolysis and insulin resistance in adipocytes,57 and hGIP(3-30) inhibits GIP-induced increases in abdominal adipose tissue blood flow and decreases adipose tissue triacylglyceride uptake.58,59 Furthermore, knowledge that highly effective bariatric weight loss surgeries are, in part, linked to surgical removal of GIP-secreting K cells and compromised GIP secretion13,14 strongly suggests translatable benefits of GIPR antagonists for human obesity. Notably, the dose of GIPR antagonists employed for the current study is well beyond normal circulating levels of GIP,60 implying that such regimens would effectively annul the biological actions of endogenously released GIP.
In conclusion, the current studies support the concept of species-specific activity of GIP in different mammalian systems.21,30,32,34 Given the potential therapeutic application of peptide-based GIPR antagonists in human obesity and diabetes,61 the origin of such GIP peptides needs to be carefully considered. In this regard, we present mGIP(3-30) as a highly effective molecule to inhibit GIPR activity in mice. Utilisation of mGIP(3-30) for murine studies should better reflect the expected impact of GIPR inhibition with human GIP sequences, such as hGIP(3-30), in the human setting.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a PhD studentship (awarded to RAP) from the Department for the Economy (DfE) Northern Ireland and University of Ulster strategic research funding.
Declaration of Conflicting Interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: NI, VAG and PRF conceived the study, drafted the manuscript and revised it critically for intellectual content. RAP, SLC and MTN participated in the analysis and interpretation of data, drafted the manuscript and revised it critically for intellectual content. All authors approved the final version of the manuscript.
ORCID iD: SL Craig  https://orcid.org/0000-0001-7871-1468
https://orcid.org/0000-0001-7871-1468
References
- 1. Seino Y, Fukushima M, Yabe D. GIP and GLP-1, the two incretin hormones: similarities and differences. J Diabetes Investig. 2010;1:8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Al-Sabah S. Molecular pharmacology of the incretin receptors. Med Princ Pract. 2016;25:15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Szalowska E, Meijer K, Kloosterhuis N, Razaee F, Priebe M, Vonk RJ. Sub-chronic administration of stable GIP analog in mice decreases serum LPL activity and body weight. Peptides. 2011;32:938-945. [DOI] [PubMed] [Google Scholar]
- 4. Mroz PA, Finan B, Gelfanov V, et al. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol Metab. 2019;20:51-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nasteska D, Harada N, Suzuki K, et al. Chronic reduction of GIP secretion alleviates obesity and insulin resistance under high-fat diet conditions. Diabetes. 2014;63:2332-2343. [DOI] [PubMed] [Google Scholar]
- 6. Boylan MO, Glazebrook PA, Tatalovic M, Wolfe MM. Gastric inhibitory polypeptide immunoneutralization attenuates development of obesity in mice. Am J Physiol Endocrinol Metab. 2015;309:E1008-E1018. [DOI] [PubMed] [Google Scholar]
- 7. Killion EA, Wang J, Yie J, et al. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci Transl Med. 2018;10:eaat3392. [DOI] [PubMed] [Google Scholar]
- 8. Norregaard PK, Deryabina MA, Tofteng Shelton P, et al. A novel GIP analogue, ZP4165, enhances glucagon-like peptide-1-induced body weight loss and improves glycaemic control in rodents. Diabetes Obes Metab. 2018;20:60-68. [DOI] [PubMed] [Google Scholar]
- 9. Irwin N, McClean PL, Patterson S, Hunter K, Flatt PR. Active immunisation against gastric inhibitory polypeptide (GIP) improves blood glucose control in an animal model of obesity-diabetes. Biol Chem. 2009;390:75-80. [DOI] [PubMed] [Google Scholar]
- 10. Thomsen C, Rasmussen O, Lousen T, et al. Differential effects of saturated and monounsaturated fatty acids on postprandial lipemia and incretin responses in healthy subjects. Am J Clin Nutr. 1999;69:1135-1143. [DOI] [PubMed] [Google Scholar]
- 11. Creutzfeldt W, Ebert R, Willms B, Frerichs H, Brown JC. Gastric inhibitory polypeptide (GIP) and insulin in obesity: increased response to stimulation and defective feedback control of serum levels. Diabetologia. 1978;14:15-24. [DOI] [PubMed] [Google Scholar]
- 12. Irwin N, Flatt PR. Therapeutic potential for GIP receptor agonists and antagonists. Best Pract Res Clin Endocrinol Metab. 2009;23:499-512. [DOI] [PubMed] [Google Scholar]
- 13. Flatt PR. Effective surgical treatment of obesity may be mediated by ablation of the lipogenic gut hormone gastric inhibitory polypeptide (GIP): evidence and clinical opportunity for development of new obesity-diabetes drugs. Diab Vasc Dis Res. 2007;4:151-153. [DOI] [PubMed] [Google Scholar]
- 14. Mingrone G, Nolfe G, Gissey GC, et al. Circadian rhythms of GIP and GLP1 in glucose-tolerant and in type 2 diabetic patients after biliopancreatic diversion. Diabetologia. 2009;52:873-881. [DOI] [PubMed] [Google Scholar]
- 15. Xiong SW, Cao J, Liu XM, Deng XM, Liu Z, Zhang FT. Effect of modified roux-en-Y gastric bypass surgery on GLP-1, GIP in patients with type 2 diabetes mellitus. Gastroenterol Res Pract. 2015;2015:625196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coskun T, Sloop KW, Loghin C, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol Metab. 2018;18:3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frias JP, Nauck MA, Van J, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet. 2018; 392:2180-2193. [DOI] [PubMed] [Google Scholar]
- 18. Bergmann NC, Lund A, Gasbjerg LS, et al. Effects of combined GIP and GLP-1 infusion on energy intake, appetite and energy expenditure in overweight/obese individuals: a randomised, crossover study. Diabetologia. 2019;62: 665-675. [DOI] [PubMed] [Google Scholar]
- 19. Kerr BD, Flatt AJ, Flatt PR, Gault VA. Characterization and biological actions of N-terminal truncated forms of glucose-dependent insulinotropic polypeptide. Biochem Biophys Res Commun. 2011;404:870-876. [DOI] [PubMed] [Google Scholar]
- 20. Pathak V, Gault VA, Flatt PR, Irwin N. Antagonism of gastric inhibitory polypeptide (GIP) by palmitoylation of GIP analogues with N- and C-terminal modifications improves obesity and metabolic control in high fat fed mice. Mol Cell Endocrinol. 2015;401:120-129. [DOI] [PubMed] [Google Scholar]
- 21. Hansen LS, Sparre-Ulrich AH, Christensen M, et al. N- and C-terminally truncated forms of glucose-dependent insulinotropic polypeptide are high-affinity competitive antagonists of the human GIP receptor. Br J Pharmacol. 2015;173:826-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O’Harte FP, Hunter K, Gault VA, et al. Antagonistic effects of two novel GIP analogs, (Hyp3)GIP and (Hyp3)GIPLys16PAL, on the biological actions of GIP and longer-term effects in diabetic ob/ob mice. Am J Physiol Endocrinol Metab. 2007;292:E1674-E1682. [DOI] [PubMed] [Google Scholar]
- 23. Gault VA, Hunter K, Irwin N, et al. Characterisation and glucoregulatory actions of a novel acylated form of the (Pro3)GIP receptor antagonist in type 2 diabetes. Biol Chem. 2007;388:173-179. [DOI] [PubMed] [Google Scholar]
- 24. McClean PL, Irwin N, Cassidy RS, Holst JJ, Gault VA, Flatt PR. GIP receptor antagonism reverses obesity, insulin resistance, and associated metabolic disturbances induced in mice by prolonged consumption of high-fat diet. Am J Physiol Endocrinol Metab. 2007;293:E1746-E1755. [DOI] [PubMed] [Google Scholar]
- 25. McClean PL, Irwin N, Hunter K, Gault VA, Flatt PR. (Pro(3))GIP[mPEG]: novel, long-acting, mPEGylated antagonist of gastric inhibitory polypeptide for obesity-diabetes (diabesity) therapy. Br J Pharmacol. 2008;155:690-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gault VA, O’Harte FP, Harriott P, Mooney MH, Green BD, Flatt PR. Effects of the novel (Pro3)GIP antagonist and exendin(9-39)amide on GIP- and GLP-1-induced cyclic AMP generation, insulin secretion and postprandial insulin release in obese diabetic (ob/ob) mice: evidence that GIP is the major physiological incretin. Diabetologia. 2003;46:222-230. [DOI] [PubMed] [Google Scholar]
- 27. Gault VA, McClean PL, Cassidy RS, Irwin N, Flatt PR. Chemical gastric inhibitory polypeptide receptor antagonism protects against obesity, insulin resistance, glucose intolerance and associated disturbances in mice fed high-fat and cafeteria diets. Diabetologia. 2007;50:1752-1762. [DOI] [PubMed] [Google Scholar]
- 28. Irwin N, McClean PL, O’Harte FP, Gault VA, Harriott P, Flatt PR. Early administration of the glucose-dependent insulinotropic polypeptide receptor antagonist (Pro3)GIP prevents the development of diabetes and related metabolic abnormalities associated with genetically inherited obesity in ob/ob mice. Diabetologia. 2007;50:1532-1540. [DOI] [PubMed] [Google Scholar]
- 29. Irwin N, Gault VA, Green BD, et al. Effects of short-term chemical ablation of the GIP receptor on insulin secretion, islet morphology and glucose homeostasis in mice. Biol Chem. 2004;385:845-852. [DOI] [PubMed] [Google Scholar]
- 30. Sparre-Ulrich AH, Hansen LS, Svendsen B, et al. Species-specific action of (Pro3)GIP – a full agonist at human GIP receptors, but a partial agonist and competitive antagonist at rat and mouse GIP receptors. Br J Pharmacol. 2016; 173:27-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Al-Sabah S, Al-Fulaij M, Ahmed HA. Selectivity of peptide ligands for the human incretin receptors expressed in HEK-293 cells. Eur J Pharmacol. 2014;741: 311-315. [DOI] [PubMed] [Google Scholar]
- 32. Sparre-Ulrich AH, Gabe MN, Gasbjerg LS, et al. GIP(3-30)NH2 is a potent competitive antagonist of the GIP receptor and effectively inhibits GIP-mediated insulin, glucagon, and somatostatin release. Biochem Pharmacol. 2017;131: 78-88. [DOI] [PubMed] [Google Scholar]
- 33. Gasbjerg LS, Christensen MB, Hartmann B, et al. GIP(3-30)NH2 is an efficacious GIP receptor antagonist in humans: a randomised, double-blinded, placebo-controlled, crossover study. Diabetologia. 2018;61:413-423. [DOI] [PubMed] [Google Scholar]
- 34. Gabe MBN, Sparre-Ulrich AH, Pedersen MF, et al. Human GIP(3-30)NH2 inhibits G protein-dependent as well as G protein-independent signaling and is selective for the GIP receptor with high-affinity binding to primate but not rodent GIP receptors. Biochem Pharmacol. 2018;150:97-107. [DOI] [PubMed] [Google Scholar]
- 35. Lafferty RA, Flatt PR, Irwin N. C-terminal degradation of PYY peptides in plasma abolishes effects on satiety and beta-cell function. Biochem Pharmacol. 2018;158:95-102. [DOI] [PubMed] [Google Scholar]
- 36. McClenaghan NH, Barnett BR, Ah-Sing E, et al. Characterization of a novel glucose-responsive insulin-secreting cell line, BRIN-BD11, produced by electrofusion. Diabetes. 1996;45:1132-1140. [DOI] [PubMed] [Google Scholar]
- 37. Flatt PR, Bailey CJ. Plasma glucose and insulin response to glucagon and arginine in Aston ob/ob mice: evidence for a selective defect in glucose-mediated insulin release. Horm Metab Res. 1982;14:127-130. [DOI] [PubMed] [Google Scholar]
- 38. Holst JJ. The incretin system in healthy humans: the role of GIP and GLP-1. Metabolism. 2019;96:46-55. [DOI] [PubMed] [Google Scholar]
- 39. Gallwitz B, Witt M, Folsch UR, Creutzfeldt W, Schmidt WE. Binding specificity and signal transduction of receptors for glucagon-like peptide-1(7-36)amide and gastric inhibitory polypeptide on RINm5F insulinoma cells. J Mol Endocrinol. 1993;10:259-268. [DOI] [PubMed] [Google Scholar]
- 40. Morrow GW, Kieffer TJ, McIntosh CHS, et al. The insulinotropic region of gastric inhibitory polypeptide; fragment analysis suggests the bioactive site lies between residues 19 and 30. Can J Physiol Pharmacol. 1996;74:65-72. [PubMed] [Google Scholar]
- 41. Hinke SA, Manhart S, Pamir N, et al. Identification of a bioactive domain in the amino-terminus of glucose-dependent insulinotropic polypeptide (GIP). Biochim Biophys Acta. 2001;1547:143-155. [DOI] [PubMed] [Google Scholar]
- 42. Fujita Y, Wideman RD, Asadi A, et al. Glucose-dependent insulinotropic polypeptide is expressed in pancreatic islet alpha-cells and promotes insulin secretion. Gastroenterology. 2010;138:1966-1975. [DOI] [PubMed] [Google Scholar]
- 43. Gault VA, Porter DW, Irwin N, Flatt PR. Comparison of sub-chronic metabolic effects of stable forms of naturally occurring GIP(1-30) and GIP(1-42) in high-fat fed mice. J Endocrinol. 2011;208:265-271. [DOI] [PubMed] [Google Scholar]
- 44. Yanagimachi T, Fujita Y, Takeda Y, et al. Pancreatic glucose-dependent insulinotropic polypeptide (GIP) (1-30) expression is upregulated in diabetes and PEGylated GIP(1-30) can suppress the progression of low-dose-STZ-induced hyperglycaemia in mice. Diabetologia. 2016;59:533-541. [DOI] [PubMed] [Google Scholar]
- 45. Deacon CF, Nauck MA, Meier J, Hucking K, Holst JJ. Degradation of endogenous and exogenous gastric inhibitory polypeptide in healthy and in type 2 diabetic subjects as revealed using a new assay for the intact peptide. J Clin Endocrinol Metab. 2000;85:3575-3581. [DOI] [PubMed] [Google Scholar]
- 46. Gault VA, Parker JC, Harriott P, Flatt PR, O’Harte FP. Evidence that the major degradation product of glucose-dependent insulinotropic polypeptide, GIP(3-42), is a GIP receptor antagonist in vivo. J Endocrinol. 2002;175:525-533. [DOI] [PubMed] [Google Scholar]
- 47. Deacon CF, Plamboeck A, Rosenkilde MM, de Heer J, Holst JJ. GIP-(3-42) does not antagonize insulinotropic effects of GIP at physiological concentrations. Am J Physiol Endocrinol Metabol. 2006;291:E468-E475. [DOI] [PubMed] [Google Scholar]
- 48. Röhrborn D, Wronkowitz N, Eckel J. DPP4 in diabetes. Front Immunol. 2015;6:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pederson RA, Schubert HE, Brown JC. The insulinotropic action of gastric inhibitory polypeptide. Can J Physiol Pharmacol. 1975;53:217-223. [DOI] [PubMed] [Google Scholar]
- 50. Cassidy RS, Irwin N, Flatt PR. Effects of gastric inhibitory polypeptide (GIP) and related analogues on glucagon release at normo- and hyperglycaemia in Wistar rats and isolated islets. Biol Chem. 2008;389:189-193. [DOI] [PubMed] [Google Scholar]
- 51. Miyawaki K, Yamada Y, Yano H, et al. Glucose intolerance caused by a defect in the entero-insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:14843-14847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Naitoh R, Miyawaki K, Harada N, et al. Inhibition of GIP signaling modulates adiponectin levels under high-fat diet in mice. Biochem Biophys Res Commun. 2008;376:21-25. [DOI] [PubMed] [Google Scholar]
- 53. Fulurija A, Lutz TA, Sladko K, et al. Vaccination against GIP for the treatment of obesity. PLoS ONE. 2008;3:e3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Irwin N, Green BD, Parker JC, Gault VA, O’Harte FP, Flatt PR. Biological activity and antidiabetic potential of synthetic fragment peptides of glucose-dependent insulinotropic polypeptide, GIP(1-16) and (Pro3)GIP(1-16). Regul Pept. 2006;135:45-53. [DOI] [PubMed] [Google Scholar]
- 55. Nakamura T, Tanimoto H, Mizuno Y, et al. Gastric inhibitory polypeptide receptor antagonist, SKL-14959, suppressed body weight gain on diet-induced obesity mice. Obes Sci Pract. 2018;4:194-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gault VA, O’Harte FP, Harriott P, Flatt PR. Characterization of the cellular and metabolic effects of a novel enzyme-resistant antagonist of glucose-dependent insulinotropic polypeptide. Biochem Biophys Res Commun. 2002;290:1420-1426. [DOI] [PubMed] [Google Scholar]
- 57. Timper K, Grisouard J, Sauter NS, et al. Glucose-dependent insulinotropic polypeptide induces cytokine expression, lipolysis, and insulin resistance in human adipocytes. Am J Physiol Endocrinol Metab. 2016;304:E1-E13. [DOI] [PubMed] [Google Scholar]
- 58. Asmar M, Asmar A, Simonsen L, et al. The gluco- and liporegulatory and vasodilatory effects of glucose-dependent insulinotropic polypeptide (GIP) are abolished by an antagonist of the human GIP receptor. Diabetes. 2017;66:2363-2371. [DOI] [PubMed] [Google Scholar]
- 59. Asmar M, Asmar A, Simonsen L, Dela F, Holst JJ, Bulow J. GIP-induced vasodilation in human adipose tissue involves capillary recruitment. Endocr Connect. 2019;8:806-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gjesing AP, Ekstrom CT, Eiberg H, et al. Fasting and oral glucose-stimulated levels of glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) are highly familial traits. Diabetologia. 2012;55:1338-1345. [DOI] [PubMed] [Google Scholar]
- 61. Irwin N, Flatt PR. Evidence for beneficial effects of compromised gastric inhibitory polypeptide action in obesity-related diabetes and possible therapeutic implications. Diabetologia. 2009;52:1724-1731. [DOI] [PubMed] [Google Scholar]