

Abstract
Objective—
Mutations in ABCC6 underlie the ectopic mineralization disorder pseudoxanthoma elasticum (PXE) and some forms of generalized arterial calcification of infancy, both of which affect the cardiovascular system. Using cultured cells, we recently showed that ATP-binding cassette subfamily C member 6 (ABCC6) mediates the cellular release of ATP, which is extracellularly rapidly converted into AMP and the mineralization inhibitor inorganic pyrophosphate (PPi). The current study was performed to determine which tissues release ATP in an ABCC6-dependent manner in vivo, where released ATP is converted into AMP and PPi, and whether human PXE ptients have low plasma PPi concentrations.
Approach and Results—
Using cultured primary hepatocytes and in vivo liver perfusion experiments, we found that ABCC6 mediates the direct, sinusoidal, release of ATP from the liver. Outside hepatocytes, but still within the liver vasculature, released ATP is converted into AMP and PPi. The absence of functional ABCC6 in patients with PXE leads to strongly reduced plasma PPi concentrations.
Conclusions—
Hepatic ABCC6-mediated ATP release is the main source of circulating PR, revealing an unanticipated role of the liver in systemic PPi homeostasis. Patients with PXE have a strongly reduced plasma PPi level, explaining their mineralization disorder. Our results indicate that systemic PPi is relatively stable and that PXE, generalized arterial calcification of infancy, and other ectopic mineralization disorders could be treated with PPi supplementation therapy.
Keywords: multidrug resistance-associated proteins, nucleotides, pathologic calcification, pyrophosphatases, vascular calcification
Pseudoxanthoma elasticum (PXE) is an autosomal recessive disease characterized by progressive ectopic mineralization of the skin, eyes, and arteries.1 Approximately 150 000 patients with PXE worldwide experience stigmatizing skin lesions, progressive loss of vision, and cardiovascular complications, against which no effective therapy exists.2
In 2000, several groups reported that PXE is caused by inactivating mutations in the ATP-binding cassette subfamily C member 6 (ABCC6) gene,3–5 and more recently, ABCC6 defects were also found to cause some forms of generalized arterial calcification of infancy (GACI),6 a severe form of arterial calcification. ABCC6 (also known as multidrug resistance protein 6) is an ATP-dependent orphan efflux transporter that is primarily expressed in the liver.7 Importantly, PXE is not caused by a lack of ABCC6 in the affected tissues but by the absence of an unknown factor in the central circulation requiring active ABCC6.8 Despite extensive research, the identity of this factor has long remained a mystery.
We recently showed that overexpression of ABCC6 in human embryonic kidney 293 (HEK293) cells induces the release of nucleoside triphosphates, predominantly ATP, in vitro.9 Secreted ATP was extracellularly converted into AMP and the mineralization inhibitor inorganic pyrophosphate (PPi) by ectonucleotide pyrophosphatase-phosphodiesterase (ENPP)–type ectonucleotidases. The in vivo relevance of these findings was demonstrated in Abcc6−/− mice, which have plasma PPi levels <40% of those found in wild-type control animals. ABCC6 is a member of the ABCC (multidrug resistance protein) family, which contains large proteins transporting a variety of organic anions.10 ABCC6 is mainly present in the sinusoidal membrane of the hepatocytes.11 Because we could not demonstrate direct ABCC6-mediated ATP transport in vitro, we postulated that ABCC6 secretes an organic anion, factor X, into the circulation that induces local ATP release in the periphery.9 The alternative possibility that the liver directly releases ATP in an ABCC6-dependent manner seemed unlikely. Secretion of ATP over the sinusoidal membrane of hepatocytes has never been described, and the extremely short half-life of ATP in the blood circulation (<1 seconds)12 does not allow PPi formation from liver-derived ATP in the periphery. The current study was performed to show that ABCC6 affects plasma PPi levels in humans and to assess whether ABCC6 directly affects hepatic ATP release or indirectly induces peripheral ATP release.
Materials and Methods
Materials and Methods are available in the online-only Supplement.
Results
HEK293 and HeLa Cells Release ATP on the Expression of ABCC6
We have previously shown that the introduction of ABCC6 in HEK293 cells results in the release of large amounts of ATP into the culture medium.9 To determine whether ABCC6-dependent ATP release is cell type-dependent, we generated HeLa cells in which the expression of rat ABCC6 could be induced by doxycycline. A luciferin/luciferase-based assay was used to follow the appearance of ATP in the cell culture medium in real-time. In the absence of rat ABCC6, cells released almost no ATP (Figure 1A and 1B). However, on induction of rat ABCC6, both 293 and HeLa cells released substantial amounts of ATP into the cell culture medium (Figure 1). These data show that ATP release is a general feature of ABCC6-containing cells and not specific for HEK293 cells.
Figure 1.
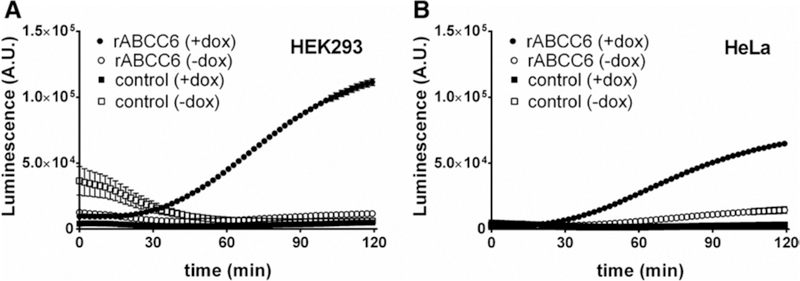
HEK293 and HeLa cells overproducing rat ATP-binding cassette subfamily C member 6 (rABCC6) release ATP. A, Flp-In T-REx 293 control (squares) or Flp-In T-REx 293 rABCC6 (circles) cells were grown in the presence (filled symbols) or absence (open symbols) of 1 μg/mL doxycycline to induce rABCC6 expression. Two days later, ATP efflux was followed in real-time for 2 hours using the ATP detection reagent BactiterGlo. B, ATP efflux from Flp-In T-REx HeLa control (squares) or Flp-In T-REx HeLa rABCC6 (circles) cells grown in the presence (filled symbols) or absence (open symbols) of 1 μg/mL doxycycline was followed for 2 hours in real-time. Data (n=12) represent mean±SEM.
Appearance of PPi in the Culture Medium of Sandwich-Cultured Hepatocytes Depends on ABCC6
ABCC6 is predominantly present in the liver.11 Next, we therefore explored in sandwich-cultured hepatocytes the possibility that hepatocytes directly release ATP over their basolateral membrane in an ABCC6-dependent manner. We were unable to detect ATP release directly in these experiments, presumably because of the high ectonucleotidase activity of hepatocytes. We, therefore, followed the appearance of the ATP metabolite PPi in the culture medium. PPi levels clearly increased in culture medium of wild-type hepatocytes over time, with substantially lower levels detected in medium of hepatocytes lacking ABCC6 (Figure 2A). These results indicate that hepatocytes release ATP over their sinusoidal membrane in an ABCC6-dependent manner and are also able to convert it to PPi. We also detected some PPi in medium from Abcc6−/− cells, which we attribute to ATP release unrelated to ABCC6, or leakage from damaged cells.
Figure 2.
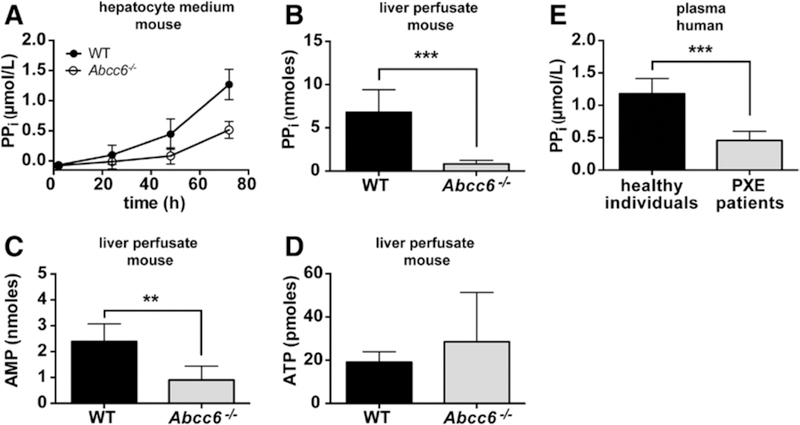
Hepatic ATP-binding cassette subfamily C member 6 (ABCC6) raises inorganic pyrophosphate (PPi) levels via ATP release. Released ATP is rapidly converted into AMP and PPi within the liver vasculature. A, PPi levels in culture medium of sandwich-cultured primary wild-type (WT) and Abcc6−/− hepatocytes (n=3 for WT, n=4 for Abcc6−/−); total amount of (B) PPi, (C) AMP, and (D) ATP in mouse liver perfusates collected from WT and Abcc6−/− livers during 30 minutes (n=5 for WT, n=6 for Abcc6−/−). E, PPi levels in platelet-free plasma samples from healthy subjects (n=14) and patients with PXE (n=12). Patient and control characteristics are given in the online-only Data Supplement. Data are presented as mean±SD. **P<0.01, ***P<0.001. Note that AMP and PPi levels are in nmoles, whereas ATP levels are in pmoles and close to background levels.
Hepatic ABCC6 Mediates the Sinusoidal Release of ATP, Which Is Converted Into AMP and PPi Within the Liver Vasculature
To assess whether ABCC6 is an important factor in hepatic ATP release in vivo, we performed liver perfusion experiments. PPi and AMP levels in the liver perfusates strongly depended on the presence of ABCC6 (Figure 2B and 2C). Interestingly, ATP levels did not differ between the 2 genotypes and were extremely low, representing <1% of the PPi and AMP levels (Figure 1D). The AMP and PPi that we detect in the liver perfusates must be derived from ATP: Enpp1−/− mice have pp. levels that are <5% of those found in wild-type mice,13 implying that also the PPi in plasma that depends on ABCC6 must come from ATP. Conversion of released ATP into AMP and PPi within the liver is fast. We calculated that during our single-pass perfusion experiments the buffer is present in the liver for ≈10 seconds (for the calculation, see the Materials and Methods section in the online-only Data Supplement). During this short period, the substantial amounts of ATP released are almost quantitatively converted into PR and AMP (Figure 2B–2D). This rapid and efficient conversion also explains why we were unable to detect ATP release from cultured wild-type hepatocytes: any released ATP is almost instantaneously converted into AMP and PPi by hepatic NPP1.
From our perfusion experiments, we calculate that ABCC6 mediates ≈90% of the hepatic nucleotide release. During 24 hours, this corresponds to ≥5% of the total hepatic adenine nucleotide pool (Figure 2B; for the calculation, see the Materials and Methods section in the online-only Data Supplement). The plasma t1/2 of PPi has been estimated to be 33 minutes, which requires a hepatic release rate of 6 nmoles pp. per hour to achieve the steady state levels of 2.3 μmol/L that we have reported for mice9 (for calculation, see the Materials and Methods section in the online-only Data Supplement). Importantly, the amount of PPi detected in liver perfusates of wild-type mice is high enough to explain these steady state PPi levels in mouse plasma.
Patients With PXE Have Strongly Reduced PPi Plasma Levels
An important question is whether our mouse results translate to human PXE patients. We have, therefore, studied a group of 12 Dutch patients with PXE with known ABCC6 mutations (Table I in the online-only Data Supplement). The plasma PPi concentrations were ≈2.5-fold lower in patients than in healthy individuals (Figure 2E). This difference did not depend on sex and is in line with the reduced plasma PPi levels we previously reported for Abcc6−/− mice.9
Discussion
PPi is a key regulator of ectopic mineralization acting by inhibiting hydroxyapatite crystal growth.14 As a result, mutations in genes encoding known PPi-regulating enzymes like ENPP1, ecto-5′-nucleotidase, progressive ankylosis protein homolog, and tissue-nonspecific alkaline phosphatase (TNAP) cause various mineralization disorders.15–18 The clinical symptoms of the mineralization disorders caused by non-functional ENPP1 (GACI) and ecto-5′-nucleotidase (arterial calcification due to deficiency of CD73) highly overlap those of PXE.19 The similarity between GACI and PXE is underlined by the recent observations that both GACI and PXE can be caused by mutations in ENPP1, as well as ABCC6.6 Our data unexpectedly falsify the factor X-hypothesis9 and show that ABCC6-mediated ATP release from the liver is the principal source of plasma PPi. A factor involved in the local release of PPi is progressive ankylosis protein homolog, a membrane protein postulated to mediate the direct release of PPi from cells. Progressive ankylosis protein homolog does, however, not substantially contribute to plasma PPi levels, which almost exclusively depend on ENPP1 activity and hence ATP release.13 Based on the currently available data, we propose the model presented in Figure 3.
Figure 3.
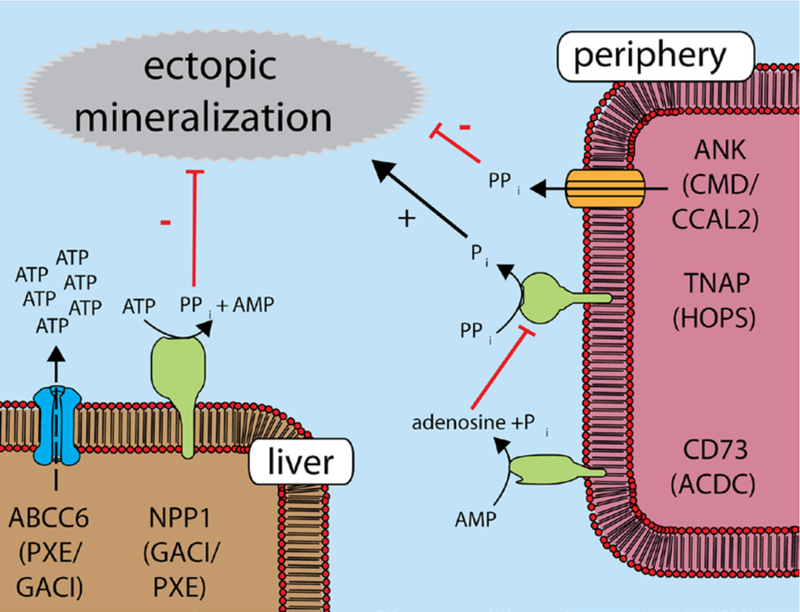
Proposed model for hepatic ATP-binding cassette subfamily C member 6 (ABCC6)–mediated pyrophosphate generation and ectopic mineralization. ATP released from the liver by an ABCC6-dependent mechanism is converted into the mineralization inhibitor pyrophosphate (inorganic pyrophosphate [PPi]) by hepatic ectonucleotide pyrophosphatase-phosphodiesterase 1 (ENPP1). In the periphery, PPi is hydrolyzed by tissue-nonspecific alkaline phosphatase (TNAP). Inactive ABCC6 classically causes pseudoxanthoma elasticum (PXE), whereas inactive ENPP1 causes generalized arterial calcification of infancy (GACI). Nonfunctional ecto-5′-nucleotidase results in arterial calcification due to deficiency of CD73 (ACDC), and inactive TNAP causes hypophosphatasia (HOPS). Local PPi levels also depend on the transmembrane protein progressive ankylosis protein homolog (ANKH), a protein postulated to be a PPi channel/efflux transporter. Mutations in ANKH can result in chon-drocalcinosis type 2 (CCAL2) or craniometaphyseal dysplasia (CMD).
Our finding that PPi generated within the liver is able to act in the periphery shows that increased systemic PPi levels are sufficient to inhibit local ectopic mineralization. Importantly, Lomashvili et al13 recently showed in Enpp1−/− mice that ectopic calcification depends on plasma PPi levels and not local PPi production. The crucial role of plasma PPi in the prevention of ectopic calcification has important therapeutic consequences: raising PPi levels in the blood circulation of patients with PXE, GACI, and arterial calcification due to deficiency of CD73 should suffice to halt ectopic mineralization. The short plasma half-life and lack of a suitable dosage form do not make PPi an attractive candidate for supplementation therapy in humans,20 but it might be possible to generate suitable PPi precursors. Alternatively, bisphosphonates, a class of metabolically stable, synthetic PPi analogs that have been used in GACI with reasonable success,21 may represent an attractive treatment strategy for PXE and arterial calcification due to deficiency of CD73.
The AMP metabolite adenosine is known to inhibit the expression of TNAP (Figure 3).16 It is, therefore, tempting to speculate that the increased TNAP activity seen in fibroblasts isolated from patients with PXE22 and Abcc6−/− mice23 is because of a reduction in the amount of released AMP. Low AMP levels might reduce local formation of adenosine and subsequent TNAP inhibition. AMP-derived adenosine might, therefore, be involved in priming of the periphery for subsequent PPi influx. This model would imply that both AMP and PPi are necessary to prevent ectopic mineralization: PPi by directly inhibiting the formation of calcium phosphate crystals and AMP after being metabolized to adenosine by inhibiting premature degradation of circulating PPi by TNAP.
In vitro, ABCC6 transports glutathione conjugates and the synthetic cyclic peptide BQ-123, suggesting that ABCC6 is a bona fide transporter.11,24 We were unable, however, to demonstrate ABCC6-mediated nucleoside triphosphate transport in vesicular transport experiments.9 Factors could be missing in vitro, however, that allows ABCC6 to transport ATP in vivo, or ABCC6 could indirectly stimulate ATP release by regulating vesicular transport or ion channels.25
Taken together, we show that ABCC6 mediates the release of ATP directly from the liver into the circulation. Within the liver vasculature, ATP is converted into AMP and PPi and represents the main source of the mineralization inhibitor PPi in plasma. This fully explains why absence of ABCC6 results in the ectopic mineralization observed in patients with PXE. Our data indicate that correcting PPi to normal levels could prevent the ectopic mineralization observed in PXE, GACI, and arterial calcification due to deficiency of CD73.
Supplementary Material
Significance.
Pseudoxanthoma elasticum is a hereditary ectopic mineralization disorder caused by the absence of functional ATP-binding cassette subfamily C member 6 that affects ≈150 000 patients worldwide. An effective therapy does not exist because the pathology underlying the disease is not well understood. Here, we show that ATP-binding cassette subfamily C member 6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation, explaining the ectopic calcification observed in patients with pseudoxanthoma elasticum. Our data indicate that correcting inorganic pyrophosphate to normal levels could prevent the ectopic mineralization observed in pseudoxanthoma elasticum and related mineralization disorders.
Acknowledgments
We thank our colleague Alfred Schinkel for critically reading the article. Pyruvate orthophosphate dikinase was kindly provided by Kikkoman Biochemifa, Tokyo, Japan.
Sources of Funding
Our work is supported by PXE international and a Hungarian Research Foundation Grant (OTKA-104227). The work of A. Váradi is also supported by NIH grant R01 AR055225.
Nonstandard Abbreviations and Acronyms
- ABCC6
ATP-binding cassette subfamily C member 6
- ENPP
ectonucleotide pyrophosphatase-phosphodiesterase
- GACI
generalized arterial calcification of infancy
- PPi
inorganic pyrophosphate
- PXE
pseudoxanthoma elasticum
- TNAP
tissue-nonspecific alkaline phosphatase
Footnotes
Disclosures
None.
References
- 1.Neldner KH. Pseudoxanthoma elasticum. Clin Dermatol. 1988;6:1–159. [DOI] [PubMed] [Google Scholar]
- 2.Uitto J, Váradi A, Bercovitch L, Terry PF, Terry SF. Pseudoxanthoma elasticum: progress in research toward treatment: summary of the 2012 PXE international research meeting. J Invest Dermatol. 2013;133:1444–1449. [DOI] [PubMed] [Google Scholar]
- 3.Le Saux O, Urban Z, Tschuch C, Csiszar K, Bacchelli B, Quaglino D, Pasquali-Ronchetti I, Pope FM, Richards A, Terry S, Bercovitch L, de Paepe A, Boyd CD. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat Genet. 2000;25:223–227. [DOI] [PubMed] [Google Scholar]
- 4.Bergen AA, Plomp AS, Schuurman EJ, Terry S, Breuning M, Dauwerse H, Swart J, Kool M, van Soest S, Baas F, ten Brink JB, de Jong PT. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat Genet. 2000;25:228–231. [DOI] [PubMed] [Google Scholar]
- 5.Ringpfeil F, Lebwohl MG, Christiano AM, Uitto J. Pseudoxanthoma elasticum: mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc Natl Acad Sci U S A. 2000;97:6001–6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nitschke Y, Rutsch F. Generalized arterial calcification of infancy and pseudoxanthoma elasticum: two sides of the same coin. Front Genet. 2012;3:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kool M, van der Linden M, de Haas M, Baas F, Borst P. Expression of human MRP6, a homologue of the multidrug resistance protein gene MRP1, in tissues and cancer cells. Cancer Res. 1999;59:175–182. [PubMed] [Google Scholar]
- 8.Jiang Q, Oldenburg R, Otsuru S, Grand-Pierre AE, Horwitz EM, Uitto J. Parabiotic heterogenetic pairing of Abcc6−/−/Rag1−/− mice and their wildtype counterparts halts ectopic mineralization in a murine model of pseudoxanthoma elasticum. Am J Pathol. 2010;176:1855–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jansen RS, Küçükosmanoglu A, de Haas M, Sapthu S, Otero JA, Hegman IE, Bergen AA, Gorgels TG, Borst P, van de Wetering K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc Natl Acad Sci U S A. 2013;110:20206–20211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–592. [DOI] [PubMed] [Google Scholar]
- 11.Madon J, Hagenbuch B, Landmann L, Meier PJ, Stieger B. Transport function and hepatocellular localization of mrp6 in rat liver. Mol Pharmacol. 2000;57:634–641. [DOI] [PubMed] [Google Scholar]
- 12.Mortensen SP, Thaning P, Nyberg M, Saltin B, Hellsten Y. Local release of ATP into the arterial inflow and venous drainage of human skeletal muscle: insight from ATP determination with the intravascular microdialysis technique. J Physiol. 2011;589(pt 7):1847–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lomashvili KA, Narisawa S, Millán JL, O’Neill WC. Vascular calcification is dependent on plasma levels of pyrophosphate. Kidney Int. 2014;85:1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fleisch H, Russell RG, Straumann F. Effect of pyrophosphate on hydroxyapatite and its implications in calcium homeostasis. Nature. 1966;212:901–903. [DOI] [PubMed] [Google Scholar]
- 15.Rutsch F, Ruf N, Vaingankar S, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003;34:379–381. [DOI] [PubMed] [Google Scholar]
- 16.St Hilaire C, Ziegler SG, Markello TC, et al. NT5E mutations and arterial calcifications. N Engl J Med. 2011;364:432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henthorn PS, Raducha M, Fedde KN, Lafferty MA, Whyte MP. Different missense mutations at the tissue-nonspecific alkaline phosphatase gene locus in autosomal recessively inherited forms of mild and severe hypo-phosphatasia. Proc Natl Acad Sci U S A. 1992;89:9924–9928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pendleton A, Johnson MD, Hughes A, et al. Mutations in ANKH cause chondrocalcinosis. Am J Hum Genet. 2002;71:933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rutsch F, Nitschke Y, Terkeltaub R. Genetics in arterial calcification: pieces of a puzzle and cogs in a wheel. Circ Res. 2011;109:578–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Neill WC, Lomashvili KA, Malluche HH, Faugere MC, Riser BL. Treatment with pyrophosphate inhibits uremic vascular calcification. Kidney Int. 2011;79:512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rutsch F, Böyer P, Nitschke Y, et al. ; GACI Study Group. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet. 2008;1:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boraldi F, Annovi G, Bartolomeo A, Quaglino D. Fibroblasts from patients affected by Pseudoxanthoma elasticum exhibit an altered PPi metabolism and are more responsive to pro-calcifying stimuli. J Dermatol Sci. 2014;74:72–80. [DOI] [PubMed] [Google Scholar]
- 23.Boraldi F, Bartolomeo A, Li Q, Uitto J, Quaglino D. Changes in dermal fibroblasts from Abcc6(−/−) mice are present before and after the onset of ectopic tissue Mineralization. J Invest Dermatol. 2014;134:1855–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iliás A, Urbán Z, Seidl TL, Le Saux O, Sinkó E, Boyd CD, Sarkadi B, Váradi A. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6). J Biol Chem. 2002;277:16860–16867. [DOI] [PubMed] [Google Scholar]
- 25.Lazarowski ER. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal. 2012;8:359–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.