

Abstract
Nonheme diiron monooxygenases make up a rapidly growing family of oxygenases that are rarely identified in secondary metabolism. Herein, we report the in vivo, in vitro, and structural characterizations of a nonheme diiron monooxygenase, PtmU3, that installs a C-5 β-hydroxyl group in the unified biosynthesis of platensimycin and platencin, two highly functionalized diterpenoids that act as potent and selective inhibitors of bacterial and mammalian fatty acid synthases. This hydroxylation sets the stage for the subsequent A-ring cleavage step key to the unique diterpene-derived scaffolds of platensimycin and platencin. PtmU3 adopts an unprecedented triosephosphate isomerase (TIM)-barrel structural fold for this class of enzymes and possesses a noncanonical diiron active site architecture with a saturated six-coordinate iron center lacking a μ-oxo bridge. This study reveals the first member of a previously unidentified superfamily of TIM-barrel fold enzymes for metal-dependent dioxygen activation, with the majority predicted to act on CoA-linked substrates, thus expanding our knowledge of nature’s repertoire of nonheme diiron monooxygenases and TIM-barrel fold enzymes.
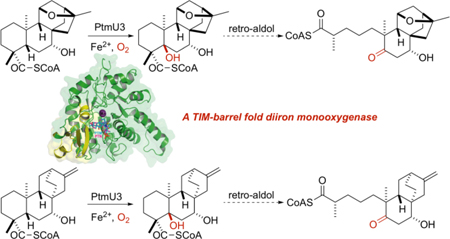
Graphical Abstarct
INTRODUCTION
Nonheme diiron monooxygenases are a growing family of oxygenases that characteristically contain a nonheme dinuclear iron center in the active site to activate molecular oxygen and play significant roles in generating diversity in bioactive small molecules. Representative enzymatic reactions catalyzed by nonheme diiron monooxygenases include methane hydroxylation, fatty acid desaturation, fatty aldehyde deformylation, and arene hydroxylation, among others.1,2 Nonheme diiron monooxygenases are also involved in the biosynthesis of secondary metabolites, albeit only rarely.3 To date, only three enzymes, acting on two natural product scaffolds, have been biochemically verified. Two of these enzymes are involved in the biosynthesis of chloramphenicol: CmlA catalyzes the β-hydroxylation of L-p-aminophenylalanine (L-PAPA) activated as a thioester that is covalently linked to the peptidyl carrier protein (PCP) CmlP, and CmlI catalyzes the 6-electron oxidation of an arylamine to the arylnitro functionality seen in the chloramphenicol scaffold (Figure 1A).4,5 The third enzyme is AurF from the aureothin biosynthetic machinery, and it resembles CmlI in that it catalyzes a 6-electron N-oxidation, though with a different substrate, p-aminobenzoate (Figure 1A).6,7 The structures of CmlI and AurF resemble the 4-helix bundle fold found in nearly every non heme diiron monooxygenase, while the structure of CmlA contains a metallo-β-lactamase protein fold unique to this family.3,6,8 The relative scarcity of known nonheme diiron monooxygenases in secondary metabolism, each of which act on very specific substrates, raises the important question of whether or not additional nonheme diiron monooxygenases are hiding in plain sight.
Figure 1. Non-heme diiron monooxygenases in secondary metabolism.

(A) Nonheme diiron monooxygenases discovered in chloramphenicol and aureothin biosynthesis. (B) Structures of PTM and PTN as well as their thioacid derivatives, thioPTM and thioPTN, produced by the engineered overproducer S. platensis SB12029. The aliphatic ketolide and 3-amino-2,4-dihydroxybenzoic acid moieties of PTM and PTN are highlighted in blue and red, respectively.
The triosephosphate isomerase (TIM)-barrel scaffold is the most common protein structural fold found in nature.9 The TIM-barrel is characterized as an α/β protein fold because the classical TIM-barrel fold consists of an eight-fold repeat of (βα) motifs. Typically, eight-stranded parallel β-sheets form the inner wall of barrel and are surrounded by α-helices. TIM-barrel enzymes are widely distributed due to their simple protein fold, but they display remarkable catalytic versatility.9 To date, at least 37 homologous superfamilies have been grouped together as TIM-barrels by CATH (Class, Architecture, Topology, and Homologous superfamily) classification.10 The majority of characterized enzymes within the superfamilies are aldolases, glycosidases, enolases, and metal-dependent hydrolases. However, no metal-dependent oxygenases, including nonheme diiron monooxygenases, have been classified in the topology level of TIM-barrel (CATH ID: 3.20.20) to date.
Platensimycin (PTM, 1) and platencin (PTN, 2) are two highly functionalized bacterial diterpenoids isolated from Streptomyces platensis.11 They are potent and selective inhibitors of bacterial and mammalian fatty acid synthases and are promising drug leads for both antibacterial and antidiabetic therapies.12,13 Structurally, PTM and PTN are composed of two distinct moieties, a 3-amino-2,4-dihydroxybenzoic acid and an aliphatic ketolide, coupled by an amide bond (Figure 1B). We previously cloned and sequenced the ptm gene cluster from a PTM and PTN dual producer S. platensis CB00739. Inactivation of the pathway-specific negative regulator ptmR1 afforded the PTM- and PTN overproducing SB12029 strain that has served as a model strain to study PTM and PTN biosynthesis (Figure S1).14,15 In light of a C-5 ketone group conserved in PTM and PTN, as well as their late-stage congeners, C-5 hydroxylation followed by a retro-aldol ring cleavage of the C-4/C-5 bond of the A-ring was proposed as one of the most intriguing structural transformations in PTM and PTN biosynthesis (Figure 2A).9,14 This ring cleavage step would be unprecedented in diterpenoid biosynthesis and therefore, the ptm gene cluster presents a unique opportunity to study this novel chemistry for both ent-kauranol (PTM) and ent-atiserene (PTN) diterpene scaffolds.
Figure 2. Characterization of PtmU3 as a C-5 β-hydroxylase.

(A) A unified pathway for PTM and PTN biosynthesis, featuring enzymes with promiscuity acting on both the ent-kauranol-derived PTM and the ent-atiserene-derived PTN scaffolds. The C-19 carboxylic acid is activated in the form of CoA esters by PtmA1 and PtmA2. PtmU3 then catalyzes C-5 β-hydroxylation (highlighted in red), which sets the stage for the ensuing A-ring cleavage at the C-4/C-5 bond via a retro-aldol reaction. In the ΔptmU3 mutant strain SB12050, the nascent products of CoA esters 7 and 11 are isolated in the free acid forms 5 and 9 due to spontaneous hydrolysis during isolation and purification. Compound 13, a precursor of 5, can also be isolated from SB12050. (B) HPLC analysis of crude extracts, with total ion current detection, of the ΔptmU3 mutant strain SB12050 (ii) with the engineered PTM and PTN overproducer SB12029 serving as a positive control (i). (C) HPLC analysis, with UV detection at 260 nm, of in vitro PtmU3-catalyzed reactions with 7 as a substrate. (D) HPLC analysis, with UV detection at 260 nm, of in vitro PtmU3-catalyzed reactions with 11 as a substrate. Std, standard.
The ptm gene cluster encodes two cytochrome P450 monooxygenases, PtmO2 and PtmO5, and two functionally redundant α-ketoglutarate-dependent dioxygenases, PtmO3 and PtmO6 (Figure S2).11,16,17 However, each has an assigned biosynthetic function, and no other genes from the ptm gene cluster could be readily predicted to be responsible for C-5 hydroxylation, thereby setting the stage to explore novel chemistry and enzymology. Herein, we describe the identification and characterization of PtmU3 as a nonheme diiron monooxygenase that plays a role in a key missing step in the biosynthesis of PTM and PTN, unveiling the first member of a new superfamily of nonheme diiron hydroxylases.
RESULTS AND DISCUSSION
Identification of Atypical Hydroxylase in ptm Gene Cluster.
In order to identify which genes were responsible for catalyzing the tailoring steps for the biosynthesis of the PTM and PTN diterpenoid scaffolds, we first set out to inactivate the remaining genes of unknown function within the ptm gene cluster in SB12029, which encodes for PTM and PTN dual production. The resultant mutant strains were fermented under our standard conditions for PTM and PTN dual production with SB12029 as a positive control.14 The timing of the C-5 hydroxylation was proposed to happen immediately following CoA thioesterification of the ketolide moieties and just prior to A-ring cleavage.11 If correct, the fermentation profile of the relevant mutant would be expected to be similar to that of the ΔptmA1 mutant, due to the hydrolysis of CoA-linked intermediates,18 i.e. fully abolishing PTM (1), PTN (2), thioPTM (3), and thioPTN (4) production,19 and instead, accumulating precursors 5 and 9, as well as 13, a precursor of 5 (Figure 2A). Upon HPLC analysis, the metabolite profile of the ΔptmU3 mutant matched this expectation (Figures 2B and S3), suggesting PtmU3 as the candidate for C-5 hydroxylation. Interestingly, ptmU3 was originally annotated as encoding a metal-dependent amidohydrolase. This superfamily is comprised of many enzymes that share a TIM-barrel structural fold.20 However, to date, no hydroxylase has been identified from this superfamily, and thus, PtmU3 likely represents the first characterized member of a new superfamily of enzymes utilizing a TIM-barrel fold.
In Vitro Characterization of PtmU3 Confirms its Role as an Iron-Dependent Enzyme Responsible for C-5 β-hydroxylation in PTM and PTN Biosynthesis.
To confirm that PtmU3 is indeed the monooxygenase responsible for C-5 hydroxylation, ptmU3 was cloned and heterologously expressed in E. coli, and the overproduced PtmU3 protein was purified to homogeneity (Figure S4A). PtmU3 is a homodimer in solution based on size-exclusion chromatography (Figure S4B). Initially, PtmU3 was incubated with platensimycin ML17 (7) or platencin ML4 (11) as substrates (Scheme S1); however, no obvious product was formed upon HPLC analysis (Figure 2C,D). Given that PtmU3 was predicted to require a metal for activity, a catalytic amount of Fe2+, with ascorbic acid as a chemical reductant, was supplemented. Using boiled PtmU3 as a control, a new peak was observed (8 or 12, Figure 2C,D) after incubation with the substrate 7 or 11, respectively.18 High-resolution mass spectrometry (HRMS) analysis revealed the molecular weights of 8 and 12 to be 1098.307 and 1082.312 Da, respectively, which are 16 Da higher than those of substrates 7 and 11, suggesting addition of a hydroxyl (Figures S5 and S6). No products were observed upon incubating PtmU3 with either the C-19 carboxylic acid, 5, or its adenylated variant, platensimycin ML16 (6), indicating that the CoA thioesters are the native substrates, i.e. PtmU3 functions immediately after the CoA ligase PtmA2 (Figure 2A). The optimized reaction conditions were determined to be 50 mM MES, pH 5.5, containing 0.1 mM Fe2+ (Figure S7A). Though Fe2+ was preferred (calculated as 100%), PtmU3 was also active with other divalent cations: Mn2+ (41.1%), Co2+ (35.4%), and Cu2+ (1.6%) (Figure S7B). No product was observed in the presence of Zn2+ or EDTA, despite Zn2+ often serving as a cofactor in the related amidohydrolase superfamily of enzymes (Figure S7B).20 To better characterize the effects of the different metals on PtmU3, pseudo-first-order kinetics were attempted; however, substrate inhibition by 7 resulted in a curve that disallowed calculation of an accurate kcat or Km (Figure S8).
To unambiguously characterize the products of PtmU3, a one-pot reaction with purified PtmA2 and PtmU3 and either 6 or 10 as a substrate, was used to prepare 8 or 12 (Scheme S2).18 The structures of products 8 and 12 were fully characterized by 1D and 2D NMR spectroscopy (Figures S9–S21). Based on the NMR data, the only difference between 7 and 8 was the presence of a hydroxyl group at C-5 (δC 78.1) in 8. HMBC correlations of Me-18, Me-20, H-3, and H-7 with C-5 supported the assignment of the hydroxyl group at C-5. In the ROESY spectrum of 8, the strong correlation of Me-18 with Hb-6, as well as no correlations of Me-18 with either H-7 or H-9 being observed, supported the β-configuration of the C-5 hydroxyl. A β-configuration of the C-5 hydroxyl in 12 was similarly assigned based on the same HMBC and ROESY correlations as those for 8 (Figure S9).
Crystal Structures of PtmU3 Reveal Unusual Diiron Binding Site in a TIM-Barrel Fold.
Given the unique chemistry catalyzed by PtmU3 and an inability to initially predict its activity bioinformatically, we set out to characterize the PtmU3 structure for a deeper understanding of its catalytic mechanism. The protein structure of PtmU3 (PDB entry: 6OMP) was solved at a resolution of 1.70 Å. Consistent with the size-exclusion chromatographic analysis, the PtmU3 structure is made up of two polypeptide chains (chains A and B) in the asymmetric unit (Figure S22). The overall structure of PtmU3 comprises of a typical TIM-barrel fold at the C-terminus (residues 1–22; 64–356) and a small N-terminal domain (residues 23–63) of a three-stranded antiparallel β-sheet flanked by two α-helices (Figure 3A). Although the TIM-barrel structure is the most common fold found in nature, making up approximately 10% of all proteins and catalyzing a wide variety of enzymatic reactions,9,21 to the best of our knowledge, PtmU3 is the first characterized TIM-barrel hydroxylase. Intriguingly, there is no significant homologue of the small N-terminal domain by DALI search, though many TIM barrel-containing enzymes do integrate additional folds.9,22 The closest structural homologue of PtmU3 in the PDB is a putative TIM-barrel metal-dependent hydrolase from Mycobacterium avium (PDB: 4DZI, ∼23% identity); however, this enzyme has yet to be biochemically characterized (Figure S23).
Figure 3. Crystal structures of PtmU3.

(A) Overall structure of PtmU3 monomer. PtmU3 is composed of a small N-terminal domain (yellow) and a C-terminal TIM-barrel fold (green). (B) The nonheme diiron center in PtmU3. Two metal ions are coordinated with the side chains of seven residues (3 His, 2 Asp, and 2 Glu) and a water molecule (yellow dashed lines). (C) Active site of PtmU3 complexed with 7. (D) Active site of PtmU3 complexed with 11. The active sites of PtmU3 complexed with 7 and 11 are very similar. The two substrates (7 and 11) align well and are depicted in cyan and magenta, respectively. The two metal ions are shown in purple. The distances between the two metals or water and C-5 of 7 and 11 are shown as orange dashed lines. Hydrogen bonds are shown as brown dashed lines. 2Fo-Fc electron density maps of substrates 7 and 11 contoured at 1σ are depicted with grey mesh.
Two metal ions (manganese in the crystal structures) are located at the top of the PtmU3 β-barrel center and are coordinated with the side chains of seven residues (Asp10, His12, His189, Glu241, Asp308, His311, and Glu313) and a water molecule (Figure 3B). The distance between the two metals is 3.8 Å (within the normal range of 2.7 – 4.8 Å for previously characterized diiron enzymes).2 Each of the metals form six coordinations with residues and/or water in a distorted octahedral geometry with distances ranging from 2.0 – 2.5 Å. Glu241 and Asp308 act as bridging ligands between the two metal centers instead of the more common μ-oxo or μ-peroxo bridging ligand. Mutagenesis studies showed that Glu 241 was essential and the E241A mutant completely lost its enzymatic activity, indicating that the metals are crucial for the function of PtmU3 (Figure S4C). To probe the saturated M1 center, the D10A, H189A, and D10A/H189A mutants were similarly generated, but no soluble proteins were obtained, putatively due to a structural rather than catalytic role played by M1. Notably, the structural homologue from M. avium (PDB: 4DZI) also has a saturated M1 center.
This saturated M1 center provides a distinction from other characterized diiron monooxygenases, whether they utilize one or both metal centers for catalysis. Detailed mechanistic studies of the diiron arylamine oxygenases AurF and CmlI have shown that they utilize both irons to activate a peroxo intermediate;23–25 however, the organization of the metal centers makes this mechanism unlikely for PtmU3. Conspicuously, there are other diiron enzymes that catalyze oxygen activation using only one of the two irons: the myo-inositol oxygenase MIOX and the organophosphonate oxidase PhnZ.26–27 However, in both of these enzymes, the second iron is utilized to bind their respective substrates, a role clearly not filled by M1 in PtmU3 (Figure 3). Therefore, the combination of mutagenesis, structural data, and literature precedent provides evidence that M1 may act in a structural role rather than a catalytic one.
Though structurally similar to characterized members of the amidohydrolase superfamily, the metal binding site of PtmU3 is distinct from that of any amidohydrolase in large part due to the saturated M1.20 While it is possible that PtmU3 or its ancestors evolved from a member of the amidohydrolase superfamily, it may be that to serve as a hydroxylase rather than a hydrolase, this new superfamily needed to shift from two metals to a single catalytic metal, as has been shown for a pair of amidohydrolases recently evolved for synthetic substrates.28
We next solved structures of PtmU3 complexed with substrates 7 and 11 (PDB entries: 6OMQ and 6OMR, respectively) to further probe the active site and the interactions between substrates and the enzyme. Both substrates adopt similar orientations in the active site, providing a structural rationale for stereospecific C-5 β-hydroxylation of both PTM and PTN (Figure 3A). Leu93 and Ile190 hydrophobically interact with the ent-kauranol and ent-atiserene moieties; however, most of the strong interactions are with the CoA moiety. The two substrate-bound structures suggest that the primary role of the novel N-terminal domain is to provide a binding site for CoA. Arg52 (located in the N-terminal domain) and Asn208 hydrogen bond with the two amide carbonyls of the β-alanine and pantoic acid regions of CoA, respectively (Figure 3C,D). This model would be consistent with the fact that the R52A mutant lost about 60% of its activity in vitro, in comparison to the native PtmU3 (Figure S4C). The adenosine 3’,5’-bisphosphate moiety is missing from the substrates in the two structures, likely attributed to its substantial flexibility due to its exposure to the solvent.
The distances between the C-5 carbon of 7 or 11 and the M2-coordinated waters in the complexed structures are 3.4 and 3.2 Å, respectively (Figure 3C,D). During catalysis, it is expected that the water molecule is replaced by molecular oxygen. Additionally, only one active site (in chain B) was occupied in each of the substrate-complexed structures. This could result from the structural differences between the two polypeptide chains during crystal packing, as the major conformational differences between the two chains in the asymmetric unit are within the small N-terminal domain (residues 23–63) and a long loop (residues 193–200) near the substrate binding site (Figure S24). Finally, we constructed the T196A mutant to probe the role of the loop rearrangement, but no change in activity was observed relative to native PtmU3 (Figures S4C and S23), a finding that would suggest that this loop rearrangement is not necessary for catalysis.
PtmU3 Homologues are Widespread in Actinobacteria.
The novel chemistry, enzymology, and structure of PtmU3 in catalyzing a key step in PTM and PTN biosynthesis prompted us to ask how widespread PtmU3-like proteins are found in nature. Remarkably, a sequence similarity network revealed 142 homologues that are broadly distributed in nature (Figure 4, based upon a survey of the NCBI database as of May 2019). While PtmU3 represents the first enzyme characterized from this cluster, a BLAST search revealed >50% protein sequence identity to PtmU3 for all homologues. Except for two homologues from Alphaproteobacteria, all remaining homologues are from Actinobacteria. The low taxonomic diversity may be a sign that PtmU3 and its homologues have evolved relatively recently, possibly from amidohydrolases. Significantly, all homologues have the seven conserved residues (3 His, 2 Asp, and 2 Glu) that are responsible for binding the diiron center as exemplified by PtmU3 (Figure S25), and the residues responsible for binding CoA also appear to be widely conserved (Arg52, Arg53, Arg200, and Asn208). Together with the fact that around half (74 of 142) of the homologues have putative CoA ligases encoded in genetic proximity (Table S6), these homologues may also prefer CoA-linked substrates. As highlighted by the set of representative gene clusters encoding PtmU3 homologues shown in Figure S26, this study should inspire future utilization of this novel class of nonheme diiron monooxygenase as a beacon to mine genomes for discovering novel natural products.
Figure 4. Broad distribution of PtmU3-like nonheme diiron monooxygenases in nature.

(A) Sequence similarity network (SSN) of PtmU3-like proteins in bacteria.29–30 A BLAST e-value threshold of 10–90 was employed. Colors represent different bacterial phylogenetic classes. The cluster containing 142 PtmU3-like nonheme diiron monooxygenases is circled. (B) A subnetwork of putative PtmU3-like nonheme diiron monooxygenases from (A) with a BLAST e-value threshold of 10–140 (median 53% sequence identity). Six clusters and four singletons were separated when the e-value threshold of 10–140 was used. The corresponding accession numbers are listed in Table S6. PtmU3 and its homologue from the PTN pathway, PtnU3, are depicted as red triangles.
CONCLUSIONS
In summary, PtmU3 has been biochemically and structurally characterized as a nonheme diiron monooxygenase that installs a C-5 β-hydroxyl group. Therefore, the stage is set for a retro-aldol reaction resulting in the key A-ring cleavage of the C-4/C-5 bond to afford the characteristic aliphatic ketolide moieties in PTM and PTN biosynthesis. We have elucidated the biosynthetic timing of this step and provided evidence of in vitro and in vivo activity of PtmU3 consistent with this role. The complex structures of PtmU3 with both substrates reveal that the C-5 carbons of both substrates are adjacent to the nonheme diiron center in the active site, thereby supporting PtmU3 as a C-5 hydroxylase for both the ent-kauranol-derived PTM and ent-atiserene-derived PTN diterpenoid scaffolds. Thus, we have also provided a molecular basis for recognition of two divergent CoA-linked substrates. Through this biochemical characterization, we have shed new light into diterpenoid biosynthesis, especially for those of bacterial origin.
Structurally, PtmU3 adopts an unprecedented TIM-barrel structural fold with a novel CoA-binding N-terminal domain, possessing a noncanonical active site architecture with one saturated iron center (M1) that lacks a μ-oxo or μ-peroxo bridge as commonly found in the family of nonheme diiron monooxygenases (Figure S27).2 Mutagenic studies were performed in parallel with crystallographic studies that demonstrate that the M1 center may play a structural role rather than a catalytic one. Thus, we have demonstrated that the divergent structure and chemistry of PtmU3 entrenches it as the first characterized member of a new superfamily of TIM-barrel fold metal-dependent oxidoreductases. Further bioinformatic analysis has shown that PtmU3-like enzymes are widespread, found primarily in Actinobacteria. With the extraordinary prevalence of the TIM-barrel fold and the specificity, as revealed from PtmU3, for CoA-linked substrates, it is likely that this type of chemistry, enzymology, and biosynthetic logic exist in other taxonomic classes for more diverse natural product pathways. Together, the PtmU3-catalyzed key step for initiation of the A-ring cleavage in PTM and PTN biosynthesis discovered in this study has provided a solid foundation, and will surely inspire further effort, to investigate the novel dioxygen activation mechanisms mediated by a noncanonical diiron center in the TIM-barrel fold.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported, in part, by the National Institutes of Health Grant GM094585 (A.J.), GM098248 (G.N.P.), and GM114353 (B.S.). Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center (SBC) at the Advanced Photon Source. SBC-CAT is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02–06CH11357. J.D.R. is supported, in part, by an Arnold O Beckman Postdoctoral Fellowship. M.-R.D. is supported, in part, by Guangdong Institute of Microbiology, Guangzhou, Guangdong, P. R. China, and a scholarship (2017GDASCX-0502) from Guangdong Academy of Science, Guangzhou, P. R. China. This is manuscript #29844 from The Scripps Research Institute.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website.
Materials, methods, detailed experimental procedures, bioinformatic analysis, in vivo, in vitro, and structural characterizations of PtmU3, and structural elucidation for 8 and 12 (PDF).
REFERENCES
- 1.Banerjee R; Jones JC; Lipscomb JD, Soluble methane monooxygenase. Annu. Rev. Biochem 2019, 88, 409–431. [DOI] [PubMed] [Google Scholar]
- 2.Jasniewski AJ; Que L, Dioxygen activation by nonheme diiron enzymes: Diverse dioxygen adducts, high-valent intermediates, and related model complexes. Chem. Rev 2018, 118, 2554–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Komor AJ; Jasniewski AJ; Que L Jr.; Lipscomb JD, Diiron monooxygenases in natural product biosynthesis. Nat. Prod. Rep 2018, 35, 646–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makris TM; Chakrabarti M; Munck E; Lipscomb JD, A family of diiron monooxygenases catalyzing amino acid beta-hydroxylation in antibiotic biosynthesis. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 15391–15396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu H; Chanco E; Zhao H, CmlI is an N-oxygenase in the biosynthesis of chloramphenicol. Tetrahedron 2012, 68, 7651–7654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi YS; Zhang H; Brunzelle JS; Nair SK; Zhao H , In vitro reconstitution and crystal structure of p-aminobenzoate N-oxygenase (AurF) involved in aureothin biosynthesis. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 6858–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He J; Hertweck C, Biosynthetic origin of the rare nitroaryl moiety of the polyketide antibiotic aureothin: Involvement of an unprecedented N-oxygenase. J. Am. Chem. Soc 2004, 126, 3694–3695. [DOI] [PubMed] [Google Scholar]
- 8.Makris TM; Knoot CJ; Wilmot CM; Lipscomb JD, Structure of a dinuclear iron cluster-containing β-hydroxylase active in antibiotic biosynthesis. Biochemistry 2013, 52, 6662–6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagano N; Orengo CA; Thornton JM, One fold with many functions: The evolutionary relationships between TIM barrel families based on their sequences, structures and functions. J. Mol. Biol 2002, 321, 741–765. [DOI] [PubMed] [Google Scholar]
- 10.Dawson NL; Lewis TE; Das S; Lees JG; Lee D; Ashford P; Orengo CA; Sillitoe I, CATH: an expanded resource to predict protein function through structure and sequence. Nucleic Acids Res 2017, 45, D289–D295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rudolf JD; Dong L-B; Shen B, Platensimycin and platencin: Inspirations for chemistry, biology, enzymology, and medicine. Biochem. Pharmacol 2017, 133, 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J; Kodali S; Lee SH; Galgoci A; Painter R; Dorso K; Racine F; Motyl M; Hernandez L; Tinney E; Colletti SL; Herath K; Cummings R; Salazar O; Gonzalez I; Basilio A; Vicente F; Genilloud O; Pelaez F; Jayasuriya H; Young K; Cully DF; Singh SB, Discovery of platencin, a dual FabF and FabH inhibitor with in vivo antibiotic properties. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 7612–7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J; Soisson SM; Young K; Shoop W; Kodali S; Galgoci A; Painter R; Parthasarathy G; Tang YS; Cummings R; Ha S; Dorso K; Motyl M; Jayasuriya H; Ondeyka J; Herath K; Zhang C; Hernandez L; Allocco J; Basilio A; Tormo JR; Genilloud O; Vicente F; Pelaez F; Colwell L; Lee SH; Michael B; Felcetto T; Gill C; Silver LL; Hermes JD; Bartizal K; Barrett J; Schmatz D; Becker JW; Cully D; Singh SB, Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 2006, 441, 358–361. [DOI] [PubMed] [Google Scholar]
- 14.Rudolf JD; Dong L-B; Huang T; Shen B, A genetically amenable platensimycin- and platencin-overproducer as a platform for biosynthetic explorations: a showcase of PtmO4, a long-chain acyl-CoA dehydrogenase. Mol. BioSyst 2015, 11, 2717–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hindra; Huang T; Yang D; Rudolf JD; Xie P; Xie G; Teng Q; Lohman JR; Zhu X; Huang Y; Zhao L-X; Jiang Y; Duan Y; Shen B, Strain prioritization for natural product discovery by a high-throughput real-time PCR method. J. Nat. Prod 2014, 77, 2296–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong L-B; Zhang X; Rudolf JD; Deng M-R; Kalkreuter E; Cepeda AJ; Renata H; Shen B, Cryptic and stereospecific hydroxylation, oxidation, and reduction in platensimycin and platencin biosynthesis. J. Am. Chem. Soc 2019, 141, 4043–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudolf JD; Dong L-B; Zhang X; Renata H; Shen B, Cytochrome P450-catalyzed hydroxylation initiating ether formation in platensimycin biosynthesis. J. Am. Chem. Soc 2018, 140, 12349–12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang N; Rudolf JD; Dong L-B; Osipiuk J; Hatzos-Skintges C; Endres M; Chang C-Y; Babnigg G; Joachimiak A; Phillips JGN; Shen B, Natural separation of the acyl-CoA ligase reaction results in a non-adenylating enzyme. Nat. Chem. Biol 2018, 14, 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong L-B; Rudolf JD; Kang D; Wang N; He CQ; Deng Y; Huang Y; Houk KN; Duan Y; Shen B, Biosynthesis of thiocarboxylic acid-containing natural products. Nat. Commun 2018, 9, 2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seibert CM; Raushel FM, Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry 2005, 44, 6383–6391. [DOI] [PubMed] [Google Scholar]
- 21.Goldman AD; Beatty JT; Landweber LF, The TIM barrel architecture facilitated the early evolution of protein-mediated metabolism. J. Mol. Evol 2016, 82, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holm L; Laakso LM, Dali server update. Nucleic Acids Res 2016, 44, W351–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park K; Li N; Kwak Y; Srnec M; Beli CB; Liu LV; Wong SD; Yoda Y; Kitao S; Seto M; Hu M; Zhao J; Krebs C; Bollinger JM; Solomon EI, Peroxide activation for electrophilic reactivity by the binuclear non-heme iron enzyme AurF. J. Am. Chem. Soc 2017, 139, 7062–7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jasniewski AJ; Komor AJ; Lipscomb JD; Que L, Unprecedented (μ−1,1-peroxo)diferric structure for the ambiphilic orange peroxo intermediate of the nonheme N-oxygenase CmlI. J. Am. Chem. Soc 2017, 139, 10472–10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang C; Chen H, Convergent theoretical prediction of reactive oxidant structures in diiron arylamine oxygenases AurF and CmlI: peroxo or hydroperoxo? J. Am. Chem. Soc 2017, 139, 13038–13046. [DOI] [PubMed] [Google Scholar]
- 26.Brown PM; Caradoc-Davies TT; Dickson JMJ; Cooper GJS; Loomes KM; Baker EN, Crystal structure of a substrate complex of myo-inositol oxygenase, a di-iron oxygenase with a key role in inositol metabolism. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 15032–15037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Staalduinen LM; McSorley FR; Schiessl K; Séguin J; Wyatt PB; Hammerschmidt F; Zechel DL; Jia Z, Crystal structure of PhnZ in complex with substrate reveals a di-iron oxygenase mechanism for catabolism of organophosphonates. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 5171–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sugrue E; Fraser NJ; Hopkins DH; Carr PD; Khurana JL; Oakeshott JG; Scott C; Jackson CJ, Evolutionary expansion of the amidohydrolase superfamily in response to synthetic compounds molinate and diuron. Appl. Environ. Microbiol 2015, 81, 2612–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerlt JA; Bouvier JT; Davidson DB; Imker HJ; Sadkhin B; Slater DR; Whalen KL, Enzyme function initiative-enzyme similarity tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shannon P; Markiel A; Ozier O; Baliga NS; Wang JT; Ramage D; Amin N; Schwikowski B; Ideker T, Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 2003, 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.