

Abstract
The yeast high-osmolarity glycerol (HOG) stress-activated protein kinase Hog1 is activated in response to hyperosmotic stress, inducing the production and retention of glycerol to restore osmotic balance. Hog1 promotes retention of glycerol through closure of the plasma-membrane glycerol channel Fps1. Treatment of yeast with the toxic trivalent metalloid arsenite (As(III)) also activates Hog1 as part of a protective response in which Hog1 closes Fps1, the main entry port for As(III). In this study, we investigated how cells treated with As(III) avoid creating a new stress caused by the accumulation of glycerol in the absence of hyperosmotic stress conditions. We found that As(III) treatment did not induce glycerol accumulation and, in fact, blocked the accumulation of glycerol induced by constitutive Hog1 activity. We demonstrated that As(III) blocks glycerol production indirectly after its metabolic activation to methylarsenite (MAs(III)), which is a potent inhibitor of glycerol-3-phosphate dehydrogenase. Finally, we used a biotinylated arsenic probe to establish that Cys306 of yeast Gpd1, a highly conserved residue within the active site, is the key target of MAs(III). Conservative mutations at this residue greatly diminished Gpd1 activity. This study offers insight into mechanisms by which SAPK outputs are tailored to specific stressors.
INTRODUCTION
Arsenic is the most prevalent toxin in the environment (Rosen and Liu, 2009). Its ubiquitous presence has driven the evolution of arsenic resistance mechanisms, which exist in nearly every organism (Yang and Rosen, 2016). Human exposure to arsenic is mainly through food, water, and air, and contamination of groundwater is a worldwide health problem (Smedley and Kinniburgh, 2002; Naujokas et al., 2013). Inorganic aqueous arsenic exists mainly as oxyanions of trivalent arsenite (As(III)) and pentavalent arsenate (As(V)). Chronic exposure to inorganic arsenic is associated with cardiovascular disease and hypertension, diabetes mellitus, neurological disorders, and various forms of cancer (Abernathy et al., 2003; Beane Freeman et al., 2004; Naujokas et al., 2013). Despite these health effects, As(III) is in current use as a highly effective treatment for acute promyelocytic leukemia (Rehman and Naranmandura, 2012; Kozono et al., 2018). Therefore, it is important to understand the cellular responses mobilized by arsenic-induced stress.
In mammalian cells, As(III) enters through the aquaglyceroporins and the glucose permeases (Maciaszczyk-Dziubinska et al., 2012). It can be transported out of the cell directly, or after conjugation with glutathione, by various ABC family transporters. Additionally, As(III) is metabolized in mammals by As(III) S-adenosylmethionine (SAM) methyltransferase (AS3MT), which catalyzes the transfer of methyl groups from SAM to As(III) to produce methylarsenite (MAs(III)) and dimethylarsenite (DMAs(III)) (Ren et al., 2011; Cullen, 2014; Dheeman et al., 2014; Dong et al., 2015), both of which are more toxic than inorganic As(III) (Dong et al., 2015).
In the yeast Saccharomyces cerevisiae, As(III) enters the cell principally through the aquaglyceroporin Fps1, a bidirectional channel that normally functions to transport glycerol (Wysocki et al., 2001), and secondarily through the hexose permeases (Maciaszczyk-Dziubinska et al., 2012). As(III) is actively transported out of the yeast cell through the plasma membrane metalloid/H+ antiporter Acr3 (Maciaszczyk-Dziubinska et al., 2011). Alternatively, As(III) can be conjugated to glutathione and sequestered in the yeast vacuole through ABC family transporters Ycf1 and Vmr1 (Maciaszczyk-Dziubinska et al., 2012). Metabolic conversion of As(III) to MAs(III) by the dimeric methyltransferase Mtq2:Trm112 was demonstrated recently (Lee and Levin, 2018).
With regard to signaling, As(III) stimulates the mammalian stress-activated protein kinase (SAPK) p38 (Elbirt et al., 1998; Verma et al., 2002). The yeast SAPK Hog1 of the high-osmolarity glycerol (HOG) pathway is similarly activated in response to As(III) treatment and plays an important role in tolerance for As(III) (Sotelo and Rodríguez-Gabriel, 2006; Thorsen et al., 2006). Activation of Hog1 by As(III) occurs through an indirect route that involves its metabolic activation to MAs(III) (Lee and Levin, 2018). MAs(III) inhibits the tyrosine-specific protein phosphatases Ptp2 and Ptp3, which normally maintain Hog1 in a low-activity state. Active Hog1 protects cells from As(III) toxicity in large part by inducing closure of Fps1 to prevent its entry (Thorsen et al., 2006). This occurs through Hog1 phosphorylation of regulators of the glycerol channel, Rgc1 and Rgc2, which drives their displacement from Fps1 (Lee et al., 2013; Lee and Levin, 2018).
This study was motivated by our desire to understand the mechanisms by which various stress signals that activate a common SAPK elicit stress-specific outputs under the control of the activated protein kinase. We expected that comparison of hyperosmotic stress and As(III) stress, both of which activate Hog1, would provide an instructive example. Under conditions of hyperosmotic stress, Hog1 is activated by signals from the cell surface. Its major role under these conditions is to restore osmotic balance by driving the production and accumulation of intracellular glycerol as an osmolyte. Hog1 controls glycerol accumulation in several ways, including transcriptional induction of genes for glycerol production and import (e.g., GPD1, GPP1, GPP2, and STL1), posttranslational modification of enzymes important for glycerol biosynthesis (e.g., Gpd1 and Pfk2), and closure of Fps1 (Hohmann, 2009; Lee et al., 2013; Brewster and Gustin, 2014). However, glycerol accumulation in the absence of high external osmolarity increases turgor, which causes cell wall stress (Beese et al., 2009). Here, we address the question of how cells avoid creating cell wall stress from glycerol accumulation during their response to As(III). We found that As(III) activation of Hog1, unlike hyperosmotic stress, does not induce the accumulation of glycerol. Glycerol accumulation on As(III) treatment, after its conversion to MAs(III), is prevented through direct inhibition of glycerol-3-phosphate dehydrogenase, the first step in glycerol biosynthesis from glycolysis intermediates.
RESULTS AND DISCUSSION
Arsenite treatment blocks Hog1-driven glycerol accumulation
Yeast cells restrict entry of As(III) by closure of Fps1 (Thorsen et al., 2006; Lee and Levin, 2018). However, mutants that are impaired for glycerol efflux through Fps1 accumulate high levels of intracellular glycerol, which causes considerable cell wall stress (Beese et al., 2009). Thus, we were interested in understanding how the cell addresses the problem of preventing As(III) entry while not creating cell wall stress through the accumulation of glycerol.
We first examined the effect of As(III) treatment on glycerol accumulation. Unlike treatment of cells with a brief hyperosmotic shock (1 M NaCl for 10 min), treatment with 1 mM As(III) for 10 or 30 min did not detectably elevate intracellular glycerol (Figure 1), despite observations that Hog1 is activated and Fps1 is closed rapidly in response to As(III) treatment (Lee and Levin, 2018). This finding suggested that As(III) may play an important role in the prevention of glycerol accumulation. To test this hypothesis, we examined the effect of As(III) treatment on glycerol accumulation in a ptp2Δ ptp3Δ mutant, which displays constitutively high Hog1 activity (Wurgler-Murphy et al., 1997; Mattison and Ota, 2000) and consequent Fps1 closure and glycerol accumulation (E.V. Laz and D.E.L., unpublished data). We chose this mutant because As(III) treatment activates Hog1 through indirect inhibition of Ptp2 and Ptp3 tyrosine phosphatase activity against Hog1 (Lee and Levin, 2018). Thus, a ptp2Δ ptp3Δ mutant mimics the activation of Hog1 by As(III). We found that As(III) treatment reduced the level of intracellular glycerol in a ptp2Δ ptp3Δ mutant to nearly wild-type levels within 30 min (Figure 1), supporting the hypothesis that As(III) blocks Hog1-driven glycerol accumulation. It should be noted that this experiment required the use of higher doses of As(III) than would be used for wild-type cells (4 mM rather than 1 mM), because its entry is less efficient when Fps1 is closed (Lee and Levin, 2018).
FIGURE 1:

Impact of As(III) treatment on intracellular glycerol accumulation. Wild-type cells (DL2772) were treated for 10 or 30 min with 1 mM As(III) or 10 min with 1 M NaCl, or were untreated (Cont.), and intracellular glycerol levels were measured. A ptp2Δ ptp3Δ strain (DL4299) was used to test the impact of 4 mM As(III) treatment for the indicated times on the elevated basal level of intracellular glycerol. A high concentration of As(III) was used in the ptp2Δ ptp3Δ strain because its uptake is impaired by the constitutive closure of Fps1 in this mutant. Values are means and SD from three independent cultures.
Methylarsenite binds to Gpd1
Trivalent arsenicals bind covalently to cysteine thiols in proteins (Shen et al., 2013). One way in which As(III) treatment might prevent glycerol accumulation is through the direct binding of As(III) to an enzyme required for glycerol biosynthesis. In yeast, glycerol is produced in two steps from the glycolytic intermediate dihydroxyacetone phosphate (DHAP). The glycerol-3-phosphate dehydrogenases (Gpd1 and Gpd2) catalyze the first reaction, converting DHAP to glycerol-3-phosphate. The glycerol-3-phosphate phosphatases (Gpp1 and Gpp2) then convert glycerol-3-phosphate to glycerol (Hohmann, 2009). A study of trivalent arsenic binding to human proteins identified 360 target proteins (Zhang et al., 2015). Although not pursued in that study, human GPD1 was identified as among the proteins binding most strongly to an arsenite–biotin conjugate probe (biotinylated p-aminophenyl arsenic acid; As–biotin; Figure 2A). Therefore, we tested yeast Gpd1 binding to As–biotin. We expressed Gpd1-TAP (tandem-affinity purification) in wild-type cells and treated them in vivo with As–biotin with, or without, a 10-min pretreatment with As(III). Extracts were subjected to a pull down with streptavidin–agarose beads, followed by immunoblot analysis for Gpd1-TAP. We found that As–biotin bound to Gpd1-TAP and that pretreatment with As(III) attenuated binding (Figure 2B).
FIGURE 2:

MAs(III) binds to Gpd1. (A) The structure of the As–biotin conjugate used. (B) In vivo binding of As–biotin to Gpd1-TAP is diminished by pretreatment with As(III). A wild-type strain (DL3187) transformed with a plasmid expressing Gpd1-TAP under the inducible control of the GAL1 promoter (p3467) was pretreated for 10 min with, or without, 1 mM As(III) before a 10-min treatment with 10 μM As–biotin. Extracts were subjected to affinity pull down with streptavidin agarose (SA) beads before SDS–PAGE and immunoblot analysis for Gpd1-TAP. Molecular mass markers (in kDa) are on the right. (C) In vivo binding of As–biotin to Gpd1-TAP in the absence of MTQ2 is blocked by pretreatment with MAs(III), but not As(III). An mtq2Δ strain (DL4313) transformed with p3467 was pretreated for 10 min with either 1 mM As(III) or 0.5 mM MAs(III) before As–biotin treatment, as above. (D) Comparison of As–biotin binding to Gpd1-TAP in wild type and mtq2Δ mutant. The same strains as above were used to compare relative pull down of Gpd1-TAP by As–biotin. (E) As–biotin treatment activates Hog1. The same strains as above were used to test the ability of As–biotin to activate Hog1. Cells were treated with the indicated concentrations of As–biotin for 10 min before SDS–PAGE and immunoblot analysis of extracts for active Hog1 (P-Hog1) and total Hog1. Molecular mass markers (in kDa) are on the right.
Because As(III) is metabolized to MAs(III), and we have found that MAs(III), but not As(III), inhibits the Ptp2 and Ptp3 phosphatases (Lee and Levin, 2018), we asked whether the in vivo interaction of Gpd1 with As(III) was direct, or similarly required its conversion to MAs(III). To address this question, we conducted another As–biotin pull down of Gpd1-TAP in an mtq2Δ mutant strain, which is blocked for conversion of As(III) to MAs(III) (Lee and Levin, 2018). In this setting, As(III) pretreatment failed to block in vivo As–biotin binding to Gpd1-TAP (Figure 2C). However, MAs(III) pretreatment attenuated the binding of As–biotin to Gpd1-TAP, suggesting both that MAs(III) is the biologically active molecule that binds to Gpd1 and that the phenylarsine of As–biotin mimics MAs(III), at least with regard to its ability to bind Gpd1.
It is possible that As–biotin is modified in vivo to a methylated form by Mtq2. However, we found that an As–biotin pull down of Gpd1-TAP gave equivalent signals from wild-type cells and the mtq2Δ mutant (Figure 2D), suggesting that the phenylarsine itself, rather than a methylated metabolite, mimics MAs(III). As a further test of the ability of As–biotin to mimic MAs(III), we examined its ability to activate Hog1 in vivo, which is activated in an mtq2Δ mutant by MAs(III), but not by As(III) (Lee and Levin, 2018). We found that treatment with As–biotin activated Hog1 to comparable levels in both wild-type cells and the mtq2Δ mutant (Figure 2E), indicating that As–biotin is capable of inhibiting the Ptp2 and Ptp3 phosphatases similarly to MAs(III), and that no metabolism of As–biotin by Mtq2 is required for this activity. Thus, As–biotin mimics the behavior of MAs(III) with regard to inhibition of the Ptp phosphatases and binding to Gpd1-TAP.
Gpd1 activity is inhibited by MAs(III)
Because MAs(III) and As–biotin bound to Gpd1, we tested the ability of these toxins to inhibit Gpd1 activity. We overexpressed Gpd1-TAP under the inducible control of the GAL1 promoter in a gpd1Δ gpd2Δ strain and measured glycerol-3-phosphate dehydrogenase activity in crude extracts (Gancedo et al., 1968). Robust Gpd1 activity was detected from this construct, whereas virtually no activity was detected in extracts of the gpd1Δ gpd2Δ strain bearing empty vector. We found that As(III) was a poor inhibitor of Gpd1 activity—it displayed little inhibition at 10 μm and required 1 mM to inhibit 85% of the activity (Table 1). In contrast to this, MAs(III) inhibited Gpd1 activity nearly completely at 10 μM, revealing that MAs(III) is at least 100-fold more effective than As(III) at inhibiting this enzyme. As–biotin at 1 μM also inhibited Gpd1 activity nearly completely. These results are consistent with our findings that these agents bind to Gpd1, whereas As(III) binding was not detected. Because these enzyme assays were conducted in crude extracts, the weak inhibition observed by As(III) might be the result of in vitro production of MAs(III) from As(III). This was tested using an mtq2Δ mutant background, which is blocked for production of MAs(III). The weak inhibition of Gpd1 activity by As(III) remained in extracts of the mtq2Δ mutant, indicating that the observed inhibition is not the consequence of metabolic conversion to MAs(III) (Table 1).
TABLE 1:
Inhibition of glycerol-3-phosphate dehydrogenase activity by As(III), MAs(III), and As–biotin.
| Activity (%) | ||
|---|---|---|
| Inhibitor | gpd1Δgpd2Δ | mtq2Δ |
| None | 100 | 100 |
| As(III) 10 μM | 81 ± 5.3 | NT |
| As(III) 100 μM | 51 ± 3.4 | 65 ± 3.6 |
| As(III) 1 mM | 15 ± 1.2 | 9.0 ± 1.0 |
| MAs(III) 1 μM | 55 ± 3.7 | NT |
| MAs(III) 5 μM | 23 ± 1.1 | NT |
| MAs(III) 10 μM | 4.0 ± 0.3 | NT |
| As–biotin 1 μM | 2.7 ± 0.3 | NT |
Glycerol-3-phosphate dehydrogenase activity was measured in a gpd1Δ gpd2Δ strain (DL4285) or an mtq2Δ strain (DL4313) expressing Gpd1-TAP borne on a plasmid (p3467) and under the control of the inducible GAL1 promoter. NT: not tested. Values are the mean and SD from three independent experiments.
We next assessed which cysteine residues within Gpd1 were targets of trivalent arsenicals. Yeast Gpd1 has three cysteine residues that are conserved in human GPD1 (Cys134, Cys241, and Cys306; yeast numbering). One of these (Cys306) is also conserved in yeast Gpd2. Five additional Cys residues are conserved between yeast Gpd1 and Gpd2 (Cys150, Cys176, Cys252, Cys344, and Cys354; Gpd1 numbering), but not in human GPD1. We created Cys-to-Ser mutations at each of these cysteine residues in the Gpd1-TAP construct and tested the effect of these mutations on As–biotin binding. All of the mutant forms were expressed at similar levels. The strongest effect was observed for the Gpd1-C306S form, for which As–biotin binding was nearly completely eliminated (Figure 3A). It is interesting to note that this is the only cysteine residue that is conserved among yeast Gpd1, Gpd2, and human GPD1. However, the Gpd1-C134S form also displayed a reproducible decrease in As–biotin binding. Therefore, we combined these two mutations. Binding of the Gpd1-C134S, C306S form to As–biotin was below the level of detection (Figure 3B), suggesting that Cys306 is the principal target of trivalent arsenic and that Cys134 is also a target. Mutations at the Cys residues conserved between yeast Gpd1 and Gpd2, but not in human GPD1 did not display detectable changes in As–biotin binding (Supplemental Figure S1).
FIGURE 3:
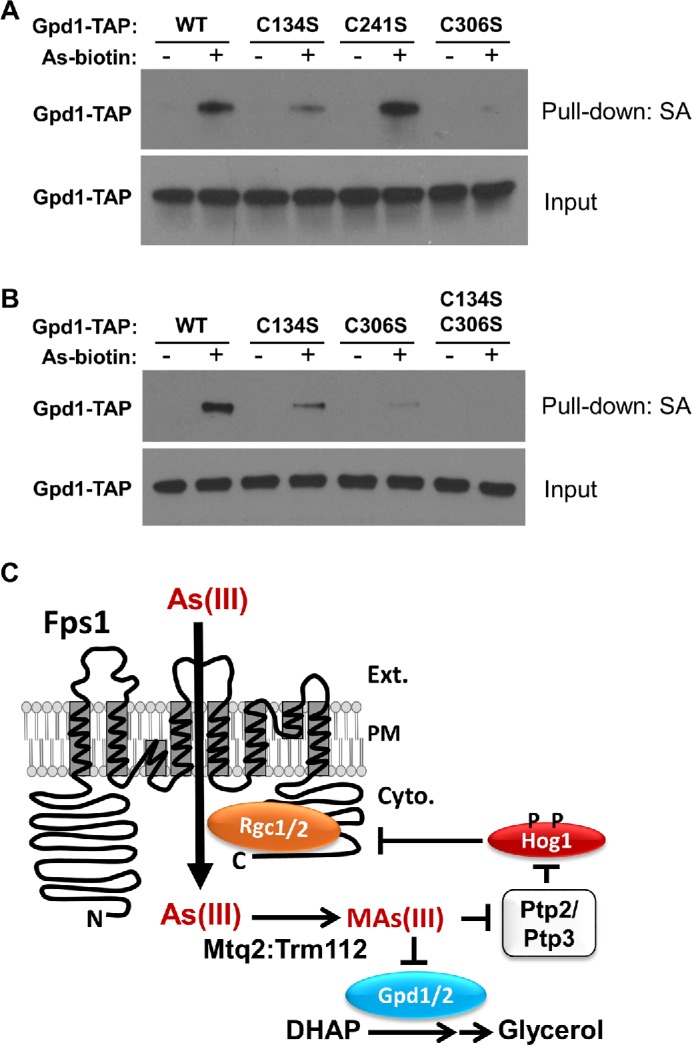
As–biotin pull down of Gpd1 Cys–Ser mutants. (A) A gpd1Δ gpd2Δ mutant (DL4285) transformed with plasmids expressing Gpd1-TAP forms under the inducible control of the GAL1 promoter (WT, p3467; C134S, p3494; C241S, p3495; C306S, p3496) was treated for 10 min with 10 μM As–biotin. Extracts were subjected to affinity pull down with streptavidin agarose (SA) beads before SDS–PAGE and immunoblot analysis for Gpd1-TAP. (B) The same strain expressing Gpd1-TAP forms was treated as described, including the C134S C306S double mutant (p3498). (C) Model for the As(III)-induced mechanism by which the cell coordinately closes the glycerol channel (Fps1) to restrict As(III) entry while blocking glycerol production through inhibition of the glycerol-3-phosphate dehydrogenases (Gpd1 and Gpd2).
We tested the Gpd1-C134S and Gpd1-C306S mutant forms for glycerol-3-phosphate dehydrogenase activity. The C134C mutation displayed only a modest decrease in catalytic activity (Table 2). However, the C306S mutation reduced Gpd1 activity by 95% as compared with the wild type. We also tested another conservative mutation at this site, Gpd1-C306A, which reduced activity by 83%, revealing that even conservative alterations to this residue have a great impact on catalytic activity. The activity of the double mutant form Gpd1-C134S, C306S was reduced by 97%. These results suggest that modification of Gpd1 by MAs(III) at Cys306, and to a lesser extent at Cys134, inhibits its activity.
TABLE 2:
Glycerol-3-phosphate dehydrogenase activity of Gpd1 mutants.
| Allele | Activity (%) |
|---|---|
| GPD1 | 100 |
| gpd1-C134S | 73 ± 1.3 |
| gpd1-C306S | 5.2 ± 0.5 |
| gpd1-C306A | 16.8 ± 0.3 |
| gpd1-C134S, C306S | 3.3 ± 0.5 |
| Vector | 0.6 ± 0.2 |
Glycerol-3-phosphate dehydrogenase activity was measured in a gpd1Δ gpd2Δ strain (DL4285) expressing mutant forms of Gpd1-TAP borne on plasmids and under the control of the inducible GAL1 promoter. Values are the mean and SD from three independent experiments.
Cys306 is highly conserved and is analogous to Cys265 of human GPD1, for which there are x-ray crystallographic data (Ou et al., 2006). Although not implicated in catalysis, Cys265 resides within a highly conserved alpha helix that forms the active site of human GPD1 (Ou et al., 2006). In fact, the crystal structure of human GPD1 in ternary complex with DHAP and NAD revealed that Asp260, Thr264, Gly268, Arg269, and Asn270 all make direct contact with DHAP. These data, combined with our observation that conservative mutations of Cys306 in yeast Gpd1 greatly diminished catalytic activity, suggest that modification of this residue by MAs(III) is likely to be responsible for the observed inhibition of Gpd1 by this reactive toxin. Although we did not examine yeast Gpd2, the paralogue of Gpd1, we assume that the analogous residue (Cys355) would similarly be targeted by MAs(III). Thus, MAs(III) coordinately activates Hog1 to close the glycerol channel and inhibits glycerol-3-phosphate dehydrogenase activity to prevent inappropriate glycerol accumulation (Figure 3C).
As–biotin as a probe for MAs(III) binding
As–biotin is used as a probe for proteins that are targets of trivalent arsenic (e.g., Donoghue et al., 2000; Zhang et al., 2007, 2015; Heredia-Moya and Kirk, 2008; Shen et al., 2013; Beauchamp et al., 2015; Kumar et al. 2016; Kozono et al. 2018). It has been viewed to date as essentially a proxy for As(III). However, we found by As–biotin pull-down experiments that As(III) and MAs(III) display different target specificities and that As–biotin mimics MAs with regard to both inhibition of Gpd1 and activation of Hog1 (through inhibition of Ptp2/3). This is a novel observation that expands our understanding of the different biochemical effects of As(III) and MAs(III). It seems likely that As–biotin also mimics As(III) in some settings. For example, As–biotin binds to Escherichia coli–expressed yeast transcription factor Acr1 (Yap8), and pretreatment of extracts with As(III) diminished this binding (Kumar et al., 2016). Although some bacterial species possess arsenic methyltransferases (Yang and Rosen, 2016), E. coli does not appear to have this enzyme, suggesting that As(III) is the biologically active arsenical in this instance. Thus, care must be taken in the use of As–biotin as a probe, particularly in crude extracts, to distinguish between As(III)-binding and MAs(III)-binding. We have shown that the biologically active form of trivalent arsenic responsible for binding any particular yeast protein can be ascertained from As–biotin pull-down experiments using an mtq2Δ mutant.
Gpd1 is among a small number of trivalent arsenic targets identified that have been shown to be bound specifically by MAs(III), rather than by As(III). Rehman et al. (2012) demonstrated that MAs(III), but not As(III), bound to and inhibited mammalian tyrosine phosphatases PTPB1 and CD45 through attack on their catalytic cysteine residues. Similarly, we found that yeast tyrosine phosphatases, Ptp2 and Ptp3, were inhibited specifically by MAs(III) (Lee and Levin, 2018). Although it is not clear why some proteins are targets of MAs(III), but not As(III), we speculate that the organic moiety of MAs(III) may be needed to access target cysteine residues that are buried within active sites, whereas As(III) may target surface cysteine residues. In any case, our work suggests a multifaceted requirement for metabolic activation of inorganic As(III) to MAs(III) in the response to arsenic. It is thought that conversion of As(III) to MAs(III) in humans assists in its excretion in the urine (Dong et al., 2015). However, it is becoming increasingly clear that production of MAs(III) from As(III) is also an activation step required to mobilize a coherent cellular response to As(III). There appear to be a large number of presumptive As(III) targets in mammalian cells (Zhang et al. 2015), many of which may require metabolism of As(III) to one or another of its methylated forms.
Mechanisms of regulating SAPK output
Previously, we suggested two ways in which the output of SAPKs activated in response to different stresses might be controlled. The first is through the specific mechanism responsible for SAPK activation, which might alter the protein complexes in which the SAPK engages (Lee and Levin, 2018; Lee et al., 2019). For example, in contrast to Hog1 activated by hyperosmotic stress through signaling that activates the HOG pathway protein kinase cascade, Hog1 activated intracellularly by MAs(III) does not involve activation of the kinase cascade (Lee and Levin, 2018). Moreover, Hog1 dissociates from the tyrosine phosphatase, Ptp3, in response to As(III) treatment, but not under hyperosmotic stress (Lee et al., 2019). These differences in activation mechanism might allow Hog1 interactions that are specific to As(III) activation and may direct the output of Hog1. A second way that a stressor might alter the output of a SAPK is through direct interaction with specific targets of the active SAPK. We have provided an example of this mechanism here in the inhibition of glycerol-3-phosphate dehydrogenase activity by MAs(III). This is in stark contrast to hyperosmotic stress, which induces an increase in this activity through multiple Hog1-dependent pathways. In conclusion, direct inhibition of glycerol-3-phosphate dehydrogenase activity provides the cell with an elegant way to address As(III) stress through activation of selected Hog1-dependent functions (e.g., closure of Fps1) while not creating a new stress associated with an osmotic imbalance caused by glycerol accumulation.
MATERIALS AND METHODS
Strains, growth conditions, transformations, and gene deletions
The S. cerevisiae strains used in this study were all derived from Research Genetics background S288c (Research Genetics, Huntsville, AL). Yeast cultures were grown in YPD (1% Bacto yeast extract, 2% Bacto peptone, 2% glucose) or minimal selective medium, SDM (0.67% yeast nitrogen base, 2% glucose), supplemented with the appropriate nutrients to select for plasmids. Yeast cultures were transformed according to Gietz et al. (1995).
Chromosomal deletion of the GPD2 gene was carried out by homologous recombination. The hygromycin-resistance gene HPHMX4 from pAG32 (Goldstein and McCusker, 1999) was amplified by high-fidelity PCR (Phusion; ThermoFisher) using primers containing the upstream region (40 base pairs immediately before the starting ATG) and the downstream region (40 base pairs immediately after the stop codon) of GPD2. The PCR product was integrated into the genome of a gpd1∆::KanMX strain (DL3222; Research Genetics) by homologous recombination. Integrants were selected on plates containing hygromycin B, yielding gpd1∆::KanMX gpd2∆::HPHMX4 (DL4285). Gene replacement was validated by PCR analysis across both integration junctions. The ptp2Δ::KanMX ptp3Δ::HPHMX4 strain (DL4299) and mtq2Δ::HPHMX4 strain (DL4313) used were described previously (Lee and Levin, 2018).
Chemicals
Sodium arsenite (NaAsO2; As(III)), β-nicotinamide adenine dinucleotide (NADH; N8129), and dihydroxyacetone phosphate (DHAP; D7137) were purchased from Sigma. Methylarsenite (CH5AsO2; Mas(III)) was purchased from ChemCruz, and N-biotinyl p-aminophenyl arsenic acid (As–biotin) was purchased from Toronto Research Chemicals.
GPD1 mutant plasmids
Point mutations at cysteine residues of GPD1 were generated by Quick Change mutagenesis (Agilent Technologies), and DNA sequences were confirmed across the entire coding region. The plasmids used in this study are listed in Supplemental Table S1.
As–biotin affinity binding experiments
Cultures for As–biotin affinity binding experiments with Gpd1-TAP were grown in selective medium containing 2% glucose, transferred to selective medium containing 2% raffinose, and grown to mid-log phase. Galactose was added to these cultures at a final concentration of 2% to induce expression of Gpd1-TAP for 2 h. Cultures were treated with 10 µM As–biotin for 10 min before preparation of extracts. For blocking experiments, cultures were pretreated with 1 or 0.5 mM MAs(III) for 10 min and then treated with 10 µM As–biotin for an additional 10 min. Protein extracts (100 µg of protein) were incubated with streptavidin agarose beads (Thermo Scientific) for 1 h at 4°C. Samples were washed three times with IP buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% Triton) and boiled in SDS–PAGE buffer.
Protein extraction
Protein extraction for As–biotin pull down of Gpd1-TAP was by bead beating in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% Triton plus protease inhibitors (cOmplete, Sigma), or using the rapid boiling method (Kushnirov, 2000) for direct immunoblot experiments of Hog1.
SDS–PAGE electrophoresis and immunoblot analysis
Proteins were separated by SDS–PAGE (7.5% gels) followed by immunoblot analysis with goat polyclonal α-Hog1 (yC-20; Santa Cruz) at a dilution of 1:10,000, or rabbit polyclonal α-phospho-p38 (T180/Y182, Cell Signaling) at a dilution of 1:2000 to detect Hog1 and phosphorylated Hog1, respectively. Secondary donkey anti-goat (Santa Cruz) antibodies were used at a dilution of 1:10,000 and donkey anti-rabbit (Amersham) was used at a dilution of 1:2000. Detection of Gpd1-TAP was by polyclonal rabbit peroxidase α-peroxidase (PAP; p1291; Sigma) at a dilution of 1:10,000. All results involving immunoblot analyses were replicated at least once, and representative blots are shown.
Glycerol-3-phosphate dehydrogenase assay
Gpd1 activity assays were carried out as described previously (Gancedo et al., 1968). Yeast strains (gpd1∆ gpd2∆, DL4285, or mtq2∆, DL4313) bearing plasmids with GPD1-TAP were grown as described under As–biotin affinity binding experiments to induce expression of Gpd1-TAP. Cells were lysed in extraction buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% Triton) containing protease inhibitors (cOmplete, Sigma). Extract (10 μg) was added to reaction buffer containing 20 mM imidazole (pH 7.0), and reactions were initiated by addition of NADH (0.1 mM) and DHAP (1.2 mM) to a final volume of 250 μl in microtiter plates. Arsenicals were added to the reaction buffer 10 min before addition of NADH and DHAP. Gpd1 activity was measured spectrophotometrically for 20 min by following oxidation of NADH at A340 using a SPECTRA max plus 384 (Molecular Devices). Gpd1 activity was calculated after subtraction of a low level of background oxidation of NADH in the absence of DHAP.
Measurement of intracellular glycerol concentrations
Intracellular glycerol concentrations were measured in whole cells grown in YPD and centrifuged briefly to remove the culture supernatant. Enzymatic assays for glycerol were carried out using a kit from Boehringer Mannheim and normalized to A600 of the initial culture.
Notes on reproducibility
All Gpd1 activity assays were reproduced at least once in independent experiments, conducted in triplicate. Similarly, all immunoblots and As–biotin pull downs were reproduced at least once in independent experiments with representative images shown.
Supplementary Material
Acknowledgments
This work was supported by a grant to D.E.L. from the National Institutes of Health (R01 GM48533).
Abbreviations used:
- ABC
ATP-binding cassette
- As(III)
arsenite
- As-biotin
N-biotinyl p-aminophenyl arsenic acid
- DHAP
dihydroxyacetone phosphate
- DMAs(III)
dimethylarsenite
- HOG
high osmolarity glycerol
- IP
immunoprecipitation
- MAs(III)
methylarsenite
- NADH
β-nicotinamide adenine dinucleotide
- SA
streptavidin agarose
- SAM
S-adenosylmethionine
- SAPK
stress-activated protein kinase
- SDM
synthetic defined medium
- TAP
tandem-affinity purification
- YPD
yeast extract/peptone/dextrose
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-04-0228) on May 29, 2019.
REFERENCES
- Abernathy CO, Thomas DJ, Calderon RL. (2003). Health effects and risk assessment of arsenic. J Nutr , 1536S–15388S. [DOI] [PubMed] [Google Scholar]
- Beane Freeman LE, Dennis LK, Lynch CF, Thorne PS, Just CL. (2004). Toenail arsenic content and cutaneous melanoma in Iowa. Am J Epidemiology , 679–687. [DOI] [PubMed] [Google Scholar]
- Beauchamp EM, Kosciuczuk EM, Serrano R, Nanavati D, Swindell EP, Viollet B, O’Halloran TV, Altman JK, Platanias LC. (2015). Direct binding of arsenic trioxide to AMPK and generation of inhibitory effects on acute myeloid leukemia precursors. Mol Cancer Ther , 202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beese SE, Negishi T, Levin DE. (2009). Identification of positive regulators of the yeast Fps1 glycerol channel. PLoS Genetics , e1000738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster JL, Gustin MC. (2014). Hog1: 20 years of discovery and impact. Sci Signal , 10.1126/scisignal.2005458. [DOI] [PubMed] [Google Scholar]
- Cullen WR. (2014). Chemical mechanism of arsenic biomethylation. Chem Res Toxicol , 457–461. [DOI] [PubMed] [Google Scholar]
- Dheeman SD, Packianathan C, Pillai JK, Rosen BP. (2014). Pathway of human AS3MT arsenic methylation. Chem Res Toxicol , 1979–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Madegowda M, Nefzi A, Houghten RA, Giulianotti MA, Rosen BP. (2015). Identification of small molecule inhibitors of human As(III) S-adenosylmethionine methyltransferase (AS3MT). Chem Res Toxicol , 2419–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue N, Yam PTW, Jiang X-M, Hogg PJ. (2000) Presence of closely spaced protein thiols on the surface of mammalian cells. Prot Sci , 2436–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbirt KK, Whitmarsh AJ, Davis RJ, Bonkovsky HL. (1998). Mechanism of sodium arsenite-mediated induction of heme oxygenase-1 in hepatoma cells. Role of mitogen-activated protein kinases. J Biol Chem , 8922–8931. [DOI] [PubMed] [Google Scholar]
- Gancedo C, Gancedo JM, Sols A. (1968). Glycerol metabolism in yeasts. Eur J Biochem , 165–172. [DOI] [PubMed] [Google Scholar]
- Gietz D, St. Jean A, Woods RA, Schiestl RH. (1995). Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast , 355–360.7785336 [Google Scholar]
- Goldstein AL, McCusker JH. (1999). Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae . Yeast , 1541–1553. [DOI] [PubMed] [Google Scholar]
- Heredia-Moya J, Kirk KL. (2008). An improved synthesis of arsenic–biotin conjugates. Bioorg Med Chem , 5743–5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann S. (2009). Control of high osmolarity signaling in the yeast Saccharomyces cerevisiae . FEBS Lett , 4025–4029. [DOI] [PubMed] [Google Scholar]
- Kozono S, Lin Y-M, Seo H-S, Pich B, Lian X, Qiu C, Herbert MK, Chen C-H, Tan L, Gao ZJ, et al. (2018). Arsenic targets Pin1 and cooperates with retinoic acid to inhibit cancer-driving pathways and tumor-initiating cells. Nat Commun , 3069–3-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar NV, Yang J, Pillai JK, Rawat S, Solano C, Kumar A, Grøtli M, Stemmler TL, Rosen BP, Tamás MJ. (2016). Arsenic directly binds to and activates the yeast AP-1-like transcription factor Yap8. Mol Cell Biol , 913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnirov VV. (2000). Rapid and reliable protein extraction from yeast. Yeast , 857–860. [DOI] [PubMed] [Google Scholar]
- Lee J, Levin DE. (2018). Intracellular mechanism by which arsenite activates yeast stress MAPK Hog1. Mol Biol Cell , 1904–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Liu L, Levin DE. (2019). Stressing out or stressing in: intracellular pathways for SAPK activation. Curr Genet , 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Reiter W, Dohnal I, Gregori C, Beese-Sims S, Kuchler K, Ammerer G, Levin DE. (2013). MAPK Hog1 closes the S. cerevisiae glycerol channel Fps1 by phosphorylating and displacing its positive regulators. Genes Dev , 2590–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciaszczyk-Dziubinska E, Migocka M, Wysocki R. (2011). Acr3p is a plasma membrane antiporter that catalyzes As(III)/H+ and Sb(III)/H+ exchange in Saccharomyces cerevisiae . Biochim Biophys Acta , 1855–1859. [DOI] [PubMed] [Google Scholar]
- Maciaszczyk-Dziubinska E, Wawrzycka D, Wysocki R. (2012). Arsenic and antimony transporters in eukaryotes. Int J Mol Sci , 3527–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattison CP, Ota I. (2000). Two protein tyrosine phosphatases, Ptp2 and Ptp3, modulate the subcellular localization of the Hog1 MAP kinase in yeast. Genes Dev , 1229–1235. [PMC free article] [PubMed] [Google Scholar]
- Naujokas MF, Anderson B, Ahsan H, Aposhian HV, Graziano JH, Thompson C, Suk WA. (2013). The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem. Env Health Perp , 295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou X, Ji C, Han X, Zhao X, Li X, Mao Y, Wong L-L, Bartlam M, Rao Z. (2006). Crystal structures of human glycerol 3-phosphate dehydrogenase 1 (GPD1). J Mol Biol , 858–869. [DOI] [PubMed] [Google Scholar]
- Rehman K, Chen Z, Wang WW, Wang YW, Sakamoto A, Zhang YF, Naranmandura H, Suzuki N. (2012). Mechanisms underlying the inhibitory effects of arsenic compounds on protein tyrosine phosphatase (PTP). Toxicol Appl Pharmacol , 273–280. [DOI] [PubMed] [Google Scholar]
- Rehman K, Naranmandura H. (2012). Arsenic metabolism and thioarsenicals. Metallomics , 881–892. [DOI] [PubMed] [Google Scholar]
- Ren X, Aleshin M, Jo WJ, Dills R, Kalman DA, Vulpe CD, Smith MT, Zhang L. (2011). Involvement of N-6 adenine-specific DNA methyltransferase (N6AMT1) in arsenic biomethylation and its role in arsenic-induced toxicity. Env Health Persp , 771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen BP, Liu Z. (2009). Transport pathways for arsenic and selenium: a minireview. Environ Int , 512–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Li X-F, Cullen WR, Weinfeld M, Le XC. (2013). Arsenic binding to proteins. Chem Rev , 7769–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley PL, Kinniburgh DG. (2002). A review of the source, behavior and distribution of arsenic in natural waters. Applied Geochem , 517–568. [Google Scholar]
- Sotelo J, Rodríguez-Gabriel MA. (2006). Mitogen-activated protein kinase Hog1 is essential for the response to arsenite in Saccharomyces cerevisiae . Eukaryot Cell , 1826–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsen M, Di Y, Tängemo C, Morillas M, Ahmadpour D, Van der Does C, Wagner A, Johansson E, Boman J, Posas F, et al. (2006). The MAPK Hog1p modulates Fps1p-dependent arsenite uptake and tolerance in yeast. Mol Biol Cell , 4400–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A, Mohindru M, Deb DK, Sassano A, Kambhampati S, Ravandi F, Minucci S, Kalvakolanu DV, Platanias LC. (2002). Activation of Rac1 and the p38 mitogen-activated protein kinase pathway in response to arsenic trioxide. J Biol Chem , 44988–44995. [DOI] [PubMed] [Google Scholar]
- Wurgler-Murphy SM, Maeda T, Witten EA, Saito H. (1997). Regulation of the Saccharomyces cerevisiae HOG1 mitogen-activated protein kinase by the PTP2 and PTP3 protein tyrosine phosphatases. Mol Cell Biol , 1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocki R, Chéry CC, Wawrzycka D, van Hulle M, Cornelis R, Thevelein JM, Tamás MJ. (2001). The glycerol channel Fps1p mediates the uptake of arsenite and antimonite in Saccharomyces cerevisiae . Mol Micro , 1391–1401. [DOI] [PubMed] [Google Scholar]
- Yang H-C, Rosen BP. (2016). New mechanisms of bacterial arsenic resistance. Biomed J , 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yang F, Ling J, Czajkowsky DM, Wang J-F, Zhang X-W, Zhou Y-M, Ge F, Yang M, Xiong Q. (2015). Systematic identification of arsenic-binding proteins reveals that hexokinase-2 is inhibited by arsenic. Proc Natl Acad Sci USA , 15084–15089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yang F, Shim J-Y, Kirk KL, Andersen DE, Chen X. (2007). Identification of arsenic-binding proteins in human breast cancer cells. Cancer Lett , 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.