

Abstract
Macrophage fusion resulting in the formation of multinucleated giant cells occurs in a variety of chronic inflammatory diseases, yet the mechanism responsible for initiating this process is unknown. Here, we used live cell imaging to show that actin-based protrusions at the leading edge initiate macrophage fusion. Phase-contrast video microscopy demonstrated that in the majority of events, short protrusions (∼3 µm) between two closely apposed cells initiated fusion, but occasionally we observed long protrusions (∼12 µm). Using macrophages isolated from LifeAct mice and imaging with lattice light sheet microscopy, we further found that fusion-competent protrusions formed at sites enriched in podosomes. Inducing fusion in mixed populations of GFP- and mRFP-LifeAct macrophages showed rapid spatial overlap between GFP and RFP signal at the site of fusion. Cytochalasin B strongly reduced fusion and when rare fusion events occurred, protrusions were not observed. Fusion of macrophages deficient in Wiskott-Aldrich syndrome protein and Cdc42, key molecules involved in the formation of actin-based protrusions and podosomes, was also impaired both in vitro and in vivo. Finally, inhibiting the activity of the Arp2/3 complex decreased fusion and podosome formation. Together these data suggest that an actin-based protrusion formed at the leading edge initiates macrophage fusion.
INTRODUCTION
Cell to cell fusion is an essential event in several biological processes such as fertilization, embryonic development, skeletal muscle and placenta formation, bone remodeling, and stem cell differentiation (Aguilar et al., 2013; Podbilewicz, 2014). Furthermore, cell–cell fusion has been observed in a number of pathological conditions. In particular, macrophage fusion resulting in the formation of multinucleated giant cells (MGCs) is associated with numerous chronic inflammatory diseases including granulomatous infection, the foreign body reaction to implanted biomaterials, atherosclerosis, amyotrophic lateral sclerosis, cancer, and others (Anderson et al., 2008; Helming and Gordon, 2008, 2009). MGCs are formed from blood monocytes recruited from the circulation to sites of inflammation where they differentiate into macrophages that undergo fusion as inflammation progresses to the chronic state. The T-helper 2 cytokine interleukin-4 (IL-4) promotes macrophages fusion in vivo (Kao et al., 1995) and when applied in cell culture can be used to study this process (McInnes and Rennick, 1988; McNally and Anderson, 1995). Although this in vitro cell system has proven invaluable to our understanding of the molecular mediators that orchestrate macrophage fusion (McNally and Anderson, 2002; Helming and Gordon, 2007; Jay et al., 2007; Milde et al., 2015), there is little information regarding the morphological changes that macrophages undergo to initiate fusion as well as the cellular mechanisms that govern this process.
MGC formation is thought to be a multistage process involving adhesion of cells to the substrate, the induction of a fusion-competent state, cellular motility, cell–cell interaction, cytoskeletal rearrangements, and subsequent membrane fusion (Helming and Gordon, 2009). Most, if not all, of the steps involved in macrophage fusion appear to rely on contractile networks formed by the actin cytoskeleton. It has been shown that the fungal toxins cytochalasin B and D, which both prevent actin polymerization, inhibit MGC formation in a concentration-dependent manner (DeFife et al., 1999). The importance of the actin cytoskeleton has been further corroborated by studies indicating that IL-4 activated the Rac-1 signaling pathway (Jay et al., 2007). Rac-1 is known to reorganize actin networks resulting in the formation of membrane ruffles and extension of lamellipodia. Abrogation of Rac-1 activation by chemical and genetic approaches inhibited lamellipodia formation and attenuated MGC formation (Jay et al., 2007).
Several types of plasma membrane protrusions, including lamellipodia, filopodia, and invadosomes can form and coexist at the leading edge of migrating cells (for a review, see Ridley, 2011). The formation of these protrusions is a result of actin polymerization mediated by actin nucleation-promoting factors. The primary mediators of actin polymerization that induce the formation of branched networks in lamellipodia are the members of the Wiscott-Aldrich syndrome protein (WASp) family that activate the Arp2/3 complex (for a review, see Takenawa and Suetsugu, 2007). The Arp2/3 complex and WASp have also been implicated in filopodia formation (Takenawa and Suetsugu, 2007; Lee et al., 2010; Yang and Svitkina, 2011). Recent studies in myoblasts, cells that undergo fusion in arthropods and vertebrates, have revealed many proteins that participate in Arp2/3-mediated actin polymerization and are required for fusion (Chen, 2011; Aguilar et al., 2013). In these cells, Arp2/3-mediated actin polymerization is responsible for the formation of F-actin-enriched structures protruding from one cell into another cell at the site of fusion (Sens et al., 2010; Haralalka et al., 2011; Shilagardi et al., 2013). The size and the molecular composition of these protrusions (Sens et al., 2010; Chen, 2011) clearly distinguish them from filopodia and lamellipodia (Mattila and Lappalainen, 2008). Based on the presence of an actin core with a surrounding ring of adhesive proteins and their protrusive nature, the protrusions in fusing myoblasts have been called “podosome-like structures” (PLS) (Onel and Renkawitz-Pohl, 2009; Sens et al., 2010). Podosomes and related structures invadopodia, collectively known as invadosomes, are ventral protrusions that form contacts with the extracellular matrix that have been identified in a variety of cell types (Linder et al., 2011; Murphy and Courtneidge, 2011). Podosomes are especially prominent in cells of the monocytic lineage, including macrophages and dendritic cells, where they have been associated with cell adhesion, migration, and matrix degradation. A defining feature of podosomes is a core of actin filaments nucleated by Arp2/3 complex (Linder et al., 2000; Kaverina et al., 2003) surrounded by adhesive plaque proteins such as talin, vinculin, integrins, and others (Zambonin-Zallone et al., 1989; Pfaff and Jurdic, 2001). In addition to activators of Arp2/3 complex, the endocytic protein dynamin plays an essential role in regulating actin polymerization in podosomes (Ochoa et al., 2000; Destaing et al., 2013) and has been shown to be involved in osteoclast fusion (Shin et al., 2014). Despite a requirement for actin polymerization in macrophage fusion, little is known about the role of actin-based protrusions during macrophage fusion.
In the present study, we reveal the existence of an actin-based protrusion that initiates IL-4–mediated macrophage fusion. Phase-contrast video microscopy demonstrated that short phase-dense protrusions originating at the leading edge initiated ∼90% of the fusion events with the remaining events having been initiated by long protrusions. Using macrophages isolated from LifeAct mice and imaging with lattice light sheet microscopy (LLSM), we observed short actin-based protrusions originating from regions enriched in podosomes prior to macrophage fusion. Inducing fusion in mixed populations of GFP- and mRFP-LifeAct macrophages showed rapid spatial overlap between GFP and RFP signals. Inhibiting actin polymerization with cytochalasin B impaired fusion, and when rare fusion events occurred, we observed no protrusions. Furthermore, Cdc42- and WASp-deficient macrophages fused at very low rates both in vitro and in vivo, and video analysis of fusion with these cells showed no clear evidence of protrusions. Finally, inhibiting the Arp2/3 complex not only reduced fusion but also rare fusion events did not appear to be dependent on protrusions.
RESULTS
Phase-dense protrusions at the leading edge precede macrophage fusion
We recently developed optical-quality glass surfaces that enabled the first time-resolved views of IL-4–induced macrophage fusion and MGC formation (Faust et al., 2017, 2018). Using these surfaces, we showed that macrophage fusion occurred between the intercellular margins of macrophages. Furthermore, we observed a founder population of mononuclear macrophages that initiates fusion with neighboring mononuclear macrophages (type 1 fusion). Early multinucleated cells then fuse with neighboring mononuclear macrophages (type 2 fusion), the most abundant event leading to MGC formation. Finally, MGCs fuse with surrounding MGCs to form syncytia (type 3 fusion). However, due to the low-magnification views required to visualize the formation of large MGCs, the mechanism underlying this process remained obscure.
To visualize structures between the intercellular margins of fusing macrophages in detail, we initially used phase-contrast video microscopy with intermediate magnification objectives. In this series of experiments, we used primary macrophages isolated from the inflamed mouse peritoneum (Helming and Gordon, 2007; Podolnikova et al., 2016) in order to avoid robust cell division observed in cultures of macrophage cell lines. Analyses of type 1 (n = 33), type 2 (n = 93), and type 3 (n = 36) fusion events revealed the existence of phase-dense protrusions immediately preceding macrophage fusion. For the majority of events (n = 148), short protrusions (2.8 ± 0.8 µm) initiated fusion (Supplemental Figure S1 for type 1 fusion and Figures 1A and 2A, and Supplemental Video S1 for type 2 fusion). However, we rarely observed long protrusions (11.6 ± 6 µm; n = 14) (Figures 1B and 2A and Supplemental Video S2). No association of long protrusions with a specific type of fusion was found. Similar to short protrusions, long protrusions were observed in all three types of fusion. In a given cell, either a short or a long protrusion initiated fusion; the coexistence of protrusions was not observed. Within each type of fusion, the lengths of protrusions, both short and long, were similar (Table 1). Furthermore, as shown in Figure 2A, there was no overlap between the distribution of lengths of short and long protrusions, further pointing to the existence of two populations of protrusions. Analyses of type 1 and type 2 of fusion showed no significant difference between spreading of mononuclear cells that fused via short (n = 116) and long (n = 10) protrusions (Figure 2B). Moreover, since type 1 fusion occurs between mononuclear cells, which are poorly spread round cells, whereas type 2 and type 3 fusion events involve multinucleated cells, which are large well-spread cells (Table 1), spreading does not seem to influence the length of protrusion. Although a percentage of long protrusions slightly increased in the population of type 3 fusion, short protrusions remained the dominant structures (Table 1). As shown in Figure 2C, the time required from first intercellular contact until full nuclear integration between two macrophages that was mediated by short protrusions was similar for all three types of fusion (59 ± 31 min, 48 ± 22 min, and 68 ± 48 for type 1, type 2, and type 3, respectively), although the time for type 2 fusion tended to be shorter. The fusion times mediated by long protrusions were significantly shorter for type 2 and tended to be shorter for type 1 and type 3 fusion events than those mediated by short protrusions (Table 1).
FIGURE 1:

Phase-dense protrusions initiate macrophage fusion. (A) Live imaging of macrophages undergoing type 2 fusion. Macrophages were isolated from the mouse peritoneum 3 d after TG injection and plated on a 35-mm Fluorodish, and fusion was induced by IL-4. Mononuclear macrophage (Cell 1) extends a short phase-dense protrusion (white arrow) toward MGC (Cell 2) immediately before fusion. The bottom panel is a diagram of frames at 1:30, 3:30, and 7:30 min illustrating morphological aspects of the fusion process. In each micrograph, time is shown in minutes:seconds. The scale bar is 10 μm. See also Supplemental Video S1. (B) Macrophage undergoing type 2 fusion extends a long protrusion (white arrow) to initiate fusion. The bottom panels show diagrams of frames at 11:00, 15:30, and 17:00 min. The scale bar is 10 μm. See also Supplemental Video S2.
FIGURE 2:

Quantification of the length of fusion-competent protrusions, fusion time, and spreading of fusing macrophages. (A) Length distribution in the populations of short and long protrusions. Type 1, 2, and 3 fusion events were pooled. (B) Spreading of mononuclear macrophages mediating fusion through short and long protrusions in the population of cells undergoing type 1 and type 2 fusion. (C) The time required from first intercellular contact until full nuclear integration between two macrophages during fusion mediated by short protrusions.
TABLE 1:
Parameters of fusion-competent protrusions.
| Type 1 fusion | Type 2 fusion | Type 3 fusion | |
|---|---|---|---|
| Number of analyzed protrusions | |||
| Short | 31 (94%) | 85 (91%) | 32 (89%) |
| Long | 2 (6%) | 8 (9%) | 4 (11%) |
| Length of protrusions, µm | |||
| Short | 2.6 ± 0.7 | 2.8 ± 1.2 | 2.6 ± 0.7 |
| Long | 10.8 ± 3.3 | 11.9 ± 7.9 | 11.2 ± 4.1 |
| Total fusion time from the first cell–cell contact to full integration, min | |||
| Short | 59 ± 31 | 48 ± 22 | 68 ± 48 |
| Long | 23 ± 0.7a | 22 ± 11b | 37 ± 24c |
| Cell spreading, µm2 | |||
| Mononuclear | 262 ± 114 | 252 ± 83 | − |
| Multinuclear | − | 1400 ± 910 | 1760 ± 1280 |
a,b,cp = 0.057, p = 0.001, and p = 0.056 (long vs. short protrusions, respectively).
Video S1.
A phase-dense protrusion initiates macrophage fusion. Short phase-dense protrusions at the leading edge initiate fusion.
Video S2.
A long phase-dense protrusion initiates macrophage fusion. Occasionally (∼10 %) long protrusions initiate fusion.
Several patterns of macrophage fusion emerged. Among type 1 events, fusion most frequently (34%) occurred between the leading edge of one cell and the cell body of another, followed by fusion from the leading edge to the rear edge (23%) and between the leading edges of two closely apposed cells (20%) (Table 2 and Supplemental Videos S3–S5). Similar patterns of fusion were observed in the type 2 group; however, fusion between the two leading edges was a dominant pattern (Table 2). The other patterns included fusion between the cell–cell bodies and fusion between the rear edges, among others. We also characterized fusion of macrophages using Permanox (n = 52), a permissive plastic surface that is widely used to induce macrophage fusion (Helming and Gordon, 2007; Milde et al., 2015). Both short and long protrusions were detected, with short protrusions being the main type (49 vs. 3). The length of protrusions and the time of fusion were also similar to those determined for macrophages undergoing fusion on the optical-quality glass (Supplemental Table S1). In addition, fusion involving the leading edge was the main pattern on Permanox (Supplemental Table S2).
TABLE 2:
Patterns of macrophage fusion.
| Pattern mediated by short protrusionsa | Schematic showing cell polarity | Type 1 fusion | Type 2 fusion |
|---|---|---|---|
| Leading edge to the cell body | 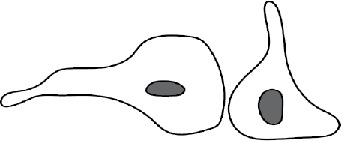 |
34% | 31% |
| Leading edge to rear edge | 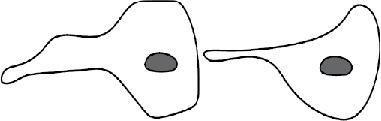 |
23% | 18% |
| Leading edge to leading edge | 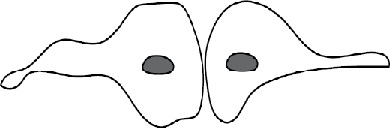 |
20% | 45% |
| Other | 23% | 6% |
aSee also Supplemental Videos S1–S5.
Video S3.
Fusion between the leading edge and cell body.
Video S4.
Fusion between the leading edge and cell rear.
Video S5.
Fusion between leading edges.
An actin-based protrusion initiates macrophage fusion
Membrane protrusions at the leading edge are actin-based structures. Furthermore, F-actin is known to be important for the formation of MGCs (DeFife et al., 1999; Jay et al., 2007). To determine if the protrusions we observed in phase-contrast micrographs were actin-based structures, we examined macrophages isolated from the inflamed peritoneum of LifeAct mice using LLSM (Chen et al., 2014). We first confirmed that LifeAct faithfully reported the distribution of F-actin in macrophages by comparing the distribution of eGFP-LifeAct and Alexa 568–conjugated phalloidin (Supplemental Figure S2). Using LLSM, we reveal waves of eGFP-LifeAct puncta emanating from the center of the cell to the cell periphery prior to fusion (Supplemental Video S6). At the time of apparent fusion, one wave of LifeAct puncta advanced into a neighboring cell (Figure 3A and Supplemental Video S6). In fixed specimens, actin puncta at cellular margins contained rings of vinculin and talin circumscribing a central actin core in both mononuclear macrophages and MGCs (Supplemental Figure S3), suggesting that structures we observed in living cells were podosomes. Furthermore, the size of individual podosomes determined in fixed samples (both GFP-LifeAct and phalloidin-labeled) and in living cells (Supplemental Figure S4) were similar. When we analyzed the site of fusion (Figure 3B, 42:14–42:41 min; boxed areas in Figure 3A) with the maximum temporal resolution achievable with LLSM under the conditions of our experiments (∼1.5 s per image for several hours), we observed a finger-like enrichment of LifeAct that extended into the neighboring cell during fusion (Figure 3B, 42:14–42:24 min; yellow arrowhead). This protrusive structure fanned from the initial point of contact as fusion proceeded (Figure 3B, 42:41 min; yellow arrowheads).
FIGURE 3:

An actin-based protrusion precedes macrophage fusion. (A) LLSM of IL-4–induced fusion of macrophages expressing eGFP-LifeAct. All images shown are maximum-intensity projections. The scale bar is 10 μm. See also Supplemental Video S6. (B) Enlarged images of events occurring between 42:14 and 42:41 min (boxed regions in A). Yellow arrowheads point to the site of fusion. The scale bar is 10 μm. (C) En face isosurface renders of LLSM data from A. The boxed region at 25:07 min corresponds to the subsequent micrographs showing the fusion progress (42:14–42:41 min). Note numerous protrusions (outlined by white dashes) formed between apposing cells. The scale bar is 10 µm. See also Supplemental Video S7. (D) Surface renders of LLSM data from another area of fusing macrophages showing contact of a fusion-competent protrusion (a single white arrowhead; 37:27–37:51 min) followed by its apparent expansion (two white arrowheads; 37:54–37:58 min). The scale bar is 5 µm. The boxed area at 36:16 min (scale bar, 10 µm) corresponds to the subsequent micrographs (37:24–37:58 min). Time in each micrograph is shown as minutes:seconds. See also Supplemental Video S8.
Video S6.
Lattice light sheet microscopy of fusing macrophages. High spatiotemporal view of a MGC undergoing multiple fusion events is shown. A wave of eGFP-LifeAct puncta (white) emanate from the interior of the MGC and enrich at the cell periphery before fusion.
To visualize the dynamics of the actin cytoskeleton prior to fusion and observe the interface between fusing cells, we used a maximum intensity isosurface render of eGPF-LifeAct applied to the area shown in Figure 3A (Supplemental Video S7). We observed numerous thin protrusions between two interacting cells after initial contact (Figure 3C, 22:20 and 25:07 min). The protrusions appeared to contact apposing cells by rounds of extension and retraction, which continued until seconds before the cells fused (Figure 3C, 42:14–42:55 min, individual protrusions are outlined; and Supplemental Video S7). Close apposition of these cells in diffraction-limited space precluded visualizing the role of protrusions during the fusion process. Analysis of another area also revealed the presence of protrusions extending and retracting between two cells (Figure 3D, white outline; and Supplemental Video S8). In this case, however, the separation between the cells made it possible to observe an actin-based protrusion that initiated fusion (Figure 3D, 37:27–37:51 min; white arrowheads). The gradual expansion of the actin network that followed at this site could potentially be attributed to the local expansion of the protrusion, the formation of a fusion pore, or both (Figure 3D, 37:54–37:58 min; arrowheads).
Video S7.
Maximum intensity isosurface render of eGFP-LifeAct. Surface render show actinbased protrusions during the fusion process.
Video S8.
Maximum intensity isosurface render of eGFP-LifeAct. When there is adequate space between fusion macrophages fusion-competent protrusions can be observed. The protrusion appear to become stabilized and subsequently expand as fusion progresses.
F-actin incorporation is asymmetric during macrophage fusion
To determine how actin is integrated during the fusion process, we mixed equal numbers of eGFP-LifeAct and mRFP-LifeAct macrophages, induced fusion, and imaged the process with LLSM (Supplemental Video S9). When we visualized cells in this mixing assay at early time points, we observed no overlap of GFP and RFP emission in diffraction limited space (Figure 4A, 5:57 min; two adjacent mononuclear cells in the right bottom quadrant are outlined). However, at the time of apparent fusion (Figure 4A, 06:14 min), we observed overlap of GFP and RFP in overlays at the cell margins, which became more apparent as fusion proceeded (Figure 4A, 6:31–7:22 min). Further, we observed reorganization of actin in cells undergoing fusion that first clearly appeared at 11:20 min and was completed by 23:48 min, suggesting mixing of the cytoplasm.
FIGURE 4:

Mixing macrophages expressing eGFP-LifeAct and mRFP-LifeAct shows asymmetric actin integration. (A) Macrophage fusion in a mixed population of eGFP/mRFP-LifeAct macrophages. Two mononuclear cells expressing eGFP-LifeAct and mRFP-LifeAct undergoing fusion are outlined in the image taken at 5:57 min. A site of eGFP- and mRFP-LifeAct integration is indicated by white arrowheads at 6:14 and 6:48 min. (B) Split-channel view of the fusion event shown in A. The boxes outlined in white dashed lines in the images taken at 6:14–6:48 min correspond to the areas where integration of mRFP-LifeAct into the eGFP-LifeAct-containing cells is observed. The enlarged images of the same areas are shown as insets. The scale bars are 5 μm. See also Supplemental Videos S9 and S10.
Video S9.
Lattice light sheet microscopy of mixed populations of eGFP- and mRFP-LifeAct. Integration of LifeAct signal appears to be asymmetric.
Separating GFP and RFP emission and analyzing fusion in the two cells outlined in green and red revealed an asymmetry in the fusion process (Figure 4B and Supplemental Video S10). Prior to fusion (5:57 min), we observed no GFP signal in the cell outlined in red and vice versa. However, at 6:14 min, the signal from RFP appeared in the green outline traversing the entire length of the cell (Figure 4B; magnified inset shows the outline of the eGFP-LifeAct macrophage). At the same time, we were unable to detect a green signal within the outline of the red cell. Only after an additional 17 s (6:31 min) did we begin to see low levels of GFP signal spatially overlapping boundaries of the cell outlined in red, which appeared to enrich as time progressed (6:48 min). Thus, it appears that one of the two cells more actively integrates cytoplasm than the other does.
Video S10.
Lattice light sheet microscopy of mixed populations of eGFP- and mRFP-LifeAct. eGFP and mRFP channels are separated to observe integration of eGFP- and mRFP-LifeAct.
Organizers of actin-based protrusions are critical for macrophage fusion in vitro
Since protrusions precede macrophage fusion, we sought to determine whether these actin-based structures are causal mediators of macrophage fusion. As a first step, we examined the role of F-actin, the cytoskeleton underlying membrane protrusions, in macrophage fusion. Consistent with previous data (DeFife et al., 1999), treatment of cells with cytochalasin B reduced MGC formation. Quantification of the fusion index indicated that at a concentration as low as 2.5 µM, cytochalasin B decreased fusion by approximately threefold from 29 ± 10% to 10 ± 5% for untreated versus treated cells, respectively (Figure 5, A and B). Furthermore, the majority of treated cells had a bulbous shape compared with a flattened shape of untreated cells (Figure 5A, bottom panels). Importantly, when we recorded fusion in the presence of cytochalasin B using phase-contrast video microscopy, we were unable to observe phase-dense protrusions preceding fusion. Rather, some cells in close apposition appeared to passively undergo fusion (Figure 5C).
FIGURE 5:

Cytochalasin B treatment reduces macrophage fusion. (A) Confocal micrographs of control (untreated and DMSO-treated) as well as cytochalasin B–treated (2.5 µM) macrophages 24 h after incubation in the presence of IL-4. The cells were labeled with Alexa Fluor 488–conjugated phalloidin (white) and DAPI (teal). The bottom panels show the x-z sections of control untreated (left), control DMSO-treated (middle), and cytochalasin B–treated (right) MGCs. The scale bars are 50 and 10 µm in the top and bottom panels, respectively. (B) Quantification of the fusion index in the population of untreated and cytochalasin B–treated macrophages. DMSO was used as vehicle control for cytochalasin B treatment. Three to five random 20× fields per sample were used to count nuclei (100–150 nuclei/field; total ∼1500 nuclei). Results shown are mean ± SD from three independent experiments. *p < 0.01. (C) Single cytochalasin B–treated macrophages that undergo fusion do not form protrusions. The cells that fuse are outlined. The scale bars are 10 μm.
It is well known that Cdc42 orchestrates filopodia formation. In addition, Cdc42 and PIP2 activate WASp to trigger downstream Arp2/3-mediated actin polymerization to form lamellipodia, podosomes, and other protrusions (Mattila and Lappalainen, 2008; Campellone and Welch, 2010; Linder et al., 2011). If actin-based protrusions are involved in macrophage fusion, then perturbing the function of these upstream regulatory proteins should inhibit macrophage fusion. To test this prediction, we isolated macrophages from a WASp-/- mouse and examined IL-4–induced macrophage fusion at various time points. Figure 6, A and B, shows that fusion of WASp-deficient macrophages was strongly impaired. Compared to wild-type (WT) macrophages, the degree of fusion of WASp-deficient macrophages at every time point tested (24, 48, and 72 h) was approximately sixfold less. WASp deficiency also inhibited the formation of podosomes in fusing macrophages (Figure 6C). Gross morphology and the degree of macrophage adhesion after 2.5 h in culture did not appear to be significantly different from WT macrophages (Figure 6A; t = 0). Using phase-contrast video microscopy performed during the first 24 h after IL-4 addition, we were able to observe rare fusion events, which appear to have occurred by a protrusion-independent mechanism (Figure 6D).
FIGURE 6:

WASp is required for macrophage fusion in vitro. (A) Fusion of WT and WASp-deficient macrophages at various time points after the addition of IL-4. After 24, 48, and 72 h, cells were fixed and labeled with Alexa Fluor 488–conjugated phalloidin (white) and DAPI (teal). The scale bars are 50 μm. (B) The time-dependent fusion indices for WT and WASp-deficient macrophages. Results shown are mean ± SD from three independent experiments. Three to five random 20× fields per sample were used to count nuclei (100–150 nuclei/field; total 1500 nuclei). ***p < 0.001. (C) The time-dependent podosome formation in fusing macrophages. The fraction of cells with >10 podosomes for each time point was calculated. Four random 20× fields each containing ∼200–300 cells were used to count podosomes. **p < 0.01, ***p < 0.001. (D) Live imaging of IL-4–treated WASp-deficient macrophages. In each micrograph, time is shown in hours:minutes. A rare fusion event detected in the population consisting of ∼1200 macrophages is shown.
We next examined whether Cdc42 is required for macrophage fusion using macrophages isolated from myeloid cell-specific Cdc42-/- mice (Figure 7, A and B). As shown in Figure 7, C and D, at 24–72 h, we observed an approximately twofold decrease in fusion of Cdc42-deficient macrophages compared with control Cdc42loxP/loxP counterparts. Similar to WASp, Cdc42 deficiency also strongly reduced the formation of podosomes (Figure 7E).
FIGURE 7:

Loss of Cdc42 in macrophages results in impaired fusion in vitro. (A) Schematic diagram of the generation of a myeloid cell-specific Cdc42-/- mouse. Conditional gene-targeted mice with exon 2 of Cdc42 gene flanked by a pair of loxP sequences (Yang et al., 2007) were crossbred with LysMcre mice to allow Cdc42 gene excision in myeloid cells. (B) Cdc42 deletion in isolated macrophages was examined by SDS–PAGE (11% gel) followed by Western blotting using anti-Cdc42 rabbit monoclonal antibody (ab187643; Abcam). (C) Fusion of WT and Cdc42-deficient macrophages at various time points after the addition of IL-4. Cells were fixed after 24, 48, and 72 h and labeled with Alexa Fluor 488–conjugated phalloidin (white) and DAPI (teal). The scale bars are 50 μm. (D) Time-dependent fusion indices of WT and Cdc42-deficient macrophages. Results shown are mean ± SD from three independent experiments. Four to five random 20× fields per sample were used to count nuclei (300 nuclei/field; total ∼4050 nuclei). *p < 0.05, ***p < 0.001. (E) The time-dependent podosome formation in fusing macrophages. The fraction of cells with >10 podosomes for each time point was calculated. Four random 20× fields each containing ∼200–300 cells were used to count podosomes. ***p < 0.001.
We next determined whether inhibition of Arp2/3 results in impaired macrophage fusion. As shown in Figure 8, A and C, the Arp2/3-specific inhibitors CK-636 and CK-548 (Nolen et al., 2009) blocked macrophage fusion in a dose-dependent manner. The IC50 values for CK-636 and CK-548 inhibition were 13 ± 0.5 and 15 ± 0.7 µM, respectively, for 72-h cultures. In addition, consistent with previous reports (Nolen et al., 2009), inhibition of Arp2/3 decreased the number of podosomes with both inhibitors exerting similar effects (IC50 = 9.5 ± 0.5 µM for CK548 and 12 ± 0.5 µM for CK636; Figure 8, B and C). Although ∼40% fusion was observed in the presence of 15 µM CK-636, MGCs did not account for the majority of fusion events. Rather, binucleated cells were the predominant cell type, which formed by a protrusion-independent mechanism (Figure 8D).
FIGURE 8:

Inhibition of actin assembly by CK-636 and CK-548 decreases macrophage fusion and podosome formation. (A) Different concentrations of the Arp2/3 inhibitors CK-636 and CK-548 were added to macrophages at the onset of fusion induction with IL-4. Control cells were treated with DMSO. The fusion rates were determined after 72 h from confocal images of samples labeled with Alexa Fluor 488–conjugated phalloidin and DAPI. Results shown are mean ± SD from three independent experiments. Three to five random 20× fields per sample were used to count nuclei (250–300 nuclei/field; total ∼3600 nuclei). Fusion of control (DMSO-treated) cells was assigned a value of 100%. (B) Effect of inhibitors (each at 15 µM) on podosome formation. The fraction of cells with >10 podosomes was calculated and normalized to DMSO control. Results shown are mean ± SD from three independent experiments with three to five random 20× fields used per sample to count cells (100–150 cells/field). (C) Representative confocal micrographs of control (DMSO-treated) and CK 548-treated (15–60 µM) macrophages 72 h after incubation in the presence of IL-4. The cells were labeled with Alexa Fluor 488–conjugated phalloidin (white) and DAPI (teal). (D) Live imaging of macrophages treated with 15 µM Arp2/3 inhibitor CK-548. In each micrograph, time is shown in minutes:seconds. A single fusion event detected is shown.
WASp- and conditional Cdc42-deficient mice do not initiate a robust foreign body response to implanted materials
Further evidence for the involvement of WASp and Cdc42 in macrophage fusion was obtained by in vivo experiments. Macrophage fusion leading to the formation of MGCs is a hallmark of the foreign body reaction that follows the implantation of vascular grafts and other engineered devices (Anderson et al., 2008; McNally and Anderson, 2011). To confirm that WASp and Cdc42 are important for the formation of MGCs in vivo, we implanted polychlorotrifluoroethylene (PCTFE) into the peritoneum of WT, WASp-/-, and conditional Cdc42-deficient mice and retrieved the implants 14 d later. Visualization of cells covering the surface of explants revealed a large number of MGCs on materials implanted into WT, but not WASp-/- mice (Figure 9, A and B). We found an approximately fivefold difference in the fusion index between WT and WASp-deficient macrophages (36 ± 6% vs. 7 ± 4% for WT and WASp-deficient cells, respectively) (Figure 9B). No significant difference in the number of cells recruited into the peritoneum of WT and WASp-/- mice (Figure 9C) as well as in the number of cells attached to the retrieved implants was found (Figure 9D). Conditional deletion of Cdc42 also strongly impaired multinucleation with an approximately sixfold difference between Cdc42loxP/loxP and Cdc42-/- cells (33% ± 12 vs. 5.9% ± 4.0) (Figure 9, E and F). The number of cells in the peritoneum 14 d after implantation (Figure 9G) and the number of cells on explants (Figure 9H) were not significantly different between the two strains of mice. Together, these data indicate that WASp and Cdc42 are essential in vivo for a robust foreign body reaction.
FIGURE 9:
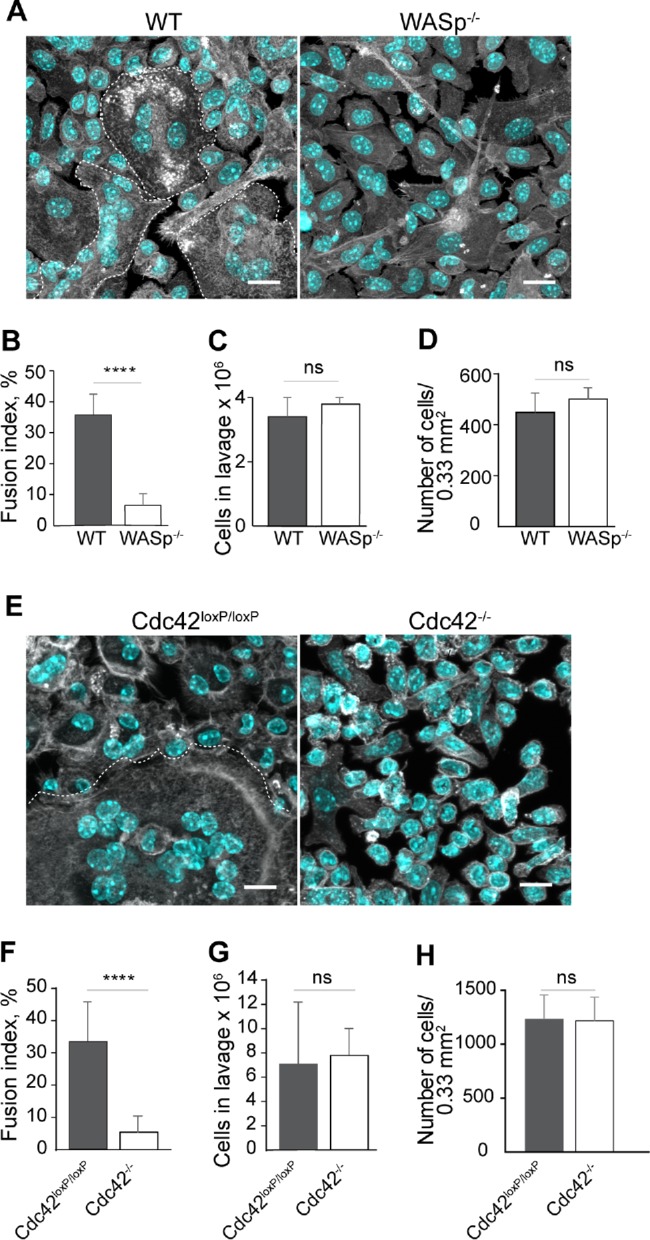
Fusion of WASp- and Cdc42-deficient macrophages is severely impaired in vivo. (A) Micrographs of macrophages on PCTFE surfaces retrieved 14 d after implantation in WT and WASp-/- mice. Three MGCs in the left panel are outlined. The scale bars are 7.5 μm. (B) Quantification of the fusion index of macrophages on PCTFE surfaces retrieved from WT and WASp-/- mice. Results shown are mean ± SD from three independent experiments. Six to eight random 20× fields per sample were used to count nuclei (250–300 nuclei/field; total 6000 nuclei). ****p < 0.0001. (C) A number of cells collected from the peritoneum of WT and WASp-/- mice before implants were retrieved. Results shown are mean ± SD from three independent experiments. No significant difference was observed. (D) A number of cells on the surface of explants retrieved from WT and WASp-/- mice. Results shown are mean ± SD from three independent experiments. No significant difference was observed. (E) Micrographs of macrophages on PCTFE surfaces retrieved 14 d after implantation in Cdc42loxP/loxP mice and mice with Cdc42-deficiency in myeloid cells. A single MGC in the left panel is outlined. The scale bars are 7.5 µm. (F) Quantification of the fusion index of macrophages on surfaces implanted into Cdc42-/- mice. Results shown are mean ± SD from three independent experiments. Six to eight random 20× fields per sample were used to count nuclei (300 nuclei/field; total 6600 nuclei). ****p < 0.0001. (G) A number of cells collected from the peritoneum of WT and Cdc42-/- mice before implants were retrieved. Results shown are mean ± SD from three independent experiments. Three to five random 20× fields were used per sample to count cells. No significant difference was observed. (H) A number of cells on the surface of explants retrieved from Cdc42loxP/loxP mice and mice with Cdc42-deficient macrophages. Results shown are mean ± SD from three independent experiments. No significant difference was observed.
DISCUSSION
Despite the long history of research on MGCs highlighted by the fact that these cells are often observed in many diseases, the molecular and cellular mechanisms of macrophage fusion remain poorly understood. While previous studies focused mainly on the identification of fusion effector molecules in macrophages (Helming and Gordon, 2009; Vignery, 2011), little effort has been expended to elucidate the involvement of the actin cytoskeleton, which has been shown to be a driving force in other types of cells undergoing fusion (Aguilar et al., 2013; Podbilewicz, 2014). Here, we used live cell imaging of macrophages to visualize actin-based structures formed at the site of contact between two fusing cells. We show for the first time that IL-4–induced macrophage fusion is initiated by a single protrusion that more often (∼90% events) extends from the leading edge of one cell in contact with another cell. The majority (∼90%) of fusion-competent protrusions are short although long structures were also observed.
Recently, a number of studies have reported the involvement of membrane extensions during osteoclastogenesis (Oikawa et al., 2012; Soe et al., 2015; Wang et al., 2015). Using RAW264.7 macrophages differentiated into osteoclasts by M-CSF and RANKL, Oikawa et al demonstrated that the majority of fusion events was mediated by membrane protrusions referred to as “podosome-related” that emerged from the sealing belt, a structure formed in large multinucleated osteoclasts from a ring of “circumferential” podosomes (Oikawa et al., 2012). Less frequently, “filopodia-like” protrusions that formed after a transient expansion of podosome-like protrusions and “long” protrusions were observed. Wang et al. (2015) have also observed finger-like extensions (∼2.5 µm) in RAW264.7 cultures, which they termed fusopods (Wang et al., 2015). In that investigation, fusopods were the predominant structures, although some cells reportedly fused at contacts between broad surfaces. Thus, both IL-4–mediated fusion of natural mouse macrophages and RANKL-mediated osteoclastogenesis in RAW264.7 cultures are initiated by a protrusion.
We demonstrated that macrophage fusion depends on F-actin and regulators of actin polymerization Cdc42 and its downstream effector WASp, inasmuch as the ability of macrophages to fuse was strongly impaired in the presence of cytochalasin B and in macrophages derived from WASp- and conditional Cdc42-deficient mice (Figures 5–7). In addition, inhibiting Arp2/3 activity also inhibited macrophage fusion (Figure 8). The decrease in fusion observed in cytochalasin B– and Arp2/3-treated as well as WASp- and Cdc42-deficient macrophages correlated with a lack of visible protrusions. Interestingly, a small portion of macrophages continued to fuse through a seemingly protrusion-independent way, the mechanism of which remains to be determined. Importantly, fusion of WASp- and Cdc42-deficient macrophages was reduced not only in vitro but also in vivo, suggesting that protrusion-mediated fusion is not an artifact observed exclusively in cultured cells. It is known that macrophages derived from Wiskott-Aldrich syndrome patients are defective in chemotaxis and phagocytosis (Thrasher, 2002). The finding that WASp is also essential for fusion extends its known roles in macrophages.
Our finding that the majority of fusion events in macrophages involved the leading edge of at least one cell (Table 2) and the observation of numerous thin protrusions at the site of contact between apposed cells (Supplemental Videos S7 and S8) are consistent with filopodia formation at this location. Indeed, thin finger-like filopodia are found at the leading edge of migrating cells and are an extension of the branched lamellipodia network (Svitkina et al., 2003). However, these protrusions did not seem to initiate fusion. Although extension and retraction of filopodia persisted for a prolonged period, fusion occurred only after podosomes concentrated at the leading edge. At that point one protrusion initiated fusion. Therefore, it may be possible to distinguish two types of protrusions during macrophage fusion: bona fide filopodia as prefusion protrusions and a fusion-competent protrusion that assembles in a region enriched in podosomes. Fusion-competent protrusions appear to have a different morphology being wider and shorter than filopodia (Figure 3D). Although the molecular composition and organization of the F-actin network within the two types of protrusions remain to be defined, our data suggest that several types of actin-based protrusions that have different functions may be involved in macrophage fusion.
Fusion-competent short protrusions appear to originate from cells that become enriched in peripheral podosomes. In these cells, podosomes emanate from the interior and move in a wave-like manner to the periphery where they accumulate at the site destined for fusion (Figure 3, A and B, and Supplemental Video S6). Before the arrival of podosomes, the interface between two apposing cells is filled with thin protrusions that undergo rounds of extension and retraction (Figure 3, C and D; Supplemental Video S7; and schematically shown in Figure 10A). This activity continues for some time until podosomes in the cell that initiates fusion align along the plasma membrane (Figure 10B). Shortly after, one of the protrusions initiates fusion (Figure 10, C and D). The association of podosomes with long protrusions is presently unclear. Because of their paucity, we were unable to detect long protrusions in our LLSM experiments using LifeAct-containing macrophages. Nonetheless, although still constituting a small portion, long connections between two distant macrophages were detected by phase-contrast video microscopy, which revealed that the pattern of fusion was rather different from cells that fuse through short protrusions. Interestingly, the long connections seemed to form by merging the tips of two protrusions arising from two macrophages (Supplemental Video S2). A similar mode of joining of two protrusions was observed in RAW264.7-derived osteoclasts (Oikawa et al., 2012). After establishing the long connection, fusion was accompanied by shortening and widening of this bridge, followed by fusion (Supplemental Video S2). At present, the differences between short and long protrusions and their origin remain to be defined.
FIGURE 10:

Model for macrophage fusion initiated by an actin-based protrusion. (A) The interface between two closely apposing macrophages contains numerous thin actin-based protrusions. A wave of podosomes moves from the interior of the cell that initiates fusion to the site of cell–cell contact. (B) Once podosomes arrive and align at the site of contact with the acceptor macrophage, membrane fusion is initiated by a single protrusion and podosomes from the donor macrophage advance into the acceptor macrophage. (C) The actin network at the site of fusion rapidly reorganizes to expand the initial pore. (D) The actin cytoskeleton and nuclei undergo directional integration.
Previous studies have shown that Cdc42, WASp, and Arp2/3 have an important role in the formation of podosomes in macrophages (Linder et al., 1999, 2011; Dovas et al., 2009). In line with these investigations, our results demonstrated fewer podosomes in macrophages derived from WASp-/- and conditional Cdc42-/- mice (Figures 6 and 7). In addition, the number of podosomes was significantly decreased in macrophages treated with Arp2/3 inhibitors (Figure 8). Although podosomes were enriched at the site of fusion and abolishing the activity of critical podosomal proteins impaired fusion, the mechanistic link between fusion-competent protrusions and podosomes remains to be established. Podosomes are formed at the ventral side of the cell and typically protrude vertically into the substrate (Labernadie et al., 2010, 2014; Proag et al., 2015; Linder and Wiesner, 2016). Our current data suggest that the force driving the formation of fusion-competent protrusions may be delivered laterally toward the apposing cell in order to initiate fusion. It is possible that podosomes may directly or indirectly generate protrusive force during the fusion process. We speculate that when podosomes arrive at the leading edge, they anchor the ventral actin network while allowing the lateral actin network to continue extending forward. Under these conditions, the cell would be unable to protrude along the entire leading edge and thus may focus the protrusive force to a limited region. Since the apposing macrophage presents an obstacle for the elongation of the protrusion, the protrusion may generate a pushing force and penetrate into the adjacent cell. It is presently unclear why only certain macrophages display the directional movement of podosomes and how it is associated with the formation of a fusion-competent protrusion.
Long fusion-competent protrusions that we occasionally observed in our experiments (Figure 1 and Supplemental Figure S1) are visually reminiscent of tunneling nanotubes (TNTs). TNTs are long, thin membranous tubes with diameters of 50–800 nm connecting two cells that have been reported in numerous cell types, including macrophages (Rustom et al., 2004; Onfelt et al., 2006; Kimura et al., 2012; Hanna et al., 2017). Formation of TNTs requires F-actin and, as recently shown in macrophages, depends on the activity of Rac1, Cdc42, and WASp (Hanna et al., 2017). Despite the general requirement for F-actin and the activators of actin polymerization, there seem to be clear distinctions between TNTs and fusion-competent protrusions. In particular, we observed that contact initiated by a long protrusion with a neighboring macrophage was invariably followed by fusion. In contrast, TNTs that also form by extending long protrusions remain stable structures that connect two cells. Furthermore, while TNTs have been reported to form in short 4-h cultures of RAW/LR5 macrophages (Hanna et al., 2017), fusion of peritoneal macrophages begins 9 h after the addition of IL-4 (Faust et al., 2017). Finally, as revealed in our studies, short rather than long protrusions predominantly initiate macrophage fusion. Nevertheless, the requirement for F-actin and actin nucleation-promoting factors of both TNTs and long fusion-competent protrusions is intriguing and suggests that two phenomena may be connected by a general mechanism.
The requirement for actin-based protrusions as initiators of cell–cell fusion seems to emerge as a unifying principle in several model systems, including fusion in osteoclasts (Oikawa et al., 2012; Shin et al., 2014; Wang et al., 2015) and fusion of muscle cells in flies, zebrafish, and mice (Chen, 2011; Abmayr and Pavlath, 2012; Gruenbaum-Cohen et al., 2012; Aguilar et al., 2013). In Drosophila, muscle fibers are formed through rounds of fusion between a myotube (founder cell) and a fusion-competent myoblast (FCM). The fusion interface between these cells was found to contain an F-actin enrichment referred to as a “focus,” which develops in FCM and then invades the myoblast with one or several finger-like protrusions (Abmayr and Pavlath, 2012; Aguilar et al., 2013). Numerous actin regulatory proteins, including WASp, WIP, SCAR/WAVE, and others have been found at the fusion site (Chen, 2011; Abmayr and Pavlath, 2012; Aguilar et al., 2013). Based on their invasiveness, size and the presence of the actin core with a surrounding ring of adhesive proteins, these structures were called PLS (Sens et al., 2010). While the molecular composition of the fusion-competent protrusions in macrophages and myoblasts may differ, their formation nevertheless requires Cdc42, WASp, and Arp2/3. Furthermore, although we did not observe stable F-actin foci in macrophages, fusion was initiated from sites where podosome clustered in large numbers (Figure 2, A and B). Recent studies have shown that actin-based protrusions, reconstituted together with adhesion molecules in Drosophila cells that normally do not undergo fusion, failed to recapitulate cell–cell fusion (Shilagardi et al., 2013). Expression of authentic fusion proteins, in addition to cell–cell and cell-matrix adhesion molecules, was necessary to induce fusion. These findings suggest that actin-based protrusions are insufficient on their own to promote cell–cell fusion. Whether force production alone is sufficient or the presence of fusion proteins is required for macrophage fusion is currently unknown. Further studies of actin dynamics may help define a link between actin-based protrusions and podosomes in macrophage fusion.
MATERIALS AND METHODS
Mice
C57BL/6J, WASp-/-(B6.129S6-Wastm1Sbs/J), and Cdc42loxP/loxP mice were purchased from The Jackson Laboratory (Bar Harbor, ME). LysMcre mice were a gift from James Lee. LifeAct mice (Riedl et al., 2010) were a gift from Janice Burkhardt and used with permission from Roland Wedlich-Söldner. The conditional Cdc42loxP/loxP mice were generated by crossing Cdc42loxP/loxP mice with LysMcre mice and screening for Cdc42 excision in myeloid leukocytes. All animals were given ad libitum access to food and water and maintained at 22°C on a 12-h light/dark cycle. Experiments were performed according to animal protocols approved by the Institutional Animal Care and Use Committees at Arizona State University and the Mayo Clinic, Arizona, and the HHMI Janelia Reseach Campus.
Macrophage isolation
Age- and sex-matched mice were injected with 0.5 ml of a sterile 4% solution of Brewer’s thioglycollate (TG) (Sigma Aldrich, St. Louis, MO). All animals were humanely killed 72 h later and macrophages were isolated by lavage with an ice-cold solution of phosphate-buffered saline (PBS, pH 7.4) supplemented with 5 mM EDTA. The cells were collected into tubes precoated with bovine serum albumin (BSA). Macrophages were counted with a Neubauer hemocytometer immediately thereafter.
IL-4–induced macrophage fusion
Macrophage fusion was induced as previously described (Faust et al., 2017, 2018). Briefly, cells were applied to various surfaces at a concentration of 5 × 106 cells/ml in DMEM/F12 supplemented with 15 mM HEPES (Cellgro, Manassas, VA), 10% fetal bovine serum (Atlanta Biological, Flowery Branch, GA), and 1% antibiotics (Cellgro, Manassas, VA) and incubated in 5% CO2 at 37°C for 30 min. Nonadherent cells were removed by washing the culture 3–5× with Hank’s balanced salt solution (HBSS; Cellgro, Manassas, VA) supplemented with 0.1% BSA. HBSS was removed and the cells were incubated in culture medium for 2 h. IL-4 (10 ng/ml; Genscript, Piscataway, NJ) was applied to cultures until the respective time points. The fusion index (McNally and Anderson, 1995) was used to determine the extent of macrophage fusion. The fusion index is defined as a fraction of nuclei within MGCs expressed as a percentage of total nuclei counted.
Phase-contrast videomicroscopy
Macrophages were cultured on Permanox plastic (Thermo Scientific, Waltham, MA), Fluorodishes (World Precision Instruments, Sarasota, FL), or surfaces adsorbed with long-chain hydrocarbons as described previously (Faust et al., 2017). Dishes were transferred from the cell culture incubator to a stage-top incubator calibrated to maintain a humidified atmosphere of 5% CO2 in air at 37°C. Phase-contrast images were collected with a 20× or 40× objective every 30 s with an EVOS FL Auto (Thermo Scientific, Waltham, MA) and transferred to ImageJ to create movies.
LLSM
The LLSM used in these experiments is housed in the Advanced Imaging Center at the Howard Hughes Medical Institute Janelia research campus. The system was configured and operated as previously described (Chen et al., 2014). Briefly, eGFP-LifeAct and/or mRFP-LifeAct peritoneal macrophages were applied to 5-mm cover glass surfaces adsorbed with long-chain hydrocarbons (Faust et al., 2017, 2018). IL-4 (10 ng/ml) was added and LLSM was conducted 8–10 h thereafter. Samples were illuminated by LLSM using 488- or 560-nm diode lasers (MPB Communications) through an excitation objective (Special Optics, 0.65 NA, and 3.74-mm WD). Fluorescent emission was collected by a detection objective (Nikon, CFI Apo LWD 25XW, 1.1 NA) and detected by an sCMOS camera (Hamamatsu Orca Flash 4.0 v2). Acquired data were deskewed as previously described (Chen et al., 2014) and deconvolved using an iterative Richardson-Lucy algorithm. Point-spread functions for deconvolution were experimentally measured using 200-nm TetraSpeck beads (Invitrogen) adhered to 5-mm glass coverslips for each excitation wavelength.
Biomaterial implantation
Animal surgery was conducted according to AVMA Guidelines by protocols approved by both the Mayo Clinic and Arizona State University. Segments (1.5 × 0.5 cm) of sterile PCTFE were implanted into the peritoneum of age- and sex-matched mice as described (Jay et al., 2007). Animals were humanely killed 14 d later and explants were analyzed for the presence of MGCs. Prior to explantation, 2 ml of PBS containing 5 mM EDTA was aseptically injected into the peritoneum and cells in the peritoneum were collected by lavage. The number of cells in the peritoneum at the time of explantation was determined by counting with a Neubauer hemocytometer. Experiments were conducted in duplicate on three independent days (i.e., total six mice per experiment).
Immunofluorescence
At the indicated time point, specimens were fixed with 2% formaldehyde in PBS for 30 min at room temperature. Samples were permeabilized with 2% formaldehyde, 0.1% Triton X-100 in PBS for 30 min, and then washed 3× with PBS containing 1% BSA (PBS-BSA). Samples were incubated overnight at 4°C with primary antibodies directed against vinculin and talin (V9131 and T3287 for anti-vinculin and anti-talin, respectively; Sigma Aldrich) and 15 nM Alexa Fluor 488–conjugated phalloidin (Thermo Scientific). The specimens were washed 3× with PBS-BSA and incubated with Alexa Fluor–conjugated secondary antibodies (Thermo Scientific) overnight at 4°C. Nuclei were labeled with DAPI according to the manufacturer’s recommendation (Thermo Scientific). Samples were mounted in Prolong Diamond (Thermo Scientific) and imaged with a Leica SP5 and Leica SP8 laser scanning confocal microscopes. The effect of CK-636 and CK-548 on podosome formation in macrophages labeled with Alexa Fluor 488–conjugated phalloidin was assessed by counting a fraction of cells with >10 podosomes normalized to DMSO control, as previously described (Nolen et al., 2009).
Statistical analyses
Unless indicated otherwise results are shown as the mean ± SD from three independent experiments. Multiple comparisons were made via ANOVA followed by Tukey’s or Dunn’s posttest using GraphPad Instat software. Samples that passed the normal distribution test were analyzed by t test. The remaining samples were analyzed by the Mann-Whitney test. Data were considered significantly different if p < 0.05.
Supplementary Material
Acknowledgments
We thank the members of the Ugarova laboratory and the ASU/Mayo Clinic Center for Metabolic and Vascular Biology for helpful discussions. We thank Satya Khuon at HHMI Janelia Research Campus for help with sample preparation for LLSM. During the preparation of this work, J.J.F. was supported by a T32 Fellowship (5T32DK007569-28). The AIC at HHMI Janelia is jointly funded by the Howard Hughes Medical Institute and the Gordon and Betty Moore Foundation. Image data were collected using a Leica TCS SP5 LSCM (National Institutes of Health [NIH] SIG award S10 RR027154) and Leica TCS SP8 LSCM (NIH SIG award S10 OD023691) housed in the W.M. Keck Bioimaging Facility at Arizona State University. This work was supported by NIH grant R01 HL-63199 to T.P.U.
Abbreviations used:
- BSA
bovine serum albumin
- FCM
fusion-competent myoblast
- HBSS
Hank’s balanced salt solution
- IL-4
interleukin-4
- LLSM
lattice light sheet microscopy
- MGC
multinucleated giant cell
- PBS
phosphate-buffered saline
- PCTFE
polychlorotrifluoroethylene
- PLS
podosome-like structures
- TG
thioglycollate
- TNT
tunneling nanotube
- WASp
Wiscott-Aldrich syndrome protein
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-01-0009) on June 26, 2019.
REFERENCES
- Abmayr SM, Pavlath GK. (2012). Myoblast fusion: lessons from flies and mice. Development , 641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar PS, Baylies MK, Fleissner A, Helming L, Inoue N, Podbilewicz B, Wang H, Wong M. (2013). Genetic basis of cell-cell fusion mechanisms. Trends Genet , 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JM, Rodriguez A, Chang DT. (2008). Foreign body reaction to biomaterials. Semin Immunol , 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campellone KG, Welch MD. (2010). A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol , 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BC, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA, III, Liu Z, et al (2014). Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science , 1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EH. (2011). Invasive podosomes and myoblast fusion. Curr Top Membr , 235–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFife KM, Jenney CR, Colton E, Anderson JM. (1999). Disruption of filamentous actin inhibits human macrophage fusion. FASEB J , 823–832. [DOI] [PubMed] [Google Scholar]
- Destaing O, Ferguson SM, Grichine A, Oddou C, De Camilli P, Albiges-Rizo C, Baron R. (2013). Essential function of dynamin in the invasive properties and actin architecture of v-Src induced podosomes/invadosomes. PLoS One , e77956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovas A, Gevrey JC, Grossi A, Park H, Abou-Kheir W, Cox D. (2009). Regulation of podosome dynamics by WASp phosphorylation: implication in matrix degradation and chemotaxis in macrophages. J Cell Sci , 3873–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust JJ, Christenson W, Doudrick K, Heddleston J, Chew TL, Lampe M, Balabiyev A, Ros R, Ugarova TP. (2018). Fabricating optical-quality glass surfaces to study macrophage fusion. J Vis Exp , 56866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust JJ, Christenson W, Doudrick K, Ros R, Ugarova TP. (2017). Development of fusogenic glass surfaces that impart spatiotemporal control over macrophage fusion: Direct visualization of multinucleated giant cell formation. Biomaterials , 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenbaum-Cohen Y, Harel I, Umansky KB, Tzahor E, Snapper SB, Shilo BZ, Schejter ED. (2012). The actin regulator N-WASp is required for muscle-cell fusion in mice. Proc Natl Acad Sci USA , 11211–11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna SJ, McCoy-Simandle K, Miskolci V, Guo P, Cammer M, Hodgson L, Cox D. (2017). The role of Rho-GTPases and actin polymerization during macrophage tunneling nanotube biogenesis. Sci Rep , 8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haralalka S, Shelton C, Cartwright HN, Katzfey E, Janzen E, Abmayr SM. (2011). Asymmetric Mbc, active Rac1 and F-actin foci in the fusion-competent myoblasts during myoblast fusion in Drosophila. Development , 1551–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helming L, Gordon S. (2007). Macrophage fusion induced by IL-4 alternative activation is a multistage process involving multiple target molecules. Eur J Immunol , 33–42. [DOI] [PubMed] [Google Scholar]
- Helming L, Gordon S. (2008). The molecular basis of macrophage fusion. Immunobiology , 785–793. [DOI] [PubMed] [Google Scholar]
- Helming L, Gordon S. (2009). Molecular mediators of macrophage fusion. Trends Cell Biol , 514–522. [DOI] [PubMed] [Google Scholar]
- Jay SM, Skokos E, Laiwalla F, Krady MM, Kyriakides TR. (2007). Foreign body giant cell formation is preceded by lamellipodia formation and can be attenuated by inhibition of Rac1 activation. Am J Pathol , 632–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao WJ, McNally AK, Hiltner A, Anderson JM. (1995). Role for interleukin-4 in foreign-body giant cell formation on a poly(etherurethane urea) in vivo. J Biomed Mater Res , 1267–1275. [DOI] [PubMed] [Google Scholar]
- Kaverina I, Stradal TE, Gimona M. (2003). Podosome formation in cultured A7r5 vascular smooth muscle cells requires Arp2/3-dependent de-novo actin polymerization at discrete microdomains. J Cell Sci , 4915–4924. [DOI] [PubMed] [Google Scholar]
- Kimura S, Hase K, Ohno H. (2012). Tunneling nanotubes: emerging view of their molecular components and formation mechanisms. Exp Cell Res , 1699–1706. [DOI] [PubMed] [Google Scholar]
- Labernadie A, Bouissou A, Delobelle P, Balor S, Voituriez R, Proag A, Fourquaux I, Thibault C, Vieu C, Poincloux R, et al (2014). Protrusion force microscopy reveals oscillatory force generation and mechanosensing activity of human macrophage podosomes. Nat Commun , 5343. [DOI] [PubMed] [Google Scholar]
- Labernadie A, Thibault C, Vieu C, Maridonneau-Parini I, Charriere GM. (2010). Dynamics of podosome stiffness revealed by atomic force microscopy. Proc Natl Acad Sci USA , 21016–21021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Gallop JL, Rambani K, Kirschner MW. (2010). Self-assembly of filopodia-like structures on supported lipid bilayers. Science , 1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder S, Higgs H, Hufner K, Schwarz K, Pannicke U, Aepfelbacher M. (2000). The polarization defect of Wiskott-Aldrich syndrome macrophages is linked to dislocalization of the Arp2/3 complex. J Immunol , 221–225. [DOI] [PubMed] [Google Scholar]
- Linder S, Nelson D, Weiss M, Aepfelbacher M. (1999). Wiskott-Aldrich syndrome protein regulates podosomes in primary human macrophages. Proc Natl Acad Sci USA , 9648–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder S, Wiesner C. (2016). Feel the force: Podosomes in mechanosensing. Exp Cell Res , 67–72. [DOI] [PubMed] [Google Scholar]
- Linder S, Wiesner C, Himmel M. (2011). Degrading devices: invadosomes in proteolytic cell invasion. Annu Rev Cell Dev Biol , 185–211. [DOI] [PubMed] [Google Scholar]
- Mattila PK, Lappalainen P. (2008). Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol , 446–454. [DOI] [PubMed] [Google Scholar]
- McInnes A, Rennick DM. (1988). Interleukin 4 induces cultured monocytes/macrophages to form giant multinucleated cells. J Exp Med , 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally AK, Anderson JM. (1995). Interleukin-4 induces foreign body giant cells from human monocytes/macrophages. Differential lymphokine regulation of macrophage fusion leads to morphological variants of multinucleated giant cells. Am J Pathol , 1487–1499. [PMC free article] [PubMed] [Google Scholar]
- McNally AK, Anderson JM. (2002). Beta1 and beta2 integrins mediate adhesion during macrophage fusion and multinucleated foreign body giant cell formation. Am J Pathol , 621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally AK, Anderson JM. (2011). Macrophage fusion and multinucleated giant cells of inflammation. Adv Exp Med Biol , 97–111. [DOI] [PubMed] [Google Scholar]
- Milde R, Ritter J, Tennent GA, Loesch A, Martinez FO, Gordon S, Pepys MB, Verschoor A, Helming L. (2015). Multinucleated giant cells are specialized for complement-mediated phagocytosis and large target destruction. Cell Rep , 1937–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DA, Courtneidge SA. (2011). The “ins” and “outs” of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol , 413–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD, Hartman J, Sakowicz R, Pollard TD. (2009). Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature , 1031–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa GC, Slepnev VI, Neff L, Ringstad N, Takei K, Daniell L, Kim W, Cao H, McNiven M, Baron R, De Camilli P. (2000). A functional link between dynamin and the actin cytoskeleton at podosomes. J Cell Biol , 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa T, Oyama M, Kozuka-Hata H, Uehara S, Udagawa N, Saya H, Matsuo K. (2012). Tks5-dependent formation of circumferential podosomes/invadopodia mediates cell-cell fusion. J Cell Biol , 553–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onel SF, Renkawitz-Pohl R. (2009). FuRMAS: triggering myoblast fusion in Drosophila. Dev Dyn , 1513–1525. [DOI] [PubMed] [Google Scholar]
- Onfelt B, Nedvetzki S, Benninger RK, Purbhoo MA, Sowinski S, Hume AN, Seabra MC, Neil MA, French PM, Davis DM. (2006). Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J Immunol , 8476–8483. [DOI] [PubMed] [Google Scholar]
- Pfaff M, Jurdic P. (2001). Podosomes in osteoclast-like cells: structural analysis and cooperative roles of paxillin, proline-rich tyrosine kinase 2 (Pyk2) and integrin alphaVbeta3. J Cell Sci , 2775–2786. [DOI] [PubMed] [Google Scholar]
- Podbilewicz B. (2014). Virus and cell fusion mechanisms. Annu Rev Cell Dev Biol , 111–139. [DOI] [PubMed] [Google Scholar]
- Podolnikova NP, Kushchayeva YS, Wu Y, Faust J, Ugarova TP. (2016). The role of integrins alphaMbeta2 (Mac-1, CD11b/CD18) and alphaDbeta2 (CD11d/CD18) in macrophage fusion. Am J Pathol , 2105–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proag A, Bouissou A, Mangeat T, Voituriez R, Delobelle P, Thibault C, Vieu C, Maridonneau-Parini I, Poincloux R. (2015). Working together: spatial synchrony in the force and actin dynamics of podosome first neighbors. ACS Nano , 3800–3813. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. (2011). Life at the leading edge. Cell , 1012–1022. [DOI] [PubMed] [Google Scholar]
- Riedl J, Flynn KC, Raducanu A, Gartner F, Beck G, Bosl M, Bradke F, Massberg S, Aszodi A, Sixt M, Wedlich-Soldner R. (2010). Lifeact mice for studying F-actin dynamics. Nat Methods , 168–169. [DOI] [PubMed] [Google Scholar]
- Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH. (2004). Nanotubular highways for intercellular organelle transport. Science , 1007–1010. [DOI] [PubMed] [Google Scholar]
- Sens KL, Zhang S, Jin P, Duan R, Zhang G, Luo F, Parachini L, Chen EH. (2010). An invasive podosome-like structure promotes fusion pore formation during myoblast fusion. J Cell Biol , 1013–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilagardi K, Li S, Luo F, Marikar F, Duan R, Jin P, Kim JH, Murnen K, Chen EH. (2013). Actin-propelled invasive membrane protrusions promote fusogenic protein engagement during cell-cell fusion. Science , 359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin NY, Choi H, Neff L, Wu Y, Saito H, Ferguson SM, De Camilli P, Baron R. (2014). Dynamin and endocytosis are required for the fusion of osteoclasts and myoblasts. J Cell Biol , 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soe K, Hobolt-Pedersen AS, Delaisse JM. (2015). The elementary fusion modalities of osteoclasts. Bone , 181–189. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Bulanova EA, Chaga OY, Vignjevic DM, Kojima S, Vasiliev JM, Borisy GG. (2003). Mechanism of filopodia initiation by reorganization of a dendritic network. J Cell Biol , 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenawa T, Suetsugu S. (2007). The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol , 37–48. [DOI] [PubMed] [Google Scholar]
- Thrasher AJ. (2002). WASp in immune-system organization and function. Nat Rev Immunol , 635–646. [DOI] [PubMed] [Google Scholar]
- Vignery A. (2011). Macrophage fusion: the making of a new cell. In: Cell Fusions: Regulation and Control, ed. Larsson L-I, New York: Springer, 219–231. [Google Scholar]
- Wang Y, Brooks PJ, Jang JJ, Silver AS, Arora PD, McCulloch CA, Glogauer M. (2015). Role of actin filaments in fusopod formation and osteoclastogenesis. Biochim Biophys Acta , 1715–1724. [DOI] [PubMed] [Google Scholar]
- Yang C, Svitkina T. (2011). Filopodia initiation: focus on the Arp2/3 complex and formins. Cell Adh Migr , 402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Wang L, Geiger H, Cancelas JA, Mo J, Zheng Y. (2007). Rho GTPase Cdc42 coordinates hematopoietic stem cell quiescence and niche interaction in the bone marrow. Proc Natl Acad Sci USA , 5091–5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambonin-Zallone A, Teti A, Grano M, Rubinacci A, Abbadini M, Gaboli M, Marchisio PC. (1989). Immunocytochemical distribution of extracellular matrix receptors in human osteoclasts: a beta 3 integrin is colocalized with vinculin and talin in the podosomes of osteoclastoma giant cells. Exp Cell Res , 645–652. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.