

Abstract
Mitochondrial decline is a hallmark of aging, and cells are equipped with many systems to regulate mitochondrial structure and function in response to stress and metabolic alterations. Here, using budding yeast, we identify a proteolytic pathway that contributes to alterations in mitochondrial structure in aged cells through control of the mitochondrial fusion GTPase Fzo1. We show that mitochondrial fragmentation in old cells correlates with reduced abundance of Fzo1, which is triggered by functional alterations in the vacuole, a known early event in aging. Fzo1 degradation is mediated by a proteolytic cascade consisting of the E3 ubiquitin ligases SCFMdm30 and Rsp5, and the Cdc48 cofactor Doa1. Fzo1 proteolysis is activated by metabolic stress that arises from vacuole impairment, and loss of Fzo1 degradation severely impairs mitochondrial structure and function. Together, these studies identify a new mechanism for stress-responsive regulation of mitochondrial structure that is activated during cellular aging.
INTRODUCTION
Mitochondria play a central role in cellular metabolism. They produce energy through oxidative phosphorylation, and promote the synthesis and/or breakdown of amino acids, lipids, nucleotides, iron–sulfur clusters, and heme (Rutter and Hughes, 2015). Mitochondria are also involved in cellular signaling, apoptosis, and innate immunity (Chandel, 2014). The execution of these activities requires coordination between hundreds of mitochondrial resident proteins (Calvo and Mootha, 2010), and dynamic communication between mitochondria and other organelles, including vacuoles/lysosomes, peroxisomes, and the endoplasmic reticulum (ER; Eisenberg-Bord and Schuldiner, 2017). Because mitochondria are highly connected with other organelles and perform many functions, they are quite susceptible to functional alterations, and their decline is a hallmark of aging and a multitude of human pathologies (Wallace, 2005; Nunnari and Suomalainen, 2012).
To maintain mitochondrial health, cells employ a variety of mechanisms to alter the structure, function, and proteome of mitochondria in response to a number of stressors and metabolic alterations. These strategies include regulation of dynamins and mitofusins involved in mitochondrial fission and fusion to control mitochondrial structure (Labbe et al., 2014), modulation of interorganelle contacts between mitochondria and other organelles (Murley and Nunnari, 2016), regulation of mitochondrial protein import (Harbauer et al., 2014), maintenance of the mitochondrial proteome via activation of the mitochondrial unfolded protein response (UPRmt; Shpilka and Haynes, 2018), and selective turnover of mitochondrial resident proteins by internal mitochondrial proteases (Quiros et al., 2015), the ubiquitin proteasome system (UPS; Karbowski and Youle, 2011), mitochondrial-derived vesicles (Sugiura et al., 2014) and compartments (Hughes et al., 2016), and mitophagy (Pickles et al., 2018).
Our understanding of the relationships mitochondria forge with other organelles and the quality control pathways that operate to maintain mitochondrial function is deep. However, how these interorganelle connections impact mitochondria during the physiological context of aging, and how and when quality control systems operate to maintain mitochondrial health over the course of an organism’s lifespan is less clear. To gain insight into these questions, we and others have been analyzing mitochondrial function and quality control in the context of budding yeast replicative aging, which is defined as the number of times an individual Saccharomyces cerevisiae cell asymmetrically divides to produce a daughter (Steinkraus et al., 2008). To date, these studies have demonstrated that mitochondrial structure and function is impaired with age in yeast, similar to other organisms (Scheckhuber et al., 2007; McFaline-Figueroa et al., 2011; Hughes and Gottschling, 2012).
We recently showed that mitochondrial dysfunction in aged yeast is driven in part by alterations in a complex metabolic relationship between lysosomes (vacuoles in yeast) and mitochondria (Hughes and Gottschling, 2012). Vacuoles are acidic organelles that mediate protein degradation, pH maintenance, metabolite storage, and metabolic signaling (Klionsky et al., 1990). Most functions of the vacuole require a proton gradient generated by the evolutionarily conserved vacuolar H+-ATPase (V-ATPase) across the vacuole membrane (Kane, 2006). Our previous studies demonstrated that the vacuolar proton gradient declines early in aging, resulting in amino acid–dependent metabolic stress that causes mitochondrial impairment and fragmentation through an unknown mechanism (Hughes and Gottschling, 2012). These results are consistent with prior studies showing that V-ATPase mutants are respiratory deficient and exhibit altered mitochondrial morphology (Ohya et al., 1991; Dimmer et al., 2002). Moreover, a growing body of literature indicates that lysosomes and mitochondria are functionally and physically linked across organisms (Elbaz-Alon et al., 2014; Honscher et al., 2014; Wong et al., 2018), and that interdependent functional alterations of these organelles underlie the development of numerous age-related and metabolic disorders (Terman et al., 2010; Kim et al., 2013; Colacurcio and Nixon, 2016; de la Mata et al., 2016; Plotegher and Duchen, 2017; Audano et al., 2018). How mitochondria and vacuoles are functionally linked, and why changes in vacuole function cause mitochondrial fragmentation in the context of aging remains unclear.
Here, we sought to address these questions, and uncovered a new proteolytic pathway that regulates mitochondrial structure in response to changes in vacuole function. Specifically, we show that vacuole impairment in aged cells triggers rapid, proteasome-dependent destruction of the yeast mitofusin, Fzo1 (Hermann et al., 1998). Fzo1 degradation requires redundant activity of the E3 ubiquitin ligases SCFMdm30 (Fritz et al., 2003) and Rsp5 (Hein et al., 1995). Destruction of Fzo1 appears to be triggered by nutrient cues coming from the vacuole, and loss of Fzo1 regulation severely impairs mitochondrial health. These results suggest that cells alter the shape of the mitochondrial network in response to vacuole-induced nutrient stress, and provide insight into molecular mechanisms that regulate mitochondrial structure during the aging process.
RESULTS
Mitochondrial fusion declines in aged yeast cells
We previously demonstrated that budding yeast mitochondria fragment and aggregate with increasing replicative age, and that these structural alterations are triggered by changes in vacuole function (Hughes and Gottschling, 2012). How vacuole impairment causes mitochondrial fragmentation is unknown, and we wanted to identify the molecular mechanism underlying this phenomenon. We first confirmed that mitochondria fragment in old yeast cells by visualizing a GFP-tagged version of the mitochondrial outer membrane protein Tom70-GFP expressed from its endogenous locus (Figure 1A). Mitochondrial morphology is maintained through a balance of fission and fusion (Chan, 2012), and the fragmented mitochondrial network in old cells indicates that the fission/fusion equilibrium shifts toward fission with increasing age. Consistent with this idea, deletion of Dnm1, a dynamin-related GTPase essential for mitochondrial fission (Bleazard et al., 1999), prevents age-induced mitochondrial fragmentation, as previously observed (Figure 1A; Scheckhuber et al., 2007).
FIGURE 1:

Mitochondrial fusion is reduced in aged yeast cells. (A) Representative single Z-plane images of wild-type (WT) and dnm1Δ yeast of the indicated age expressing Tom70-GFP. n = 30 cells. For A–C, age (number of cell divisions) identified by calcofluor (Calc) staining of bud scars. Age ranges: wild-type (WT), young = 0–3, old = 16–22; dnm1Δ, young = 0–3, old = 15–21. DIC = differential interference contrast. (B, C) Representative single Z-plane fluorescent images of WT (B) or dnm1Δ (C) zygotes derived from mating of Tom70-GFP and Tom70-mCherry expressing haploid cells of the indicated age. n = 50 cells for both B and C. (D) Whole cell extracts from young (Y) and old (O) cells expressing the indicated proteins were analyzed by Western blot with anti-HA, anti-Ugo1, and anti-Pgk1 antibodies. Age ranges (n = 30 cells): Fzo1-HA, Y = 0–3 divisions, O = 6–10; Mgm1-HA, Y = 0–3, O = 6–10; Ugo1, Y = 0–3, O = 12–15.
To directly test the fusion capability of aged cells, we utilized a previously described in vivo mitochondrial fusion assay (Nunnari et al., 1997). This assay measures fusion as the degree of mitochondrial content mixing in zygotes produced by mating two haploid yeast strains containing either red or green fluorescently labeled mitochondria, respectively. As expected, the mitochondrial network in zygotes derived from young haploid cells was uniformly both green and red, indicating that fusion is normal in these cells (Figure 1B). Conversely, zygotes derived from aged haploids contained nonoverlapping green and red mitochondria, indicating that fusion is reduced in old cells (Figure 1B). Mitochondrial fusion was also impaired in zygotes derived from a combination of young and old cells, indicating that the old cell phenotype is dominant (Supplemental Figure 1). Consistent with our observation that deletion of Dnm1 suppressed mitochondrial fragmentation in aged cells, mitochondrial fusion was restored in zygotes derived from aged dnm1Δ cells (Figure 1C). Altogether, these data indicate that mitochondrial fusion is reduced in aged yeast, causing fragmentation of the mitochondrial network.
The mitofusin Fzo1 is reduced in aged cells
Mitochondrial fusion in budding yeast is mediated by three proteins: Mgm1, a GTPase that promotes mitochondrial inner membrane fusion (Wong et al., 2000, 2003); Fzo1, a GTPase that mediates fusion of mitochondrial outer membranes (Hermann et al., 1998); and Ugo1, a modified carrier protein that localizes to the mitochondrial outer membrane (Sesaki and Jensen, 2001). We monitored protein abundance of endogenous Ugo1 and functional epitope-tagged versions of Mgm1-HA (Wong et al., 2000) and Fzo1-HA (Supplemental Figure 2) in young and aged cells via Western blotting. Mgm1 and Ugo1 were unchanged with age (Figure 1D and Supplemental Figure 3). However, steady-state levels of Fzo1 were reduced in old cells compared with young cells (Figure 1D and Supplemental Figure 3). This change was not caused by the HA epitope tag, as steady-state levels of endogenous, untagged Fzo1 also declined with age (Supplemental Figure 4). It remains unclear whether reduced Fzo1 is sufficient to cause mitochondrial fragmentation in aged cells because our attempts to increase Fzo1 abundance before or during the aging process caused aberrant mitochondrial aggregation. Nevertheless, the decline in mitochondrial fusion in old yeast cells correlates with reduced levels of Fzo1.
Loss of vacuole acidity triggers mitochondrial fragmentation and reduced Fzo1 protein abundance
We previously showed that mitochondrial fragmentation in aged cells is caused by loss of vacuolar acidity, and that treatment of young cells with the specific V-ATPase inhibitor concanamycin A (ConcA; Drose et al., 1993) recapitulates age-induced changes in mitochondria (Hughes and Gottschling, 2012). We confirmed that V-ATPase inhibition caused fragmentation of the mitochondrial network in young cells (Figure 2, A and B). To test whether V-ATPase inhibition is sufficient to reduce steady-state concentrations of Fzo1, we monitored the abundance of fusion proteins in young yeast cells treated with ConcA. Indeed, acute V-ATPase inhibition triggered Fzo1 reduction, without altering levels of Mgm1 and Ugo1 (Figure 2C). These results mirror our observations in aged cells, and indicate that loss of vacuole function triggers alterations in mitochondrial structure that correlate with a reduction in the mitofusin Fzo1.
FIGURE 2:

Vacuole impairment induces mitochondrial fragmentation and Fzo1 depletion. (A) Representative maximum-intensity projection images of wild-type cells expressing Tom70-GFP treated with DMSO or concanamycin A (ConcA) for 6 h. Scale bar = 2 μm. (B) Quantification of mitochondrial phenotypes (tubular or fragmented) from panel A. Error bars show mean ± SE of three replicates. n = 100 cells/replicate. (C) Whole cell extracts from yeast cells expressing the indicated protein grown in the absence or presence of concanamycin A (ConcA) for 4 h were analyzed by Western blot with anti-HA, anti-Ugo1, and anti-Pgk1 antibodies.
Fzo1 decline in vacuole-impaired cells is proteasome and Doa1-dependent
We sought to elucidate the molecular basis of Fzo1 decline in aged and vacuole-impaired cells. Prior studies have shown that Fzo1 is a frequent target of the proteasome in nonstressed cells through a mechanism that requires the AAA+-ATPase Cdc48 and its cofactor Doa1, two factors that promote extraction and delivery of proteins to the proteasome (Fritz et al., 2003; Neutzner and Youle, 2005; Cohen et al., 2008; Anton et al., 2013; Wu et al., 2016; Simoes et al., 2018). On this basis, we tested whether vacuole stress triggered Fzo1 decline through a similar mechanism. Treatment of young cells with the proteasome inhibitor MG-132 prevented vacuole-stress–induced Fzo1 decline (Figure 3A), whereas impairment of autophagy (atg5Δ; Feng et al., 2014) and vacuole-dependent protein degradation (pep4Δ or pep4Δ prb1Δ; Zubenko et al., 1983) had no effect (Figure 3B and Supplemental Figure 5). Furthermore, deleting the Cdc48 cofactor Doa1 prevented Fzo1 loss caused by acute ablation of vacuole function (Figure 3C), indicating that Doa1 coordinates with the proteasome to mediate ConcA-induced Fzo1 turnover. Finally, we tested whether age-induced decline of Fzo1 was mediated by this same mechanism. Although we were unable to age cells successfully in the presence of proteasome inhibitor, deletion of Doa1 prevented age-induced loss of Fzo1 (Figures 1D and 3D). These results implicate the proteasome and Doa1 as major players in vacuole and age-induced turnover of the mitofusin Fzo1.
FIGURE 3:
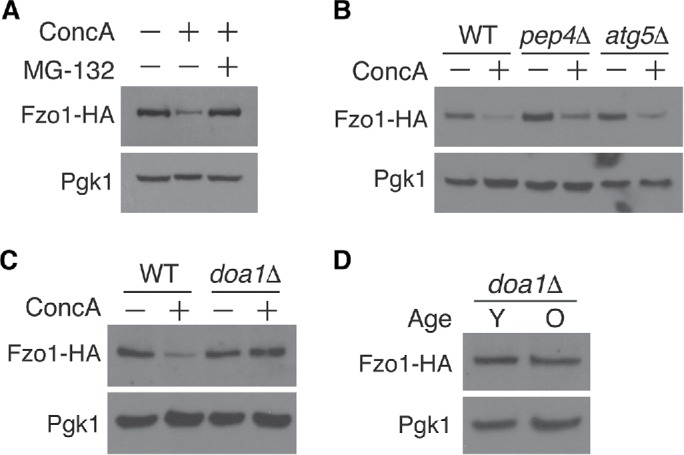
Fzo1 depletion triggered by aging and vacuole impairment is proteasome and Doa1-dependent. (A) Whole cell extracts from yeast expressing Fzo1-HA grown in the absence or presence of concanamycin A (ConcA) or proteasome inhibitor MG-132 for 4 h were analyzed by Western blot with anti-HA and anti-Pgk1 antibodies. (B, C) Whole cell extracts from wild-type (WT), atg5Δ, and pep4Δ (B) or WT and doa1Δ (C) yeast expressing Fzo1-HA grown in the absence or presence of concanamycin A for 4 h were analyzed as in A. (D) Whole cell extracts from young (Y) and old (O) doa1Δ yeast expressing Fzo1-HA protein were analyzed as in A. Age ranges (n = 30 cells): Y = 0–3 divisions; O = 6–10 divisions.
SCF Mdm30 and Rsp5 mediate vacuole-stress–dependent Fzo1 degradation
We next wanted to identify the E3 ubiquitin ligase(s) that promotes Fzo1 degradation in response to vacuole dysfunction. Prior studies showed that ubiquitin–proteasome-dependent Fzo1 destruction in nonstressed cells is mediated by the SCFMdm30 E3 ubiquitin ligase complex through a highly regulated cascade controlled in part by the action of two deubiquitylating enzymes, Ubp2 and Ubp12 (Fritz et al., 2003; Escobar-Henriques et al., 2006; Cohen et al., 2008, 2011; Anton et al., 2013). Turnover of Fzo1 by this system promotes mitochondrial fusion by clearing Fzo1 dimer complexes formed after engagement of two mitochondria. In addition to this fusion promoting pathway, these previous studies proposed the existence of an unidentified, E3 ubiquitin ligase that antagonizes mitochondrial fusion by promoting Fzo1 degradation. Consistent with this idea, we found that steady-state Fzo1 levels are elevated in mdm30Δ cells, but still decline upon vacuole inhibition with ConcA, indicating that another ubiquitin ligase promotes vacuole stress-induced Fzo1 turnover (Figure 4A).
FIGURE 4:

Fzo1 degradation induced by vacuole dysfunction is Rsp5- and Mdm30-dependent. (A) Whole cell extracts from wild-type (WT) and mdm30Δ yeast expressing Fzo1-HA grown in the absence or presence of concanamycin A for 4 h were analyzed by Western blot with anti-HA and anti-Pgk1 antibodies. (B) Whole cell extracts from wild-type (WT), mdm30Δ, doxycycline-repressible RSP5 (tetp-RSP5), and mdm30Δ tetp-RSP5 yeast expressing Fzo1-HA grown in the absence or presence of concanamycin A for 4 h were analyzed as in A. All cells were treated with 20 μg/ml doxycycline for 16 h before ConcA addition to inhibit RSP5 expression. (C) Schematic of Fzo1 showing the location of the heptad repeats (HRN, HR1, and HR2), GTPase domain, and PY motif (LPHY). (D) Whole cell extracts from wild-type (WT) and mdm30Δ yeast expressing wild-type Fzo1-HA or PY motif mutated Fzo1-HA (fzo1-PY) grown in the absence or presence of concanamycin A for 4 h were analyzed by Western blot as in A.
Emerging evidence suggests that the HECT-domain E3 ubiquitin ligase, Rsp5, well known for its role in the turnover of nutrient transporters at the cell surface (Lauwers et al., 2010), also acts on mitochondrial outer membrane proteins in nonstressed cells (Wu et al., 2016; Belgareh-Touze et al., 2017). On the basis of these observations, we tested whether Rsp5 regulates Fzo1 protein abundance in response to vacuole impairment. Because Rsp5 is essential, we utilized a yeast strain in which Rsp5 expression is under control of a doxycycline-repressible promoter (Mnaimneh et al., 2004). Upon doxycycline-dependent Rsp5 depletion, Fzo1 still declined in response to vacuole impairment (Figure 4B). However, combined loss of both Mdm30 and Rsp5 prevented Fzo1 depletion in the presence of the V-ATPase inhibitor ConcA, indicating that Rsp5 and SCFMdm30 act redundantly to regulate Fzo1 levels in response to vacuole dysfunction (Figure 4B).
A previous study implicated Rsp5 in regulation of Fzo1 protein levels under steady-state conditions, but concluded that regulation was indirect (Cavellini et al., 2017). To test the role of Rsp5 in vacuole dysfunction-induced Fzo1 degradation more directly, we analyzed turnover of a mutant version of Fzo1 lacking a putative PY motif. PY motifs, PPxY or LPxY (where x represents any amino acid), are well-characterized Rsp5 binding sequences present in numerous Rsp5 substrates (Rotin and Kumar, 2009). In most cases, ablation of this motif prevents Rsp5 binding, ubiquitylation, and subsequent proteasomal degradation. Fzo1 contains an LPxY motif at amino acids 414–417 in its cytoplasmic domain, which is accessible to the soluble, cytoplasmic localized Rsp5 (Figure 4C; De Vecchis et al., 2017). We mutated this motif on endogenous Fzo1 to AAAA, and tested the impact of this mutation on vacuole-dysfunction–induced Fzo1 degradation. Importantly, mutating the PY motif did not impact Fzo1 function (Figure 6B). Furthermore, we found that Fzo1 levels still declined in cells expressing an Fzo1-PY mutant alone, but were stabilized in an mdm30Δ/Fzo1-PY–mutant strain (Figure 4D; Supplemental Figure 6), indicating a direct role for Rsp5 in regulating the levels of Fzo1 in response to vacuole impairment. Taken together, these data suggest that vacuole-dysfunction–induced Fzo1 destruction is mediated by redundant actions of SCFMdm30 and Rsp5.
FIGURE 6:
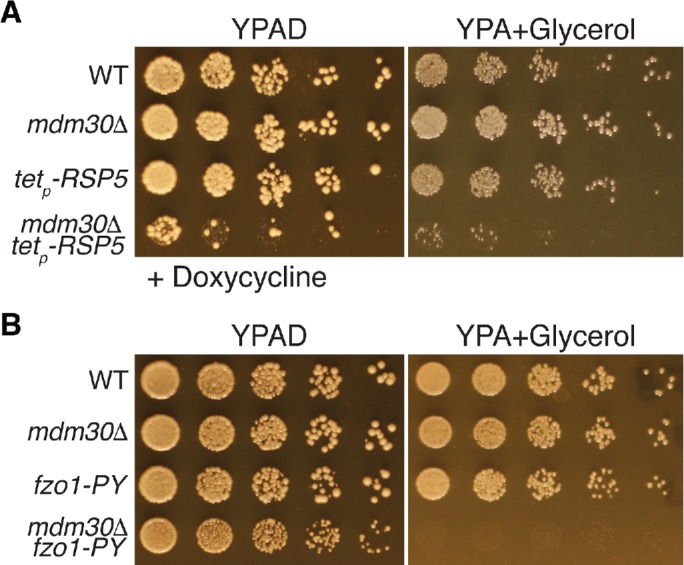
Dual regulation of Fzo1 by Rsp5 and Mdm30 is required for normal mitochondrial function. (A, B) Serial dilutions of wild-type (WT) and the indicated mutant yeast strains plated on rich medium containing glucose (YPAD) or glycerol (YPA + glycerol). Plates in panel A contain 20 μg/ml doxycycline to inhibit expression of RSP5.
Loss of Mdm30- and Rsp5-dependent regulation of Fzo1 impairs mitochondrial structure and function
A prediction of our data is that age- and vacuole-dysfunction–induced mitochondrial fragmentation is driven by the Fzo1 proteolytic pathway outlined above. To test this, we examined mitochondrial morphology in aged doa1Δ cells, which maintain Fzo1 levels with age (Figure 3D). Despite the fact that age-induced decline of Fzo1 is blunted in doa1Δ cells, mitochondria fragmentation was not suppressed in these mutants (Figure 5A). The same was true in mdm30Δ/Fzo1-PY–mutant cells (Figure 5B). This lack of suppression was not restricted to age-induced phenotypes—mitochondrial fragmentation caused by the V-ATPase inhibitor ConcA was also not suppressed in cells lacking DOA1 or a combination of RSP5 and MDM30 (Figure 5, C and D), despite restored Fzo1 levels in these mutants (Figures 3C and 4B).
FIGURE 5:

Preventing destruction of Fzo1 does not restore mitochondrial morphology in aged or vacuole-impaired cells. (A) Representative single Z-plane images of wild-type (WT) and doa1Δ yeast expressing Tom70-GFP of the indicated age. n = 50 cells. For A and B, age (number of cell divisions) determined by calcofluor staining (calc.). Age ranges: WT, Y = 0–2, O = 12–14; doa1Δ, Y = 0–2, O = 11–14. DIC = differential interference contrast. Scale bar = 2 μm. (B) Representative single Z-plane images of wild-type (WT) and mdm30Δ fzo1-PY–mutant yeast expressing Tom70-GFP of the indicated age. n = 50 cells. Age ranges: WT, Y = 0–2, O = 11–14; mdm30Δ fzo1-PY, Y = 0–2, O = 12–14. Scale bar = 2 μm. (C) Representative maximum-intensity projection images of wild-type (WT), mdm30Δ, doxyclycline-repressible RSP5 (tetp-RSP5), and mdm30Δ tetp-RSP5 yeast expressing Tom70-GFP grown in the absence or presence of concanamycin A for 6 h. All cells were treated with 20 μg/ml doxycycline for 16 h before ConcA addition to inhibit RSP5 expression. Scale bar = 2 μm. (D) Quantification of mitochondrial phenotypes (tubular or fragmented) from panel C. Error bars show mean ± standard error of three replicates. n = 100 cells/replicate.
Two possible interpretations of these results are 1) that mitochondrial fragmentation in response to vacuole impairment is not caused by Fzo1 reduction, or 2) that fragmentation is also impacted by other unknown factors in addition to Fzo1 decline. We currently cannot rule out either of these possibilities. However, our results also raise an alternative explanation: that mitochondrial morphology is not restored in strains lacking Fzo1 proteolytic machinery because total loss of Fzo1 regulation is catastrophic for mitochondrial structure and function. Consistent with this latter idea, young cells lacking Fzo1 proteolytic machinery exhibited severely altered mitochondrial morphology, even in the absence of vacuole stress (Figure 5, C and D). Furthermore, combined loss of Mdm30 and Rsp5 reduced growth of yeast on the fermentable carbon source glucose, and severely reduced growth on the nonfermentable carbon source glycerol (Figure 6A). The same was true in mdm30Δ strains expressing a PY-mutant version of Fzo1, indicating that the growth phenotypes resulted from loss of Fzo1 regulation, and not another function of Rsp5 (Figure 6B). Collectively, these results suggest that dual regulation of Fzo1 by Rsp5 and SCFMdm30 is important for maintaining mitochondrial structure and function.
Vacuole dysfunction-induced Fzo1 degradation occurs in response to metabolic alterations
Finally, we wanted to identify the signal arising from dysfunctional vacuoles that triggers Fzo1 degradation. Vacuole impairment causes mitochondrial depolarization, oxidative stress, blunted protein degradation, and amino acid–induced toxicity (Milgrom et al., 2007; Li and Kane, 2009; Hughes and Gottschling, 2012; Hughes et al., 2016). The latter phenotype arises from a breakdown in the ability of faulty vacuoles to effectively compartmentalize amino acids, leading to their cytoplasmic elevation (Kitamoto et al., 1988). Of all the stresses caused by V-ATPase impairment, we previously showed that elevated amino acid stress is the primary inducer of mitochondrial dysfunction with age, and that this toxicity can be overcome by overexpressing a vacuolar amino acid importer, Avt1, which transports neutral amino acids from the cytoplasm into the vacuole lumen (Russnak et al., 2001; Hughes and Gottschling, 2012; Tone et al., 2015). Consistent with our previous observations that Avt1 overexpression prevents age-induced mitochondrial fragmentation, Fzo1 decline was blunted in aged cells overexpressing Avt1 (Figure 7A). Furthermore, we found that Fzo1 regulation by the Doa1/Mdm30/Rsp5 proteolytic pathway was specific to vacuole-induced metabolic stress. Ablating vacuole protein degradation by deleting the master vacuole protease Pep4 did not trigger Fzo1 degradation (Figure 3B). Additionally, although mitochondrial depolarization with carbonyl cyanide m-chlorophenyl hydrazine (CCCP) and H2O2-mediated oxidative stress reduced steady-state levels of Fzo1 (Figure 7, B and C), this effect was independent of Doa1.
FIGURE 7:

Vacuole-dysfunction–induced Fzo1 degradation occurs in response to elevated metabolic stress. (A) Whole cell extracts from young (Y) and old (O) wild-type or AVT1 overexpressing Fzo1-HA cells were analyzed by Western blot with anti-HA and anti-Pgk1 antibodies. Age ranges (n = 50 cells): WT, Y = 0–2, O = 7–9; AVT1 OE, Y = 0–2, O = 7–10. (B, C) Whole cell extracts from wild-type (WT) and doa1Δ yeast expressing Fzo1-HA grown in the absence or presence of carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (B) or hydrogen peroxide (H2O2) (C) for 4 h were analyzed by Western blot with anti-HA and anti-Pgk1 antibodies. (D) Illustration showing that Fzo1 is degraded by a proteolytic cascade requiring Doa1 and redundant actions of SCFMdm30 and Rsp5 in response to vacuole impairment.
Collectively, these results suggest that turnover of Fzo1 mediated by Rsp5 and SCFMdm30 is triggered by vacuole-induced metabolic stress. The specific nature of this stress and how it triggers Fzo1 decline remains to be determined, as genetic inhibition of common nutrient sensing pathways, including the PKA pathway (gpa2Δ and gpr1Δ; Rubio-Texeira et al., 2010) and the SSY amino acid sensing system (ssy1Δ; Poulsen et al., 2005), as well as pharmacological inhibition of the nutrient responsive mechanistic target of rapamycin (mTOR) pathway with rapamycin (Saxton and Sabatini, 2017), did not prevent Conc A–mediated Fzo1 decline (Supplemental Figure 7, A and B). In fact, inhibiting mTOR with rapamycin actually triggered Fzo1 depletion in a Doa1-independent manner (Supplemental Figure 7, B and C). This effect was similar to the decline caused by CCCP and H2O2, suggesting that in addition to vacuole dysfunction, multiple forms of stress converge to regulate Fzo1, through a variety of mechanisms.
DISCUSSION
Alterations in mitochondrial structure and function are hallmarks of aging across species. However, our understanding of the driving forces behind these changes is incomplete. Here, we sought to elucidate the molecular basis of age-induced mitochondrial fragmentation using budding yeast as a model system. Our results confirmed previous observations that age-induced mitochondrial fragmentation is triggered by decline in the function of the vacuole, and uncovered a new proteolytic pathway that regulates mitochondrial structure in response to changes in vacuole function. As outlined in Figure 7D, we show that loss of vacuole function, which is an early event in yeast aging (Hughes and Gottschling, 2012), triggers proteasomal degradation of the mitochondrial fusion GTPase Fzo1. Degradation requires the redundant actions of the SCFMdm30 and Rsp5 E3 ubiquitin ligases, as well as the Cdc48 cofactor, Doa1. Fzo1 degradation in this context appears to be specifically triggered by metabolic cues coming from dysfunctional vacuoles, and loss of this pathway impairs mitochondrial respiration.
Our study provides new insight into the molecular basis for mitochondrial fragmentation during aging, and indicates that in yeast, age-induced mitochondrial fragmentation is an adaptive, proteolytic response activated by functional decline of the yeast vacuole. Despite the fact that loss of this regulatory system is detrimental for respiratory growth, it remains an open question whether age-induced mitochondrial fragmentation is beneficial or harmful. Prior reports in yeast and Caenorhabditis elegans indicated that a hyperfused mitochondrial network is linked to longevity (Scheckhuber et al., 2007; Houtkooper et al., 2013; Chaudhari and Kipreos, 2017). However, other recent studies in Drosophila melanogaster suggest the opposite—that mitochondrial fragmentation is beneficial during aging (Rana et al., 2013, 2017). A possible explanation for these conflicting observations is that the relationship between mitochondrial network shape and aging/lifespan is highly context dependent. This hypothesis is supported by a recent study in C. elegans that showed that mitochondrial fission and fusion are both necessary for reaping the longevity benefits associated with dietary restriction, and that combined loss of fusion and fission extends lifespan (Weir et al., 2017). Clearly, additional studies are required to fully elucidate the contributions of mitochondrial dynamics to the maintenance of cellular lifespan.
Our discovery expands the repertoire of regulatory pathways that control mitochondrial shape in response to a variety of stressors and changes in cellular metabolism. These systems, which have been extensively reviewed elsewhere, utilize a variety of pre- and posttranslational mechanisms, including transcription, phosphorylation, ubiquitylation, redox modifications, acetylation, and more to control the levels and/or activity of key fusion and fission factors, including the fusion GTPases Opa1 and Mfn1/2 (yeast Mgm1 and Fzo1, respectively) and the fission GTPase Drp1 (yeast Dnm1; Chan, 2012; Youle and van der Bliek, 2012; Elgass et al., 2013; Mishra and Chan, 2014; Roy et al., 2015). To our knowledge, this report is the first to describe a regulatory cascade that alters mitochondrial dynamics in response to changes in the function of the vacuole/lysosome. This raises an important question: why do cells adjust mitochondrial structure upon V-ATPase impairment? On the basis of our prior work, and observations described in Figure 7, we speculate that mitochondrial fragmentation in this context is triggered by metabolic alterations that occur upon loss of V-ATPase activity. Vacuoles carry out numerous functions in cells, including protein degradation, pH control, nutrient sensing, and ion and amino acid homeostasis (Li and Kane, 2009; Perera and Zoncu, 2016). We previously showed that structural and functional changes that occur in mitochondria upon vacuole impairment are driven by a breakdown in the compartmentalization of cellular amino acids, leading to acute amino acid toxicity (Hughes and Gottschling, 2012). Our current data are consistent with this model, and support the hypothesis that mitochondrial fragmentation during aging may be a specific response to elevated nutrient stress.
Consistent with this idea, prior reports showed that mitochondrial shape in eukaryotes is quite responsive to changes in cellular nutrient status. During starvation, the mitochondrial network hyperfuses (Hailey et al., 2010; Gomes et al., 2011), which likely protects it from degradation and boosts ATP production. Likewise, studies in insulin-producing beta cells showed that elevated supply of glucose and fatty acids promotes mitochondrial fragmentation (Molina et al., 2009). The reason for this is not entirely clear, but fragmentation may enable cells to better utilize these metabolic substrates as fuel sources, or cope with stress associated with their metabolism. In our current study, it remains unclear whether mitochondrial fragmentation benefits the yeast cell during times of vacuole-induced metabolic stress. Like the situation in beta cells, it may enable yeast to better metabolize the excess amino acids that arise when amino acid sequestration in vacuoles is compromised. Alternatively, mitochondrial fragmentation by Rsp5/Mdm30 may protect mitochondria from stress associated with high rates of metabolism, or help prime mitochondria for autophagy-dependent turnover. It is also possible that the purpose of modulating Fzo1 levels in response to vacuole dysfunction is not to control mitochondrial dynamics, but rather, to regulate interorganelle contacts between mitochondria and peroxisomes. A recent study showed that Fzo1 may function as a tether between these organelles (Shai et al., 2018), but how mitochondria peroxisome tethering is regulated is currently unknown.
Moving forward, it will be important to define how Rsp5 and SCFMdm30 coordinate with one another to mediate Fzo1 degradation, and how specific signals are relayed to this system. Numerous prior studies have demonstrated that Mdm30 ubiquitylates Fzo1 (Fritz et al., 2003; Neutzner and Youle, 2005; Durr et al., 2006; Escobar-Henriques et al., 2006; Cohen et al., 2008, 2011; Anton et al., 2013). Current models predict that this ubiquitylation is important for promoting mitochondrial fusion through a complex cascade that is regulated by the deubiquitylating enzymes Ubp2 and Ubp12 (Anton et al., 2013). These studies also leave open the possibility of other roles for Mdm30. Our data are consistent with two potentially separable functions for Mdm30: one that ubiquitylates Fzo1 to promote fusion under steady-state conditions, and another that promotes mitochondrial fragmentation. How these potential opposing roles of SCFMdm30 are achieved is currently unclear.
Like Mdm30, prior studies have demonstrated a role for both Rsp5 and Doa1 at the mitochondrial surface, promoting steady-state turnover of mitochondrial outer membrane proteins (Wu et al., 2016; Belgareh-Touze et al., 2017). Many of these proteins contain PY motifs like Fzo1 that are recognized by Rsp5, and it has been demonstrated for some mitochondrial substrates that ablation of this motif impairs their turnover. Our current results confirm the importance of Rsp5 and Doa1 in the maintenance of mitochondrial homeostasis, and demonstrate that they are also capable of promoting regulated degradation at the mitochondrial surface in response to stress. This raises an important question as to how signals are relayed to Rsp5 and SCFMdm30 to promote Fzo1 degradation in the context of vacuole stress. Although we do not yet have an answer to this question, we speculate that the mechanism probably exhibits parallels with well-characterized nutrient-dependent protein degradation performed by Rsp5 in other contexts, including turnover of nutrient transporters at the plasma and vacuole membranes (MacGurn et al., 2012; Li et al., 2015). In these other contexts, nutrient cues trigger Rsp5 recruitment to its substrates either directly or through adaptor proteins of the arrestin family (Lin et al., 2008; Nikko and Pelham, 2009). Recruitment is triggered by several mechanisms, including alterations in protein structure that expose PY motifs, or phosphoregulation of substrates and/or adaptors by well-characterized nutrient signaling pathways (Lauwers et al., 2010; MacGurn et al., 2011). Whether mitochondria structure is tightly coordinated with Rsp5-dependent regulation of cellular nutrient import remains to be determined, but it is an exciting possibility moving forward.
MATERIALS AND METHODS
Reagents
Chemicals were obtained from the following sources: Concanamycin A (ConcA) from Santa Cruz Biotechnology (SC-202111C); dimethyl sulfoxide (DMSO; D2650), CCCP (C2920), and doxycycline from Sigma Aldrich (D9891); MG-132 (10012628) from Cayman Chemical; hydrogen peroxide (H2O2; H325-500) from Fisher Scientific; and rapamycin (R-5000) from LC Laboratories. Source and catalogue numbers for plasmids, antibodies, other reagents, and yeast strains are described in the appropriate sections below. Final reagent concentrations in all experiments were as follows: ConcA = 500 nM; DMSO = 0.4%; CCCP = 10 μM; doxycycline = 20 μg/ml; MG-132 = 55 μM; H2O2 = 1 mM; and rapamycin = 1 μg/ml.
Yeast strains, oligos, and plasmids
All yeast strains (Table 1) are derivatives of S. cerevisiae S288C (BY background) unless otherwise noted (Brachmann et al., 1998). Deletion strains were created by one-step PCR-mediated gene replacement using the oligos listed in Table 1 and plasmid templates pRS306 and pRS400-HygMX (Brachmann et al., 1998). Strains expressing fluorescently tagged Tom70 (yeGFP or mCherry) or 3XHA-tagged Fzo1 from their native loci were created using one-step PCR-mediated C-terminal endogenous epitope tagging using standard techniques and the oligos listed in Table 1 (Shaner et al., 2004; Sheff and Thorn, 2004). Plasmid templates for epitope insertion at the C-terminus of TOM70 were pKT127 (yeGFP) and pKT127-mCherry (mCherry; Sheff and Thorn, 2004). Plasmid templates for 3XHA epitope insertion at the C-terminus of FZO1 were pFA6a-3HA-KamnMX6 and pFA6a-3HA-HIS3MX6 (Wach et al., 1997; Longtine et al., 1998). Correct integrations and gene deletions were confirmed by colony PCR across the chromosomal insertion site and, where appropriate, microscopy visualizing the correct subcellular localization of the fusion protein. The yeast strain overexpressing AVT1 and its empty vector control (AHY7091 and AHY7089) were constructed by digesting pAG306 GPD-AVT1_chrI and pAG306 GPD-empty_chrI with NotI and integrating into an empty region of Chr I as previously described (Hughes and Gottschling, 2012). Strains for regulated expression of RSP5 by the tetracycline promoter were derived from the Hughes yeast tetracycline promoters strain collection (Mnaimneh et al., 2004).
TABLE 1:
Yeast strains and oligos used in this study.
| Strain | Parent and genotype |
|---|---|
| BY4741 | MATa his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 |
| BY4742 | MATα his3Δ1 leu2Δ0 ura3Δ0 lys2Δ0 |
| BY4743 | MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 ura3Δ0/ura3Δ0 met15Δ0/+lys2Δ0/+ |
| AHY1443 | BY4741 TOM70-yeGFP:KanMX |
| AHY3417 | BY4741 TOM70-yeGFP:KanMX doa1Δ::URA3 |
| AHY1919 | BY4743 TOM70-yeGFP:KanMX/+ |
| AHY3324 | BY4741 FZO1-3xHA:KanMX |
| AHY3720 | BY4741 FZO1-3xHA:KanMX pdr5Δ::URA3 |
| AHY3716 | BY4741 FZO1-3xHA:KanMX atg5Δ::URA3 |
| AHY3714 | BY4741 FZO1-3xHA:KanMX pep4Δ::URA3 |
| AHY8285 | BY4741 FZO1-3xHA:KanMX pep4Δ::URA3 prb1Δ::LEU2 |
| AHY3619 | BY4741 FZO1-3xHA:KanMX doa1Δ::URA3 |
| AHY4758 | BY4741 FZO1-3xHA:KanMX mdm30Δ::HygMX |
| AHY3450 | BY4741 fzo1-PY (LPHY→AAAA)-3xHA:KanMX |
| AHY3453 | BY4741 fzo1-PY (LPHY→AAAA)-3xHA:KanMX mdm30Δ::HygMX |
| AHY7089 | BY4741 FZO1-3HA:KanMX ChrI nt199459::GPDp-URA3 |
| AHY7091 | BY4741 FZO1-3HA:KanMX ChrI nt199459::GPDp-AVT1-URA3 |
| AHY8286 | BY4741 FZO1-3xHA:KanMX gpr1Δ::LEU2 |
| AHY8288 | BY4741 FZO1-3xHA:KanMX gpa2Δ::LEU2 |
| AHY8290 | BY4741 FZO1-3xHA:KanMX ssy1Δ::LEU2 |
| AHY4605 | MATα his3Δ1 leu2Δ0 ura3Δ0 fzo1-PY (LPHY→AAAA) TOM70-yeGFP:KanMX mdm30Δ::HygMX |
| R1158 | BY4741 URA3::CMV-tTA |
| AHY3850 | R1158 FZO1-3xHA:His3MX |
| AHY3852 | R1158 RSP5p::kanR-tet07-TATA FZO1-3xHA:His3MX |
| AHY3910 | R1158 FZO1-3xHA:His3MX mdm30Δ::HygMX |
| AHY3911 | R1158 RSPp5::kanR-tet07-TATA FZO1-3xHA:His3MX mdm30Δ::HygMX |
| AHY1421 | R1158 TOM70-yeGFP:spHIS5MX |
| AHY1422 | R1158 RSP5p::kanR-tet07-TATA TOM70-yeGFP:spHIS5MX |
| AHY1424 | R1158 TOM70-yeGFP:spHIS5MX mdm30Δ::HygMX |
| AHY1430 | R1158 RSP5p::kanR-tet07-TATA TOM70-yeGFP:spHIS5MX mdm30Δ::HygMX |
| UCC8773 | MATa his3Δ1 leu2Δ0 ura3Δ0 lys2Δ0 hoΔ::SCW11p-Cre-EBD78-NatMX loxP-CDC20-Intron-loxP-HphMX loxP-UBC9-loxP-LEU2 |
| UCC895 | MATa his3Δ1 leu2Δ0 ura3Δ0 lys2Δ0 hoΔ::SCW11p-Cre-EBD78-NatMX loxP-CDC20-Intron-loxP-HphMX loxP-UBC9-loxP-LEU2 TOM70-yeGFP:KanMX |
| AHY490 | MATα his3Δ1 leu2Δ0 ura3Δ0 trp1Δ63 hoΔ::SCW11p-Cre-EBD78-NatMX loxP-CDC20-Intron-loxP-HphMX loxP-UBC9-loxP-LEU2 TOM70-mCherry:KanMX |
| AHY923 | MATa his3Δ1 leu2Δ0 ura3Δ0 lys2Δ0 hoΔ::SCW11p-Cre-EBD78-NatMX loxP-CDC20-Intron-loxP-HphMX loxP-UBC9-loxP-LEU2 TOM70-yeGFP:KanMX dnm1Δ::URA3 |
| AHY937 | MATα his3Δ1 leu2Δ0 ura3Δ0 trp1Δ63 hoΔ::SCW11p-Cre-EBD78-NatMX loxP-CDC20-Intron-loxP-HphMX loxP-UBC9-loxP-LEU2 TOM70-mCherry:KanMX dnm1Δ::URA3 |
| AHY546 | MATa/α his3Δ1/hisΔ1 leu2Δ0/leu2Δ0 ura3Δ0/ura3Δ0 lys2Δ0/+ trp1Δ63/+hoΔ::SCW11p-Cre-EBD78-NatMX/hoΔ::SCW11p-Cre-EBD78-NatMX loxP-CDC20-Intron-loxP-HphMX/loxP-CDC20-Intron-loxP-HphMX loxP-UBC9-loxP-LEU2/loxP-UBC9-loxP-LEU2 |
| AHY951 | MATa/α his3Δ1/hisΔ1 leu2Δ0/leu2Δ0 ura3Δ0/ura3Δ0 lys2Δ0/+ trp1Δ63/+hoΔ::SCW11p-Cre-EBD78-NatMX/hoΔ::SCW11p-Cre-EBD78-NatMX loxP-CDC20-Intron-loxP-HphMX/loxP-CDC20-Intron-loxP-HphMX loxP-UBC9-loxP-LEU2/loxP-UBC9-loxP-LEU2 TOM70-yeGFP-KanMX/+ VPH1-mCherry-KanMX/+ |
| AHY995 | MATa/α his3Δ1/hisΔ1 leu2Δ0/leu2Δ0 ura3Δ0/ura3Δ0 lys2Δ0/+ trp1Δ63/+hoΔ::SCW11p-Cre-EBD78-NatMX/hoΔ::SCW11p-Cre-EBD78-NatMX loxP-CDC20-Intron-loxP-HphMX/loxP-CDC20-Intron-loxP-HphMX loxP-UBC9-loxP-LEU2/loxP-UBC9-loxP-LEU2 TOM70-yeGFP:KanMX/+ VPH1-mCherry:KanMX/+ dnm1Δ::URA3/ dnm1Δ::URA3 |
| JSY2519 | MATa ura3-52 his3Δ200 trp1Δ63 ura3Δ met15Δ MGM1:3xHA (W303) |
| Oligo name | Oligo sequence |
|---|---|
| FZO1 pFA6a tag F | TCAAAAATTGATGGTGGAAGAAATAAATTTAGACATCGATCGGATCCCCGGGTTAATTA |
| FZO1 pFA6a tag R | TATATTGATTTGAAAAGACCTCATATATTTACAAGAATATGAATTCGAGCTCGTTTAAAC |
| FZO1 tag check | CCCGTGCCTTACCAATGAATCTTTCC |
| HA tag check R | ACGTCATATGGATAGGATCC |
| TOM70 pKT tag F | TCAAGAAACTTTAGCTAAATTACGCGAACAGGGTTTAATGGGTGACGGTGCTGGTTTA |
| TOM70 pKT tag R | TTTGTCTTCTCCTAAAAGTTTTTAAGTTTATGTTTACTGTTCGATGAATTCGAGCTCG |
| VPH1 pKT tag F | GGAAGTCGCTGTTGCTAGTGCAAGCTCTTCCGCTTCAAGCGGTGACGGTGCTGGTTTA |
| VPH1 pKT tag R | AGTACTTAAATGTTTCGCTTTTTTTAAAAGTCCTCAAAATTCGATGAATTCGAGCTCG |
| PDR5 KO F | AAGTTTTCGTATCCGCTCGTTCGAAAGACTTTAGACAAAAAGATTGTACTGAGAGTGCAC |
| PDR5 KO R | TCTTGGTAAGTTTCTTTTCTTAACCAAATTCAAAATTCTACTGTGCGGTATTTCACACCG |
| PDR5 KO check | CGGAACTCTTCTACGCCGTGGTACGATATC |
| URA check | ACCTAATGCTTCAACTAACTCCAG |
| PEP4 KO F | ATTTAATCCAAATAAAATTCAAACAAAAACCAAAACTAACGATTGTACTGAGAGTGCAC |
| PEP4 KO R | GGCAGAAAAGGATAGGGCGGAGAAGTAAGAAAAGTTTAGCCTGTGCGGTATTTCACACG |
| PEP4 KO check | GCCTAGTGACCTAGTATTTAATCC |
| ATG5 KO F | GGTTCTAGAAGAACGGAGATAGGAAACCTATGATGTAAGTGATTGTACTGAGAGTGCAC |
| ATG5 KO R | GATATTTGAATGACACTTTTAAATGCGTATCACACCGTATAACAGCTCCTGTGCGGTATT |
| ATG5 KO check | CACTAAATAATGAAGTAGCATGCTCAG |
| DOA1 KO F | TCATGTGTGATAGTAAGGTGTAGAGCAGCAGATTTGGAGTAGATTGTACTGAGAGTGCAC |
| DOA1 KO R | ATCTAGACATTATGTGTTTTATATGATTGCTGTAAAAGTACTGTGCGGTATTTCACACCG |
| DOA1 KO check | ATGCAACTCCAGTAGCTTGATATATTCGGC |
| MDM30 KO F | ACAATTTTTCGGTATTAGTTACTAAAAGGCTCACATATACAGATTGTACTGAGAGTGCACC |
| MDM30 KO R | TGTGTCAGGATGCTACTTTTGGAAACCTCCTTAAATATGACTGTGCGGTATTTCACACCG |
| MDM30 KO Check | GTTTATGTTACATAATAAGTGAAAG |
| DNM1 KO F | TTAAGTAGCTACCAGCGAATCTAAATACGACGGATAAAGAGATTGTACTGAGAGTGCACC |
| DNM1 KO R | TGTTGAAGTAAGATCAAAAATGAGATGAATTATGCAATTACTGTGCGGTATTTCACACCG |
| DNM1 KO Check | GTGGGAACAATAAATAAGAGGGTAGG |
| Hyg Check | TATGCTCCGCATTGGTCTTG |
| URA3 mid R | CCACGGTTCTATACTGTTGAC |
| Ura3 mid F | GCTACATATAAGGAACGTGCTG |
| FZO1 PY Adapt 1 | CGGTGTGAAATACCGCACAGGCTGCTGCTGCTCATAATGAAAATGACAATGAAGATCATG |
| FZO1 PY Adapt 2 | GGTGCACTCTCAGTACAATCTAATCGATGTCTAAATTTATTTCTTCCACC |
| FZO1 PY Mutamer | GTTTCCAAAAACGGAGATGAAGCTGCTGCTGCTCATAATGAAAATGACAATG |
| pRS306 RS2 R | CTGTGCGGTATTTCACACCG |
| pRS306 RS1 F | ATTGTACTGAGAGTGCACC |
| pRS306 RS1 R | GGTGCACTCTCAGTACAAT |
| PRB1 KO F | CAATAAAAAAACAAACTAAACCTAATTCTAACAAGCAAAGCTGTGCGGTATTTCACACCG |
| PRB1 KO R | AAGAAAAAAAAAAGCAGCTGAAATTTTTCTAAATGAAGAAAGATTGTACTGAGAGTGCAC |
| PRB1 KO Check | AGATGATGTTATTGTTGCGG |
| SSY1 KO F | GTTATAAACTGCTTAAAAATAGGGAAGTTCCTTGAGGAATAGATTGTACTGAGAGTGCAC |
| SSY1 KO R | AATACTAACAATAATAATACTAATAATAGTACATATAACCCTGTGCGGTATTTCACACCG |
| SSY1 KO Check | TTAATCCTGTTACTCCTGTGCTAGATGGGG |
| GPA2 KO F | TTGTTACAGCACAAATCACGCGTATTTTCAAGCAAATATCAGATTGTACTGAGAGTGCAC |
| GPA2 KO R | AGAAGAGGCATGCAGTTTTGTCTCTGTTTTAGCTGTGCATCTGTGCGGTATTTCACACCG |
| GPA2 KO Check | GATATACCATATATTACGATCCCTGCGCCC |
| GPR1 KO F | ATCCGAAGTGTGACGAATAAAGCAAACTCTCCAACTCAAAAGATTGTACTGAGAGTGCAC |
| GPR1 KO R | TTCCTTACTTTCCATTTTCAAACATCGCGATACAAAAACTCTGTGCGGTATTTCACACCG |
| GPR1 KO Check | CACAAGCTGACCCTAATTACCAAGGGACCT |
Yeast strain AHY3450, in which the wild-type FZO1 gene has been replaced by a mutant version of FZO1 lacking its PY amino acid motif (LPHY→AAAA) was created using a previously described fusion PCR URA3 counterselection strategy (Reid et al., 2002). Briefly, a 3′ segment of the FZO1 gene containing the mutant PY motif (3′ fzo1-PY) was amplified from yeast genomic DNA via PCR with oligos FZO1 PY Adapt 1 and FZO1 PY Adapt 2 and subsequently fused in two separate reactions to both the 5′ and 3′ ends of the yeast URA3 gene that had been amplified via PCR using oligo pairs URA3 mid F/pRS306 RS2 R and URA3 mid R/pRS306 RS1 F, respectively. The two fusion PCR products (3′ fzo1-PY-5′ URA3 and 3′ URA3-3′ fzo1-PY) were transformed into yeast strain BY4741, for recombination/integration at the endogenous FZO1 locus to create FZO1:URA3:3′ fzo1-PY. Finally, strains with fzo1 alleles containing the mutant PY motif were generated via recombination-mediated removal of the URA3 gene by counterselection on synthetic defined media containing 1 mg/ml 5-fluoro-orotic acid (5-FOA). Cells able to grow on 5-FOA were sequenced to verify the loss of URA3, the presence of the fzo1-PY motif mutation, and the lack of any additional disruptions to the FZO1 locus.
Yeast cell culture and media
Yeast cells were grown exponentially overnight to a density of 4 × 106 cells/ml before initiating any treatments. For all experiments, cells were cultured in YPAD medium (1% yeast extract, 2% peptone, 0.005% adenine, 2% glucose) and were not allowed to reach densities higher than 1 × 107 cells/ml during experimental treatment. Drug treatments were carried out for the amount of time indicated in each figure legend, and final concentrations of compounds utilized in all experiments are listed above in the Reagents section. YPA + glycerol medium for serial dilution growth assays contained 1% yeast extract, 2% peptone, 0.005% adenine, and 3% glycerol. Selection of auxotrophic markers during yeast strain construction was performed on synthetic defined media with glucose (SD) lacking the appropriate media component for selection (0.67% yeast nitrogen base without amino acids, 2% glucose, supplemented nutrients 0.074 g/l each adenine, alanine, arginine, asparagine, aspartic acid, cysteine, glutamic acid, glutamine, glycine, histidine, myo-inositol, isoleucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine, uracil, and valine, 0.369 g/l leucine, 0.007 g/l para-aminobenzoic acid).
Yeast serial dilution growth assays
Fivefold serial dilutions of exponentially growing yeast cells were prepared in YPAD or YPA + glycerol medium, and 3 μl of each dilution of cells was spotted on YPAD or YPA + glycerol agar. Cells were grown at 30°C for 2 (YPAD) or 3 (YPA + glycerol) days before imaging plates. Total cells plated in each dilution spot: 5000, 1000, 200, 40, and 8.
Isolation of aged yeast cells
Yeast cells were aged and purified for downstream analysis as previously described with slight modifications (Hughes and Gottschling, 2012). Briefly, 2.5 × 107 cells from a 15-h YPAD exponentially growing log-phase culture were washed twice in phosphate-buffered saline (PBS), pH 7.4, and resuspended in PBS containing 3 mg/ml sulfa-NHS-LC-biotin (Pierce) at a final concentration of 2.5 × 107 cells/ml. Cells were incubated for 30 min at RT (23°C) and then washed twice in PBS and once in YPAD. Biotinylated cells were then seeded in YPAD at a final density of 1 × 104 cells/ml and cultured at 30°C for 15 h. Cell densities of these cultures never surpassed 1 × 107 cells/ml. At this stage, cells were either reseeded in YPAD at 1 × 104 and grown for an additional 12 h, or immediately subjected to purification. For live-cell purification after either one or two rounds of culturing (15 or 27 h total), cells were collected by centrifugation, washed twice in PBS, resuspended at a density of 2 × 108 cells/ml in 500 μl PBS, and incubated for 30 min at RT with 50 μl streptavidin-coated magnetic beads (MicroMACS; Miltenyi Biotec). After incubation, cells were washed twice in PBS, resuspended in 8 ml of PBS, and loaded onto a LS MACS column (Miltenyi Biotec) equilibrated with 5 ml of PBS. After gravity flow-through of unlabeled cells, columns were washed twice with 8 ml of PBS. Columns were then removed from the magnetic field and aged cells were eluted by gravity flow with 8 ml of PBS. After elution, cells were either pelleted and flash-frozen in liquid nitrogen for immunoblotting analysis, or pelleted and resuspended in YPAD for a 1-h recovery incubation at 30°C before microscopy and/or fusion assay analysis. Young cell fractions were obtained by collecting the flow-through of unlabeled “young” cells that eluted from the LS MACS column on the first pass.
Determination of yeast cell age
Age of purified yeast cells was determined by calcofluor staining of bud scars as previously described using fluorescent brightener 28 (Sigma Aldrich; Hughes and Gottschling, 2012). For microscopy experiments (including fusion assays), calcofluor staining was conducted just before imaging, and age ranges of cells scored as “young” or “old” for each experiment, as well as number of cells quantified are listed in the figure legends. For Western blot experiments, calcofluor staining was conducted on a small aliquot of purified cells, and age ranges of cells scored as “young” or “old” are included in the figure legends. The percent purity of old cells in the final samples was calculated for Western blot experiments. Purity of old cells ranged from 68% to 82% across experiments when available.
Mitochondrial fusion assays
To assess mitochondrial fusion competency of old and young yeast cells, equal quantities of young, old, or young and old haploid yeast of opposite mating types expressing Tom70-yeGFP or Tom70-mCherry were mixed together in a small volume of YPAD and spotted onto a YPAD agar plate. Cells were incubated for 3 h at 30°C to promote mating and zygote formation. Young and old cells utilized in the fusion assays had been aged and isolated via biotinylation/streptavidin purification as described above. After 3 h, cells were removed from the plate, resuspended in a small volume of imaging buffer (10 mM HEPES, pH 7.6, + 5% glucose), stained with calcofluor as described above, and examined via widefield microscopy. Fusion competency of haploids was scored as yes/normal if green and red mitochondrial signals completely overlapped in zygotes, and no/compromised if complete mitochondrial content mixing did not occur or was incomplete.
Microscopy
Single optical fluorescent Z-sections of live yeast cells for experiments in Figure 1 (and its supplements) were acquired using a Nikon Eclipse E800 equipped with a CoolSNAP HQ2 CCD camera (Photometrics) and 60× oil-immersion objective. Widefield images were acquired and processed using Metamorph version 7.1.1.0 imaging software, Fiji, and Adode Photoshop. For experiments in Figures 2–7, 200 nM optical Z-sections of live yeast cells were acquired with an AxioImager M2 (Carl Zeiss) equipped with an Axiocam 506 monochromatic camera (Carl Zeiss) and 100× oil-immersion objective (Carl Zeiss, Plan Apochromat; NA 1.4). Widefield images were acquired with ZEN software (Carl Zeiss), processed with Fiji, and presented as maximum-intensity projections or individual Z-slices as indicated in the figure legends.
Mitochondrial morphology quantification
Mitochondrial morphology was assessed via microscopy as described above. Cells with tubular, well-connected mitochondrial morphologies were scored as “tubular.” Cells with fragmented, aggregated, or spherical mitochondrial morphologies were scored as “fragmented” or “fragmented and aggregated.” The number of cells and experimental replicates are indicated in the figure legends. Note that the percentage of cells exhibiting a fragmented mitochondrial network upon ConcA treatment differed between the strain backgrounds used in Figures 2B (AHY1919) and 5D (AHY1421).
Protein isolation and immunoblotting
Total protein lysates were prepared from exponentially growing yeast cells as previously described (Hughes et al., 2016). Briefly, 2 × 107 total cells were harvested by centrifugation, resuspended in 0.1 M NaOH, and incubated at room temperature for 5 min. Samples were then centrifuged at 21,000 × g for 10 min at 4°C. Resulting pellets were resuspended in 100 μl SDS lysis buffer (10 mM Tris, pH 6.8, 100 mM NaCl, 1% SDS, 1 mM EDTA, 1 mM EGTA) plus protease inhibitors (Sigma Aldrich; 11697498001). Loading dye (25 μl, 150 mM Tris HCl, pH 6.8, 15% SDS, 25% glycerol, 600 uM bromophenol blue, 35 mM β-mercaptoethanol) was added and lysates were incubated at 95°C for 10 min or 65°C for 30 min.
For Western blot analysis, protein lysates were run on polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were processed as previously described (Hughes et al., 2016). Primary antibodies were anti-HA 12CA5 (Santa Cruz; Sc-57592, 1:5000), anti-Pgk1 22C5D8 (Abcam; ab113687, 1:1000), anti-Ugo1 (gift from Jodi Nunnari, University California, Davis; 1:500), anti-Fzo1 (gift from Jodi Nunnari, University California, Davis; 1:1000), and anti-carboxypeptidase Y 10A5 (Invitrogen; A6428, 1:1000). Secondary antibodies conjugated to horseradish peroxidase (HRP) were from Jackson Immunoresearch. Proteins were detected using Supersignal enhanced chemiluminescence solution (dura or pico; catalogue numbers 34076 or 34580, respectively; ThermoFisher) and either film or a BioRad ChemiDoc MP system.
Immunoblot quantification
All immunoblot-based experiments were conducted at least two independent times to ensure reproducibility. For immunoblot quantification, integrated density analysis of protein bands was performed using the image processing software Fiji/ImageJ. Quantification was conducted on raw, unaltered blot images obtained from a BioRad ChemiDoc MP system. Quantification was carried out on three to five technical replicates. The abundance of each protein band was normalized to the intensity of a Pgk1 loading control. The average of the replicates was taken and an unpaired t test was used to assess significance.
Supplementary Material
Acknowledgments
We thank Janet Shaw (University of Utah School of Medicine) and members of the Shaw and Hughes labs for insightful discussions and critical review of the manuscript and J. Nunnari (University of California, Davis) for Anti-Fzo1 and anti-Ugo1 antiserum. Research was supported by National Institutes of Health Grant no. AG-023779 to D.E.G.; Grants no. AG-043095, no. GM-119694, no. AG-061376, and no. AG-055648 to A.L.H.; and Grant no. T32 DK-091317 to J.M.G. and T.K.C. A.L.H. was further supported by an American Federation for Aging Research Junior Research Grant, a United Mitochondrial Disease Foundation Early Career Research Grant, a Searle Scholars Award, and a Glenn Foundation for Medical Research Award.
Abbreviations used:
- ConcA
concanamycin A
- SCF
SKP1-cullin-1-F-box.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-02-0094) on May 29, 2019.
REFERENCES
- Anton F, Dittmar G, Langer T, Escobar-Henriques M. (2013). Two deubiquitylases act on mitofusin and regulate mitochondrial fusion along independent pathways. Mol Cell , 487–498. [DOI] [PubMed] [Google Scholar]
- Audano M, Schneider A, Mitro N. (2018). Mitochondria, lysosomes, and dysfunction: their meaning in neurodegeneration. J Neurochem , 291–309. [DOI] [PubMed] [Google Scholar]
- Belgareh-Touze N, Cavellini L, Cohen MM. (2017). Ubiquitination of ERMES components by the E3 ligase Rsp5 is involved in mitophagy. Autophagy , 114–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM. (1999). The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol , 298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast , 115–132. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Mootha VK. (2010). The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet , 25–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavellini L, Meurisse J, Findinier J, Erpapazoglou Z, Belgareh-Touze N, Weissman AM, Cohen MM. (2017). An ubiquitin-dependent balance between mitofusin turnover and fatty acids desaturation regulates mitochondrial fusion. Nat Commun , 15832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. (2012). Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet , 265–287. [DOI] [PubMed] [Google Scholar]
- Chandel NS. (2014). Mitochondria as signaling organelles. BMC Biol , 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari SN, Kipreos ET. (2017). Increased mitochondrial fusion allows the survival of older animals in diverse C. elegans longevity pathways. Nat Commun , 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MM, Amiott EA, Day AR, Leboucher GP, Pryce EN, Glickman MH, McCaffery JM, Shaw JM, Weissman AM. (2011). Sequential requirements for the GTPase domain of the mitofusin Fzo1 and the ubiquitin ligase SCFMdm30 in mitochondrial outer membrane fusion. J Cell Sci , 1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MMJ, Leboucher GP, Livnat-Levanon N, Glickman MH, Weissman AM. (2008). Ubiquitin-proteasome-dependent degradation of a mitofusin, a critical regulator of mitochondrial fusion. Mol Biol Cell , 2457–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colacurcio DJ, Nixon RA. (2016). Disorders of lysosomal acidification—the emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res Rev , 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Mata M, Cotan D, Villanueva-Paz M, de Lavera I, Alvarez-Cordoba M, Luzon-Hidalgo R, Suarez-Rivero JM, Tiscornia G, Oropesa-Avila M. (2016). Mitochondrial dysfunction in lysosomal storage disorders. Diseases , E31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vecchis D, Cavellini L, Baaden M, Henin J, Cohen MM, Taly A. (2017). A membrane-inserted structural model of the yeast mitofusin Fzo1. Sci Rep , 10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer KS, Fritz S, Fuchs F, Messerschmitt M, Weinbach N, Neupert W, Westermann B. (2002). Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Mol Biol Cell , 847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drose S, Bindseil KU, Bowman EJ, Siebers A, Zeeck A, Altendorf K. (1993). Inhibitory effect of modified bafilomycins and concanamycins on P- and V-type adenosinetriphosphatases. Biochemistry , 3902–3906. [DOI] [PubMed] [Google Scholar]
- Durr M, Escobar-Henriques M, Merz S, Geimer S, Langer T, Westermann B. (2006). Nonredundant roles of mitochondria-associated F-box proteins Mfb1 and Mdm30 in maintenance of mitochondrial morphology in yeast. Mol Biol Cell , 3745–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg-Bord M, Schuldiner M. (2017). Mitochatting—If only we could be a fly on the cell wall. Biochim Biophys Acta , 1469–1480. [DOI] [PubMed] [Google Scholar]
- Elbaz-Alon Y, Rosenfeld-Gur E, Shinder V, Futerman AH, Geiger T, Schuldiner M. (2014). A dynamic interface between vacuoles and mitochondria in yeast. Dev Cell , 95–102. [DOI] [PubMed] [Google Scholar]
- Elgass K, Pakay J, Ryan MT, Palmer CS. (2013). Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta , 150–161. [DOI] [PubMed] [Google Scholar]
- Escobar-Henriques M, Westermann B, Langer T. (2006). Regulation of mitochondrial fusion by the F-box protein Mdm30 involves proteasome-independent turnover of Fzo1. J Cell Biol , 645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, He D, Yao Z, Klionsky DJ. (2014). The machinery of macroautophagy. Cell Res , 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz S, Weinbach N, Westermann B. (2003). Mdm30 is an F-box protein required for maintenance of fusion-competent mitochondria in yeast. Mol Biol Cell , 2303–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L. (2011). During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol , 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. (2010). Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell , 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C. (2014). The protein import machinery of mitochondria—a regulatory hub in metabolism, stress, and disease. Cell Metab , 357–372. [DOI] [PubMed] [Google Scholar]
- Hein C, Springael JY, Volland C, Haguenauer-Tsapis R, Andre B. (1995). NPl1, an essential yeast gene involved in induced degradation of Gap1 and Fur4 permeases, encodes the Rsp5 ubiquitin-protein ligase. Mol Microbiol , 77–87. [DOI] [PubMed] [Google Scholar]
- Hermann GJ, Thatcher JW, Mills JP, Hales KG, Fuller MT, Nunnari J, Shaw JM. (1998). Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J Cell Biol , 359–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honscher C, Mari M, Auffarth K, Bohnert M, Griffith J, Geerts W, van der Laan M, Cabrera M, Reggiori F, Ungermann C. (2014). Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev Cell , 86–94. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature , 451–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Gottschling DE. (2012). An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature , 261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Hughes CE, Henderson KA, Yazvenko N, Gottschling DE. (2016). Selective sorting and destruction of mitochondrial membrane proteins in aged yeast. eLife , e13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane PM. (2006). The where, when, and how of organelle acidification by the yeast vacuolar H+-ATPase. Microbiol Mol Biol Rev , 177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ. (2011). Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol , 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Mendiratta S, Kim J, Pecot CV, Larsen JE, Zubovych I, Seo BY, Kim J, Eskiocak B, Chung H, et al. (2013). Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell , 552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto K, Yoshizawa K, Ohsumi Y, Anraku Y. (1988). Dynamic aspects of vacuolar and cytosolic amino acid pools of Saccharomyces cerevisiae. J Bacteriol , 2683–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Herman PK, Emr SD. (1990). The fungal vacuole: composition, function, and biogenesis. Microbiol Rev , 266–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe K, Murley A, Nunnari J. (2014). Determinants and functions of mitochondrial behavior. Annu Rev Cell Dev Biol , 357–391. [DOI] [PubMed] [Google Scholar]
- Lauwers E, Erpapazoglou Z, Haguenauer-Tsapis R, Andre B. (2010). The ubiquitin code of yeast permease trafficking. Trends Cell Biol , 196–204. [DOI] [PubMed] [Google Scholar]
- Li SC, Kane PM. (2009). The yeast lysosome-like vacuole: endpoint and crossroads. Biochim Biophys Acta , 650–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Rong Y, Chuang YS, Peng D, Emr SD. (2015). Ubiquitin-dependent lysosomal membrane protein sorting and degradation. Mol Cell , 467–478. [DOI] [PubMed] [Google Scholar]
- Lin CH, MacGurn JA, Chu T, Stefan CJ, Emr SD. (2008). Arrestin-related ubiquitin-ligase adaptors regulate endocytosis and protein turnover at the cell surface. Cell , 714–725. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast , 953–961. [DOI] [PubMed] [Google Scholar]
- MacGurn JA, Hsu PC, Emr SD. (2012). Ubiquitin and membrane protein turnover: from cradle to grave. Annu Rev Biochem , 231–259. [DOI] [PubMed] [Google Scholar]
- MacGurn JA, Hsu PC, Smolka MB, Emr SD. (2011). TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell , 1104–1117. [DOI] [PubMed] [Google Scholar]
- McFaline-Figueroa JR, Vevea J, Swayne TC, Zhou C, Liu C, Leung G, Boldogh IR, Pon LA. (2011). Mitochondrial quality control during inheritance is associated with lifespan and mother-daughter age asymmetry in budding yeast. Aging Cell , 885–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milgrom E, Diab H, Middleton F, Kane PM. (2007). Loss of vacuolar proton-translocating ATPase activity in yeast results in chronic oxidative stress. J Biol Chem , 7125–7136. [DOI] [PubMed] [Google Scholar]
- Mishra P, Chan DC. (2014). Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol , 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mnaimneh S, Davierwala AP, Haynes J, Moffat J, Peng WT, Zhang W, Yang X, Pootoolal J, Chua G, Lopez A, et al. (2004). Exploration of essential gene functions via titratable promoter alleles. Cell , 31–44. [DOI] [PubMed] [Google Scholar]
- Molina AJ, Wikstrom JD, Stiles L, Las G, Mohamed H, Elorza A, Walzer G, Twig G, Katz S, Corkey BE, Shirihai OS. (2009). Mitochondrial networking protects β-cells from nutrient-induced apoptosis. Diabetes , 2303–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murley A, Nunnari J. (2016). The emerging network of mitochondria-organelle contacts. Mol Cell , 648–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neutzner A, Youle RJ. (2005). Instability of the mitofusin Fzo1 regulates mitochondrial morphology during the mating response of the yeast Saccharomyces cerevisiae. J Biol Chem , 18598–18603. [DOI] [PubMed] [Google Scholar]
- Nikko E, Pelham HR. (2009). Arrestin-mediated endocytosis of yeast plasma membrane transporters. Traffic , 1856–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Marshall WF, Straight A, Murray A, Sedat JW, Walter P. (1997). Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol Biol Cell , 1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. (2012). Mitochondria: in sickness and in health. Cell , 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya Y, Umemoto N, Tanida I, Ohta A, Iida H, Anraku Y. (1991). Calcium-sensitive cls mutants of Saccharomyces cerevisiae showing a Pet− phenotype are ascribable to defects of vacuolar membrane H+-ATPase activity. J Biol Chem , 13971–13977. [PubMed] [Google Scholar]
- Perera RM, Zoncu R. (2016). The lysosome as a regulatory hub. Annu Rev Cell Dev Biol , 223–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles S, Vigie P, Youle RJ. (2018). Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol , R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotegher N, Duchen MR. (2017). Crosstalk between lysosomes and mitochondria in Parkinson’s disease. Front Cell Dev Biol , 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen P, Wu B, Gaber RF, Ottow K, Andersen HA, Kielland-Brandt MC. (2005). Amino acid sensing by Ssy1. Biochem Soc Trans , 261–264. [DOI] [PubMed] [Google Scholar]
- Quiros PM, Langer T, Lopez-Otin C. (2015). New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol , 345–359. [DOI] [PubMed] [Google Scholar]
- Rana A, Oliveira MP, Khamoui AV, Aparicio R, Rera M, Rossiter HB, Walker DW. (2017). Promoting Drp1-mediated mitochondrial fission in midlife prolongs healthy lifespan of Drosophila melanogaster. Nat Commun , 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A, Rera M, Walker DW. (2013). Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc Natl Acad Sci USA , 8638–8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid RJ, Lisby M, Rothstein R. (2002). Cloning-free genome alterations in Saccharomyces cerevisiae using adaptamer-mediated PCR. Methods Enzymol , 258–277. [DOI] [PubMed] [Google Scholar]
- Rotin D, Kumar S. (2009). Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol , 398–409. [DOI] [PubMed] [Google Scholar]
- Roy M, Reddy PH, Iijima M, Sesaki H. (2015). Mitochondrial division and fusion in metabolism. Curr Opin Cell Biol , 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Texeira M, Van Zeebroeck G, Voordeckers K, Thevelein JM. (2010). Saccharomyces cerevisiae plasma membrane nutrient sensors and their role in PKA signaling. FEMS Yeast Res , 134–149. [DOI] [PubMed] [Google Scholar]
- Russnak R, Konczal D, McIntire SL. (2001). A family of yeast proteins mediating bidirectional vacuolar amino acid transport. J Biol Chem , 23849–23857. [DOI] [PubMed] [Google Scholar]
- Rutter J, Hughes AL. (2015). Power(2): the power of yeast genetics applied to the powerhouse of the cell. Trends Endocrinol Metab , 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM. (2017). mTOR signaling in growth, metabolism, and disease. Cell , 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheckhuber CQ, Erjavec N, Tinazli A, Hamann A, Nystrom T, Osiewacz HD. (2007). Reducing mitochondrial fission results in increased life span and fitness of two fungal ageing models. Nat Cell Biol , 99–105. [DOI] [PubMed] [Google Scholar]
- Sesaki H, Jensen RE. (2001). UGO1 encodes an outer membrane protein required for mitochondrial fusion. J Cell Biol , 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shai N, Yifrach E, van Roermund CWT, Cohen N, Bibi C, Lodewijk IJLST, Cavellini L, Meurisse J, Schuster R, Zada L, et al. (2018). Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat Commun , 1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol , 1567–1572. [DOI] [PubMed] [Google Scholar]
- Sheff MA, Thorn KS. (2004). Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast , 661–670. [DOI] [PubMed] [Google Scholar]
- Shpilka T, Haynes CM. (2018). The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol , 109–120. [DOI] [PubMed] [Google Scholar]
- Simoes T, Schuster R, den Brave F, Escobar-Henriques M. (2018). Cdc48 regulates a deubiquitylase cascade critical for mitochondrial fusion. eLife , e30015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinkraus KA, Kaeberlein M, Kennedy BK. (2008). Replicative aging in yeast: the means to the end. Annu Rev Cell Dev Biol , 29–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A, McLelland GL, Fon EA, McBride HM. (2014). A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J , 2142–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. (2010). Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal , 503–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tone J, Yoshimura A, Manabe K, Murao N, Sekito T, Kawano-Kawada M, Kakinuma Y. (2015). Characterization of Avt1p as a vacuolar proton/amino acid antiporter in Saccharomyces cerevisiae. Biosci Biotechnol Biochem , 782–789. [DOI] [PubMed] [Google Scholar]
- Wach A, Brachat A, Alberti-Segui C, Rebischung C, Philippsen P. (1997). Heterologous HIS3 marker and GFP reporter modules for PCR-targeting in Saccharomyces cerevisiae. Yeast , 1065–1075. [DOI] [PubMed] [Google Scholar]
- Wallace DC. (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet , 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir HJ, Yao P, Huynh FK, Escoubas CC, Goncalves RL, Burkewitz K, Laboy R, Hirschey MD, Mair WB. (2017). Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab , 884–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ED, Wagner JA, Gorsich SW, McCaffery JM, Shaw JM, Nunnari J. (2000). The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion competent mitochondria. J Cell Biol , 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ED, Wagner JA, Scott SV, Okreglak V, Holewinske TJ, Cassidy-Stone A, Nunnari J. (2003). The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J Cell Biol , 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Ysselstein D, Krainc D. (2018). Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature , 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Li L, Jiang H. (2016). Doa1 targets ubiquitinated substrates for mitochondria-associated degradation. J Cell Biol , 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. (2012). Mitochondrial fission, fusion, and stress. Science , 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubenko GS, Park FJ, Jones EW. (1983). Mutations in PEP4 locus of Saccharomyces cerevisiae block final step in maturation of two vacuolar hydrolases. Proc Natl Acad Sci USA , 510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.