

Abstract
As one of the four fundamental building blocks of life, carbohydrates assume varied and expansive roles in biological contexts. More in-depth understanding of carbohydrates and their interactions, however, is often restricted by our inability to synthesize and subsequently functionalize them in a site-selective manner. This review will summarize recent advances in the site-selective functionalization of carbohydrates using organocatalysts, including achiral catalysts, chiral nucleophilic bases, chiral N-heterocyclic carbenes, and chiral phosphoric acids, with an emphasis on the catalytic nature in each case. As in many endeavors, taking an alternative approach can often lead to success, and selected examples of these achievements will be highlighted as well.
Keywords: Carbohydrate, organocatalysis, site-selective functionalization
Graphical Abstract

1.1. Introduction
As one of the four macromolecules of life, carbohydrates have a vital role in a multitude of biological processes, including cellular communication, disease progression, and metabolism. Carbohydrate scaffolds can be found in commercially available therapeutics and vaccines in various stages of pharmaceutical development,[1] and the evolving methods for carbohydrate synthesis continue to influence the drug discovery landscape [2]. Considering the widespread role that carbohydrates play in daily life, scientists constantly seek to learn more about their structure-function relationships and how they influence natural systems. In essence, further progress towards understanding the role of carbohydrates is often limited by synthetic success, or lack thereof.
One of the most challenging aspects of working with carbohydrates revolves around their physical structures. While there are less than a dozen mammalian monosaccharides, the different types of isomers and anomers all present their own challenges in terms of synthesis, and the existence of multiple hydroxyls in similar environments makes site-selective functionalization even more daunting. These challenges coupled with the ability to link monosaccharides in a linear or branched manner at multiple attachment points makes producing a relatively small oligosaccharide quite the formidable task.
Other macromolecules, such as proteins, nucleic acids, and lipids, have less varied structures and more general synthetic routes, which has led to successful automation platforms for the quick, efficient syntheses of these molecules. While automation in carbohydrate chemical synthesis is becoming more common thanks to efforts from synthetic [3] and chemoenzymatic groups,[4] challenges still remain for accessing diverse monosaccharide building blocks and oligosaccharides. For example, it is difficult to perform reliable large scale chemical synthesis of carbohydrates, obtain carbohydrates in homogenous forms, and site-selectively functionalize carbohydrates. With these issues at hand, the importance of innovation in regard to synthetic methodology cannot be understated. From an environmental impact point of view, continually increasing reaction efficiency in terms of step counts and reagents via catalysis remains attractive and is the subject of this review.
1.1.1. This review
A brief review such as this cannot possibly cover all pertinent publications related to the site-selective functionalization of carbohydrates. A comprehensive review of methods for the regioselective acylation, alkylation, silylation, and glycosylation of monosaccharides was published in 2016 [5]. The goal of this review is to provide a more targeted update focusing on recent advances in organocatalytic reactions that highlights new angles and perspectives taken since 2015. Site-selective chemical methods prior to this date have been previously covered [1,6] as have enzymatic methods [7,8] This review is organized by the different types of organocatalysts, including achiral catalysts, chiral nucleophilic bases, chiral N-heterocyclic carbenes, and chiral phosphoric acids, with an emphasis on the catalytic nature in each case as appropriate. Looking ahead to the future of the field and the need for untraditional approaches to longstanding problems, recent publications in boron and photoredox chemistry will also be presented. Much of the work presented here comes from research groups that are not solely devoted to carbohydrate chemistry, and it is encouraging to see how various new ideas and strategies were introduced to the field of carbohydrate research from chemists with varied expertise. The intent of this review is to provide the reader with the resources to obtain a strong understanding of current trends in organocatalysis for the site-selective functionalization of carbohydrates and an idea of where this field is heading in the future.
2.1. Achiral catalysts
The initial catalysts to be examined are achiral catalysts, N,N-diisopropylethylamine (DIPEA) and 4-dimethylaminopyridine (DMAP), and their use in site-selective transformations. Dong and Ren recently reported the self-catalyzed site-selective acylation of methyl pyranosides using a catalytic amount DIPEA in the presence of an anhydride to functionalize the C3 and/or C6 hydroxyl of various monosaccharides [9]. Initial studies on methyl 6-O-(tert-butyldimethylsilyloxy)-α-D-mannopyranoside 1.1 were carried out in the presence of DIPEA, anhydride, and an anticipated required metal catalyst, Fe(dibm)3, which afforded the C3-acylated product 1.2 in 92% yield. Further studies with different metal catalysts, all produced the C3-acylated product leading the authors to conclude that the metal catalyst was not involved in the reaction. This hypothesis was confirmed when the same result was obtained in the absence of any metal catalyst, enhancing the method’s appeal since one less reagent is needed. Bases other than DIPEA were examined, but lower yields and poor selectivity resulted. Overall, DIPEA exhibited the best combination of steric hindrance and base strength compared with other amines or K2CO3. The use of AcCl as the acylating reagent resulted in poor yield and selectivity, and anhydrides were shown to be the superior acylating reagent. Furthermore, the reaction with Ac2O in the absence of DIPEA yielded less than 10% of the acylated product indicating the necessity of the DIPEA.
The authors’ proposed catalytic cycle is shown in Scheme 1. The cycle begins with DIPEA reacting with acetic anhydride forming the acylammonium ion 1.3 while releasing the carboxylate ion 1.4. The mechanism hinges on the carboxylate ion forming dual hydrogen bonds with a 1,2-diol, as shown in 1.6. These hydrogen bonds activate the alcohol groups for acylation (either by way of 1.7 or 1.8) and result in the formation of 1.9, which frees DIPEA to react with another equivalent of anhydride. The catalytic cycle was investigated using variable temperature NMR and by examining the change in chemical shift of the hydroxyl protons in a 1,2-diol. In the presence of a carboxylate ion, the hydroxyl protons shifted approximately 0.3 ppm downfield while in the presence of DIPEA alone, there is essentially no change in chemical shift. Following the reaction by NMR, there is approximately a 0.2 ppm downfield shift after 20 minutes. This data, along with the fact that reactions using AcCl instead of Ac2O resulted in poor yields and selectivity, strongly supports the formation of a dual hydrogen bonded complex with the diol. In conjunction with an achiral catalyst and anhydride, the authors have developed an operationally simple protocol for an organic amine self-catalyzed site-selective acylation of manno-, gluco- and galactopyranosides that supplements other protocols currently in use.
Scheme 1:

Dong and Ren’s use of DIPEA and an anhydride to self-catalyze site-selective acylation.
There are a number of methods for the site-selective acylation of the equatorial hydroxyl groups in cis-1,2-diols, yet methods for functionalizing the axial hydroxyl group remain sparse. In 2016 Schmidt published a method for the selective acylation of the axial hydroxyl group in cis-1,2-diols in appropriately protected pyranosides using BzCN in the presence of a catalytic amount of DMAP [10]. Specifically, methoxyphenyl 2,6-di-O-benzyl-β-D-galactopyranoside 2.1 yielded only the axial C4-benzoylated product 2.2 in 90% yield. Conversely, using Bz2O, BzCl, or BzF as the acylating agent produced the C3-benzoylated isomer 2.3 as the major or exclusive product. The authors put forth the “cyanide effect” as the source of site-selectivity with BzCN (Scheme 2).
Scheme 2:
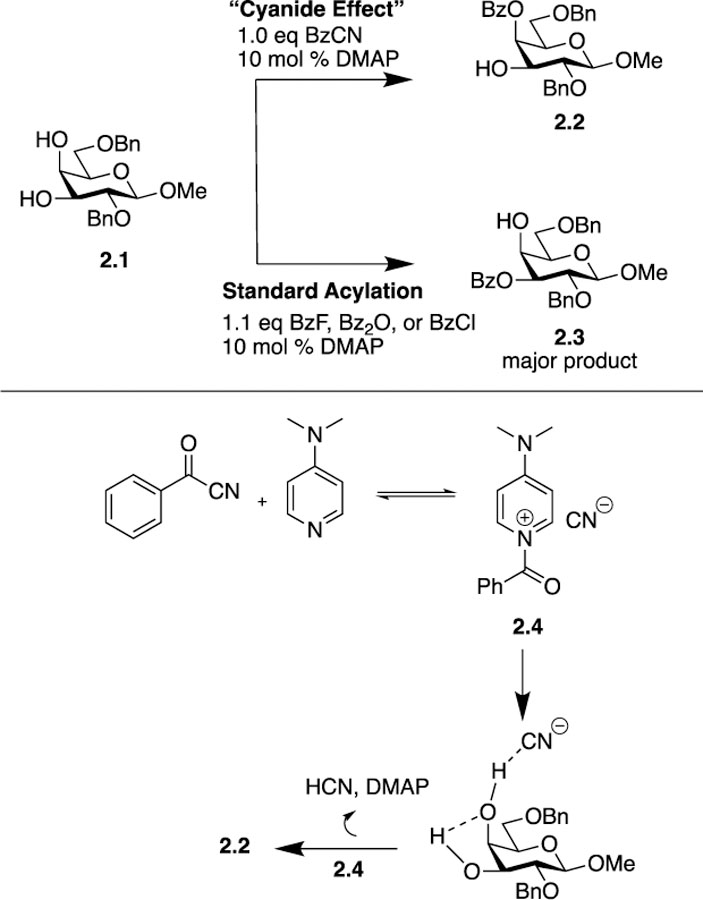
Schmidt’s use of benzoyl cyanide (BzCN) to acylate the axial hydroxyl group in cis-1,2-diols.
By using BzCN in conjunction with DMAP, cationic adduct 2.4 is formed leaving the negatively charged cyanide ion as a counterion. The cyanide ion is relatively basic compared to other counterions, such as chloride or carboxylate, which increases the rate of reaction and allows acylation to occur at lower temperatures. The cyanide ion is also sterically small and capable of forming hydrogen bonds with hydroxyl groups at either its carbon or nitrogen atom. Cyanide’s base strength and size, in conjunction with the greater acidity of the C4-OH versus the C3-OH in galactopyranosides, provides the environment for cyanide to preferentially hydrogen bond to the C4-OH activating it for acylation relative to the C3-OH at low temperatures. Furthermore, the activation of the axial C4-OH hydroxyl group by cyanide is strong enough to cause this secondary C4-OH group to react faster than the primary C6-OH group with a 2-O-benzyl galactopyranoside substrate. The method was then tested on a series of protected galactopyranosides to determine if either sterics or electronics would influence the site of functionalization. The authors increased the steric bulk of the protecting group at the C6-OH (TBDPS) and the electron withdrawing ability of the protecting groups at the C2-OH and C6-OH (Bz and Ac), but in each case the axial C4-OH was selectively benzoylated (using BzCN, 84 – 92% isolated yields) or acetylated (using AcCN, 77 – 87% isolated yields).
Computational studies were done using allyl 4,6-O-benzylidene-α-D-mannopyranoside as the model compound to examine the energies of the intermediates formed when the cyanide ion hydrogen bonds to the axial C2-OH and the equatorial C3-OH. Results showed that when the carbon atom of the cyanide ion was involved in hydrogen bonding with the designated hydroxyl group, the resulting structure was more stable than if the nitrogen atom of the cyanide ion was involved. The hydrogen bonded structure with the cyanide carbon associated with the more acidic axial C2-OH was 1.0 kcal/mol lower in energy than when the cyanide carbon was associated with the equatorial C3-OH. The deprotonation of the more acidic C2-OH by the cyanide ion is facilitated and stabilized by intramolecular hydrogen bonding from the C3-OH to form a five-membered ring, thus leading to the activation of the C2-OH towards acylation relative to the C3-OH. Studies using fluoride rather than cyanide as the counter ion/base showed the fluoride ion hydrogen bonded to both hydroxyl groups to be the most stable structure. With no clear preference for C2-OH coordination/deprotonation, the C3-OH acylated product results from the preferred reaction pathway. The “cyanide effect” provides an avenue for the selective acylation of the axial hydroxyl group of cis-1–2-diols via reagent control. The reaction of BzCN with a catalytic amount of DMAP complements the use of other acylating reagents (Bz2O, BzF) in affording selective acylation at either the axial or equatorial hydroxyl groups in cis-1,2-diols.
Schmidt and Peng later extended their 2016 work by reporting the direct formation of 3-O-α-galacto- and fucopyranosides in a single pot benzoylation with either a 6-O-protected or unprotected pyranoside as the starting compound and using a catalytic amount of DMAP in conjunction with DIPEA [11]. Initial studies were carried out on methyl 6-O-tert-butyldiphenylsilyl-α-D-galactopyranoside 3.1 to examine the relative reactivities of the C2, C3, and C4 hydroxyl groups under various acylation reaction conditions with the goal of leaving the C3-OH unfunctionalized. Theoretically, the equatorial C2-OH, being adjacent to the axial anomeric alkoxy group, should be functionalized quickly with BzCN due to the “alkoxy mediated diol effect”, and previous studies demonstrated the kinetic selectivity of the C4-OH to be functionalized over the C3-OH due to the “cyanide effect”, thus leaving the C3-OH unprotected. The authors determined that benzoylation of the 6-O-protected galactopyranoside did in fact produce the 2,4-di-O-benzoyl-α-D-galactopyranoside product 3.2 in 60% yield.
Since undesired mono- and dibenzoylated products attributed to the modest 60% yield of 3.2, attempts to increase the selectivity of the reaction and enhance the yield were carried out using different amines as catalysts; however, each produced a lower yield and selectivity. Changes in solvent composition, including using CH2Cl2 with MeCN, DMF, or THF as a co-solvent, did not improve selectivity either. The addition of chloroform (CH2Cl2:CHCl3, 1:3) or chloroform alone, however, did improve the yield of the desired product to 70% and 69%, respectively. The addition of 2.0 equivalents of DIPEA to neutralize the acid formed in the reaction also increased the selectivity. Thus, the optimal reaction conditions were: BzCN (2.1 eq.), DMAP (10 mol %), DIPEA (2.0 eq.), DCM:CHCl3 (1:3) at −78°C which yielded 3–2 in 76% yield. The method was examined on a series of α−galactopyranosides where the C6-OH protecting group and anomeric group varied while the C2, C3, and C4 hydroxyl groups remained unprotected (Scheme 3). All of the glycosides studied yielded the 2,4-di-O-benzoyl-α-D-galactopyranosides products in 70 – 76% yield.
Scheme 3:

Schmidt and Peng’s one-pot synthesis of unprotected C3-OH monosaccharides via site-selective acylation.
This work demonstrates the relative reactivities of the hydroxyl groups at carbons 2, 3 and 4 to be C2-OH > C3-OH >> C4-OH in the first acylation, then C4-OH >> C3-OH in the second acylation for a variety of α-D-galactopyranosides providing the 2,4-O-dibenzoylated product in good yield. The “alkoxy group mediated diol effect” and “cyanide effect” both play important roles in this work and led to the synthesis of both linear and branched trisaccharides, foreshadowing their use in more complex oligosaccharide synthesis down the road.
DMAP and DIPEA both function as relatively simple, achiral organocatalysts for the site-selective functionalization of carbohydrates. The centrality of these organocatalysts will be evident with their use in conjunction with other catalysts or in modified versions as they form the basis of more complex catalysts.
2.2. Chiral nucleophilic bases
As a small molecule organocatalyst, benzotetramisole (BTM) has emerged as a promising catalyst for the site-selective acylation of carbohydrates. While originally developed by Birman for the kinetic resolution of alcohols,[12] in 2017 the Tang group used the commercially-available paired chiral BTM catalysts to differentiate the prevalent trans-1,2-diol motif in various pyranoses, whether they were simple monosaccharides or more complex disaccharides [13]. Using 4.3 as a model substrate and reacting it with (R)-BTM 4.1 and isobutyric anhydride, the authors obtained the C3-acylated product 4.5 in 92% yield with greater than 20:1 selectivity, while the (S)-BTM 4.2 catalyst afforded C2-acylated product 4.4, again with good yield and selectivity. Using DMAP as an achiral catalyst to investigate the intrinsic selectivity, the authors determined there was no preference for the formation of either of the two products.
Upon obtaining experimental data and running Density Functional Theory (DFT) calculations, the working model shown in Scheme 4 was proposed. The origin of site-selectivity is a transition state stabilizing cation-lone pair interaction between the positively-charged acylated BTM catalyst and the lone pair on the alkoxy or hydroxyl group of the substrate. The proposed catalytic cycle begins with the nucleophlic attack of the BTM catalyst on the anhydride to form the positively-charged acylated BTM catalyst 4.9. Then, a carbohydrate, such as 4.3, associates with the catalyst, allowing the lone pair of the C4 oxygen to interact with the acylated catalyst species. This stabilizing interaction between the lone pair and the electron deficient acylated catalyst allows the C3 hydroxyl group of 4.3 to abstract the acyl group from the catalyst, as shown in 4.10, leading to the formation of 4.4. Conversely, the cation-lone pair interaction between the C1-OMe group of 4.3 and R-BTM 4.1 was not available due to the improper axial orientation of the OMe group. When (S)-BTM catalyst 4.2 was used, the C2 acylated product was favored because the cation-lone pair interaction between the C4-OR lone pair and the catalyst was available. Although the cation-lone pair interaction was also available between the C2-OH group and catalyst, this interaction was significantly weakened by steric repulsion between the axial 1-OMe group and the catalyst. This model was used in predicting the site-selective acylation of many other 1,2-trans-diols and polyols in carbohydrates, including disaccharide 4.6, tetrol 4.7 derived from trehalose, and natural product derived 4.8. In the future, the authors hope to use similar types of interactions to direct the site-selective functionalization of other carbohydrate species.
Scheme 4:

Tang’s use of chiral benzotetramisole (BTM) catalysts to differentiate trans-1,2-diols for acylation.
In 2017, Kawabata reported the C4-acylation of β-glucopyranosides with a chiral pyrrolidinopyridine (PPy) catalyst and an acid chloride in the presence of a carboxylic acid and base [14]. Both catalyst loading and reaction times were substantially decreased from previously published results leading to a significantly increased catalytic efficiency for this process [15]. The authors previously used anhydrides to site-selectively acylate the C4-hydroxyl, but the reaction time ranged from 12–24 h depending on the anhydride and amount of catalyst used. In contrast, the most recent work using an acid chloride and carboxylic acid takes less than an hour in most cases, regardless of the amount of catalyst used.
To begin their work, the authors used PPy catalyst 5.1 and an acid chloride or anhydride to study the formation of acylpyridinium salts and their subsequent reaction with 1-phenyl-2,2-dimethylethanol to produce the acylated product. Results showed the formation of the acylpyridinium salts was fast and complete with the acid chlorides, while the anhydrides showed low conversions into the desired acylpyridinium salt. However, when in situ counter anion exchange provided the acylpyridinium carboxylates, these species were 800–1300 times more active than the corresponding acylpyridinium chlorides in the acylation of 1-phenyl-2,2-dimethylethanol. This significant increase in activity then led the authors to try their system with a carbohydrate substrate. The anticipated result was realized in the acylation of octyl β-D-glucopyranoside 5.2 to selectively produce the octyl C4-acyl glucopyranoside 5.3 (C4-acylation:C3-acylation ratio 98:2) in 99% yield in just five minutes using 0.1 mol % catalyst and isobutyryl chloride. Almost identical results were achieved in 25 minutes with 0.02 mol% catalyst (C4-acylation:C3-acylation ratio 96:4, 96% yield). The reaction with benzoyl chloride and 0.1 mol % catalyst led to an 87% yield of monobenzoylation products in 15 minutes with high site-selectivity (C4-benzoylation: C3-benzoylation:C6-benzoylation, 95:4:1). Reducing the catalyst loading to 0.02 mol% maintained site-selectivity but required a 12-hour reaction time to give a 76% yield of monobenzoylated products (C4-benzoylation:C3-benzoylation, 95:5).
The first step of the catalytic cycle is the reaction between chiral catalyst 5.1 and the acid chloride to form the acylpyridinium chloride 5.4 (Scheme 5). Next, an in situ counter anion exchange occurs to form the more reactive acylpyridinium carboxylate 5.5, which results in an increased acylation rate. The carboxylate ion, being both a stronger base than chloride and being able to coordinate to the C2-hydrogen of the acylpyridinium ion and a carbohydrate hydroxyl group via hydrogen bonds, promotes the reactivity of the acylpyridinium ion in the following acylation step of the catalytic cycle. Introduction of 5.6 and subsequent acyl group delivery, depicted in 5.7, then leads to formation of product 5.8 and regeneration of the catalytic species.
Scheme 5:

Kawabata’s use of in situ counter anion exchange to promote quick site-selective acylation.
Kawabata’s work prior to this paper achieved selective C4-acylation using 1 or 10 mol % catalyst with 1.1 equivalents of acylating agent and 1.5 equivalents of collidine over a 12–24 h period. The performance of PPy catalyst 5.1 is significantly increased by generating the acylpyridinium ion with an acid chloride in the presence of a carboxylate ion. The current procedure matches C4-OH selectivity and yield while significantly reducing both reaction time and catalyst loading (0.02 or 0.1 mol%). Thus, the identity of the counter anion is the key to achieving high catalytic activity within this system while retaining the same chiral catalyst used previously.
Continuing the prevalent trend of using peptide catalysts for site-selective functionalization, [16] in 2016 Kirsch and co-workers reported an instance where DMAP-based small molecular peptides were synthesized using click chemistry to tether DMAP to a peptide side chain [17]. DMAP, being one of the most well-studied acylation catalysts, was the catalytically active unit, while the peptide unit influenced the site-selective outcome. Using solid-phase peptide synthesis, a variety of peptide scaffolds were synthesized to form a library of DMAP-based catalysts. Upon screening the catalyst library, the authors determined that substrate 6.2 could undergo C2-acylation to form product 6.3 with excellent site-selectivity (96%) and yield (91%) using peptide catalyst 6.1 (Scheme 6). When DMAP was used to determine intrinsic reactivity, only 51% of the C2-acylated product was formed, along with 18% of the C3-acylated product and 31% bisacylated product making the peptide catalyst’s result even more impressive. Overall, catalyst 6.1 provided improvement in terms of site-selectivity, however, the fact that peptide catalysts are highly substrate-specific still remains as the major limitation. Different peptide catalysts must be screened for different monosaccharide substrates or even the same monosaccharide with different protecting groups. When looking to functionalize a specific target compound with multiple hydroxyl groups there is often a need for substrate-specific catalysts, and peptide catalysts can provide a solution.
Scheme 6:
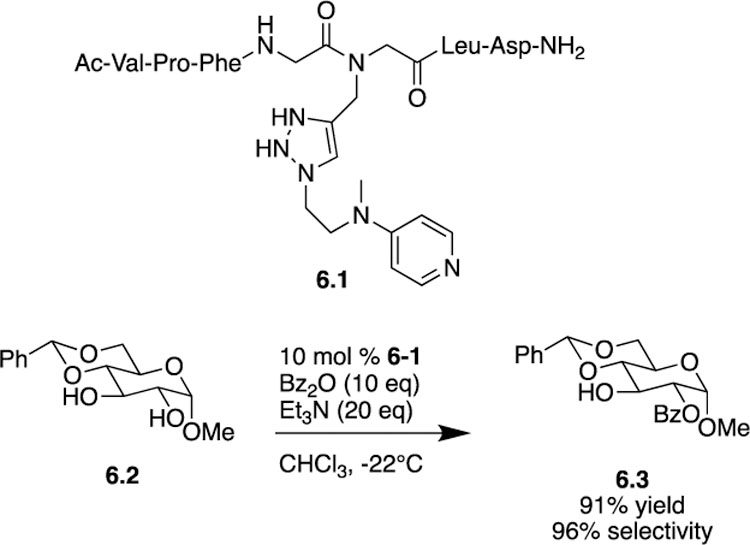
Kirsch’s use of a DMAP-based amino acid catalyst for site-selective acylation.
As a follow up to their initial peptide catalysis work, in 2017 Kirsch discovered that increased site-selectivity and an easier purification process could be achieved when they attached the peptide catalysts to solid supports [18]. By attaching the DMAP-peptide unit to a Boc-glycine Merrifield resin, the catalytic units could be made via automated synthesis. Additionally, the solid-supported catalysts also provided a simplified purification procedure of the product from the reaction mixture, and the recovery of the catalysts for additional future use is also possible. The authors questioned whether the resin alone was the reason for site-selectivity, but upon screening catalysts with subtle variations in the peptide sequence, they determined that catalyst structure undoubtedly influenced reaction selectivity. Their eventual conclusion was that the solid-supported catalysts made of resin could influence substrate-specificity via diffusion and polarity effects. Although different catalysts were still required for different substrates, the addition of the solid-supported catalysts allowed the entire process to be more cost-effective than their previous protocol. Furthermore, this work is limited by the fact that the authors did not rationally design the peptide structures. Instead, the amino acid sequence was randomly selected. Perhaps future studies will subject rationally designed peptide structures to the above conditions and allow for greater insight into how various catalysts contribute to site-selectivity. This work is just one of many harnessing the power of peptides in recent years, and undoubtedly more catalysts of this type or similar strategies will be seen in the future.
2.3. Chiral N-heterocyclic carbenes
In recent years the Sundén and Studer groups have taken a different approach to site-selective functionalization by employing the use of N-hetereocyclic carbenes (NHCs). In 2016 the Sundén group used dialkylimidazolium-based ionic liquids (ILs) to generate the NHCs, and since ILs are generally considered to be safe and recyclable solvents, [19] their methods embrace the intentions of the current green chemistry movement while also providing an example of the innovation that will keep carbohdrate chemistry moving forward. Since sterics often affect reactivity patterns, Sundén envisioned that sterically encumbered IL-derived acylazolium compounds could be used to site-selectively functionalize carbohydrates by discriminating against the incoming polyol nucleophile in a regioselective fashion, thereby directing the nucleophile to a desired hydroxyl group [20].
Using ionic liquid 1-ethyl-3-methylimidazolium acetate (EMIMAc) 7.1, DBU, cinnamaldehyde 7.3, and a chalcone 7.4, the first step towards product formation is the construction of the acylazolium compound 7.5, which occurs in a highly disastereoselective manner as a 1:1 ratio of the two anti-diastereomers was formed (Scheme 7). Using this IL-derived NHC complex can assist in dissolving polar substrates, such as 7.2, and subsequent attack by the primary hydroxyl on the acylazolium complex leads to functionalization of the primary alcohol 7.6 in each case. Alternatively, the authors discovered that using non-IL derived NHC complexes were unsuccessful in forming the desired product.
Scheme 7:
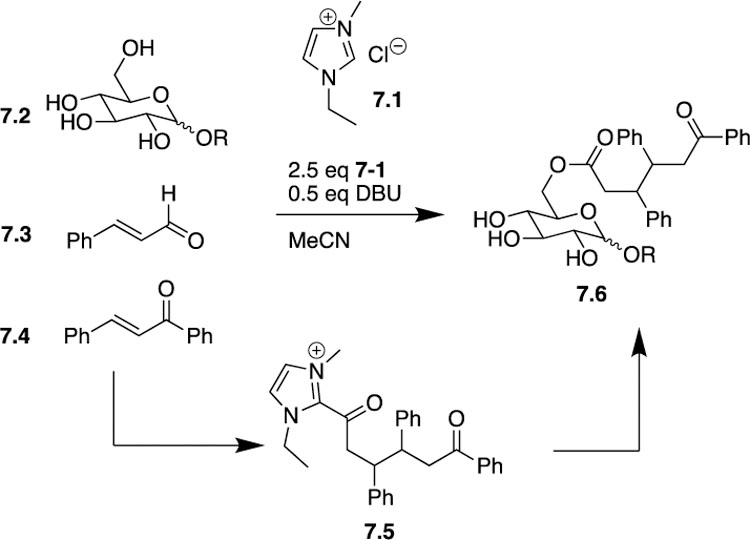
Sundén’s use of ionic liquids to promote a NHC-catalyzed site-selective functionalization of carbohydrates.
DFT calculations rationalize the greather than 20:1 preference for functionalization of the primary hydroxyl over any possible secondary hydroxyls by considering the conformation of the intermediate in conjunction with solvation effects. When the C6-OH attacks the acylazolium complex, the intermediate forms a rather compact structure that minimizes contact between the hydrophobic regions of the molecule and the polar acetonitrile solvent while simultaneously exposing the imidazolium and carbohydrate backbone to the solvent. Conversely, when the authors performed calculations that used the C4-OH attacking the acylazolium complex, a more elongated intermediate formed to relieve steric stress at the expense of losing stabilization due to solvation. Overall, the most stable intermediate formed by C6-OH attack was favored by 6 kJ/mol over the most stable intermediate formed by C4-OH attack, which is in agreement with experimental results.
Products resulting from this method could be isolated without column chromatography, and although an excess of the ionic liquid 7.1 was used, the authors consistently recycled the ionic liquid and used it in further runs without sacrificing yields, hence its inclusion in this section. Additionally, the reactions were shown to be successful on the gram scale, able to be run in one-pot, and amenable to various aromatic substituents on the chalcone, making this method a promising approach to introduce large functional groups to the primary hydroxyl.
Also in 2016, Studer and co-workers emplyed chiral NHCs for the site-selective acylation of diols and triols in various carbohydrates [21]. Since the actual acylating reagent in oxidative NHC catalysis is an acylazolium ion formed from an aldehyde, modifying the original aldehyde or the actual NHC allows for a fair amount of flexibility in terms of tuning reagents. Substrate 8.2 when reacted with 8.3, 8.5, and chiral NHC catalyst (S)-8.1, yielded the C2-acylated product 8.4 in 85% yield with 89% site-selectivity. Because their previous work [22] indicated that a second equivalent of the free NHC catalyst was required in the acylation step, the authors expected a nonlinear effect on the site-selectivity of the reaction. Upon screening different ratios of (S)-8.1 and (R)-8.1, they did observe this non-linear trend and found that when the ratio between (S)- and (R)-8.1 was 3:1, 99% site-selectivity for product 8.4 was obtained. These results indicated that a cooperative process was involved in this reaction. During the acylating step, one enantiomer of the catalyst was part of acylating reagent, while the other enantiomer of the catalyst likely activated the alcohol for acylation through hydrogen bonding. Compound 8.5 was used as a two-electron-oxidant to oxidize key Breslow intermediate 8.7 through radical cation 8.8 and radical 8.9 intermediates, resulting in the formation of 8.4 and 8.6 (Scheme 8).
Scheme 8:

Studer’s use of oxidative NHC catalysis to perform site-selectively acylation.
In this second example of NHC catalysis, Studer was able to selectivity functionalize carbohydrates under mild conditions. Even more notable is that these carbohydrates can contain either cis- or trans-2,3-diols for the glucose, galactose, and mannose monosaccharides, or the substrates can be complex aminosugars where acylation occurs but leaves the amino functional group untouched.
2.4. Chiral phosphoric acids
While most of the material presented thus far has focused on site-selective functionalizaton of carbohydrates, equally important, however, is site-selective glycosylation. Completing a late-stage glycosylation of a complex molecule, natural product, or polyol with multiple free hydroxyl groups without additional protection and deprotection steps is essential. In these situations, site-selective glycosylations are highly valuable, and methods to accomplish the desired result are imperative. To this end, chiral phosphoric acids have recently been used to increase the site- and stereoselectivity in glycosylations [23].
Just as peptide catalysts are used to discriminate substrates, chiral catalysts can be used to discriminate glycosyl donors by changing their surrounding environments. The Nagorny group has been one team to tackle site-selective glycosylations, and their strategy of choice is to employ a chiral phosphoric acid to functionalize complex polyols, such as oleandomycin-derived macrolactones [24]. In order to demonstrate the ability of chiral catalysts for promoting site-selective glycosylation of complex molecules, Nagorny chose triol, 6-deoxyerythronolide B (6-dEB), as the target molecule for his work (Scheme 9). Not only were the authors able to functionalize all three hydroxyl groups individually, but they also completed the mechanistic studies to explain the selectivity.
Scheme 9:

Nagorny’s use of chiral phosphoric acids to site-selectively glycosylate 6-deoxy-erythronolide B.
After control experiments were done to determine intrinsic reactivity of the C3, C5, and C11 hydroxyl groups, the authors determined that BINOL based (S)-9.1 was superior at promoting the glycosylation at C5 leading to 9.6 (9.6:9.7 99:1, 98% yield). Structurally similar catalyst (R)-9.2 was able to promote a majority of C3-glycosylated product, but there was definitely room for selectivity gains and yield improvement. By screening SPINOL-based catalysts, which boast a less flexible backbone, the authors were able to obtain the C3-glycosylated product 9–7 in 82% yield with a 73:27 ratio of 9.7:9.6 using (S)-9.3. With 2 of the 3 hydroxyl groups able to be differentiated, the authors began to tackle the functionalization of the C11 hydroxyl group. Having 1,3-syn hydroxyl groups at C3 and C5 led the authors to hypothesize the formation of a transient cyclic boronic ester protecting group 9.8 followed by glycosylation at the C11 hydroxyl and subsequent removal of the boronic ester. While this type of protecting group strategy has gained attention recently, its potential for success on a complex molecule was unknown. The authors’ plan was successful and C11-glycosylated product 9.9 was obtained in 62% yield (99:1 r.r., 3.7:1 β:α).
To further rationalize observed glycosylation selectivities, the authors ran computational experiments on a simplified model system. Their data suggested that covalently linked anomeric phosphates 9.10 were the reactive intermediates instead of traditional oxocarbenium ion pairs. Since these results were not consistent with results from similar systems, the authors sought experimental evidence to support the computations, which they obtained via NMR studies, and the authors intend to perform additional mechanistic investigations to determine the origin of much of the observed selectivities. While this review has primarily focused on chiral organocatalysts promoting the site-selective functionalization of monosaccharides, Nagorny has shown that chiral organocatalysts can be employed for the site-selective glycosylation of complex chiral polyols in lieu of enzymatic approaches.
Recognizing the inherent potential of chiral phosphoric acids, in 2017 the Galan group reported the use of a chiral phosphoric acid in conjunction with a thiourea catalyst to promote the stereoselective formation of deoxyglycosides from glycals [25]. Galan previously used thiourea catalysts to prepare 2-deoxygalactosides, but long reaction time and a limited substrate scope prompted the development of a more successful method [26]. Using thiourea catalyst 10.1, the authors determined that it had a significant effect on the reaction rate and yield, while chiral phosphoric acid 10.2 was the predominant species affecting the stereoselectivity of the reaction (Scheme 10). Furthermore, the absence of 10.2 resulted only in starting material, reinforcing the synergistic nature of both catalysts. Using 10.4 as a model substrate, the reaction was shown to proceed fairly quickly (<6 h) to yield the desired products in good yield and greater than 30:1 α:β selectivity while also tolerating acetal, ether, ester, and carbamate protecting groups. Since the scope of the glycosyl donor in the authors’ previous work was limited, they now turned to various galactals, glucals, and rhamnals with diverse protecting groups to investigate the generality of their method. The authors were pleased to find that using a model secondary acceptor 10.3 resulted in good yields and greather than 30:1 α:β selectivity in a majority of cases.
Scheme 10:

Galan’s synergistic catalysis using thiourea and a chiral phosphoric acid to promote stereoselective glycosylations.
The authors subjected a 3:1 α:β mixture of 10.8 to the reaction conditions to determine if the high degree of α selectivity resulted from anomerization, but no change in the anomeric ratio was observed. Using deuterated galactal 10.4, the authors employed NMR spectroscopy and analyzed proton shifts leading to the formation of 10.5, in which the newly formed bonds are cis to each other, indicating that the C-H and C-O bond forming events occur in a syn-diastereoselective fashion. Thus, the authors propose that a hydrogen-bond-mediated complex 10.6 forms from the association of 10.1 and 10.2. The phosphoric acid proton from 10.6 then adds to the more accessible face of the deuterated enol ether to form a transient oxocarbenium intermediate 10.7. 10.7 is then trapped by the corresponding nucleophile, in this case 10.3, and subsequently activated by the phosphate intermediate generated in situ to form 10.5. With this cycle in mind, the authors rationalize why the chirality of the phosphoric acid affects the stereoselectivity of the glycosylation.
The authors synthesized various disaccharides, glycosyl-amino acids, and glycoconjugates using their cooperative Brønsted-acid/thiourea synergistic catalysis system for direct glycosylation and hope to apply it to other important glycosides in the future. Their method was amenable to a host of glycosyl donors and various nucleophile acceptors while leading to a high degree of selectivity for α-anomers of each product.
3.1. Stoichiometric boronic esters as temporary protecting groups or activating groups
To this point, we have focused on recent examples of organocatalysts that have been used for site-selective reactions. Boron is typically considered as metalloid, a type of chemical element with properties between metals and nonmetals. Taylor’s group has developed a series of reactions using boron-based catalysts for the site-selective functionalization of carbohydrates by forming cyclic boronic ester intermediates [27]. This section will focus on their recent work since 2015 and related work from others, most of which actually employ stoichiometric amount of boronic acids. Although the boron reagents are not catalytic, the ability to recover them prompted us to include them in this review, especially as we continue to develop nontraditional methods for site-selective reactions.
In 2017, the Taylor group made two key contributions regarding the use of boronic esters as activating and protecting groups for carbohydrate functionalization. In the first, phenylboronic acids were used as protective intermediates for cis-1,2- or cis-1,3-diols to site-selectively install a variety of functional groups, including acyl groups, silyl ethers, and para-methoxybenzyl (PMB) ethers to furanoside and pyranoside substrates [28]. Using methyl α-D-glucopyranoside 11.1 as a model substrate, the authors aimed to acylate a variety of monosaccharides and then recover the boronic acid 11.3 in addition to the desired product 11.4. Using a simple three step sequence: protection with phenylboronic acid, acylation, and deprotection, the authors were able to generate the cyclic boronic ester and form the acylated product (Scheme 11). The utility of the boronic acid hinges upon the formation of the cyclic boronic ester that temporarily masks the C4 and C6 hydroxyl groups. Once functionalization of the C2 and C3 hydroxyl groups is complete, facile removal of the boronic ester allows for any remaining transformations to be completed. While protection and acylation proceeded as expected, hydrolysis of the boronic ester, however, proved more challenging than anticipated. After screening various hydrolysis methods with little success, Taylor envisioned that using Hall’s “phase-switching” method [29] could alleviate the deprotection issue by using a liquid-liquid extraction method. This method was successful for producing bisacylated compound 11.4 in 80% yield while also allowing recovering 77% of the phenylboronic acid 11.3 used. The authors used this three-step sequence for the synthesis of a variety of mono- and bisacylated products and were able to scale up the procedure leading to isolation of products on the gram scale.
Scheme 11:
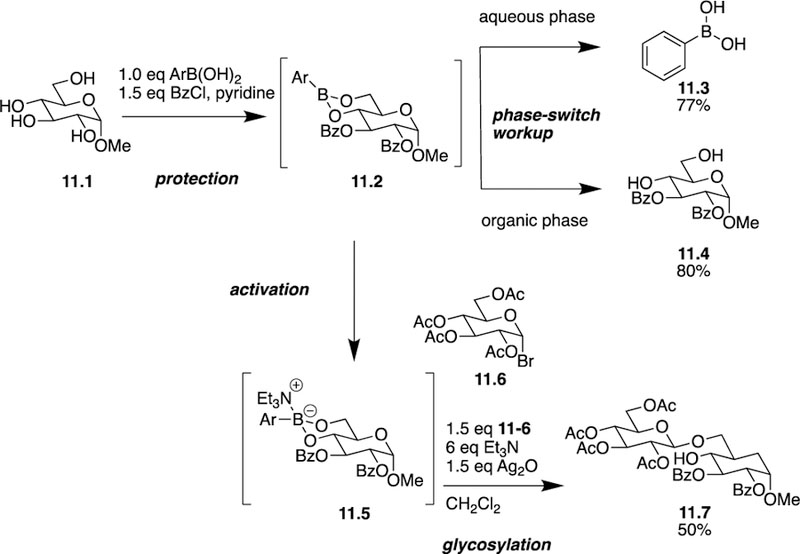
Taylor’s use of boronic acid esters as transient protecting groups and activators for site-selective glycosylation.
Not only did this procedure lend itself well to acylation, but silylation and the less common alkylation of hydroxyl groups were reported as well. In a relatively simple substitution, chlorotriethylsilane was used in place of benzoyl chloride under the standard reaction conditions to give a variety of partially silylated pyranosides. Furthermore, hydroxyl group alkylation was possible after some adjustments to previous reaction conditions. Using para-methoxybenzyl trichloroacetimidate in conjunction with 4-(trifluoromethyl)phenylboronic acid, lanthanum (III) trifluoromethanesulfonate, and a mixture of dichloromethane/toluene allowed for the production of the para-methoxybenzyl protected disaccharides as well. Overall, all functionalizations resulting from this procedure were able to be completed in a one-pot transformation with recovery of the initial boronic acid. Considering the standard protection/deprotection sequences needed to achieve site-selective transformations, the advantage of the one-pot protocol further enhances the efficiency of this method.
In the previously described work, Taylor predominately used transient boronic esters as protecting groups then removed them to reveal the desired product. In the work that follows, however, he increases the functionality of the organoboron complex by simply switching the coordination number. Initially, the boronic ester is used as a tricoordinate protecting group to enable free hydroxyl group functionalization. What follows is the addition of a Lewis base leading to formation of a tetracoordinate organoboron complex, which functions as an activating group to allow for further derivatization of a boron-bound oxygen [30]. Upon obtaining tricoordinate complex 11.2, the authors treated it with triethylamine and silver (I) oxide to form tetracoordinate activating complex 11.5. The triethylamine functioned as the Lewis basic activator, and silver (I) oxide acted as the halide scavenger and base. Of the two hydroxyl groups coordinated to the boron atom, the equatorial or primary hydroxyl group is consistently activated and functions as the glycosyl acceptor, even in the presence of deactivating acyl groups. As seen in the formation of disaccharide 11.7 (50% yield), the authors acknowledge the less than ideal overall yields of the process, but since the entire process occurs without purification of the intermediates, the operational simplicity excuses the resulting yields. Using this methodology, the authors were able to synthesize multiple disaccharides and branched trisaccharides. While boronic acids are often mentioned alongside acetals/ketals for protection of cis-1,2- and cis-1,3-diols, the work presented here highlights the differences in their fundamental reactivity profiles and represents the first time the cyclic protecting group has been removed by site-selective glycosylation.
Likewise, Taylor has been able to use boronic acids to promote the site-selective O-arylation of carbohydrates by way of a copper (II) mediated process [31]. O-arylation of carbohydrates is a difficult feat on its own, and the authors’ success completing O-arylation site-selectivity is even more impressive. The authors started with methyl α-L-rhamnopyranoside 12.1 and treated it with an arylboronic acid to form the canonical organoboron complex 12.2. Traditionally, acylation, silylation, or some other functionalization occurs at this point, but in this case, the addition of Cu(OAc)2 directly leads to formation of the C4-monoarylated product 12.5 in 72% yield with greater than 20:1 site-selectivity (Scheme 12). The site-selective outcome is in accordance with previous experimental observations where the boronic acid ester is formed with the cis-diols and the remaining free hydroxyl group is functionalized. Aside from being the transient protecting group, competition experiments also indicated that the boronic acid ester may facilitate quicker arylation of the free hydroxyl group. In an effort to better understand the reaction results, the authors set out to determine the mechanism of the reaction, and they used DFT calculations to help explain the origin of the accelerating effect of boronic ester. Computational results indicate a Lewis acid-base interaction between the cyclic boronic ester and the acetate ligand in 12.3 and subsequent formation of the copper alkoxide in 12.4 may be responsible for the previously mentioned accelerating effect, but additional mechanistic studies are needed to conclude this definitively. This method not only boasted a large substrate scope of various furanosides, pyranosides, and glucals, but also allowed for the installation of many substituted aryl ethers. Overall, using copper (II) acetate, Taylor was able to demonstrate that site-selective carbohydrate O-arylation is possible, and the resulting products can be used in applications with widespread implications in the synthetic and therapeutic arenas.
Scheme 12:
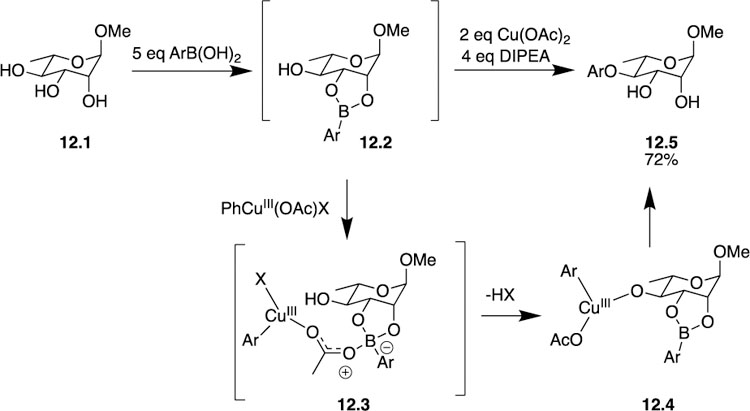
Taylor’s use of boronic acid esters to promote copper (II) mediated O-arylation of carbohydrates.
While Taylor has worked extensively with phenylboronic acids for site-selective functionalization of carbohydrates, the Makino group has also employed phenylboronic acids albeit for site-selective sulfation in a similar sequence. Considering the importance of sulfated carbohydrates in biological events and pharmaceuticals agents, it should come as no surprise that novel methods for the site-selective sulfation of carbohydrates have come out of the pipeline. Makino’s new sulfation method, which boasts tin-free conditions and the use of unprotected substrates, permitted the authors to use phenylboronic acid alongside a 2,2,2-trichloroethyl-protected (TCE) sulfurylimidazolium salt 13–3 to successfully perform sulfation in a one-pot protocol [32]. Traditionally, sulfur trioxide-amine complexes are used as sulfating reagents, but conversion and purification issues often ensue. Using the TCE protected sulfurylimidazolium salt alleviates the traditional issues while allowing for further functionalization of the sulfated carbohydrates. Additionally, one would expect that unprotected carbohydrates would have poor solubility in organic solvents, but the presence of the cyclic boronic ester actually increases the solubility of these compounds to allow reactions to proceed smoothly in various organic solvents. In a given monosaccharide, such as methyl α-L-fucopyranoside 13.1, the boronic ester 13.2 is formed, addition of 13.3 and 1,2-dimethylimidazole (1,2-dMeIm) yields 13.4, and subsequent deprotection with pinacol yields monosulfated product 13.5 in 93% yield (Scheme 13). Obviously, if only one free hydroxyl group remains, it will clearly be the site of sulfation. In the case of glucose and galactose, however, two free hydroxyl groups remain at C2 and C3. Glucose is sulfated exclusively at the C2 hydroxyl, while galactose is sulfated at the C3 hydroxyl. The reason for this revolves around steric considerations and the necessity for adequate space around the reaction site. When two free hydroxyls remain unprotected, sulfation will preferentially occur at the equatorial hydroxyl group whose adjacent neighbor is oriented axially, which provides the increased space. Using this protocol Makino was able to site-selectively perform sulfation on a variety of unprotected monosaccharides and more complex molecules like D-trehalose. Disulfated compounds could also be obtained by simple scaling up the appropriate reagents. In the future, the authors hope to apply this methodology to the synthesis of more complicated sulfoglycolipids, but for the time being their sulfation method employing phenylboronic acid as a temporary protecting group is a step away from excessive manipulations in the past and a step towards a more efficient process.
Scheme 13:
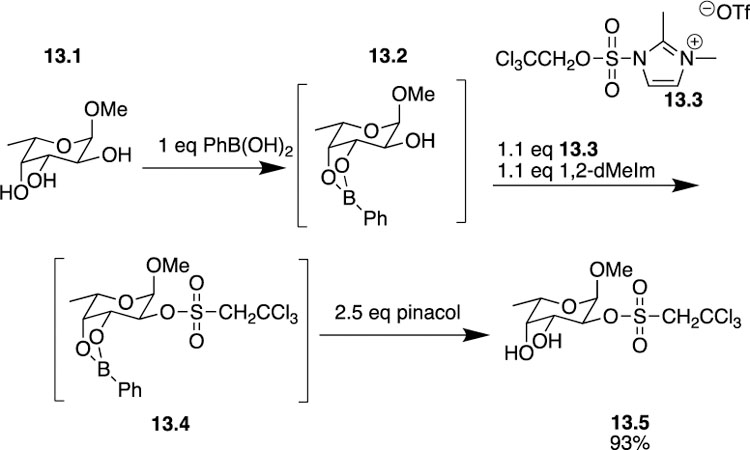
Makino’s use of boronic acid esters for site-selective sulfation.
As most of the previous work covered the functionalization of groups with cis-1,2-diols, a knowledge gap remains of how to efficiently site-selectively functionalize glucose. In an attempt to realize this need, Moitessier uses arylboronic acids in conjunction with DMAP and an acylating reagent to site-selectively functionalize methyl-α-D-glucopyranoside 14.1 and extends the method to include its galactose and mannose counterparts (Scheme 14) [33]. After some extensive optimizations, experimental results showed that phenylboronic acid was the superior boronic acid, and even trace amounts of water present were detrimental to the reaction so the authors used a Dean-Stark apparatus to avoid moisture in the reactions. Overall, smaller electrophiles, such as acetic anhydride, led to a majority of the C3-acylated product 14.6, while larger electrophiles, such as pivaloyl chloride and benzoyl chloride preferentially formed the C2-acylated product 14.5. Furthermore, the authors demonstrated the viability of this method to be successful on the gram scale while completing all of the necessary reactions in one-pot, which adds to the green and efficient nature of the method.
Scheme 14:

Moitessier’s use of boronic acid esters to site-selectively acylate unprotected carbohydrates
On one occasion in 2016 the O’Doherty group used a combination of boron and palladium catalysts to promote glycosylations necessary to synthesize the mezzettiasides, a family of natural products consisting of partially acetylated rhamnose monosaccharide units [34]. Prior to this study, the authors noticed that different acetylation patterns with cleistriosides and cleistetrosides significantly affected their biological activity, [35] and they decided that a natural continuation of that work was to access the mezzettiasides. As the O’Doherty group has a history using the de novo asymmetric approach to synthesize natural products, including those with carbohydrate motifs, they continued with the de novo strategy in this work while incorporating a dual B-/Pd-catalyzed glycosylation using the popular boron reagent, 2-aminoethyl diphenylborinate 15.3, developed by Taylor [36]. After condition optimization, the authors were able to obtain a 6:1 ratio of C3:C2 isomers in a 77% yield. O’Doherty surmises that the anionic character of the borinate complex 15.3 makes it a suitable coupling partner for the cationic Pd-π-allyl complex shown in 15.4. Furthermore, they observed that smaller ester groups on the C4 position led to even better regioselectivity in the glycosylation. Overall, the innovation of this dual catalysis strategy and its iterative use allowed for the first synthesis of all 10 members of the mezzettiaside family of natural products and structure-activity studies that came as a result of this synthetic strategy discovered previously unknown antibacterial activities of the mezzettiasides [37]. O’Doherty’s use of chemical methods to answer biological questions is exactly the type of critical thinking needed as researchers work on more significant challenges at the chemistry-biology interface.
4.1. Conclusion
The highly varied structures of carbohydrates make developing universal methods for their modification a formidable challenge, and until this challenge is met, the site-selective functionalization of carbohydrates will remain an active area of research. Many of the chemists working on the site-selective carbohydrate functionalization come from varied backgrounds far outside the carbohydrate chemistry field, and their unique experiences and skill sets will be instrumental in addressing current carbohydrate problems from different angles. Furthermore, while this review has focused on chemical methods utilizing organocatalysts, nature remains the ultimate synthetic chemist. Thus, enzymatic and chemoenzymatic methods constitute a complementary strategy to promote site-selective transformations. While many of the modifications presented here focused on acylation or alkylation, site-selective sulfation, phosphorylation, and glycosylation will be important to address in the future as well. The subset of mammalian monosaccharides is relatively small, yet immenses challenges result when trying to modify this small group of molecules. As the field becomes more competent in manipulating mammalian monosaccharides, scientists will likely turn an eye towards the synthesis and funtionalization of bacterial and/or rare sugars. With hundreds of monosaccharides comprising these groups, the need for site-selective methods will continue and be greater than ever.
Scheme 15:
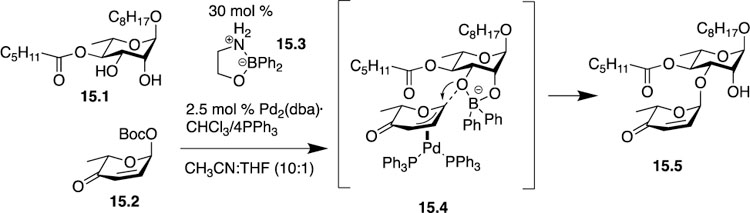
O’Doherty’s use of a dual B-/Pd- catalyzed glycosylation to synthesize the mezzettiasides.
Acknowledgements
We would like to thank the University of Wisconsin-Madison and NIH (U01GM125290) for financial support. S.A.B. was supported in part by the NIH Chemistry–Biology Interface Training Grant (T32 GM008505).
References
- [1].Wang H-Y, Blaszczyk SA, Xiao G, Tang W, Chiral reagents in glycosylation and modification of carbohydrates, Chem. Soc. Rev 47 (2017) 681–701. [DOI] [PubMed] [Google Scholar]
- [2].Seeberger PH, Automated synthesis of oligosaccharides as a basis for drug discovery, Nat. Rev. Drug Discov 4 (2005) 751–763. [DOI] [PubMed] [Google Scholar]
- [3].(a) Panza M, Pistorio SG, Stine KJ, Demchenko AV, Automated chemical oligosaccharide synthesis: novel approach to traditional challenges, Chem. Rev 118 (17) (2018) 8105–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hsu CH, Hung S-C, Wu C-Y, Wong C-H, Toward automated oligosaccharide synthesis, Angew. Chem. Int. Ed 50 (2011) 11872–11939. [DOI] [PubMed] [Google Scholar]; (c) Seeberger PH, Automated oligosaccharide synthesis, Chem. Soc. Rev 37 (2008) 19–28. [DOI] [PubMed] [Google Scholar]
- [4].Wen L, Edmunds G, Gibbons C, Zhang J, Gadi MR, Zhu H, Fang J, Liu X, Kong Y, Wang PG, Towards automatic enzymatic synthesis of oligosaccharides, Chem. Rev 118 (17) (2018) 8151–8187. [DOI] [PubMed] [Google Scholar]
- [5].Lawandi J, Rocheleau S, Moitessier N, Regioselective acylation, acylation, silylation, and glycosylation of monosaccharides, Tetrahedron 72 (2016) 6283–6319. [Google Scholar]
- [6].(a) Taylor MS, Catalyst-controlled, regioselective reactions of carbohydrate derivatives, Top. Curr. Chem 372 (2016) 125–156. [DOI] [PubMed] [Google Scholar]; (b) Williams R, Galan MC, Recent advances in organocatalytic glycosylations. Eur. J. Org. Chem 42 (2017) 6247–6264. [Google Scholar]; (c) Song W, Zheng N, Chiral catalyst-directed site-selective functionalization of hydroxyl groups in carbohydrates. J. Carbohydr. Chem 36, (2017) 143–161. [Google Scholar]
- [7].(a) Baba A, Yoshioka T, Complementary and synergistic roles in enzyme-catalyzed regioselective and complete hydrolytic deprotection of O-acetylated beta-D-glucopyranosides of N-arylacetohydroxamic acids, J. Org. Chem 77 (2012) 1675–1685. [DOI] [PubMed] [Google Scholar]; (b) Gridley JJ, Hacking AJ, Osborn HMI, Spackman DG, Regioselective lipase-catalysed acylation of 4,6-O-benzylidene-α- and-β-d-pyranoside derivatives displaying a range of anomeric substituents, Tetrahedron 54 (1998) 14925–14946. [Google Scholar]
- [8].Mastihubová M, Mastihuba V, Donor specificity and regioselectivity in Lipolase mediated acylations of methyl α-D-glucopyranoside by vinyl esters of phenolic acids and their analogues, Bioorg. Med. Chem. Lett 23 (2013) 5389–5392. [DOI] [PubMed] [Google Scholar]
- [9].Ren B, Gan L, Zhang L, Yan N, Dong H, Diisopropylethylamine-triggered, highly efficient, self-catalyzed regioselective acylation of carbohydrates and diols, Org. Biomol. Chem 16 (2018) 5591–5597. [DOI] [PubMed] [Google Scholar]
- [10].Peng P, Linseis M, Winter R, Schmidt R, Regioselective acylation of diols and triols: the cyanide effect, J. Am. Chem. Soc 138 (2016) 6002–6009. [DOI] [PubMed] [Google Scholar]
- [11].Li T, Li T, Cui T, Sun Y, Wang F, Cao H, Schmidt R, Peng P, Regioselective one-pot benzoylation of triol and tetraol arrays in carbohydrates, Org. Lett 20 (2018) 3862–3865. [DOI] [PubMed] [Google Scholar]
- [12].(a) Birman VB, Li X, Benzotetramisole: a remarkably enantioselective acyl transfer catalyst, Org. Lett 8 (2006) 1351–1354. [DOI] [PubMed] [Google Scholar]; (b) Birman VB, Uffman EW, Jiang H, Li X, Kilbane CJ, 2,3-dihydroimidazo[1,2-a]pyridines: a new class of enantioselective acyl transfer catalysts and their use in kinetic resolution of alcohols, J. Am. Chem. Soc 126 (2004) 12226–12227. [DOI] [PubMed] [Google Scholar]
- [13].Xiao G, Cintron-Rosado GA, Glazier DA, Xi B-M, Liu C, Liu P, Tang W, W, Catalytic site-selective acylation of carbohydrates directed by cation-n interaction, J. Am. Chem. Soc 139 (2017) 4346–4349. [DOI] [PubMed] [Google Scholar]
- [14].Yanagi M, Imayoshi A, Ueda Y, Furuta T, Kawabata T, Carboxylate anions accelerate pyrrolinopyridine (PPy)-catalyzed acylation: catalytic site-selective acylation of carbohydrate by in situ counteranion exchange, Org. Lett 19 (2017) 3099–3102. [DOI] [PubMed] [Google Scholar]
- [15].Kawabata T, Muramatsu W, Nishio T, Shibata T, Schedel H, A catalytic one-step process for the chemo- and regioselective acylation of monosaccharides, J. Am. Chem. Soc 129 (2007) 12890–12895. [DOI] [PubMed] [Google Scholar]
- [16].Giuliano M, Miller SJ, Site-selective reactions with peptide-based catalysts, Top. Curr. Chem 372 (2016) 157–201. [DOI] [PubMed] [Google Scholar]
- [17].Huber F, Kirsch SF, Site-selective acylations with tailer-made catalysts, Chem. Eur. J 22 (2016) 5914–5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tong ML, Huber F, Taghuo Kaptouom ES, Cellnik T, Kirsch SF, Enhanced site-selectivity in acylation reactions with substrate-optimized catalysts on solid supports, Chem. Commun 53 (2017) 3086–3089. [DOI] [PubMed] [Google Scholar]
- [19].Welton T, Room-temperature ionic liquids. Solvents for synthesis and catalysis, Chem. Rev 99 (1999) 2071–2084. [DOI] [PubMed] [Google Scholar]
- [20].Axelsson A, Ta L, Sundén H, Direct highly regioselective functionalization of carbohydrates: a three-component reaction combining the dissolving and catalytic efficiency of ionic liquids, Eur. J. Org. Chem 20 (2016) 3339–3343. [Google Scholar]
- [21].Cramer DL, Bera S, Studer A, Exploring cooperative effects in oxidative NHC catalysis: regioselective acylation of carbohydrates, Chem. Eur. J 22 (2016) 7403–7407. [DOI] [PubMed] [Google Scholar]
- [22].(a) Samanta RC, De Sarkar S, Frçhlich R, Grimme S, Studer A, N-heterocyclic carbene (NHC) catalyzed chemoselective acylation of alcohols in the presence of amines with various acylating reagents, Chem. Sci 4 (2013) 2177–2184. [Google Scholar]; (b) De Sarkar S, Grimme S, Studer A, NHC catalyzed oxidations of aldehydes to esters: chemoselective acylation of alcohols in presence of amines, J. Am. Chem. Soc 132 (2010) 1190–1191. [DOI] [PubMed] [Google Scholar]
- [23].(a) Cox DJ, Smith MD, Fairbanks AJ, Glycosylation catalyzed by a chiral Brønsted acid, J. Org. Lett 12 (2010) 1452–1455. [DOI] [PubMed] [Google Scholar]; (b) Liu D, Sarrafpour S, Guo W, Goulart B, Bennett CS, Matched/mismatched interactions in chiral Brønsted acid-catalyzed glycosylation reactions with 2-deoxy-sugar trichloroacetimidate donors, J. Carbohydr. Chem 33 (2014) 423–434. [Google Scholar]
- [24].Tay J-H, Argüelles AJ, DeMars MD, Zimmerman PD, Sherman DH, Nagorny PJ, Regiodivergent glycosylations of 6-deoxy-Erythronolide B and Oleandomycin-derived macrolactones enabled by chiral acid catalysis, J. Am. Chem. Soc 139 (2017) 8570–8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Palo-Nieto C, Sau A, Williams R, Galan MC, Cooperative Brønsted acid-type organocatalysis for the stereoselective synthesis of deoxyglycosides, J. Org. Chem 82 (2017) 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Balmond EI, Coe DM, McGarrigle EM, Galan MC, α-selective organocatalytic synthesis of 2-deoxygalactosides, Angew. Chem. Int. Ed 51 (2012) 9152–9155. [DOI] [PubMed] [Google Scholar]
- [27].Taylor MS, Catalysis based on reversible covalent interactions of organoboron compounds, Acc. Chem. Res 48 (2015) 295–305. [DOI] [PubMed] [Google Scholar]
- [28].Mancini RS, Lee JB, Taylor MS, Boronic esters as protective groups in carbohydrate chemistry: processes for acylation, silylation, and alkylation of glycoside-derived boronates, Org. Biomol. Chem 15 (2017) 132–143. [DOI] [PubMed] [Google Scholar]
- [29].Mothana S, Grassot J-M, Hall DG, Multistep phase-switch synthesis by using liquid-liquid partitioning of boronic acids: productive tags with an expanded repertoire of compatible reactions, Angew. Chem. Int. Ed 49 (2010) 2883–2887. [DOI] [PubMed] [Google Scholar]
- [30].Mancini RS, Lee JB, Taylor MS, Sequential functionalizations of carbohydrates enabled by boronic esters as switchable protecting/activating groups, J. Org. Chem 82 (2017) 8777–8791. [DOI] [PubMed] [Google Scholar]
- [31].Dimakos V, Garrett GE, Taylor MS, Site-selective, copper-mediated O-arylation of carbohydrate derivatives, J. Am. Chem. Soc 139 (2017) 15515–15521. [DOI] [PubMed] [Google Scholar]
- [32].Fukuhara K, Shimada N, Nishino T, Kaji E, Makino KE, Regioselective, tin-free sulfation of unprotected hexopyranosides by using phenylboronic acid, Eur. J. Org. Chem 2016, 902–905.
- [33].Rocheleau S, Pottel J, Huskić I, Moitessier N, Highly regioselective monoacylation of unprotected glucopyranoside using transient directing-protecting groups, Eur. J. Org. Chem 2017, 646–656.
- [34].Bajaj SO, Sharif EU, Akhmedov NG, O’Doherty GA, De novo asymmetric synthesis of the mezzettiaside family of natural products via the iterative use of a dual B-/Pd-catalyzed glycosylation, Chem. Sci 5 (2016) 2230–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wu D, Li M, O’Doherty GA, Synthesis of several cleistriosides and cleistetrosides natural products via a divergent de novo asymmetric approach, Org. Lett 12 (2010) 5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lee D, Taylor MS, Borinic acid-catalyzed regioselective acylation of carbohydrate derivatives, J. Am. Chem. Soc 133 (2011) 3724–3727. [DOI] [PubMed] [Google Scholar]
- [37].Bajaj SO, Shi P, Beuning PJ, O’Doherty GA, Structure activity relationship study of mezzettiasides natural products and their four new disaccharide analogues for anticancer/antibacterial activity, Med. Chem. Commun 5 (2014) 1138–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]