

Abstract
目的
探讨钙/钙调蛋白依赖的蛋白激酶Ⅱ(CaMKⅡ)在离体心脏缺血再灌注(IR)损伤中的作用和可能机制。
方法
采用大鼠离体心脏缺血再灌注(IR 45 min/120 min)模型,将40只大鼠随机分为对照组(Control)、KN-93药物对照组(2.5 μmol/L KN-93)、IR组和CaMKⅡ特异性抑制剂KN-93干预缺血再灌注组(KN-93+IR),以左室心脏功能、冠脉流出液中乳酸脱氢酶(LDH)活性和心肌肌钙蛋白(cTnI)含量、心肌梗死面积评价心脏损伤程度,Western-blot检测CaMKⅡ磷酸化(p-CaMKⅡ)、CaMKⅡ氧化(ox-CaMKⅡ)和PLN磷酸化(p-PLN)蛋白的表达,试剂盒检测线粒体超氧化物歧化酶(SOD)的活性和丙二醛(MDA)的含量。
结果
与Control组相比,KN-93组各项指标均无明显变化;IR组心脏功能、线粒体SOD的活性降低(P < 0.01),而心肌梗死面积、冠脉流出液中LDH活性和cTnI含量、p-CaMKⅡ、ox-CaMKⅡ和p-PLN的表达、线粒体MDA的含量均明显升高(P < 0.01);KN-93干预IR组可明显改善心脏功能(P < 0.01),增加线粒体SOD活性,减小心肌梗死面积、LDH活性、cTnI含量、p-CaMKⅡ、ox-CaMKⅡ和p-PLN的表达以及线粒体MDA含量(P < 0.01)。
结论
CaMKⅡ参与离体心脏缺血再灌注损伤,抑制CaMKⅡ可通过降低线粒体氧化应激进而减轻离体心脏缺血再灌注损伤。
Keywords: 钙/钙调蛋白依赖的蛋白激酶Ⅱ, 心脏缺血再灌注损伤, 线粒体, 超氧化物歧化酶, 丙二醛
Abstract
Objective
To investigate the role of calcium/calmodulin-dependent protein kinase Ⅱ (CaMKⅡ) in myocardial ischemia-reperfusion (IR) injury in isolated perfused rat heart and explore the underlying mechanisms.
Methods
An ischemiareperfusion (IR) model was prepared using isolated rat hearts perfused with Krebs-Henseleit solution were randomly divided into control group, 2.5 μmol/L KN-93 group, IR (induced by ischemia for 45 min followed by reperfusion for 120 min) group and KN-93 + IR group. The myocardial performance was evaluated by assessing the left ventricular pressure. Lactate dehydrogenase (LDH) activity and cTnI content in the coronary flow and the infarct size were determined to evaluate the myocardial injury. The phosphorylation of CaMKⅡ (p-CaMKⅡ) and PLN (p-PLN) and oxidation of CaMKⅡ (ox-CaMKⅡ) were measured with Western blotting. The activity of mitochondrial superoxide dismutase (SOD) and the content of malondialdehyde (MDA) were determined using ELISA.
Results
Compared with the control group, KN-93 treatment at 2.5 μmol/L produced no significant effects on cardiac function or performance in rat hearts without IR injury. Myocardial IR injury significantly decreased myocardial performance and mitochondrial SOD activity in the perfused hearts (P < 0.01) and caused significantly increased infarct size, LDH activity, cTnI content, expressions of p-CaMKⅡ, ox-CaMKⅡ and p-PLN, and also increased mitochondrial MDA content (P < 0.01). KN-93 treatment at 2.5 μmol/L administered before ischemia and before reperfusion markedly attenuated such changes induced by ischemia and reperfusion (P < 0.01).
Conclusion
CaMKⅡ participates in myocardial IR injury in isolated rat heart, and inhibiting CaMKⅡ can alleviate myocardial injury by relieving mitochondrial oxidation stress.
Keywords: calcium/calmodulin-dependent protein kinase Ⅱ, myocardial ischemia reperfusion injury, mitochondria, superoxide dismutase, malondialdehyde
心肌缺血再灌注是急性心梗溶栓、冠脉血管成形术和心脏停跳手术等心血管疾病手术治疗中不可避免的过程[1]。目前关于心肌缺血再灌注损伤机制的研究主要集中在钙超载、氧化应激、细胞凋亡、炎症反应、线粒体功能障碍以及自噬[2-8]。尽管有大量文献报道了心肌缺血再灌注损伤的机制,但临床转化有限,应用价值不高,治疗效果并不理想。
钙/钙调蛋白依赖的蛋白激酶Ⅱ(CaMKⅡ)是一种多功能的丝氨酸/苏氨酸蛋白激酶,有α、β、δ和γ 4种亚型,其中心脏上主要分布δ亚型[9-11]。CaMKⅡ通过自身磷酸化或活性氧(ROS)氧化M281/282位点激活[12-14]。激活的CaMKⅡ可磷酸化L型钙通道、肌浆网雷诺丁受体(RyR)和钙泵(SERCA2a)调节心肌细胞内钙变化[15-18]。
CaMKⅡ的特异性抑制剂KN-93,可与钙调蛋白(CaM)竞争性结合CaMKⅡ的结合区,抑制CaMKⅡ的活性[1]。根据以往文献的报道,CaMKⅡ参与心肌损伤的机制主要是调控L型钙通道和肌质网功能加重胞内钙超载[15-18],而对线粒体功能特别是在调控线粒体内氧化还原反应中所发挥的作用还未见报道。本研究采用离体心脏缺血再灌注模型,观察KN-93抑制CaMKⅡ后对线粒体氧化应激损伤的影响。
1. 材料和方法
1.1. 实验动物和试剂
40只健康雄性SD大鼠购买于第四军医大学实验动物中心,体质量250~300 g。氯化三苯基四氮唑(TTC)购买于Sigma,KN-93购自Tocris,依据文献确定其浓度[19],乳酸脱氢酶试剂盒、SOD试剂盒和MDA试剂盒均购于南京建成生物公司,Cell Signaling Technology公司购买磷酸化CaMKⅡ、PLN、CaMKⅡ和GAPDH抗体,Badrilla购买PLN-Thr17位点磷酸化抗体,氧化CaMKⅡ抗体购自Millipore。正常灌流液Krebs-Henseleit(KH solution)的成分和含量(mmol/L):NaCl 118,KCl 4.7,KH2PO4 1.2,MgSO4 1.2,CaCl2 1.25,Glucose 11,NaHCO3 25。实验开始前45 min用混合气体(95% O2/5% CO2, v/v)向正常KH液中通气至实验结束,灌流液pH维持在7.35~7.45,温度保持在37 ℃。
1.2. 方法
1.2.1. 实验分组和模型的制备
实验时,SD大鼠快速腹腔注射戊巴比妥钠(40 mg/kg)和肝素(500 U/kg),麻醉好后迅速沿剑突两侧剪开胸腔,暴露心脏,并立即取下心脏放置于预冷的4 ℃ KH液中。用0号线将保留的主动脉根部快速结扎在Langendorff灌流管口,灌流液从主动脉口逆行灌注,灌注压维持在80 mmHg。将乳胶球囊刺破二尖瓣插入左心室,通过Labchart 7软件记录心脏功能。控制球囊内液体量使左室舒张末压保持在0~5 mmHg,使心脏稳定15 min。待其平衡后,随机分为4组(n=10):对照组(Control)、KN-93药物对照组(KN-93)、缺血再灌注组(IR)和KN-93干预缺血再灌注组(KN-93+IR)。Control组正常灌注165 min;KN-93组用含2.5 μmol/L KN-93的KH液灌注6 min,恢复正常KH液灌注159 min;IR组缺血45 min,再灌注120 min;KN-93+IR组先用含2.5 μmol/L KN-93的KH液处理1 min,缺血45 min,再用含2.5 μmol/L KN-93的KH液处理5 min,恢复KH液灌注114 min。
1.2.2. 心脏功能的监测
采用Labchart 7软件,整个实验过程中监测左室内压(LVP)、左室收缩峰压(LVPSP)和左室舒张末压(LVEDP)的变化,左室发展压(LVDP)的变化采用LVPSP和LVEDP的差值表示。LVDP和LVEDP作为评价心脏功能的指标。
1.2.3. 心肌梗死面积的检测
再灌注结束后,立刻将心脏冻存于-80 ℃冰箱60 min;垂直于心脏纵轴方向切成连续6片厚度约为1 mm的切片,放入盛有1% TTC溶液的24孔板中,37 ℃恒温水浴避光孵育15 min;4%多聚甲醛固定2 d;采用数码相机拍照,Image J软件计算心肌梗死面积。白色区代表梗死心肌,红色区代表正常心肌,心肌梗死比例=(梗死区面积/心肌总面积)×100%。
1.2.4. LDH活性和cTnI的检测
收集各组心脏再灌注前10 min内的冠脉流出液,严格按照南京建成生物公司提供的LDH试剂盒和cTnI检测试剂盒说明书操作,读取各组冠脉流出液的吸光值并换算成相应的LDH活性。
1.2.5. Western blot
剪取各组大鼠左室相同重量的心肌组织,加入含蛋白酶抑制剂、磷酸酶抑制剂和RIPA裂解液(强)的混合液中冰上裂解30 min,12 000 g,4 ℃离心20 min,取上清,BCA蛋白定量;配制5 %浓缩胶和10%分离胶,恒压90 V湿转70 min将蛋白转移至PVDF膜;用TBST配制5%脱脂奶粉室温封闭90 min,分别用p-CaMKⅡ、ox-CaMKⅡ、CaMKⅡ、p-PLN、PLN(1:1000)以及GAPDH(1:4000)一抗4 ℃孵育过夜;第2天,TBST洗膜6次,5 min/次,用HRP标记的二抗(1:5000)室温孵育90 min,TBST洗膜6次,5 min/次;ECL显色,曝光后扫描,用Image J图像分析软件对各组条带进行灰度值分析并统计,以GAPDH作为内参。
1.2.6. 线粒体SOD活性和MDA含量检测
再灌流结束后,分别提取各组心肌线粒体,方法如下:各组分别称取40 mg心肌组织,放入盛有500 μL预冷的PBS的1.5 mL离心管中,在冰上剪碎,800 g 4 ℃离心60 s,弃上清,沉淀中加入500 μL含PMSF的线粒体分离试剂,冰上匀浆,1000 g 4 ℃离心5 min,上清转移至另一离心管中,12 000 g 4 ℃离心15 min,所得沉淀即为线粒体。按照南京建成生物公司提供的线粒体MDA和SOD试剂盒说明书操作,检测MDA含量(nmol/mg)和SOD活性(U/mg)。
1.3. 统计学分析
结果采用均数±标准差表示,使用SPSS 13.0统计软件进行数据分析,单因素方差分析用于各组间差异比较,两组之间的比较采用Tukey检验,P < 0.05表示差异具有统计学意义。
2. 结果
2.1. KN-93可改善离体心脏缺血再灌注心脏功能
如图 1所示,Control组和KN-93组在整个灌注期LVDP和LVEDP均无明显变化,且两组之间差异无统计学意义(P > 0.05);与Control组相比,IR组心脏缺血45 min后再灌注,LVDP显著降低,LVEDP显著升高(P < 0.01);而与IR组心脏相比,KN-93+IR组心脏再灌注后LVDP明显升高,LVEDP显著降低(P < 0.01)。
1.
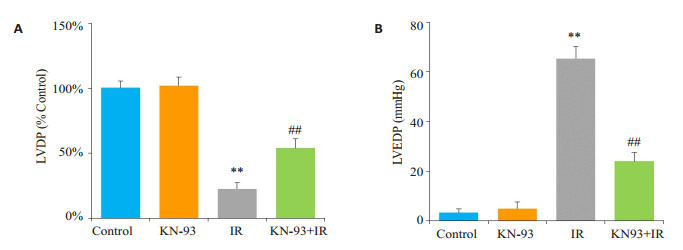
缺血再灌注末左室功能的变化
Left ventricular function of isolated rat hearts at the end of ischemia-reperfusion (IR) in each group. A: Recovery of left ventricular developed pressure (LVDP) at the end of reperfusion. B: Level of left ventricular end-diastolic pressure (LVEDP) at the end of reperfusion. **P < 0.01 IR vs control, ##P < 0.01 KN-93+IR vs IR
2.2. KN-93可减少离体心脏缺血再灌注心肌梗死面积和cTnI的释放
如图 2所示,Control组和KN-93组心脏未见梗死区域,冠脉流出液中LDH活性和cTnI的含量很低;与Control组相比,IR组心脏心肌梗死面积明显增大,冠脉流出液中LDH活性和cTnI的含量明显升高(P < 0.01);而KN-93+IR组心脏心肌梗死面积明显减小,冠脉流出液中LDH活性和cTnI含量均显著降低(P < 0.01)。
2.
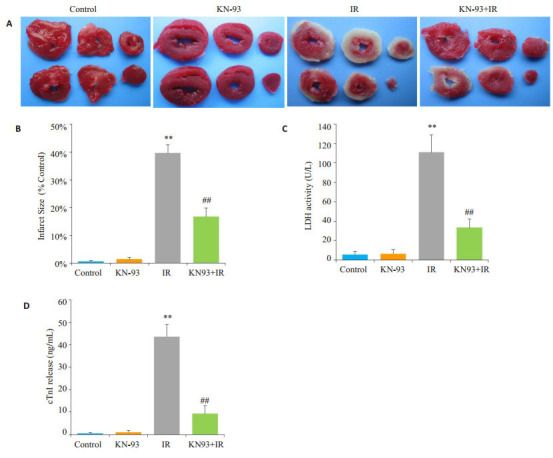
各组心肌梗死面积,LDH活性和cTnI含量
Myocardial infarct size, LDH activity and cTnI content in each group. A: TTC Staining; B: Percentage of myocardial infarct size; C: LDH activity; D: cTnI content; **P < 0.01 IR vs control, ##P < 0.01 KN-93+IR vs IR
2.3. KN-93可抑制离体心脏缺血再灌注损伤中CaMKⅡ和PLN的活性
如图 3,Western blot结果显示,与Control组相比,KN-93组ox-CaMKⅡ、p-CaMKⅡ和p-PLN的水平没有明显变化(P > 0.05);IR组心脏ox-CaMKⅡ和p-CaMKⅡ的表达明显增加,其底物PLN的磷酸化水平也明显增加(P < 0.01);而与IR组相比,KN-93 +IR组心脏oxCaMKⅡ、p-CaMKⅡ和p-PLN的表达均明显降低(P < 0.01)。
3.
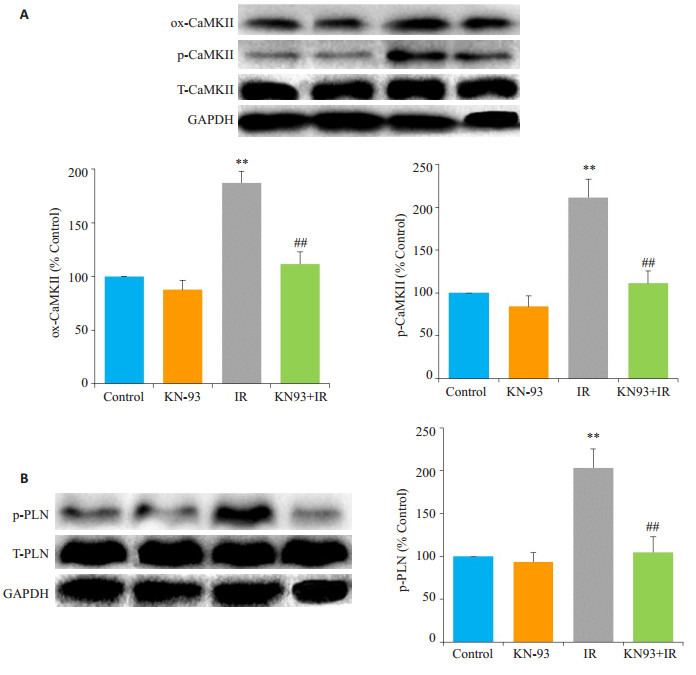
各组磷酸化CaMKⅡ和PLN的表达
Western blotting for detecting expressions of p-CaMKⅡ (A) and p-PLN (B) in each group. **P < 0.01 IR vs control, ##P < 0.01 KN-93+IR vs IR
2.4. KN-93可减轻离体心脏缺血再灌注损伤中线粒体氧化应激
如图 4所示,与Control组相比,KN-93组线粒体SOD活性和MDA含量均没有明显变化(P > 0.05);IR组线粒体SOD活性明显降低,MDA含量明显增加(P < 0.01);与IR组相比,KN-93+IR组可明显增加线粒体SOD活性,降低MDA含量(P < 0.01)。
4.
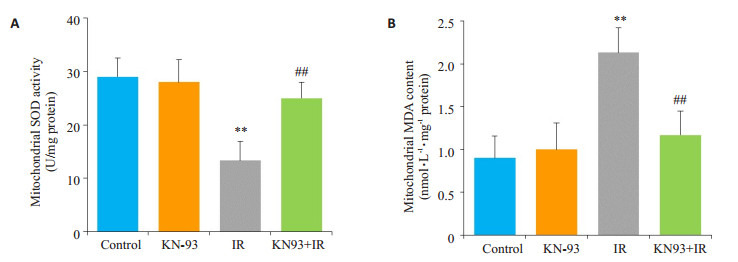
线粒体SOD活性和MDA含量
Mitochondrial superoxide dismutase (SOD) activity (A) and malondialdehyde (MDA) content (B) in each group. **P < 0.01 IR vs control, ##P < 0.01 KN-93+IR vs IR
3. 讨论
缺血心肌再灌注是目前治疗缺血性心脏病的最有效方法,然而,再灌注本身会对缺血心肌造成损伤,增加细胞凋亡和坏死,引起心脏功能障碍和恶性心律失常,严重影响患者的预后。近年来,CaMKⅡ在心血管疾病中发挥的重要作用已经成为研究的热点[20-22]。大量研究报道CaMKⅡ参与心肌缺血再灌注损伤、心肌梗死、心力衰竭和糖尿病心肌病等病理性过程的发生、发展[23-27],其机制主要集中在CaMKⅡ可增加心肌细胞内钙超载引起损伤,而对于心肌内氧化应激的作用报道较少。
本研究采用离体心脏缺血再灌注模型,观察了临床上反映心脏功能的相关指标,如心脏舒缩功能、心肌梗死面积、冠脉流出液中LDH活性和cTnI的含量以评价心肌损伤程度,并检测了CaMKⅡ磷酸化和氧化的表达及其底物PLN磷酸化水平的变化来反映CaMKⅡ活性,进而评价CaMKⅡ在心脏缺血再灌注过程中的作用。实验结果证实,KN-93干预IR后,可明显改善心脏功能,减小心肌梗死面积,降低冠脉流出液中LDH活性和cTnI的含量,结果与以往的大量文献报道一致[4, 19],这说明抑制CaMKⅡ可显著减轻心肌损伤。同时,IR组可明显增加CaMKⅡ磷酸化和氧化表达,其底物PLN的磷酸化水平也明显增加,而KN-93抑制CaMKⅡ后,CaMKⅡ磷酸化和氧化表达均明显降低,PLN磷酸化水平也随之降低,这说明KN-93可同时抑制CaMKⅡ的两种激活方式减轻IR引起的心肌损伤。
线粒体作为心肌细胞内产生能量的主要场所,在心肌细胞能量代谢过程中发挥至关重要的作用。心肌缺血再灌注破坏了线粒体结构和氧化磷酸化过程,增加了线粒体氧化应激损伤,导致氧自由基大量堆积,ATP合成减少,膜通透性转换孔开放,细胞凋亡的启动[28-29]。因此,抑制线粒体氧化应激可有效减轻心脏缺血再灌注损伤。SOD活性可反映线粒体清除氧自由基的能力,而MDA含量可间接反映脂质过氧化的程度。本实验结果表明,离体心脏缺血再灌注后,线粒体SOD活性明显降低,MDA含量却显著升高,说明线粒体氧化应激程度明显增加,而KN-93干预IR后可增加线粒体SOD活性,降低MDA含量,这说明抑制CaMKⅡ可减轻线粒体氧化应激。
综上所述,本实验进一步明确CaMKⅡ参与了心脏缺血再灌注损伤,并证明KN-93抑制CaMKⅡ磷酸化和氧化激活后可通过降低线粒体氧化应激改善心脏损伤,这为CaMKⅡ参与心脏缺血再灌注损伤提供了新的实验依据。后续工作还需进一步阐明CaMKⅡ磷酸化和氧化两种激活方式在介导线粒体氧化应激中的不同作用及分子机制,这将为心脏缺血再灌注保护提供新的思路。
Biography
孔令恒,在读博士研究生,讲师,E-mail: lhkong1984@163.com
Funding Statement
陕西省教育厅重点实验室科研计划项目(16JS098);陕西省缺血性心血管疾病重点实验室开放基金(2017ZDKF10);陕西省基础医学优势学科建设经费(陕教位[2014]3号-1001);陕西省中医管理局中医药基础研究项目(JCMS023);西安医学院校级重点学科(神经-内分泌生理)
Contributor Information
孔 令恒 (Lingheng KONG), Email: lhkong1984@163.com.
苏 兴利 (Xingli SU), Email: suxingli@126.com.
References
- 1.孔 令恒, 刘 哲, 张 建英, et al. 心肌缺血-再灌注-钙超载损伤的基础与临床研究. https://www.wenkuxiazai.com/doc/e65f7f0a5fbfc77da369b17a.html. 中国体外循环杂志. 2015;13(4):253–6. [孔令恒, 刘哲, 张建英, 等.心肌缺血-再灌注-钙超载损伤的基础与临床研究[J].中国体外循环杂志, 2015, 13(4):253-6.] [Google Scholar]
- 2.Garcia-Dorado D, Ruiz-Meana M, Inserte JA, et al. Calciummediated cell death during myocardial reperfusion. Cardiovasc Res. 2012;94(2):168–80. doi: 10.1093/cvr/cvs116. [Garcia-Dorado D, Ruiz-Meana M, Inserte JA, et al. Calciummediated cell death during myocardial reperfusion[J]. Cardiovasc Res, 2012, 94(2):168-80.] [DOI] [PubMed] [Google Scholar]
- 3.Madamanchi NR, Runge MS. Redox signaling in cardiovascular health and disease. https://www.sciencedirect.com/science/article/pii/S089158491300141X. Free Radic Biol Med. 2013;61(0):473–501. doi: 10.1016/j.freeradbiomed.2013.04.001. [Madamanchi NR, Runge MS. Redox signaling in cardiovascular health and disease[J]. Free Radic Biol Med, 2013, 61(0):473-501.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salas MA, Valverde CA, Sanchez G, et al. The signaling pathway of CaMKⅡ-mediated apoptosis and necrosis in the ischemia/reperfusion injury. https://www.sciencedirect.com/science/article/pii/S0022282809005598. J Mol Cell Cardiol. 2009;48(6):1298–306. doi: 10.1016/j.yjmcc.2009.12.015. [Salas MA, Valverde CA, Sanchez G, et al. The signaling pathway of CaMKⅡ-mediated apoptosis and necrosis in the ischemia/reperfusion injury[J]. J Mol Cell Cardiol, 2009, 48(6):1298-306.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gray CB, Suetomi T, Xiang S, et al. CaMKⅡ delta subtypes differentially regulate infarct formation following ex vivo myocardial ischemia/reperfusion through NF-kappa B and TNFalpha. J Mol Cell Cardiol. 2017;103(103):48–55. doi: 10.1016/j.yjmcc.2017.01.002. [Gray CB, Suetomi T, Xiang S, et al. CaMKⅡ delta subtypes differentially regulate infarct formation following ex vivo myocardial ischemia/reperfusion through NF-kappa B and TNFalpha[J]. J Mol Cell Cardiol, 2017, 103(103):48-55.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ong SB, Hausenloy DJ. Mitochondrial morphology and cardiovascular disease. Cardiovasc Res. 2010;88(1):16–29. doi: 10.1093/cvr/cvq237. [Ong SB, Hausenloy DJ. Mitochondrial morphology and cardiovascular disease[J]. Cardiovasc Res, 2010, 88(1):16-29.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu J, Qin XH, Cai XQ, et al. Mitochondrial JNK activation triggers autophagy and apoptosis and aggravates myocardial injury following ischemia/reperfusion. Biochimica et Biophysica ActaMol Basis Dis. 2015;1852(2, SI):262–70. doi: 10.1016/j.bbadis.2014.05.012. [Xu J, Qin XH, Cai XQ, et al. Mitochondrial JNK activation triggers autophagy and apoptosis and aggravates myocardial injury following ischemia/reperfusion[J]. Biochimica et Biophysica ActaMol Basis Dis, 2015, 1852(2, SI):262-70.] [DOI] [PubMed] [Google Scholar]
- 8.Yu SY, Dong B, Zhou SH, et al. LncRNA MALAT1:A potential regulator of autophagy in myocardial ischemia-reperfusion injury. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4910607/table/T1/ Int J Cardiol. 2017;247(5):25. doi: 10.1016/j.ijcard.2017.04.011. [Yu SY, Dong B, Zhou SH, et al. LncRNA MALAT1:A potential regulator of autophagy in myocardial ischemia-reperfusion injury[J]. Int J Cardiol, 2017, 247(5):25.] [DOI] [PubMed] [Google Scholar]
- 9.Anderson ME. Pathways for CaMKⅡ activation in disease. Heart Rhythm. 2011;8(9):1501–3. doi: 10.1016/j.hrthm.2011.04.027. [Anderson ME. Pathways for CaMKⅡ activation in disease[J]. Heart Rhythm, 2011, 8(9):1501-3.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kreusser MM, Lehmann LH, Wolf N, et al. Inducible cardiomyocyte-specific deletion of CaM kinase Ⅱ protects from pressure overload-induced heart failure. https://link.springer.com/article/10.1007/s00395-016-0581-2. Basic Res Cardiol. 2016;111(6):10. doi: 10.1007/s00395-016-0581-2. [Kreusser MM, Lehmann LH, Wolf N, et al. Inducible cardiomyocyte-specific deletion of CaM kinase Ⅱ protects from pressure overload-induced heart failure[J]. Basic Res Cardiol, 2016, 111(6):10.] [DOI] [PubMed] [Google Scholar]
- 11.Perkin J, Slater R, Del Favero G, et al. Phosphorylating titin's cardiac N2B element by ERK2 or CaMKⅡ delta lowers the single molecule and cardiac muscle force. Biophys J. 2015;109(12):2592–601. doi: 10.1016/j.bpj.2015.11.002. [Perkin J, Slater R, Del Favero G, et al. Phosphorylating titin's cardiac N2B element by ERK2 or CaMKⅡ delta lowers the single molecule and cardiac muscle force[J]. Biophys J, 2015, 109(12):2592-601.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang PY. CaMKⅡ:the molecular villain that aggravates cardiovascular disease. Exp Ther Med. 2017;13(3):815–20. doi: 10.3892/etm.2017.4034. [Zhang PY. CaMKⅡ:the molecular villain that aggravates cardiovascular disease[J]. Exp Ther Med, 2017, 13(3):815-20.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monnerat G, Alarcon ML, Vasconcellos LR, et al. Macrophagedependent IL-1 beta production induces cardiac arrhythmias in diabetic mice. https://www.ncbi.nlm.nih.gov/pubmed/?term=hochman-mendez. Nat Commun. 2016;7(1):10. doi: 10.1038/ncomms13344. [Monnerat G, Alarcon ML, Vasconcellos LR, et al. Macrophagedependent IL-1 beta production induces cardiac arrhythmias in diabetic mice[J]. Nat Commun, 2016, 7(1):10.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mattiazzi A, Bassani RA, Escobar AL, et al. Chasing cardiac physiology and pathology down the CaMKⅡ cascade. Am J Physiol Heart Circ Physiol. 2015;308(10):H1177–91. doi: 10.1152/ajpheart.00007.2015. [Mattiazzi A, Bassani RA, Escobar AL, et al. Chasing cardiac physiology and pathology down the CaMKⅡ cascade[J]. Am J Physiol Heart Circ Physiol, 2015, 308(10):H1177-91.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Carlo MN, Said M, Ling HY, et al. CaMKⅡ-dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury. https://www.sciencedirect.com/science/article/pii/S0022282814001989. J Mol Cell Cardiol. 2014;74(74):274–83. doi: 10.1016/j.yjmcc.2014.06.004. [Di Carlo MN, Said M, Ling HY, et al. CaMKⅡ-dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury[J]. J Mol Cell Cardiol, 2014, 74(74):274-83.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Helms AS, Alvarado FJ, Yob J, et al. Genotype-Dependent and -Independent calcium signaling dysregulation in human hypertrophic cardiomyopathy. Circulation. 2016;134(22):1738–48. doi: 10.1161/CIRCULATIONAHA.115.020086. [Helms AS, Alvarado FJ, Yob J, et al. Genotype-Dependent and -Independent calcium signaling dysregulation in human hypertrophic cardiomyopathy[J]. Circulation, 2016, 134(22):1738-48.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li YE, Sirenko S, Riordon DR, et al. CaMKⅡ-dependent phosphorylation regulates basal cardiac pacemaker function via modulation of local Ca2+ releases. Am J Physiol Heart Circ Physiol. 2016;311(3):H532–44. doi: 10.1152/ajpheart.00765.2015. [Li YE, Sirenko S, Riordon DR, et al. CaMKⅡ-dependent phosphorylation regulates basal cardiac pacemaker function via modulation of local Ca2+ releases[J]. Am J Physiol Heart Circ Physiol, 2016, 311(3):H532-44.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Popescu I, Galice S, Mohler PJ, et al. Elevated local[Ca2+]and CaMKⅡ promote spontaneous Ca2+ release in ankyin-B-deficient hearts. Cardio Res. 2016;111(3):287–94. doi: 10.1093/cvr/cvw093. [Popescu I, Galice S, Mohler PJ, et al. Elevated local[Ca2+]and CaMKⅡ promote spontaneous Ca2+ release in ankyin-B-deficient hearts [J]. Cardio Res, 2016, 111(3):287-94.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vila-Petroff M, Salas MA, Said MA, et al. CaMKⅡ inhibition protects against necrosis and apoptosis in irreversible ischemiareperfusion injury. Cardio Res. 2007;73(4):689–98. doi: 10.1016/j.cardiores.2006.12.003. [Vila-Petroff M, Salas MA, Said MA, et al. CaMKⅡ inhibition protects against necrosis and apoptosis in irreversible ischemiareperfusion injury[J]. Cardio Res, 2007, 73(4):689-98.] [DOI] [PubMed] [Google Scholar]
- 20.Kong LH, Gu XM, Wu F, et al. CaMKⅡ inhibition mitigates ischemia/reperfusion-elicited calpain activation and the damage to membrane skeleton proteins in isolated rat hearts. Biochem Biophys Res Commun. 2017;491(3):687–92. doi: 10.1016/j.bbrc.2017.07.128. [Kong LH, Gu XM, Wu F, et al. CaMKⅡ inhibition mitigates ischemia/reperfusion-elicited calpain activation and the damage to membrane skeleton proteins in isolated rat hearts[J]. Biochem Biophys Res Commun, 2017, 491(3):687-92.] [DOI] [PubMed] [Google Scholar]
- 21.Zhong P, Quan DJ, Peng JY, et al. Role of CaMKⅡ in free fatty acid/hyperlipidemia-induced cardiac remodeling both in vitro and in vivo. http://downloads.hindawi.com/journals/ecam/2013/149363.xml. J Mol Cell Cardiol. 2017;109(109):1–16. doi: 10.1016/j.yjmcc.2017.06.010. [Zhong P, Quan DJ, Peng JY, et al. Role of CaMKⅡ in free fatty acid/hyperlipidemia-induced cardiac remodeling both in vitro and in vivo[J]. J Mol Cell Cardiol, 2017, 109(109):1-16.] [DOI] [PubMed] [Google Scholar]
- 22.Feng Y, Cheng J, Wei BZ, et al. CaMKⅡ inhibition reduces isoproterenol-induced ischemia and arrhythmias in hypertrophic mice. https://www.pubmedcentral.nih.gov/pmc/articles/PMC3314487/ Oncotarget. 2017;8(11):17504–9. doi: 10.18632/oncotarget.15099. [Feng Y, Cheng J, Wei BZ, et al. CaMKⅡ inhibition reduces isoproterenol-induced ischemia and arrhythmias in hypertrophic mice[J]. Oncotarget, 2017, 8(11):17504-9.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ling H, Gray CB, Zambon AC, et al. Ca2+/calmodulin-dependent protein kinase Ⅱ δ mediates myocardial ischemia/reperfusion injury through nuclear factor-κB. Circ Res. 2013;112(6):935–44. doi: 10.1161/CIRCRESAHA.112.276915. [Ling H, Gray CB, Zambon AC, et al. Ca2+/calmodulin-dependent protein kinase Ⅱ δ mediates myocardial ischemia/reperfusion injury through nuclear factor-κB[J]. Circ Res, 2013, 112(6):935-44.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo M, Guan XQ, Luczak ED, et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKⅡ. J Clin Invest. 2013;123(3):1262–74. doi: 10.1172/JCI65268. [Luo M, Guan XQ, Luczak ED, et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKⅡ[J]. J Clin Invest, 2013, 123(3):1262-74.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ling HY, Zhang T, Pereira L, et al. Requirement for Ca2+/calmodulin-dependent kinase Ⅱ in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119(5):1230–40. doi: 10.1172/JCI38022. [Ling HY, Zhang T, Pereira L, et al. Requirement for Ca2+/calmodulin-dependent kinase Ⅱ in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice[J]. J Clin Invest, 2009, 119(5):1230-40.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daniels L, Bell JR, Delbridge LM, et al. The role of CaMKⅡ in diabetic heart dysfunction. Heart Fail Rev. 2015;20(5):589–600. doi: 10.1007/s10741-015-9498-3. [Daniels L, Bell JR, Delbridge LM, et al. The role of CaMKⅡ in diabetic heart dysfunction[J]. Heart Fail Rev, 2015, 20(5):589-600.] [DOI] [PubMed] [Google Scholar]
- 27.Koncsos G, Varga ZV, Baranyai T, et al. Diastolic dysfunction in prediabetic male rats:Role of mitochondrial oxidative stress. Am J Physiol Heart Circ Physiol. 2016;311(4):H927–43. doi: 10.1152/ajpheart.00049.2016. [Koncsos G, Varga ZV, Baranyai T, et al. Diastolic dysfunction in prediabetic male rats:Role of mitochondrial oxidative stress[J]. Am J Physiol Heart Circ Physiol, 2016, 311(4):H927-43.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morciano G, Bonora M, Campo G, et al. Mechanistic role of mPTP in ischemia-reperfusion injury. https://link.springer.com/chapter/10.1007/978-3-319-55330-6_9/fulltext.html. Adv Exp Med Biol. 2017;982(8):169–89. doi: 10.1007/978-3-319-55330-6_9. [Morciano G, Bonora M, Campo G, et al. Mechanistic role of mPTP in ischemia-reperfusion injury[J]. Adv Exp Med Biol, 2017, 982(8):169-89.] [DOI] [PubMed] [Google Scholar]
- 29.Zhang P, Lu Y, Yu D, et al. TRAP1 provides protection against myocardial ischemia-reperfusion injury by ameliorating mitochondrial dysfunction. Cell Physiol Biochem. 2015;36(5):2072–82. doi: 10.1159/000430174. [Zhang P, Lu Y, Yu D, et al. TRAP1 provides protection against myocardial ischemia-reperfusion injury by ameliorating mitochondrial dysfunction[J]. Cell Physiol Biochem, 2015, 36(5):2072-82.] [DOI] [PubMed] [Google Scholar]