

Mouse norovirus and several other members of the Caliciviridae have been shown to have a highly unusual structure with the receptor binding protruding (P) domain only loosely tethered to the main capsid shell. Recent studies demonstrated that bile salts enhance the intrinsic P domain/receptor affinity and is necessary for cell attachment. Presented here are the high-resolution cryo-EM structures of apo MNV, MNV/bile salt, and MNV/bile salt/receptor. Bile salts cause a 90° rotation and collapse of the P domain onto the shell surface that may increase the number of available receptor binding sites. Therefore, bile salts appear to be having several effects on MNV. Bile salts shift the structural equilibrium of the P domain toward a form that binds the receptor and away from one that binds antibody. They may also cause the entire P domain to optimize receptor binding while burying a number of potential epitopes.
KEYWORDS: norovirus, murine, virion structure
ABSTRACT
Caliciviruses are single-stranded RNA viruses with 180 copies of capsid protein comprising the T=3 icosahedral capsids. The main capsid feature is a pronounced protruding (P) domain dimer formed by adjacent subunits on the icosahedral surface while the shell domain forms a tight icosahedral sphere around the genome. While the P domain in the crystal structure of human Norwalk virus (genotype I.1) was tightly associated with the shell surface, the cryo-electron microscopy (cryo-EM) structures of several members of the Caliciviridae family (mouse norovirus [MNV], rabbit hemorrhagic disease virus, and human norovirus genotype II.10) revealed a “floating” P domain that hovers above the shell by nearly 10 to 15 Å in physiological buffers. Since this unusual feature is shared among, and unique to, the Caliciviridae, it suggests an important biological role. Recently, we demonstrated that bile salts enhance cell attachment to the target cell and increase the intrinsic affinity between the P domain and receptor. Presented here are the cryo-EM structures of MNV-1 in the presence of bile salts (∼3 Å) and the receptor CD300lf (∼8 Å). Surprisingly, bile salts cause the rotation and contraction of the P domain onto the shell surface. This both stabilizes the P domain and appears to allow for a higher degree of saturation of receptor onto the virus. Together, these results suggest that, as the virus moves into the gut and the associated high concentrations of bile, the entire capsid face undergoes a conformational change to optimize receptor avidity while the P domain itself undergoes smaller conformational changes to improve receptor affinity.
IMPORTANCE Mouse norovirus and several other members of the Caliciviridae have been shown to have a highly unusual structure with the receptor binding protruding (P) domain only loosely tethered to the main capsid shell. Recent studies demonstrated that bile salts enhance the intrinsic P domain/receptor affinity and is necessary for cell attachment. Presented here are the high-resolution cryo-EM structures of apo MNV, MNV/bile salt, and MNV/bile salt/receptor. Bile salts cause a 90° rotation and collapse of the P domain onto the shell surface that may increase the number of available receptor binding sites. Therefore, bile salts appear to be having several effects on MNV. Bile salts shift the structural equilibrium of the P domain toward a form that binds the receptor and away from one that binds antibody. They may also cause the entire P domain to optimize receptor binding while burying a number of potential epitopes.
INTRODUCTION
Noroviruses are the major cause of epidemic gastroenteritis in humans, causing ∼20 million cases annually, resulting in more than 70,000 hospitalizations and 570 to 800 deaths in the United States alone (for review, see reference 1). While not often a fatal disease in areas with sufficient health care resources, worldwide there are ∼200 million cases per year in children under five, leading to an estimated 50,000 deaths per year, mostly in developing countries. Controlling the spread of norovirus outbreaks is further exacerbated by the fact that as few as ten virions are sufficient to infect an adult (2).
Caliciviruses are T=3 icosahedral particles with 180 copies of the major capsid protein (VP1; ∼58 kDa), that is divided into the N-terminal (N), shell (S), and C-terminal protruding (P) domains (3–6). The S domain forms a shell around the viral RNA genome, while the P domains dimerize to form protrusions on the capsid surface. The P domain is subdivided into P1 and P2 subdomains, with the latter containing the binding sites for cellular receptors (7, 8) and neutralizing antibodies (9–11).
The first high-resolution structure of a calicivirus was that of Norwalk virus (NV) (3). In this structure, the P domain dimers rested firmly on the shell surface similar to the Tombusviridae family. Subsequently, we determined the cryo-electron microscopy (cryo-EM) structure of mouse norovirus (MNV) in physiological, phosphate-buffered saline (PBS) (11, 12). This structure was markedly different than the crystal structure of NV in that the P domains floating more than 10 Å above the shell. At the time, we suggested that the high solute and pH conditions used for crystallization may have caused the contracted state of NV. To confirm that MNV was not an anomaly, the structure of rabbit hemorrhagic disease virus (RHDV) VLP was also determined (13). Similar to MNV-1, the electron density of the RHDV shell is well defined, but the density of the P domains was diffuse, suggesting a high degree of flexibility. The third case of a “floating P domain” was that of human Vietnam026 (GII.10) (14). The cryo-EM reconstruction of the GII.10 norovirus VLP at ∼10-Å resolution showed a number of features very similar to MNV and RHDV. The P domain appeared as a second outer shell, and a half-section through the VLP revealed that the P domain was raised off the S domain by ∼15 Å. These studies also showed that P domain flexibility may play an important role in antibody binding. The crystal structure of the P domain/Fab complex (14) showed that the Fab from a broadly neutralizing antibody (5B18) bound to a buried, conserved region of the protruding domain and yet enzyme-linked immunosorbent assay binding indicated that the 5B18 antibody was able to capture intact VLPs. The base of the P domain could only be exposed to the antibody if there is extreme flexibility in the tether region between the shell and the P domain, suggesting that the extreme flexibility of the P domain of the cryo-EM structures is biologically relevant. This was further substantiated by studies on genotypes GI.1 (15) and GII.4 (16), where various epitopes are only exposed if the P domain is allowed to lift off the shell. Studies on MNV demonstrated that a number of the antibodies raised against MNV recognized the buried shell domain and were nonneutralizing (17). Together, these results suggest that one biological purpose of the flexible P domain may be to “confuse” the immune system by presenting a wide array of epitopes only exposed in the expanded or flexible state. Alternatively, the flexible tether could be sensitive to proteases, such that the shell and P1 domains become exposed in vivo.
There is also growing evidence of marked structural flexibility within the MNV P domain. In the initial crystal structure of the MNV P domain (11), the two loops at the very top of the P2 domain (A′B′ and E′F′) displayed two conformations: a closed structure where the two loops are tightly associated and an open structure where the loops are splayed apart. In the cryo-EM structure of MNV complexed with the Fab fragment from a neutralizing A6.2 (6), it was evident that A6.2 fit better onto the open conformation than the closed, with the hydrophobic heavy chain CDR3 loop extending into the hydrophobic interface between the A′B′ and E′F′ loops.
An even more extreme case of P domain plasticity was observed in studies on a second neutralizing antibody, 2D3. Compared to A6.2, MNV1 had far more difficulty in overcoming neutralization to 2D3 (17, 18). Further, all of the MNV strains tested were neutralized to at least some degree by 2D3, whereas A6.2 was far more selective. While this suggested that the epitope on the P domain region recognized by 2D3 was significantly different and more conserved than A6.2, cryo-EM studies demonstrated that 2D3 and A6.2 contact the E′F′ loop in a very similar way (6, 11, 18). Most surprisingly, neither of the natural escape mutants to 2D3 (D348E and V339I) contact the bound antibody, and none of the escape mutations to A6.2 can escape 2D3 neutralization. To understand how these escape mutations can act in a “allosteric-like” manner, dynamic simulations were performed on the V339I P domain mutant (19). These studies suggested that the P domain itself is in a dynamic structural equilibrium composed of a montage of conformations. The V339I mutation likely blocks antibody binding by shifting this equilibrium toward the closed state.
The cell receptor for MNV has been identified as CD300lf (20). Although cell attachment does not require carbohydrates, it is greatly enhanced by serum factors, including bile salts (21). Conjugated bile salts are amphipathic molecules that form micelles in the small intestine. The micelles combine with lipases to digest lipids and are necessary for fat adsorption. Prior to secretion from the liver, bile acids are conjugated with taurine or glycine to form eight possible conjugated bile acids. In humans, the major bile salts are taurocholic acid, glycocholic acid, glycochenodeoxycholic acid (GCDCA), and taurochenodeoxycholic acid (TCA) and found in approximately equal concentrations. Humans secrete 12 to 18 g of bile acids daily, 95% of which are reabsorbed in the ileum. In the gut, the concentration of bile in the gut has been suggested to be 2.5 to 45 mM (22).
The details of the MNV-receptor interactions have been elucidated with the atomic structure of the P domain/CD300lf complex (21, 23). The CD300lf contact site is near the top of the P2 domain, between the A′B′ and D′E′ loops. The bile salts, GCDCA and lithocholic acid, bind in two deep pockets in the P domain dimer interface, distant from receptor and antibody binding sites (21). Interestingly, the conformation of the A′B′ and E′F′ loops in the receptor complex is nearly identical to that of the closed conformation. The major difference between the apo and the P domain/bile complex is that the C′D′ loop is “turned up” when bile is bound. This movement is necessary to create space for the bile salt to bind. What is particularly interesting is that one of the escape mutants to the neutralizing antibody 2D3, V339I, lies immediately adjacent to the bile salt binding site.
Presented here are the ∼3-Å cryo-EM structures of MNV in the presence or absence of bile salts and receptor. While bile salts enhance the intrinsic affinity between the receptor and P domain (21), these structures demonstrate that they also cause a collapse of the P domain onto the shell surface. This “contracted” conformation of MNV presents a stabilized P domain that allows for a tracing of the entire capsid protein, whereas only the shell domain could be built in the apo form of MNV because of its highly flexible P domain. There are more subtle differences between the apo and bile salt complex structures at the tip of the P domain that is consistent with movement of the C′D′ loop upon bile binding. This movement may represent a bile induced switch from a conformation recognized by antibodies to one favored by receptor. In this way, the flexibility of the antigenic surface of the P domain may “confuse” the immune system by presenting one surface to the immune system in the serum and another when preparing to bind to receptor. Also presented here is the ∼9.5-Å structure of the MNV/GCDCA/CD300lf complex. By comparing this structure to a hypothetical MNV/CD300lf complex in the expanded state, it appears that one biological role for the bile salt induced contraction may be to reorient the P domains so that more receptors can bind simultaneously to the capsid. Together, we propose that MNV may present two different “faces” in vivo. In the absence of serum factors such as bile salts, the C′D′ loop is in the “down” conformation allowing for flexibility in the A′B′/E′F′ loops that can be recognized by antibodies. The “expanded” P domain conformation presents an array of epitopes, either by virtue of the flexibility of the linker or from proteolytic cleavage of the linker, that are buried in the contracted state. When bile salts are present, the conformation of the P domain itself changes to one favored by receptor, not by antibodies, and the entire P domain reorients itself to optimize cell attachment. However, the true biological role of this large movement in the P domain is uncertain and will require further in vitro and in vivo analysis.
RESULTS
Image reconstructions of MNV with or without bile salts.
Several attempts were made to determine the structure of the MNV/CD300lf complex, but the bound receptor was never visible. When it was discovered that bile salts enhance receptor attachment (21), GCDCA was added to MNV alone as a control. It was immediately apparent from even the raw images that the bile salt caused contraction of the viral particle. Therefore, using the same preparation of virus, high-resolution EM data were collected with or without GCDCA (Fig. 1). As observed previously (6, 11), when MNV is in PBS the P domain “floats” by more than 10 Å above the surface of the shell domain (gray arrow). While the resolution of the apo reconstruction was calculated to be 3.1 Å, the electron density of the P domain is largely disordered. In contrast, when GCDCA was added, the P domain collapses down onto the shell surface and becomes well ordered except for the outermost loops at the tip. Previous studies showed that the P domain shows specificity for GCDCA over other bile salts such as TCA (21). Therefore, as a control, the structure of the MNV/TCA complex was also determined. Surprisingly, the virus had the same contracted structure as observed with GCDCA.
FIG 1.

Image reconstructions of apo-, GCDCA-, and TCA-bound forms of MNV. Each panel represents midsections of the image reconstructions and is colored blue to red according to radial distance. As with previous MNV image reconstructions in PBS buffer, the first panel shows that the P domain is fairly disordered and “floats” above the shell. The gray arrow denotes the gap between the shell and the P domains in the apo MNV reconstruction. In contrast, both GCDCA and TCA cause the collapse of the P domain onto the shell surface. In both cases, the quality of the P domain was of sufficient quality to trace the peptide chain.
Model building and refinement.
In the case of apo MNV, the P domain floats above the shell by more than 10 Å and is markedly disordered. As with previous cryo-EM structures (6, 11), while the diffuse P domain electron density can be used to place the known structure of the P domain, it is not of sufficient quality to trace the chain. An atomic model for the apo MNV shell domain was built using the known structure of Norwalk virus (3) as a starting model. When bile salts (GCDCA or TCA) were added to the sample, the P domain collapsed onto the shell. This stabilized the P domain sufficiently so that the full capsid could be built. COOT (24) was used to build the models into the density and the program PHENIX (25) was used for subsequent real space refinement. Real space refinement statistics are summarized in Table 1 .
TABLE 1.
Real space refinement statistics for three different image reconstructionsa
| Parameter | Apo MNV | GCDCA + mNV | TCA + mNV |
|---|---|---|---|
| PDB ID | 6P4L | 6P4J | 6P4K |
| Resolution (Å) | 3.1 | 3.0 | 3.2 |
| No. of atoms | 4,489 | 11,939 | 11,939 |
| Rwork (%) | 36 | 35 | 37 |
| CC masked | 0.83 | 0.84 | 0.84 |
| Ramachandran plot (%) | |||
| Outliers | 0 | 0.6 | 0.3 |
| Allowed | 8.5 | 12.8 | 8.4 |
| Favored | 91.5 | 86.6 | 91.3 |
| Geometry deviation | |||
| Bond (Å) | 0.01 | 0.01 | 0.01 |
| Angle (°) | 0.9 | 1.0 | 0.9 |
| Chirality (°) | 0.05 | 0.07 | 0.1 |
| Planarity (°) | 0.007 | 0.007 | 0.008 |
| Dihedral (°) | 4.0 | 7.0 | 7.1 |
| B values (Å2) | |||
| Minimum | 39 | 31 | 34 |
| Maximum | 124 | 186 | 121 |
| Mean | 63 | 100 | 58 |
Note that the Apo MNV model only represents the shell domain since the P domain was too diffuse to trace.
The electron density and a model of one subunit of the MNV/GCDCA complex are shown in Fig. 2. As shown in the top stereo image, the electron density is well defined throughout much of the atomic structure with the exception of the outermost loops. The conformations of the P domain loops were essentially the same as that observed in the crystal structure of the P domain/GCDCA complex (21), and the density for GCDCA itself was observed as noted in the figure. The quality of the map, refinement statistics, and atomic model of the MNV/TCA complex were essentially identical to the MNV/GCDCA complex. In both GCDCA and TCA there was no evidence of any additional bile salt binding sites in the electron density (e.g., at the P and S domain interface) that could explain why bile salts cause contraction of the capsid.
FIG 2.

Electron density of one of the subunits of the MNV/GCDCA complex and corresponding atomic model. (A) Stereo image of the density of one of the subunits. Note that the quality of the density is better toward the shell. (B) Ribbon diagram of one of the MNV/GCDCA subunits. The model is colored according to B values, blue to red for low to high B values, respectively. Also noted on this figure are the residues that are significantly disordered in the model.
The stereo image at the bottom of Fig. 2 shows the ribbon diagram of the subunit colored according to refined B values. The apparent mobility of the protein increases further from the shell surface. This suggests that, while the P domain position has clearly been stabilized by binding to the shell surface, it is still more mobile than the shell. Within this global flexibility of the P domain there are five loops of the P domain with some disordered residues as noted in the figure.
MNV/TCA structure.
As shown in Fig. 3, density was observed in the bile salt binding pocket (21) in both the TCA and GCDCA complexes. The local resolution in the EM maps was sufficient to observe the general location of the bound bile salts but was insufficient to detail differences in protein-ligand interactions. Notably, the refined B values for the bile salts were the same as for the surrounding protein residues. While it was unexpected to observe TCA binding in these studies, the concentration of TCA used for these studies was higher than those used for the previous binding assays (21). Further, the actual concentration of TCA on the EM grids could be significantly higher than expected if there was any dehydration during grid blotting and manipulation because of its high surface area/volume ratio. Without discernible differences between the TCA and GCDCA structures, the molecular mechanism for virion contraction is unclear. Contraction could be caused by bile salts binding between the P domains as previously described (21) and cause conformational changes that facilitate this collapse. It is also possible that bile salts bind to other locations that might enhance P domain-shell interactions. Finally, it is possible that the bile salts act similar to kosmotropic agents that enhance protein-protein interactions by stabilizing water-water interactions. Essentially, bile salts might be changing the nature of the water solvent rather than binding to specific sites on the protein. Nevertheless, the fact that TCA is able to bind and cause contraction at relatively high concentrations suggests that at the 2.5 to 45 mM concentrations of bile in the gut (22) there may not be significant differences between the effects of the different bile salts.
FIG 3.
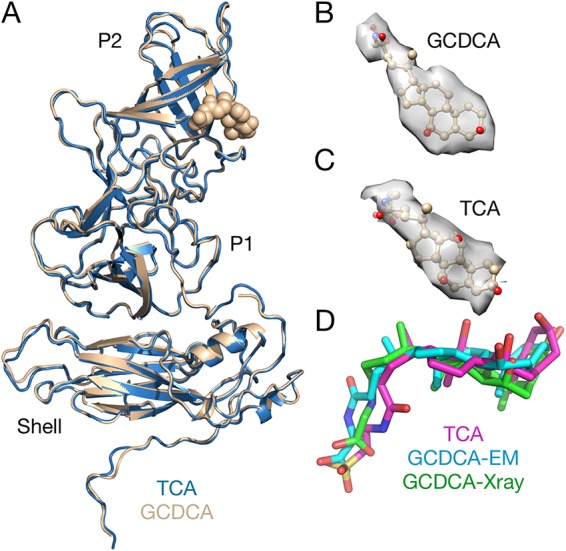
Comparison of the MNV/GCDCA and MNV/TCA complexes. (A) Overlay of MNV/GCDCA (tan) and MNV/TCA (blue) complexes. There are no significant differences between the structures of the two complexes. (B and C) Isolated electron densities of the bound bile salts. (D) Comparison of the bound bile salts as observed in the cryo-EM and crystallographic structures.
Differences between apo- and bile-bound MNV.
A comparison between the apo and MNV/bile salt forms is shown in Fig. 4. Noted on this figure are the locations of the external loops and GCDCA in the complex. When bile salts are added, the entire P domain rotates by nearly 90° and forms extensive contacts with the shell domain. The nearly linear linker region between the shell and P domains in the apo form becomes a highly compact structure in the MNV/bile salt complex. Only the shell domain of the apo form could be traced, and therefore only this domain is included in this particular PDB entry (6P4L). The root mean square difference between the shell domains of the apo- and GCDCA-bound forms is 0.4Å, and therefore there is no discernible change when bile is added to the virus and the P domain associates with the shell.
FIG 4.

Atomic models of the A/B subunit pairs for the apo (A) and GCDCA (B) MNV complexes. In these images, the A and B subunits are colored blue and tan, respectively. (A) Structure of the apo form of MNV. While the resolution of the shell domain was of sufficient quality to build an atomic model, a previously published crystal structure of the P domain (11) was used for modeling. (B) The P domain rotates by nearly 90° and drops down onto the shell in the presence of bile salts. The bound GCDCA is highlighted in yellow and red spheres. (C) Interactions between the P domains in the MNV/GCDCA complex when the capsid collapses in the presence of bile salts. The A, B, and C subunits are shown in blue, green, and red, respectively. Also shown in gray is the electron density of bound GCDCA.
To better assess the new contacts made when the P domains rotate down onto the shell surface, the capsid was analyzed using the programs COCOMAPS (26) and PDBePISA (27). For these calculations, the structure of the “contracted” capsid was used. The shell domain was considered to be residues 1 to 220, and the P domain was considered to be residues 220 to the C terminus. With regard to the P/shell contacts, since each P domain contacts more than a single shell domain, the shell domains from two adjacent, icosahedrally related capsid proteins were considered. However, the majority of P domain contacts are within the same copy of capsid protein. Table 2 summarizes all of these new contacts made when the P domain drops down onto the shell surface. The contact area between the P and shell domains is ∼1,000 Å2, which is comparable to what is typical for antibody-antigen interactions. There are relatively few specific interactions with no salt bridges being formed and only five hydrogen bonds. Approximate two-thirds of the interface is polar, and the remaining surface is nonpolar in character. While Table 2 implies that the contact is dominated by hydrophilic/hydrophobic contact residues, most of these contacts are mediated by the corresponding main-chain atoms. When the P domains rotate and drop onto the shell, the P2 domains of only the A and C domains come into proximity and make ∼260-Å2 contact (Fig. 4C and see arrow 3 in Fig. 5B). The nature of these contacts is similar to the P-S interactions: approximately two-thirds are polar and one-third are nonpolar surfaces. In this case, no salt bridges or hydrogen bonds are formed. Interestingly, PDBePISA (27) does not consider either interface to be significant types of contacts.
TABLE 2.
Summary of new P domain contacts when it collapses onto the shell surfacea
| Parameter | P domain/shell | P domains (A/C) |
|---|---|---|
| Interface surface area (Å2) | 1,047 | 262 |
| Surface (%) | ||
| Polar buried | 66 | 65 |
| Nonpolar buried | 34 | 35 |
| No. of residues | ||
| Interacting residues | 21 (shell), 15 (P domain) | 3 (chain A), 4 (chain C) |
| Hydrophilic-hydrophobic contact residues | 20 | 4 |
| Hydrophilic-hydrophilic contact residues | 4 | 0 |
| Hydrophobic-hydrophobic contact residues | 3 | 2 |
| No. of hydrogen bonds | 5 | 0 |
| No. of salt bridges | 0 | 0 |
The contacts between the A and C subunit P domains are detailed in Fig. 4.
FIG 5.

Comparison of the P domains in the presence and absence of bile salts. For panels A to D, a 2-Å Gaussian filter was applied to the electron density maps so that they could be directly compared. (A and B) Surfaces of the apo and GCDCA forms of MNV, respectively, colored according to radial distances. The mauve arrows point to the locations of the A′B′/E′F′ loops and C′D′ loops, respectively. The brown arrow denotes the A/C subunit contact detailed in the stereo figure (panel E). Note the sharp protrusion in the apo form that is a ridge in the GCDCA-bound form. (C and D) Details of one of the P domain dimers. For panel C, the atomic model is the previously published apo structure (11), with the open and closed A′B′/E′F′ loop structures colored in blue and red, respectively. For panel D, the atomic model of the P domain/GCDCA complex is shown (21). (E) Stereo image of the comparison of the atomic structures of the P domain in the presence or absence of GCDCA and resulting differences in how a neutralizing antibody would bind. The heavy and light chains of bound 3D3 antibody (19) are shown in yellow and green, respectively. The bound GCDCA is represented by cyan and red spheres. The location of the 3D3 escape mutant, V339I, is represented by black spheres. The blue ribbon structure is of the “open” conformation of the apo P domain where that A′B′/E′F′ loops are splayed apart. In contrast, the red ribbon shows the conformation of the GCDCA-bound P domain (21) and how the loops in this conformation would likely prevent binding of the neutralizing antibody.
Bile salt induced conformational changes within the P domain.
In all of the previous image reconstructions of the apo form of MNV, there were knob-like structures at the outermost tip of the P domains (6, 11). However, in the presence of bile salts, this feature appeared to morph into a more ridge-like structure. When the current apo EM map was calculated to 3 Å, the P domain was too diffuse to examine these features. Therefore, to better compare the P domain envelopes, a 2-Å Gaussian filter was applied to the apo and GCDCA maps in Chimera (28, 29), and the results are shown in Fig. 5. Figure 5A shows the surface of the apo form of MNV, and Fig. 5B shows that of the GCDCA/MNV complex. The coloration is according to radius, and therefore the “floating” P domains of the apo form, being at a longer radius, are redder than the P domains of the bile complex that has a “contracted” conformation. Note that the P domains of the two reconstructions are nearly 90° rotated to each other as shown in Fig. 5. The two arrows, mauve and brown, denote the locations of the A′B′/E′F′ and C′D′ loops, respectively, and correspond to the similarly colored arrows at the bottom of the figure. Figure 5E shows these Gaussian filtered maps overlaid onto the apo crystal structure (11) and the MNV/GCDCA complex structure determined here. Without bile bound (Fig. 5C), the C′D′ loop, highlighted in yellow, bends down toward the center of the virus. The A′B′/E′F′ loops point up and perfectly fit into the knob-like feature at the top of the P domain. In this figure, the connector between the S and P domains is denoted by the yellow arrow. When bile binds, the C′D′ loop points upward to the top of the P domain (Fig. 5D). This adds to the density of the knob and forms a ridge-like structure at the top of the P domain.
The most important point of this finding is not that the EM structures recapitulate the crystal structures but rather that the loops at the top of the P domain respond to environmental cues. This is summarized in Fig. 5E. In the absence of bile, the C′D′ loop folds down onto the P domain and likely allows for more flexibility of the A′B′ and E′F′ loops. When bile binds, The A′B′ and E′F′ loops appear to be pushed up into a more vertical conformation that is likely the preferred conformation for receptor recognition. As shown in this figure, the A′B′/E′F′ loops in the “open” conformation, where the loops are splayed apart, open a hydrophobic patch with which the heavy chain CDR3 loop of both antibodies A6.2 (6, 11, 18) and 3D3 (19) interacts. When bile salts bind, the C′D′ loop moves up, pushing the E′F′ and A′B′ loops into the “closed” conformation. There may be additional changes in the D′E′ loop that make it more conducive to receptor binding. It should be noted that in the crystal structure of apo P domain (11) that A′B′/E′F′ loops were observed in both conformations, suggesting intrinsic mobility of these loops, the conformation of which may be governed by ligands such as bile salts.
MNV/GCDCA/CD300lf complex.
To see how the receptor binds to the intact capsid, the structure of the MNV/GCDCA/CD300lf complex was determined. Figure 6A and B show models of MNV/CD300lf determined using the cryo-EM models of the apo and GCDCA forms of MNV, respectively. For these models, the atomic structure of the MNV P domain/CD300lf (21) was superimposed on the apo and GCDCA models. The yellow circles in panels A and B denote the locations of the N termini of the bound CD300lf that would extend down to the membrane in the intact protein. As we previously observed (21), in the apo/CD300lf model the termini are all clustered as trimers and are nearly on top of each other. In contrast, when the contracted GCDCA/MNV model was used, there is a more space between each receptor, except that the CD300lf (mauve colored) molecules bound to the A and C domains are fairly close to each other. Figure 6C shows the actual image reconstruction of the MNV/GCDCA/CD300lf complex with the density for the bound CD300lf molecules highlighted in mauve. As is clear in this figure, the cryo-EM density of the actual complex is nearly identical to that of the model made from the contracted conformation (Fig. 6B). Shown in the stereo figure in panel D is the cryo-EM density of the complex with the model from panel B. The model fits extremely well into the density, with the exception of the CD300lf bound to the A subunit (denoted by the orange oval). This is more than likely due to partial occupancy because of its close contact between the CD300lf molecules bound to the A and C subunits (Fig. 5).
FIG 6.

Structure of MNV complexed with the receptor, CD300lf. In panels A to C, the virus structure is colored gray, and the receptor is mauve. (A) Model showing the expanded form of MNV. The CD300lf was placed onto the surface using the atomic structure of the P domain/CD300lf complex (21). An icosahedral 5-fold axis is represented by an orange pentagon, and the C termini of some of the CD300lf molecules are highlighted with yellow circles. (B) Using the atomic model of the MNV/GCDCA complex determined here, the CD300lf molecules were placed onto the surface of the virion using the atomic structure of the P domain/CD300lf complex. (C) The 9-Å cryo-EM structure of the MNV/GCDCA/CD300lf complex. The density representing the receptor is highlighted in mauve. (D) Model used in panel B overlaid with the cryo-EM density of the MNV/GCDCA/CD300lf shown in panel C. This view is looking down on an icosahedral 3-fold axis that is denoted by the yellow triangle. Highlighted by the yellow ellipse is the density for the CD300lf bound to the A subunit. This density is notably weaker than those binding to the B and C subunits and may be due to partial occupancy. (E) Stereo diagram showing only the ribbon models for an A/B dimer that interacts with an icosahedrally related C/C dimer. The CD300lf bound to the A subunit in panel D is also highlighted with an orange oval in this figure. Note that the CD300lf bound to the A and C subunits are likely too close to be bound simultaneously.
DISCUSSION
The bile acid-induced contraction of the MNV capsid was unexpected, and the cause of this effect is unclear. One of the simplest explanations is that bile salts might be binding to the P domain-shell interface and directly facilitate association. However, the only density consistent with a bile salt was in the pocket identified in the crystal structure of the P domain complex (21). Further, the P and S domains are too tightly associated to allow for ligands to bind between them. It was also possible that the bile salts cause conformational changes that might cause the P domain collapse. The bile salt-associated conformational changes appear to be localized to the P2 domain, without any obvious links to the P1-shell interactions. However, it is interesting that the bile salts bind in close proximity to where the P2 domains of the A and C subunits interact in the contracted form. Yet, this contact area is only a quarter of that between the P and S domains, and it seems unlikely to be the driving force. It is interesting, though, that the A/C interactions break strict T=3 icosahedral symmetry in the capsid. The most likely cause for the bile salt induced contraction of the capsid is indirect, through effects on the solvent surrounding the capsid. Kosmotropic solutes increase hydrophobic effects within a solution by increasing water-water over water-protein interactions. In this case, such agents would increase the energy of interaction between the S and P domains and cause the contraction of the capsid. However, it is not clear whether bile salts are chaotropic or kosmotropic in nature. If this is the case, then it is likely that other compounds in the small intestine might also have similar effects.
From these and previous studies, it is apparent that there are at least two forms of capsid flexibility that are important for the viral life cycle: movement of the entire P domain and rearrangement within the P domain itself.
The observation that the P domain has such marked flexibility in most, if not all, of the Caliciviridae strongly suggested that this feature has some biological function. In the case of MNV, antibodies have been isolated that recognize the buried shell that could only be exposed by either the flexibility of the P domain or proteolytic removal of the P domain itself in vivo. Both are likely possible due to the extreme flexibility of the linker. In the case of human noroviruses, a number of antibodies have been shown to bind to the buried P1 domain (14, 15) and, similarly, this appears to only be possible through the flexibility of the linker region. From the structures presented here, it appears that an additional function for this linker flexibility may be to optimize receptor binding valency on the membrane surface, signaled by metabolites found in the organ where infection occurs, while burying potential epitopes only found in the expanded form.
The second set of conformational changes occur within the P domain itself. When bile salts bind, the movement of the C′D′ loop appears to push the structural equilibrium of the P domain toward the “closed” conformation of the A′B′/E′F′ loops that favors receptor binding over antibody recognition. This bile-induced structural switch may have evolved to “confuse” the immune system by presenting one face to the immune system in the serum and another when preparing to bind to receptor. Interestingly, the V339I escape mutant to the 3D3 is distal to the antibody contact region and lies in the bile salt binding pocket. Computational simulations further demonstrated that this escape mutation also shifts the structural equilibrium toward the “closed” state (19) that would block antibody binding while facilitating receptor binding.
Together, it appears that MNV presents a highly flexible, expanded structure to the immune system. These different modes of capsid flexibility represent various structural equilibria that can be shifted by enthalpic or entropic changes caused by external signals, such as bile salts and calcium binding to the protein or changing the nature of the solvent (for a review on allostery, see reference 30). When entering the gut, various compounds (e.g., bile salts and calcium) may cause the collapse of the P domain and rearrangements within the P domain that not only facilitate cell attachment but also bury some of the antigenic sites exposed in the expanded conformation. Future in vivo and in vitro studies on mutants that separate the expansion/contraction transition from bile salt binding to the P2 domain are necessary to fully understand the biological roles of these modes of MNV flexibility.
MATERIALS AND METHODS
Virus production and purification.
RAW 264.7 (RRID:CVCL_0493; https://scicrunch.org/resolver/RRID:CVCL_0493) and BV-2 (RRID:CVCL_0182; https://scicrunch.org/resolver/RRID:CVCL_0182%20) cells were used interchangeably for virus production, and a plaque-purified MNV-1 clone (GV/MNV1/2002/USA) was used as the MNV strain. The medium used to maintain cell lines in spinner cultures contained HyClone DMEM/Low Glucose (catalog no. SH30002.04) supplemented with 1 g/liter Kolliphor P188, 0.062 g/liter penicillin G, 0.4 g/liter streptomycin sulfate, and 2.2 g/liter NaHCO3, and 10 ml of NEAA and then supplemented with 10% calf serum just before use. NEAA is composed of 0.89 g/liter alanine, 1.5 g/liter asparagine, 1.33 g/liter aspartate, 0.75 g/liter glycine, 1.47 g/liter glutamate, 1.15 g/liter proline, and 1.05 g/liter serine. Cells were started in T flasks and transferred to spinner flasks placed in incubators set at 37°C and 5% CO2. The side arms to the flasks were covered with caps with 0.2-μm filters to allow for gas exchange. When the cells reached a density of ca. 0.5 × 106 to 1.0 × 106 cells/ml, ∼6 liters of the cells were harvested by centrifugation at 4,000 × g for 10 min. The cells were suspended in ∼200 ml in HEPES (AH) medium and placed into a 4-liter flask, and ∼1 × 109 PFU of MNV were added. Per liter, AH medium is composed of HyClone DMEM/Low Glucose, 10 ml of NEAA, 0.062 g of penicillin G, 0.4 g of streptomycin sulfate, 2.2 g of NaHCO3, and 5.9 g of HEPES, all adjusted to a pH of 7.2 and sterilized by filtration. The suspension was slowly shaken for 1 h at room temperature, and ∼800 ml of fresh AH medium was added; the samples were then transferred to a 37° incubator without CO2 and shaken at ∼70 rpm in the dark for 24 to 32 h. At this time, most of the infected cells have lysed.
The infected cell solution was centrifuged for 30 min at 5,000 × g, and the supernatant was collected. Dry NaCl and PEG 8000 were added to the supernatant added to yield 0.3 M and 10% concentrations, respectively. The solution was mixed at 4°C overnight and then centrifuged for 30 min at 10,000 × g, the supernatant was completely removed, and the pellet was resuspended in 50 to 80 ml of PBS. The suspension was allowed to incubate at 4°C for several hours and then centrifuged at 10,000 × g for 30 min to remove debris. Glycerol was to the supernatant added to yield a final concentration of 10% (vol/vol) glycerol as a cryoprotectant; the sample was then divided into 1.5-ml aliquots and stored at –80°C. For purification, 50 to 100 ml of this material was thawed and then centrifuged for 30 min at 10,000 × g to remove residual debris. Each supernatant was placed in the Beckman 50.2 Ti centrifuge tubes with 1 ml of a 30% sucrose cushion placed at the bottom of each tube, followed by centrifugation at 45,000 rpm for 2 h. After centrifugation, the pellets were resuspended in less than 3 ml of PBS and allowed to incubate for several hours at 4°C. The material was then centrifuged for 10 min at maximum speed and at 4°C. The supernatant was then layered onto one to two Beckman SW41 tubes containing 7.5 to 45% linear sucrose gradients made with PBS. The samples were centrifuged for 1.5 to 2.0 h at 35,000 rpm at 4°C. MNV formed a band approximately two-thirds of the way down the tube and was removed by puncturing the side of the tube with a syringe. The virus bands were pooled, diluted in PBS, and pelleted as described above. The pellets were resuspended in a minimal volume of PBS, and residual debris was removed by centrifugation.
Cryo-electron microscopy.
Purified MNV with or without additives was vitrified as previously described (31) on carbon holey film (R2 × 1 Quantifoil; Micro Tools GmbH, Jena, Germany) grids. Briefly, purified concentrated (∼1 mg/ml) suspensions of virions were applied to the holey films, blotted with filter paper, and plunged into liquid ethane. An EM-GP2 (Leica) automated plunger was used for vitrification. Frozen grids were stored under liquid nitrogen until used for microscopy.
Four different samples of MNV were analyzed: Apo-MNV, MNV/GCDCA, MNV/TCA, and MNV/GCDCA/CD300lf. Bile salts were added to MNV at room temperature due to the solubility of the bile salts and in >20-fold excess to the virus to saturate all potential binding sites. The grids were imaged in a Titan Krios (Thermo Fisher) microscope at the Stanford-SLAC Cryo-EM Facilities, as part of SLAC Regional Cryo-EM Consortium. The microscope was equipped with field emission gun (FEG) operated at 300 keV with a GIF (Gatan, Inc.) post-column energy filter. A slit width of 20 eV was used for data collection, and the image pixel size was 1.06 Å on the specimen scale. MNV/GCDCA/CD300lf grids were imaged on a JEOL 2200 FS microscope with an FEG and an omega energy filter. A DE-20 camera (Direct Electron, San Diego, CA) was used in integrating mode with a rate of 25 frames/s. The cryo-transfer holder used was a Fischione Instruments 2550 model. The microscope was operated at 200 keV, the filter slit was set to 20 eV, and the pixel size was 1.5 Å. The total dose was ∼38 electrons/Å2. Frame alignment in image stacks was done using the Direct Electron script DE_process_frames-2.8.1.py.
For data collected at the SLAC EM Facility, the overall dose was ∼56 electrons/Å2. Individual frames in each image stack were aligned using the MotionCorII script (32). Virion images were boxed from the images using template-based particle picker from cryoSPARC 2.4.6 platform (33).
The data collection statistics are summarized in Table 3.
TABLE 3.
Cryo-EM data collection statistics
| Parameter | MNV apo | MNV + CDCA | MNV + tCA | MNV + gCDCA + CD300lf |
|---|---|---|---|---|
| Instrument used | Krios Titan | Krios Titan | Krios Titan | JEM2200FS |
| No. of images | 4,077 | 4,474 | 6,332 | 70 |
| No. of particles picked | 31,292 | 40,320 | 57,201 | 839 |
| No. of particles used | 30,719 | 28,463 | 55,511 | 839 |
| Resolution (Å) | 3.1 | 3.1 | 3.2 | 9.5 |
| Software | Cryosparc | Cryosparc | Cryosparc | EMAN2 |
| EMD ID | 20252 | 20250 | 20251 | 20253 |
Image processing.
For the apo MNV sample, 4,077 micrographs were recorded, 31,292 particles were picked using the CryosparcII (33) template picker, and 30,719 particles were selected from those micrographs after using two-dimensional (2D) classification to select good classes. Subsequent image processing was done using EMAN2 (34) and Cryosparc (33).
For the MNV/GCDCA complex, 4,474 image stacks were collected. CryoSPARC 2.4.6 was used for all subsequent image processing, including contrast transfer function determination and correction (ctfind4 wrapper in cryoSPARCII), 2D classification, and 3D refinement. For this MNV/GCDCA complex, 40,320 individual images were picked from the micrographs after manual curation. After 2D classification, 28,463 particles were selected from good 2D classes and used for final 3D refinement. The effective resolution of the MNV/GCDCA complex was 3.1 Å according to the 0.143 gold standard Fourier shell correlation function (FSC) criterion (35). For the MNV/TCA complex, 6,332 image stacks were acquired, from which 57,201 particle images were selected with the cryoSPARC 2.5 platform template-based autopicker. Image processing using 55,511 particles selected from good 2D classes resulted in 3.2 Å according to the 0.143 gold standard FSC criterion.
For the MNV/GCDCA/CD300lf complex, 70 image stacks were acquired from on a DE-20 camera, and 839 particle images were picked from those using EMAN2 boxer (34). Using EMAN2 for processing, the resolution was ∼9.5 Å according to gold standard Fourier shell correlation function.
ACKNOWLEDGMENTS
This study was supported by an National Institutes of Health (NIH) grant to T.J.S. (1R01-AI141465). We acknowledge the support of the Sealy Center for Structural Biology at the University of Texas Medical Branch (UTMB). Data were collected at an NIH-supported facility operated by Wah Chiu (1U24 GM116787-01) with support from Megan Louise Mayer. We also acknowledge the Texas Advanced Computing Center at The University of Texas at Austin for providing high-performance computing resources (LONESTAR5 [http://www.tacc.utexas.edu]) that have contributed to the research results reported here. We also thank Lisa Pipper at the UTMB for all of her efforts in helping to acquire resources essential to this work.
H.W.V. is a founder of Casma Therapeutics and PierianDx; neither company funded the work here. H.W.V. is an employee and owns equity in Vir Biotechnlogy. The work here was performed at Washington University School of Medicine and was not funded by Vir.
REFERENCES
- 1.Hutson AM, Atmar RL, Estes MK. 2004. Norovirus disease: changing epidemiology and host susceptibility factors. Trends Microbiol 12:279–287. doi: 10.1016/j.tim.2004.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moe CL, Sobsey MD, Stewart PW, Crawford-Brown D. 1999. Estimating the risk of human calicivirus infection from drinking water, abstr. International Workshop on Human Caliciviruses, Atlanta, GA. [Google Scholar]
- 3.Prasad BVV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK. 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286:287–290. doi: 10.1126/science.286.5438.287. [DOI] [PubMed] [Google Scholar]
- 4.Prasad BV, Hardy ME, Jiang X, Estes MK. 1996. Structure of Norwalk virus. Arch Virol Suppl 12:237–242. [DOI] [PubMed] [Google Scholar]
- 5.Prasad BV, Matson DO, Smith AW. 1994. Three-dimensional structure of calicivirus. J Mol Biol 240:256–264. doi: 10.1006/jmbi.1994.1439. [DOI] [PubMed] [Google Scholar]
- 6.Katpally U, Wobus CE, Dryden K, Virgin HWI, Smith TJ. 2008. Structure of antibody-neutralized murine norovirus and unexpected differences from viruslike particles. J Virol 82:2079–2088. doi: 10.1128/JVI.02200-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi J-M, Hutson AM, Estes MK, Prasad B. 2008. Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc Natl Acad Sci 105:9175–9180. doi: 10.1073/pnas.0803275105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan M, Hegde RS, Jiang X. 2004. The P domain of norovirus capsid protein forms dimer and binds to histo-blood group antigen receptors. J Virol 78:6233–6242. doi: 10.1128/JVI.78.12.6233-6242.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donaldson EF, Lindesmith LC, Lobue AD, Baric RS. 2010. Viral shape-shifting: norovirus evasion of the human immune system. Nat Rev Microbiol 8:231–241. doi: 10.1038/nrmicro2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nilsson M, Hedlund KO, Thorhagen M, Larson G, Johansen K, Ekspong A, Svensson L. 2003. Evolution of human calicivirus RNA in vivo: accumulation of mutations in the protruding P2 domain of the capsid leads to structural changes and possibly a new phenotype. J Virol 77:13117–13124. doi: 10.1128/jvi.77.24.13117-13124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taube S, Rubin JR, Katpally U, Smith TJ, Kendall A, Stuckey JA, Wobus CE. 2010. High resolution X-ray structure and functional analysis of murine norovirus (MNV)-1 capsid protein protruding domain. J Virol 84:5695–5705. doi: 10.1128/JVI.00316-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katpally U, Fu T, Freed DC, Casimiro DR, Smith TJ. 2009. Antibodies to the buried N terminus of rhinovirus VP4 exhibit cross-serotypic neutralization. J Virol 83:7040–7048. doi: 10.1128/JVI.00557-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katpally U, Voss NR, Cavazza T, Taube S, Rubin JR, Young VL, Stuckey J, Ward VK, Virgin HW, Wobus CE, Smith TJ. 2010. High-resolution cryo-electron microscopy structures of murine norovirus 1 and rabbit hemorrhagic disease virus reveal marked flexibility in the receptor binding domains. J Virol 84:5836–5841. doi: 10.1128/JVI.00314-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansman GS, Taylor DW, McLellan JS, Smith TJ, Georgiev I, Tame JRH, Park S-Y, Yamazaki M, Gondaira F, Miki M, Katayama K, Murata K, Kwong PD. 2012. Structural basis for broad detection of genogroup II noroviruses by a monoclonal antibody that binds to a site occluded in the viral particle. J Virol 86:3635–3646. doi: 10.1128/JVI.06868-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruoff K, Kilic T, Devant J, Koromyslova A, Ringel A, Hempelmann A, Geiss C, Graf J, Haas M, Roggenbach I, Hansman G. 2019. Structural basis of nanobodies targeting the prototype norovirus. J Virol 93:e02005-18. doi: 10.1128/JVI.02005-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindesmith LC, Mallory ML, Debbink K, Donaldson EF, Brewer-Jensen PD, Swann EW, Sheahan TP, Graham RL, Beltramello M, Corti D, Lanzavecchia A, Baric RS. 2018. Conformational occlusion of blockade antibody epitopes, a novel mechanism of GII.4 human norovirus immune evasion. mSphere 3:e00518-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolawole AO, Xia C, Li M, Gamez M, Yu C, Rippinger CM, Yucha RE, Smith TJ, Wobus CE. 2014. Newly isolated mAbs broaden the neutralizing epitope in murine norovirus. J Gen Virol 95:1958–1968. doi: 10.1099/vir.0.066753-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolawole AO, Li M, Xia C, Fischer AE, Giacobbi NS, Rippinger CM, Proescher JB, Wu SK, Bessling SL, Gamez M, Yu C, Zhang R, Mehoke TS, Pipas JM, Wolfe JT, Lin JS, Feldman AB, Smith TJ, Wobus CE. 2014. Flexibility in surface-exposed loops in a virus capsid mediates escape from antibody neutralization. J Virol 88:4543–4557. doi: 10.1128/JVI.03685-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolawole AO, Smith HQ, Svoboda SA, Lewis MS, Sherman MB, Lynch GC, Pettitt BM, Smith TJ, Wobus CE. 2017. Norovirus escape from broadly neutralizing antibodies is limited to allosteric-like mechanisms. mSphere 2:e00334-17. doi: 10.1128/mSphere.00334-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Orchard RC, Wilen CB, Doench JG, Baldridge MT, McCune BT, Lee YC, Lee S, Pruett-Miller SM, Nelson CA, Fremont DH, Virgin HW. 2016. Discovery of a proteinaceous cellular receptor for a norovirus. Science 353:933–936. doi: 10.1126/science.aaf1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nelson CA, Wilen CB, Dai Y-N, Orchard RC, Kim AS, Stegeman RA, Hsieh LL, Smith TJ, Virgin HW, Fremont DH. 2018. Structural basis for murine norovirus engagement of bile acids and the CD300lf receptor. Proc Natl Acad Sci U S A 115:E9201. doi: 10.1073/pnas.1805797115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sjövall J. 1959. On the concentration of bile acids in the human intestine during absorption: bile acids and steroids 74. Acta Physiol Scand 46:339–345. doi: 10.1111/j.1748-1716.1959.tb01763.x. [DOI] [PubMed] [Google Scholar]
- 23.Kilic T, Koromyslova A, Malak V, Hansman GS. 2018. Atomic structure of the murine norovirus protruding domain and soluble CD300lf receptor complex. J Virol 92:e00413-18. doi: 10.1128/JVI.00413-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 25.Afonine PV, Grosse-Kunstleve RW, Adams PD. 2005. The Phenix refinement framework. CCP4 Newsl 42:contribution 8. [Google Scholar]
- 26.Vangone A, Spinelli R, Scarano V, Cavallo L, Oliva R. 2011. COCOMAPS: a web application to analyze and visualize contacts at the interface of biomolecular complexes. Bioinformatics 27:2915–2916. doi: 10.1093/bioinformatics/btr484. [DOI] [PubMed] [Google Scholar]
- 27.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 28.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera: a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 29.Goddard TD, Huang CC, Ferrin TE. 2007. Visualizing density maps with UCSF Chimera. J Struct Biol 157:281–287. doi: 10.1016/j.jsb.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 30.Motlagh HN, Wrabl JO, Li J, Hilser VJ. 2014. The ensemble nature of allostery. Nature 508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherman MB, Guenther RH, Tama F, Sit TL, Brooks CL, Mikhailov AM, Orlova EV, Baker TS, Lommel SA. 2006. Removal of divalent cations induces structural transitions in red clover necrotic mosaic virus revealing a potential mechanism for RNA release. J Virol 80:10395–10406. doi: 10.1128/JVI.01137-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, Agard DA. 2017. MotionCorII: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14:331. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Punjani A, Rubinstein J-O, Fleet DJ, Brubaker M-O. 2017. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 34.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. 2007. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 35.Scheres SH, Chen S. 2012. Prevention of overfitting in cryo-EM structure determination. Nat Methods 9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]