

Humanized mice constitute a useful model for studying the HTLV-1-associated polyclonal proliferation of CD4+ T cells and viral integration sites in the human genome. The rapid death of infected animals, however, appears to preclude the clonal selection typically observed in human ATLL, which normally develops in 2 to 5% of individuals infected with HTLV-1. Nevertheless, the expansion of multiple clones of low abundance in these humanized mice mirrors the early phase of HTLV-1 infection in humans, providing a useful model to investigate approaches to inhibit virus-induced CD4+ T cell proliferation.
KEYWORDS: CD4+ CD25+, HTLV-1, humanized mouse models, orf-I, viral replication
ABSTRACT
Human T cell leukemia virus type 1 (HTLV-1) is the ethological agent of adult T cell leukemia/lymphoma (ATLL) and a number of lymphocyte-mediated inflammatory conditions, including HTLV-1-associated myelopathy/tropical spastic paraparesis. HTLV-1 orf-I encodes two proteins, p8 and p12, whose functions in humans are to counteract innate and adaptive responses and to support viral transmission. However, the in vivo requirements for orf-I expression vary in different animal models. In macaques, the ablation of orf-I expression by mutation of its ATG initiation codon abolishes the infectivity of the molecular clone HTLV-1p12KO. In rabbits, HTLV-1p12KO is infective and persists efficiently. We used humanized mouse models to assess the infectivity of both wild-type HTLV-1 (HTLV-1WT) and HTLV-1p12KO. We found that NOD/SCID/γC−/− c-kit+ mice engrafted with human tissues 1 day after birth (designated NSG-1d mice) were highly susceptible to infection by HTLV-1WT, with a syndrome characterized by the rapid polyclonal proliferation and infiltration of CD4+ CD25+ T cells into vital organs, weight loss, and death. HTLV-1 clonality studies revealed the presence of multiple clones of low abundance, confirming the polyclonal expansion of HTLV-1-infected cells in vivo. HTLV-1p12KO infection in a bone marrow-liver-thymus (BLT) mouse model prone to graft-versus-host disease occurred only following reversion of the orf-I initiation codon mutation within weeks after exposure and was associated with high levels of HTLV-1 DNA in blood and the expansion of CD4+ CD25+ T cells. Thus, the incomplete reconstitution of the human immune system in BLT mice may provide a window of opportunity for HTLV-1 replication and the selection of viral variants with greater fitness.
IMPORTANCE Humanized mice constitute a useful model for studying the HTLV-1-associated polyclonal proliferation of CD4+ T cells and viral integration sites in the human genome. The rapid death of infected animals, however, appears to preclude the clonal selection typically observed in human ATLL, which normally develops in 2 to 5% of individuals infected with HTLV-1. Nevertheless, the expansion of multiple clones of low abundance in these humanized mice mirrors the early phase of HTLV-1 infection in humans, providing a useful model to investigate approaches to inhibit virus-induced CD4+ T cell proliferation.
INTRODUCTION
Human T cell leukemia virus type 1 (HTLV-1) is a retrovirus associated with adult T cell leukemia/lymphoma (ATLL) (1–5) and HTLV-1-associated myelopathy (HAM)/tropical spastic paraparesis (TSP) (6, 7). While the majority of individuals infected with HTLV-1 remain asymptomatic, approximately 2 to 5% develop HAM/TSP or ATLL (8). Further, HTLV-1 is associated with a number of other inflammatory conditions, including uveitis (9), Sjögren's syndrome (10, 11), broncoalveolitis and arthritis (12), and polymyositis (13). In those infected with HTLV-1, viral DNA can be found in CD4+ and CD8+ effector and memory T cells, regulatory CD4+ CD25+ T cells, and monocytes. The virus infects both monocytes and dendritic cells in vitro (14–17). A large viral DNA burden in peripheral blood mononuclear cells (PBMCs) is the only known predictive factor for HAM/TSP (18–20) or ATLL (21) development in infected individuals, but viral burden alone is not sufficient to differentiate symptomatic patients from healthy carriers, suggesting the importance of the host immune response and other factors (21–23).
The 9-kb genome of HTLV-1 is a positive, single-strand RNA genome that contains the structural and enzymatic genes gag, pro, pol, and env and encodes regulatory proteins from four partially overlapping open reading frames (ORFs). Regulatory proteins p8 and p12 (orf-I), p13 and p30 (orf-II), Rex (orf-III), Tax (orf-IV), and HBZ, encoded from the antisense viral genome, affect the extent of viral replication or transmission (24–33).
The HTLV-1 p8 and p12 proteins from orf-I, encoded by a singly spliced mRNA, affect STAT-5 activation and T cell receptor (TCR) signaling, protect infected cells from recognition by cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, increase viral transmission, and are required for the persistent infection of primary monocytes in vitro and of macaques in vivo (30, 34–43). HTLV-1 infectivity and persistence in rabbits, in contrast, do not require orf-I expression (28, 44).
More recently, the development of humanized mouse models in which the human immune system is partially reconstituted by engrafting CD34+ stem cells into immunodeficient mice has allowed for the study of several human-specific pathogens. Ultimately, differences between the available mouse strains and engraftment methods determine the optimal mouse model (45) for a given pathogen, as has been demonstrated in a variety of studies. Tezuka and colleagues developed IBMI-huNOG mice (46) by injecting human cord blood CD133+ cells into the bone marrow of 7-week-old NOD/Shi-scid/IL-2RγC−/− null mice and allowing the immune system to reconstitute for 6 to 7 months before infection of the animals with irradiated HTLV-1-infected MT-2 cells. Using this model, the authors were able to measure HTLV-1-specific adaptive responses in the form of Tax-specific CTLs and IgG to HTLV-1 antigens (46). In other studies, HTLV-1 infection of Rag1−/− γC−/− mice and bone marrow-liver-thymus (BLT) mice demonstrated persistent infection, Tax expression, and infiltration in the immune cells of the brain and spinal cord (47). Furthermore, a humanized NSG mouse model incapable of mounting an adaptive immune response demonstrated HTLV-1-associated T cell tropism and lymphoproliferation (48).
Noting the strides made by our peers, we used two humanized mouse models, engrafted with human tissue at differing times, to test the infectivity and pathogenicity of viruses differentiated by a single nucleotide affecting the orf-I expression of p8 and p12. We found that the single nucleotide mutation in HTLV-1p12KO reverted to wild-type (WT) HTLV-1 (HTLV-1WT) within 4 weeks, suggesting that orf-I expression is essential for infection in the BLT model. These data are consistent with our prior observations that orf-I expression is essential for primate infection, although the expression of this gene is not required in rabbit infection (39, 42). However, both control and infected mice in this model developed graft-versus-host disease (GvHD), which rendered the detection of leukemia/lymphoma impossible during their shortened lifespans. In the NSG-1d model, HTLV-1 infection caused rapid polyclonal proliferation of CD4+ CD25+ T cells that, by infiltrating vital organs, caused weight loss and death. The rapid onset of death, probably related to the incomplete reconstitution of a normal immune system, is a major limitation of this model, since it decreases the chances that clonal selection may have adequate time to progress as it does in humans before culminating in ATLL. However, the susceptibility of NSG-1d mice to HTLV-1 infection still provides an opportunity to identify gene families frequently targeted by HTLV-1 integration in human cells and the genetic determinants that contribute to viral persistence.
RESULTS
Epstein-Barr virus-free human primary CD4+ cells infected with HTLV-1WT and HTLV-1p12KO.
In designing this study, we sought to establish a small-animal model to investigate host determinants of virus persistence while counteracting the cost and availability constraints associated with nonhuman primates. Existing rabbit models proved inadequate for this purpose, since viral persistence in these animals is unaffected by HTLV-1 orf-I deletion (42). orf-I is crucially important in macaques (39, 42), however, and we thus turned to humanized mouse models as a more faithful re-creation of the human immune system. We studied the infectivity and pathogenicity of pAB-D26 (HTLV-1WT) and the HTLV-1 orf-I-knockout virus pAB-p12KO (HTLV-1p12KO), which carries the orf-I mutation of ATG to GTG (42) (Fig. 1A). Inoculation of NSG mice reconstituted with human PBMCs was performed with irradiated 729.6 B cells infected or not infected with HTLV-1WT, and we observed that inoculation of the uninfected, irradiated 729.6 B cells, which naturally contain Epstein-Barr virus (EBV), resulted in detectable EBV (but not HTLV-1) sequences in mouse blood (Fig. 1B, lanes 1 and 9). Blood from mice exposed to the 729.6 cell line infected with both HTLV-1WT and EBV was positive for both viruses (Fig. 1B, lanes 2 and 10). To eliminate EBV as a confounding factor (39, 42), we sorted primary CD4+ cells and cocultured them with irradiated HTLV-1WT-infected 729.6 cells. The primary CD4+ cells became infected with HTLV-1 but not EBV (Fig. 1B, lanes 4 and 12), providing an EBV-free inoculum. For subsequent experiments, we generated CD4+ cells infected with HTLV-1WT that were confirmed to be 95.4% CD4+ cells (Fig. 1C, top row), containing 1.29 copies of viral DNA per cell and producing 90.1 ng/ml of p19 Gag. We also generated primary CD4+ cells infected with HTLV-1p12KO that were confirmed to be 92.5% CD4+ (Fig. 1C, bottom row), containing 1.21 copies of viral DNA per cell and producing 69.5 ng/ml of p19 Gag.
FIG 1.

Generation of primary cell lines infected with HTLV-1WT and HTLV-1p12KO clones. (A) Schematic representation of HTLV clones used to infect primary CD4+ cells. While the nucleotide change from A to G knocks out expression of orf-I, it maintains the amino acid sequence for orf-II. LTR, long terminal repeat. (B) PCR assays for the detection of viral HTLV-1 gag, β-actin, and EBV genes in primary CD4+ cells, PBMCs from humanized mice, and HTLV-1-producing B cell lines. Genomic DNA was isolated from the PBMCs of humanized mice cocultured with irradiated uninfected 729.6 cells (lanes 1, 5, and 9), PBMCs from BLT mice cocultured with irradiated and HTLV-1WT-infected 729.6 cells (lanes 2, 6, and 10), a 729.6 cell line producing HTLV-1WT (lanes 3, 7, and 11), and primary HTLV-1WT-infected CD4+ cells 2 months following coculture with irradiated and HTLV-1WT-infected 729.6 cells (lanes 4, 8, and 12), as described in Materials and Methods. Arrows indicate the DNA fragment corresponding to the correct PCR product. (C) Flow cytometry to define the purity of CD4+ cells infected with HTLV-1WT or HTLV-1p12KO (orf-I knockout). FSC, forward scatter; SSC, side scatter.
HTLV-1p12KO reverts to HTLV-1WT in BLT mice.
Eight NSG mice were transplanted with fetal bone marrow, liver, and thymus tissue (BLT mice), as described previously (49–51). The levels of human cell engraftment were similar for all animals, with more than 30% of peripheral cells being positive for human CD45 and more than 60% of CD4+ CD45+ T cells in all mice being human (Table 1). We intraperitoneally (i.p.) inoculated 2 × 106 lethally irradiated CD4+ HTLV-1 cells producing either HTLV-1WT (mice MS12, MS13, and MS14) or HTLV-1p12KO (mice MS6, MS7, and MS8). Animals MS11 and MS15 were inoculated with saline as controls (Fig. 2A). At weeks 4, 8, and 16, HTLV-1 DNA was detected by quantitative PCR in the blood of nearly all mice, including those exposed to HTLV-1p12KO (Fig. 2B). Three of the six mice infected with HTLV-1 unexpectedly succumbed to GvHD, at weeks 5, 8, and 12. The remaining mice were sacrificed at week 16. By week 8, mice MS6 and MS7, which had been exposed to HTLV-1p12KO, had more viral DNA copies in blood than did mice MS12, MS13, and MS14, which had been exposed to HTLV-1WT (Fig. 2B and Table 2). The infection of BLT mice with HTLV-1p12KO was surprising, since prior work demonstrated that HTLV-1p12KO lacked infectivity in macaques (39). Because BLT mice do not fully reconstitute the human immune system, we concluded that there might have been a window of opportunity for the virus to replicate, thus allowing for virus variant selection.
TABLE 1.
Reconstitution of human cells and time of euthanasia of BLT mice
| Mouse | Infection | Proportion (%) |
Time of death/euthanasia (wk) | |||
|---|---|---|---|---|---|---|
| CD45+ | CD3+ CD45+ | CD3+ CD4+ CD45+ | CD3+ CD8+ CD45+ | |||
| MS6 | p12KO | 44 | 81.30 | 60.4 | 36.2 | 12 (postinfection) |
| MS7 | p12KO | 38.70 | 86.50 | 64.7 | 30.5 | 16 (euthanasia) |
| MS8 | p12KO | 39.10 | 85.60 | 61.4 | 34.25 | 5 (postinfection) |
| MS11 | Control | 31.30 | 90.50 | 69.3 | 26.77 | 16 (euthanasia) |
| MS12 | WT | 40.50 | 72.00 | 71.6 | 24.12 | 16 (euthanasia) |
| MS13 | WT | 38.80 | 89.60 | 65.6 | 31.66 | 8 (postinfection) |
| MS14 | WT | 37 | 95.10 | 64.4 | 32.75 | 16 (euthanasia) |
| MS15 | Control | 32 | 87.50 | 74.1 | 20.7 | 16 (euthanasia) |
FIG 2.

Infectivity of HTLV-1WT and HTLV-1p12KO clones in BLT mice. (A) Study design. At 8 weeks postreconstitution, three BLT mice were inoculated with CD4+ T cells producing HTLV-1WT (D26) and three were inoculated with CD4+ T cells producing HTLV-1p12KO. Two mice were used as noninfected controls. Blood was collected every 4 weeks postinoculation for genomic DNA isolation and monitoring of CD4 and CD8 phenotypes. Sp, spleen; BM, bone marrow; LN, lymph node. (B) HTLV-1 DNA copies measured in PBMC genomic DNA by Digital Droplet PCR assay at weeks 4, 8, and 16 postinfection.
TABLE 2.
Proviral load in the PBMCs of BLT micea
| Time | Control |
D26 |
p12KO |
|||||
|---|---|---|---|---|---|---|---|---|
| MS11 | MS15 | MS12 | MS13 | MS14 | MS6 | MS7 | MS8 | |
| Week 4 | 0 | 0 | 0 | 0.54 | 1.45 | 1.37 | 1.66 | 1.11 |
| Week 8 | 0 | 0 | 16.02 | 3.54 | 1.65 | 47.06 | 60.35 | Death at wk 5 |
| Week 16 | 0 | 0 | 91 | Death at wk 8 | 8.23 | Death at wk 12 | 87 | |
All values are given as percentages.
We investigated the possibility of the point mutation in HTLV-1p12KO reverting to HTLV-1WT in vivo by sequencing viral DNA obtained from PCR amplification of genomic DNA from mouse PBMCs. As expected, the amplified viral fragment from the DNA of HTLV-1WT-infected mice MS12 and MS14 carried the ATG initiation codon in orf-I at weeks 16 and 18. For the animals infected with HTLV-1p12KO, we were able to document the presence of both HTLV-1WT and HTLV-1p12KO orf-I sequences at week 4 and complete selection of the HTLV-1WT in mouse MS7 by week 16 (Table 3). Thus, we found that BLT mice could be infected with HTLV-1WT, and a robust infection with HTLV-1p12KO was also observed following its reversion and the selection of HTLV-1WT, further supporting the importance of orf-I expression in viral infectivity and persistence in the host.
TABLE 3.
orf-I DNA sequences in blood of HTLV-1-infected BLT mice
| Mouse | Wk sampleda | DNA sequenceb | Sequenced material | Proviral load (%) |
|---|---|---|---|---|
| HTLV-1p12KO group | ||||
| MS6 | 4 | (G/A)TG | Nested PCR | 1.37 |
| MS6 | 8 | (G/A)TG | Nested PCR | 47.06 |
| MS7 | 4 | (G/A)TG | Nested PCR | 1.66 |
| MS7 | 8 | ATG | 3/3 clones | 60.35 |
| MS7 | 16 | ATG | Nested PCR | 87 |
| MS8 | 4 | GTG | Nested PCR | 1.11 |
| HTLV-1WT group | ||||
| MS12 | 8 | ATG | 3/3 clones | 16.02 |
| MS12 | 16 | ATG | 3/3 clones | 91 |
| MS14 | 8 | ATG | 3/3 clones | 1.65 |
| MS14 | 16 | ATG | 3/3 clones | 8.23 |
| Parental HTLV-1p12KO-producing CD4+ cell line | NA | GTG | Nested PCR | 120 |
Mice MS7, MS12, and MS14 were sacrificed at week 16 after virus exposure. Mice MS6 and MS8 died prematurely, at week 12 and week 5, respectively, after virus exposure. NA, not applicable.
The HTLV-1p12KO molecular clone was constructed by mutating the ATG codon in orf-I to GTG. For mouse MS6, a mixture of GTG and ATG sequences, (G/A)TG, was detected at week 4 and week 8 after virus exposure. For mouse MS7, GTG and ATG sequences were detected at week 4 after virus exposure. By week 8, only the WT ATG sequence was recovered.
Proliferation of CD4+ T cells in BLT mice correlates with viral burden.
To assess whether viral infection affected the proliferation of the engrafted human cells in the BLT mice, we characterized human CD4+ CD25+ and CD8+ CD25+ T cells, classic CD14+ monocytes, and nonclassic CD16+ monocytes by flow cytometry (Fig. 3A). By week 8, we observed increases in the percentages of blood CD4+ CD45RO+ T cells (up to 70%) and CD4+ CD25+ T cells (up to 60%) but not CD3+ CD7+ cells in infected animals, with increases being especially pronounced in those with high virus levels (Fig. 3B to D and Tables 2 and 3). Indeed, the increased percentage of activated CD4+ CD25+ T cells was directly correlated with the viral DNA copies in the same compartment at 8 weeks postinfection (Fig. 3E). In contrast, no consistent changes in the levels of blood CD8+ cells expressing CD25, CD45RO, or CD7 markers were observed (Fig. 3F to H). However, an increase in CD25+ cells was observed in the HTLV-1p12KO group (Fig. 3F). Active viral replication is evidenced here by the much greater viral DNA load and point mutation reversion (Table 3). Similarly, the levels of CD14+ CD16− and CD14− CD16+ monocytes did not differ in infected animals versus controls (Fig. 3I to K).
FIG 3.

Blood cell profiles in HTLV-1-infected BLT mice. (A) Gating strategy for the identification of human cells in whole blood for a representative BLT mouse (MS12). FSC, forward scatter; SSC, side scatter. (B to D) Percentages of human CD3+ CD4+ CD25+ cells (B), CD3+ CD4+ CD45RO+ cells (C), and CD3+ CD4+ CD7+ cells (D) in blood collected from all groups at weeks 4 and 8 postinfection. A t test was used for statistical analysis. (E) Correlation of the frequency of CD3+ CD4+ CD25+ cells with viral burden (HTLV-1 DNA copies) at week 8 postinfection. The Spearman correlation was used for statistical analysis. (F to H) Percentages of human CD3+ CD8+ CD25+ cells (F), CD3+ CD8+ CD45RO+ cells (G), and CD3+ CD7+ CD8+ cells (H) in blood collected from all groups at weeks 4 and 8 postinfection. A t test was used for statistical analysis. (I and J) Percentages of human classic CD14+ CD16− monocytes (I) and nonclassic CD14− CD16+ monocytes (J) in blood collected from all groups at weeks 4 and 8 postinfection. A t test was used for statistical analysis. (K) Percentages of human CD4+ CD7+ CD25+ cells in blood collected at weeks 4 and 8 postinfection for each group.
Viral DNA was found in the bone marrow, thymus implant, and spleen of infected animals MS7, MS12, and MS14 at week 16, following euthanasia, but was not detected in control animals MS12 and MS15, as expected (Fig. 4A). The frequency of CD4+ CD25+ T cells was increased in the spleen and bone marrow of infected mice but not in the thymus implant (Fig. 4B to E). No differences in the levels of CD45RO+ memory cells and CD4+ CD7+ T cells were observed in the tissues of animals, regardless of their infection status (Fig. 4C to E). The frequency of CD8+ CD25+ T cells did not differ in the tissues of infected versus uninfected animals (Fig. 4F to H), and a correlation in the levels of CD4+ CD25+ T cells in spleen, bone marrow, and thymus with the viral burden in blood was observed only at week 16 postinfection (data not shown). Together, these results suggest that HTLV-1 infects human cells in BLT mice and is associated with the activation and expansion of CD4+ T cells. Because GvHD developed within a few months of inoculation in this model, we were unable to assess the long-term oncogenicity of HTLV-1 infection (Fig. 2A).
FIG 4.

HTLV-1WT and HTLV-1p12KO tissue infectivity in BLT mice. (A) PCR assay. Genomic DNA was isolated from spleen, thymus, bone marrow (BM), and whole blood from BLT mice at termination of the study at week 12 or 16 after infusion with irradiated HTLV-1-producing cells. Genomic DNA was used in nested PCRs to detect HTLV-1 DNA. Primers for the viral gag gene were used. ND-PBMCs, negative control; MT2 cell line, positive control; MS11 and MS15, uninfected BLT control mice; MS12 and MS14, HTLV-1WT-infected BLT mice; MS7, HTLV-1p12KO-infected BLT mouse. The first PCR gag, nested gag, and β-actin products were run on separate gels. (B) Tissue flow cytometry analysis. Spleen, bone marrow, and thymus of BLT mice (MS15, control; MS12, HTLV-1WT; MS7, HTLV-1p12KO) were analyzed 16 weeks postinfection. The percentages of human CD3+ CD4+ CD25+, CD3+ CD4+ CD45RO+, and CD3+ CD4+ CD7+ T cells from all groups were analyzed. (C to E) Percentages of human CD3+ CD4+ CD25+, CD3+ CD4+ CD45RO+, and CD3+ CD4+ CD7+ T cells from all groups analyzed in spleen tissue (C), bone marrow (D), and thymus (E) at week 16 postinfection. (F to H) Percentages of human CD3+ CD8+ CD25+, CD3+ CD8+ CD45RO+, and CD3+ CD7+ CD8+ cells from all groups analyzed in spleen tissue (F), bone marrow (G), and thymus (H) at week 16 postinfection.
HTLV-1 infection of NSG-1d mice.
We next infected nonobese diabetic (NOD) severe combined immunodeficiency (SCID) mice deficient in the common γ chain (NSG mice) that were engrafted with human fetal liver-derived hematopoietic stem cells as newborns (1 day after birth) (NSG-1d mice) (52). This model was expressly developed with the intent of minimizing GvHD development. For infection, we generated another short-term CD4+ cell line infected with HTLV-1WT, which was positive for Gag (Fig. 5A, lane 5), composed of 99.4% CD3+ CD4+ cells (Fig. 5B), and producing 0.83 ng/ml p19 Gag in the supernatant. Measurement of the HTLV-1 DNA level by real-time PCR revealed 364 copies/100 copies of the RNase P gene.
FIG 5.

CD4+-infected immortalized cell lines used for the infection of newborn (1-day-old) NOD/SCID/γC−/− c-kit+ mice with HTLV-1WT. (A) PCR assays for the detection of β-actin and viral HTLV-1 gag in generated primary CD4+ cells with HTLV-1-producing B cell lines. Genomic DNA was isolated from primary HTLV-1WT-infected CD4+ cells (lanes 1 and 5), primary uninfected CD4+ cells (lanes 2 and 6), 729.6 B cells (lanes 3 and 7), and infected HTLV-1WT-producing 729.6 cells (lanes 4 and 8). (B) Flow cytometry analysis was performed to define the purity of HTLV-1WT-infected CD4+ cells. FSC, forward scatter; SSC, side scatter. (C) Percentages of CD34 engraftment in 1-day-old NOD/SCID/γC−/− (NSG) c-kit+ mice. (D) Study design. Four mice (three female and one male) were all 8 months of age when the study began. Three mice were injected i.p. with irradiated HTLV-1WT-infected CD4+ cells, and one mock-infected mouse was injected with PBS solution. Blood was collected 3, 6, and 10 weeks after infection and then the animals were sacrificed. BM, bone marrow; LN, lymph nodes. (E) PCR assay for detection of EBV genes (EBV Bam and PO primers) in mice, primary CD4+ cells, and the HTLV-1-infected 729.6 B cell line 3 weeks postinfection. (F) HTLV-1 DNA copies measured by quantitative PCR in the whole blood of NSG c-kit+ mice at 3, 6, and 10 weeks postinfection. At week 10, all HTLV-1-infected mice (mice MS1, MS6, and MS10) had large proviral DNA loads. (G) MCT118 marker genotyping. DNA was extracted from the peripheral blood of all mice 10 weeks postinfection, and primary HTLV-1-producing CD4+ cells (CD4+ HTLV-1WT) were used for infection. The absence of residual infected CD4+ cells in the source inoculum was revealed by the distinct pattern of DNA fragments obtained by PCR in the inoculated human CD4+ cells and the DNA from mice MS1, MS3, MS6, and MS10, engrafted with cells from different human donors.
Reconstituted NSG-1d mice MS1, MS6, and MS10 (Fig. 5C) were inoculated i.p. with irradiated HTLV-1WT-infected primary human HLA.A2+ CD4+ cells, and mouse MS3 was inoculated with saline as a control (Fig. 5D). We did not detect any EBV DNA sequence in blood at 3 weeks postinjection (Fig. 5E), but we did detect the HTLV-1 gag sequence (Fig. 5F) in the mice injected with HTLV-1WT-producing cells. In order to verify that the gag sequence did not originate from the inoculum, we analyzed the genetic locus D1S58, defined by the DNA probe pMCT118 and containing a variable number of tandem repeats. The absence of residual infected CD4+ cells in the inoculum was revealed by the distinct patterns of DNA fragments obtained by PCR for the donor human CD4+ cell line and the DNA of recipient mice MS1, MS3, MS6, and MS10 engrafted with cells from different human donors (Fig. 5G).
HTLV-1 causes polyclonal expansion of CD4+ T cells in blood and tissues of NSG-1d mice.
NSG-1d mice MS1, MS6, and MS10, which were exposed to HTLV-1WT-infected cells, had marked increases in human CD4+ CD25+ CD45+ T cells, relative to the uninfected control mouse MS3 (Fig. 6A and B). These mice lost weight as the viral burden increased, and the mouse with the greatest viral burden, MS10, developed neurological symptoms, with paralysis of its hind legs. Strikingly, 40% of the CD4+ cells of mouse MS10 expressed the CD25 marker before its death. Macroscopic pathological analysis revealed a subcutaneous mass in the sacral region of its spinal cord, which explained the clinical symptoms of its lower body paralysis. Animals MS1, MS3, and MS6 were euthanized at week 11 and their tissues collected. At the time of euthanasia, levels of CD45+ and CD4+ CD25+ cells were higher in both the blood and spleen of mice MS1 and MS6, compared to the control mouse MS3 (Fig. 6C and D). HTLV-1 DNA was positive in the blood and spleen of mice MS1 and MS6 (Fig. 6E), and its levels correlated with the levels of CD4+ CD25+ T cells in both tissues (Fig. 6F). We examined multiple tissues from HTLV-1-infected mouse MS1 by immunohistochemistry and contrasted them with those of the uninfected control mouse MS3. Enlarged white pulp and multiple lymphoid follicles were present in the spleen tissue of mouse MS1. In contrast, the white pulp observed in control mouse MS3 was small and lacked well-defined follicles. Staining of tissue determined that the white pulp in mouse MS1 was populated by human lymphocytes (Fig. 7A). Throughout the hepatic parenchyma of the liver of mouse MS1, we found a moderate mononuclear inflammatory infiltrate composed predominantly of human lymphocytes, with a lesser number of macrophages, accompanied by a mild to moderate increase in granulocytic hematopoiesis (Fig. 7B). Again, these findings differed radically from those for control mouse MS3, in which we observed only minimal infiltration of inflammatory cells (Fig. 7B).
FIG 6.

Blood cell profiles in HTLV-1WT-infected 1-day-old NOD/SCID/γC−/− c-kit+ mice. (A) Measurement of the fold change of human CD45+ cells with time postinfection in the NSG c-kit+ mice. CD45+ cells increase in HTLV-1-infected mice (MS1, MS6, and MS10). (B) Measurement of the fold change of human CD4+ CD25+ cells with time postinfection in the NSG c-kit+ mice. CD4+ CD25+ cells increase in HTLV-1-infected mice (MS1, MS6, and MS10). (C) Percentages of human CD45+ cells in whole blood (10 weeks postinfection) and in spleen tissue (end of study). (D) Percentages of human CD4+ CD25+ cells in whole blood (10 weeks postinfection) and in spleen tissue (end of the study). (E) Detection of HTLV-1 DNA copies by quantitative PCR in whole blood (10 weeks postinfection) and spleen cells (end of the study) in two HTLV-1-infected mice (MS1 and MS6) and one control mouse (MS3). (F) Correlation of the proportions of CD3+ CD4+ CD25+ cells measured in blood (red) and spleen (black) with viral burden (HTLV-1 DNA copies) at week 10 postinfection (MS1, MS3, and MS6). The Spearman correlation test was used for statistical analysis.
FIG 7.
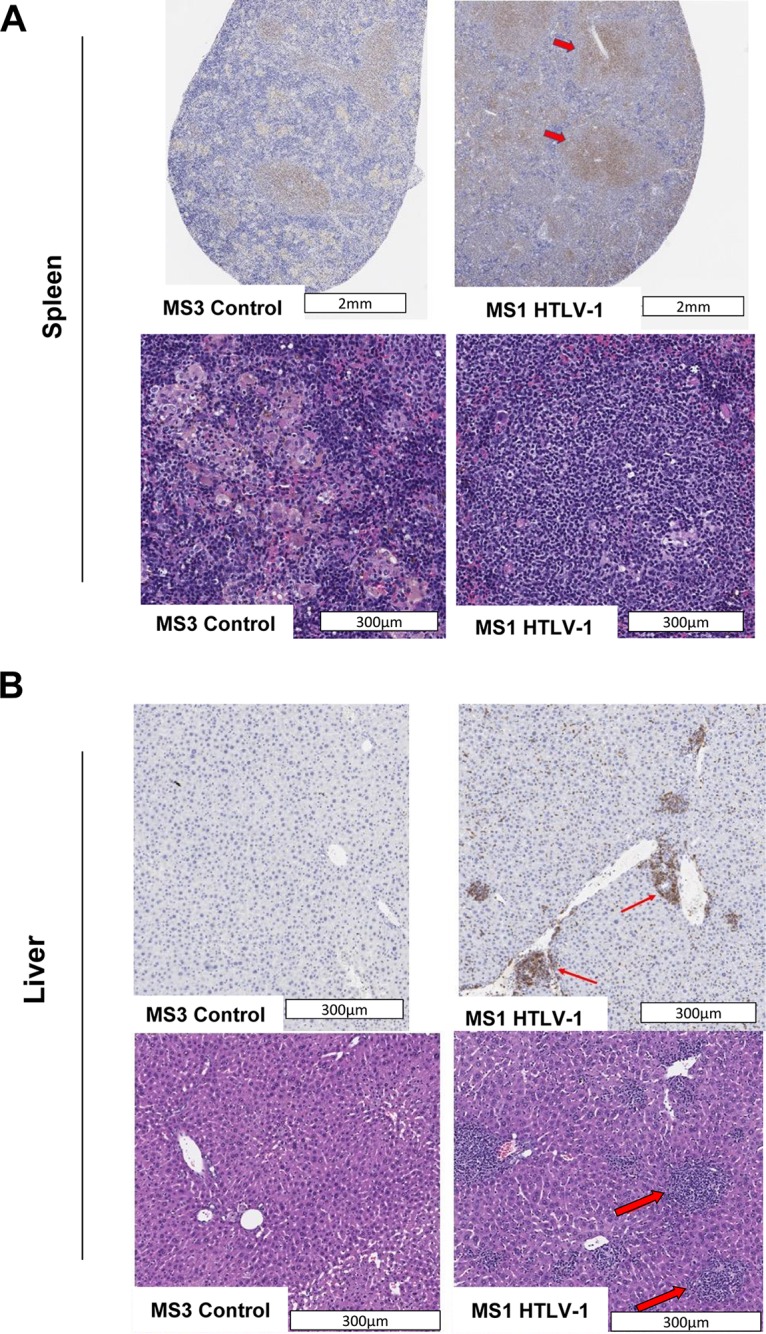
Pathology/histology analyses of spleen (A) and liver (B) tissue. (A) The spleen of a NSG-1d mouse demonstrates the absence of lymphoid follicles and diminished cellularity. The spleen of a HTLV-1-infected mouse (mouse MS1) contains an aggregate of dense cellular lymphoid infiltrate (indicated by arrows). (B) The liver exhibits multifocal accumulation of lymphoid cells in the periportal, midzonal, and centrilobular regions (indicated by arrows). The level of CD3 expression determined by immunohistochemistry in the spleen and liver was significantly higher in treated mouse MS1 than in control mouse MS3. Different stains were used for the upper and lower panels of each figure.
With the immunohistochemistry results confirming increased levels of CD3+, CD4+, and CD8+ cells in the tissues of HTLV-1-infected animals, our results indicate that humanized mice can be readily infected with HTLV-1 (53, 54). Furthermore, infection is known to result in rapid increases in the levels of CD4+ CD25+ T cells in the blood and their infiltration into tissue, followed by weight loss and finally death.
Polyclonal expansion of CD4+ T cells in humanized mice.
To explore HTLV-1 clonality in infected NSG-1d mice, we applied an optimized high-throughput sequencing (HTS) method to map viral integration sites (proviruses) in the genome of the engrafted human cells and to simultaneously measure the abundance of the corresponding clones. The method included several critical modifications in library preparation and data analysis, overcoming some of the limitations of previously published protocols (45). We examined the number of unique integration sites (corresponding to the number of independent HTLV-1-infected clones) and their abundance in PBMCs and spleen isolated from HTLV-1-infected animals at the termination of the experiments (Table 4). This revealed the presence of multiple clones of low abundance, consistent with the occurrence of a polyclonal population of HTLV-1-infected cells in both the blood and spleen of mice MS1, MS6, and MS10 (Fig. 8A to C). The observed polyclonal distribution of CD4+ T cells in humanized mice contrasts sharply with the well-established monoclonal nature of human ATLL, which mainly involves a single predominant clone (Fig. 8D) (55).
TABLE 4.
Viral integration site counts and abundances
| Mouse and sample type | Time point (wk) | DNA input (ng) | Viral burden (%)a | No. of raw reads | No. of filtered reads | Integration site count |
|---|---|---|---|---|---|---|
| MS1 | ||||||
| Blood | 6 | 86.5 | 42.63 | 1,123 | 8 | 4 |
| Blood | 10 | 1125 | 90.44 | 369,216 | 2517 | 276 |
| Spleen | 10 | 930 | 59.05 | 589,246 | 5,857 | 884 |
| MS3 | ||||||
| Blood | 6 | 153.9 | 0 | 333 | 1 | 1 |
| Blood | 10 | 334.8 | 0 | 73 | 0 | 0 |
| Spleen | 10 | 876 | NA | 152,980 | 0 | 0 |
| MS6 | ||||||
| Blood | 6 | 37.2 | 0 | 2,976 | 7 | 1 |
| Blood | 10 | 250.5 | 151.69 | 550,308 | 3,213 | 483 |
| Spleen | 10 | 1,088 | 114.95 | 476,412 | 5,513 | 1,865 |
| MS10 | ||||||
| Blood | 6 | 120.8 | 67.26 | 134,254 | 549 | 104 |
| Blood | 10 | 993.6 | 93.5 | 351,617 | 2,193? | 561 |
NA, not applicable.
FIG 8.

HTS method to map viral integration sites. Pie charts illustrate the relative abundance of HTLV-1-infected clones in representative samples of HTLV-1-infected humanized mouse spleen and blood. Each slice represents a unique integration site/clone, with the size of the slice corresponding to the relative abundance of the clone. (A) In mouse MS1, 884 clones were identified in spleen and 276 clones were identified in blood at 10 weeks postinfection. (B) In mouse MS6, 1,865 clones were retrieved from spleen and 483 clones were identified in blood at 10 weeks postinfection. (C) In mouse MS10, 104 clones were identified in blood at 6 weeks postinfection and 561 clones were identified in blood at 10 weeks postinfection. (D) An ATLL patient (ATL7) demonstrates >90% of a single dominant clone, as described previously (55).
DISCUSSION
In the present work, we explored the susceptibility of two humanized mouse models to HTLV-1WT and HTLV-1p12KO (39). In this study, we tested humanized NSG mice implanted with human fetal liver, thymus tissue, and stem cells and NSG mice engrafted with CD34+ cells precultured with interleukin 3 (IL-3), IL-6, and stem cell factor. Seven days after engraftment, anti-c-kit antibody was injected i.p. to enhance human cell engraftment. Indeed, BLT mice have reportedly demonstrated a partial ability to mount adaptive and innate immune responses (50). The inoculation of mice with lethally irradiated primary CD4+ cells producing HTLV-1WT caused an increase in circulating CD4+ CD25+ T lymphocytes, which coincided with an increase in HTLV-1 DNA levels in blood.
Surprisingly, humanized BLT mice exposed to HTLV-1p12KO had increases in circulating CD4+ CD25+ T lymphocytes and blood viral DNA levels, contrary to the results we previously observed in macaques (39, 42). Sequencing of the orf-I gene in PBMC genomic DNA isolated from infected animals revealed a reversion of the point mutation (ATG to GTG) introduced in the orf-I ATG initiation codon. This was observed as early as 4 weeks after exposure, indicating in vivo viral replication and selective pressure for orf-I expression. Unfortunately, our studies in this model ended prematurely because all mice developed GvHD by 24 weeks postreconstitution. As a result of the rapid proliferation of infected cells and the premature death of the animals, we were unable to study any possible correlation between infectivity and related diseases such as HAM/TSP and ATLL.
In the NSG-1d model, we again observed an increased frequency of CD4+ CD25+ T cells that correlated with increased viral burden. The mice in this model lost weight and developed tumors and/or masses over time, and immunohistochemistry revealed increased infiltration of human CD4+ T cells into the spleen, liver, and lymph nodes of infected mice. Mapping of viral integration sites in both the PBMCs and spleen cells of infected mice revealed high levels of polyclonal expansion reminiscent of the healthy carrier stage of HTLV-1 infection, similar to findings reported for both HTLV-1 and bovine leukemia virus (56). Together, these studies indicate that, instead of being used to study clonal ATLL, the humanized mouse model may be better suited to investigating the early phases of T cell expansion after infection with HTLV-1WT and HTLV-1 mutants.
Mouse models have already been developed to address the pathogenetic mechanisms underlying HTLV-1 infection, including transgenic and xenografted humanized mouse models (57). Early humanized mouse studies of HTLV-1 focused on the use of SCID mice as recipients in a human peripheral blood leukocyte (PBL)-SCID model in which the mice were injected with PBLs from asymptomatic HTLV-1-positive individuals or from patients with ATLL or HAM/TSP (58). Here, viral persistence and the development of lymphoblastic lymphomas in animals were observed following the injection of PBMCs from two ATLL patients (58). Experiments involving either the injection of HTLV-1-infected cell lines or ex vivo ATLL patient cells obtained from either peripheral blood or lymph nodes, coupled with daily injections of IL-2, yielded comparable results in immunodeficient SCID (CB17 scid/scid) mice (59, 60).
Human PBMC-NOG (NOD/Shi-scid/IL-2Rγnull) mice were used by Miyazato et al. to study HTLV-1 infection; the investigators first inoculated mice with human lymphocytes and then inoculated them 3 days later with mitomycin C (MMC)-treated MT-2 cells (61). The mice in that study did become infected and expressed detectable levels of HTLV-1 DNA in both CD4+ and CD8+ T cells, which continued to increase with time (61). In addition, such models have been used to explore the roles of the viral proteins Tax and HBZ in ATLL pathogenesis (58, 59, 62, 63) and to study potential therapeutic interventions (64–68). The models have only partially recapitulated ATLL, however, with the induction of large atypical lymphocytes and multiple-organ engraftment. While more work remains to be done to optimize HTLV-1 studies in humanized mouse models, the models have already demonstrated great potential as a way to understand the mechanisms underlying viral pathogenesis.
MATERIALS AND METHODS
Study design with humanized mice.
All animals used in this study were housed and handled in accordance with the standards of the Association for Assessment and Accreditation of Laboratory Animal Care International.
BLT mice were grafted as described previously (49, 69). Briefly, human fetal CD34+ cells, purified from fetal liver, were injected intravenously into recipient NSG mice (∼0.5 × 106 to 1.0 × 106 cells per recipient) following total-body irradiation at 270 rads. Next, fragments of human fetal thymus and matched fetal liver CD34+ cells were implanted under the kidney capsule of each recipient NSG mouse. Six to 8 weeks later, the level of immune reconstitution was measured via fluorescence-activated cell sorting (FACS) analysis. Eight humanized BLT mice were used in this study, with human CD45+ cells accounting for 10 to 60% of their total PBMCs. Three mice were injected i.p. with irradiated HTLV-1WT-infected CD4+ cells, three mice were injected with HTLV-1p12KO-infected CD4+ cells, and two mice were injected with phosphate-buffered saline (PBS) solution. Blood was collected every 4 weeks after infection.
Retro-orbital eye bleeds were performed with capillary tubes to collect 50 μl of blood for PCR and 100 μl for flow cytometry. Cellular fractions were frozen in 1 ml of Bambanker cell-freezing medium and stored prior to transport. BLT mice were sacrificed after 16 weeks, corresponding to initial GvHD symptoms. Tissue samples from sacrificed animals were transported overnight, subjected to Ficoll gradient centrifugation, and viably frozen.
NOD/SCID/γC−/− mice (NSG strain) were provided by Natasa Strbo (52). NSG-1d mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ [NSG], referred to as NOD/SCID/γC−/− c-kit+) (product no. 005557; The Jackson Laboratory) were irradiated with a single sublethal dose of 1 Gy in whole-body irradiation and then housed with dams. Importantly, it has been shown that newborn mice less than 3 days of age support higher transplant efficiency. Mice were injected intrahepatically with 1 × 106 to 2 × 106 precultured fetal liver CD34+ cells in a volume of 30 μl. At day 7, mice were injected i.p. with 100 μg of anti-c-kit antibody (ACK2) (40). Mice were bled at 10 weeks, and peripheral blood was analyzed for relative percentages of murine and human CD45+ cells by flow cytometry. Four animals with proportions of 5 to 50% human CD45+ cells in total PBMCs were used. Three mice were injected i.p. with irradiated HTLV-1-infected CD4+ cells, and one mock-infected mouse was injected with PBS solution. Blood was collected from the tail vein or mandible, into an EDTA-containing capillary blood collection tube, at 3, 6, and 10 weeks after infection.
Generation and characterization of HTLV-1-infected human primary CD4+ T cells.
Stable 729.6 human lymphoblastoid B cells lines producing HTLV-1 were generated as described previously (38). The 729-6 B cell lines infected with the pAB HTLV-1WT and the HTLV-1p12KO viral mutant were maintained in RPMI 1640 medium with 10% fetal bovine serum (FBS). Using negative-selection beads (StemCell, Cambridge, MA), CD4+ T cells were isolated from infected PBMCs. Stable HTLV-1-producing CD4+ T cell lines were established by cocultivation of uninfected donor primary HLA.A2+/CD4+ T cells with lethally γ-irradiated 729.6-HTLV-1-infected lines. T cells were cultured for several months in RPMI 1640 medium supplemented with 20% FBS and 100 U of IL-2. The production of HTLV-1 in the supernatants of the infected cell cultures was assessed by measuring the amount of MA (p19 Gag) protein by enzyme-linked immunosorbent assay, according to the manufacturer's instructions (Zeptometrix, Buffalo, NY). Viral genomic sequences were verified by sequencing of the ClaI-SalI fragment, as described previously (39). HTLV-1-infected CD4+ cells were stained with anti-CD3-Alexa Fluor 700, anti-CD4-peridinin chlorophyll protein complex (PerCP)-Cy5.5, anti-CD8-Brilliant Violet 650 (BV650), and anti-CD19-BV605 antibodies, all of which were obtained from BD Biosciences (San Jose, CA), and were stained with LIVE/DEAD fixable aqua dead cell stain (Molecular Probes, Eugene, OR). Samples were analyzed by flow cytometry.
Detection of HTLV-1 and EBV by PCR.
We performed PCR assays for the detection of viral HTLV-1 gag and ebv genes in primary CD4 cell and B cell lines. We also performed an assay to detect HTLV-1 genomic DNA in the inoculated mice. DNA was extracted using the DNeasy blood and tissue kit (Qiagen, Rockville, MD), according to the manufacturer’s instructions. PCR amplification was performed with 100 ng of DNA sample, 45 μl of Platinum PCR SuperMix High Fidelity (Invitrogen, Carlsbad, CA), and 0.2 μM concentrations of the primers gag-F1 (5′-GGCCAAATCCTTTCCCGTAG-3′) and gag-R1 (5′-GTTGTGGATTGTTGGCTTGG-3′). The cycle conditions were as follows: 94°C for 30 s for 1 cycle; 94°C for 30 s, 58°C for 30 s, and 68°C for 50 s for 30 cycles; and 68°C for 7 min for 1 cycle. Next, 3 μl of the first-round PCR product were added to a second-round PCR that included the sense primer gag-F2 (5′-GTCCCTCCAGTTACGATTTCC-3′) and the antisense primer gag-R2 (5′-AGGGAGGAGCAAAGGTACTG-3′), performed under the same conditions as used for the first-round PCR. To detect ebv, PCR amplification was performed using the primers EBV BAMHI-122F (5′-AGCGCATGGCCGGCATCTGAG-3′), EBV BAMHI-122R (5′-ATTCTTGCAGTGGTGCCGGAG-3′), EBV PO-F (5′-AGAGCAGGTCGGTCAGGCGTC-3′), and EBV PO-R (5′-GAGAGTTCCATAAGCACCTGG-3′). The cycle conditions for EBV BAM were as follows: 94°C for 30 s for 1 cycle; 94°C for 30 s, 55°C for 30 s, and 68°C for 50 s for 30 cycles; and 68°C for 7 min for 1 cycle. The cycle conditions for EBV PO were as follows: 94°C for 30 s for 1 cycle; 94°C for 30 s, 58°C for 30 s, and 68°C for 50 s for 30 cycles; and 68°C for 7 min for 1 cycle. Correctly sized amplicons were identified by 1% agarose gel electrophoresis.
HTLV-1 DNA assay.
HTLV-1 levels were measured in the DNA of the infected CD4+ cell inoculum and of mouse blood and tissues by a quantitative real-time PCR assay using the TaqMan probe, as described previously (70), with slight modification. Genomic DNA was extracted with the Qiagen blood minikit (Qiagen) and subjected to PCR. The HTLV-1 copy number was measured with primers for the pX region of HTLV-1. The RNase P gene, detected with the TaqMan RNase P control reagents kit (Applied Biosystems, Foster City, CA), was used as the endogenous reference in multiplex reactions. The viral levels are presented as the number of copies of HTLV-1 per 100 copies of the RNase P gene.
orf-I sequencing.
orf-I sequencing was performed as described previously (39), with slight modification. Whole blood was collected from mice at different time points postinfection, and DNA was extracted using a QIAamp DNA blood minikit (Qiagen) according to the manufacturer’s instructions. The DNA fragment including orf-I was amplified by PCR using Platinum PCR SuperMix (Invitrogen) with primers p12-F (5′-CACCTCGCCTTCCAACTG-3′) and p30-R (5′-GGAGTATTTGCGCATGGCC-3′) and 300 ng of genomic DNA. The PCR was carried out using an Eppendorf Master Cycler gradient PCR instrument for 35 cycles of 94°C for 30 s, 55°C for 30 s, and 68°C for 50 s. The PCR product was purified using a QIAquick PCR purification kit (Qiagen), and 50 ng of DNA was used with 0.64 pmol of primer (5′-CTGGACAGGTGGCCAGTA-3′) for sequencing. Sanger sequencing was carried out at the Center for Cancer Research Genomics Core at the National Cancer Institute, NIH (data not shown).
HTLV-1 inoculation of humanized mice.
CD4+ HTLV-1 cells producing HTLV-1WT or HTLV-1p12KO were irradiated with 12 Gy from a 60Co-source irradiator. Lethally irradiated HTLV-1-infected cells (2 × 106 cells) or PBS was inoculated i.p. into BLT or 1-day-old NSG-1d mice 8 weeks or 8 months, respectively, after hematopoietic stem cell engraftment. An aliquot of cells was maintained in culture to control for irradiation treatment. The study ended when the mice developed GvHD or their body weight decreased to <70% of their maximum weight. All infections were performed in a biosafety level 2 laboratory.
Flow cytometry.
Samples from the first BLT mouse study were frozen prior to flow cytometric analysis. Whole-blood samples were rapidly thawed, washed twice in Dulbecco’s PBS (DPBS), and then resuspended in 100 μl of DPBS. A 30-μl aliquot was taken for PCR analysis, and the remaining 70 μl was used for flow cytometry staining. No red blood cell lysing was required, since most of the red blood cells were lysed during thawing and CD45 gating was performed during flow cytometry data acquisition. Tissue samples were rapidly thawed, washed twice in DPBS, and resuspended in 100 μl of DPBS prior to staining. All samples were stained with LIVE/DEAD fixable aqua dead cell stain (Thermo Fisher) and the following anti-human CD antibodies: anti-CD45-allophycocyanin (APC)-Cy7 (clone 2D1), anti-CD3-Alexa Fluor 700 (clone SP34-2), anti-CD4-PerCP-Cy5.5 (clone L200), anti-CD8-BV711 (clone RPA-T8), anti-CD14-Alexa Fluor 488 (clone MφP9), anti-CD19-BV605 (clone SJ25C1), anti-CD16-V450 (clone 3G8), anti-CD45RO-APC (clone UCHL1), anti-CD45RA-phycoerythrin (PE)-Cy7 (clone HI100), anti-CD7-PE-Cy5 (clone M-T701), anti-CD2-PE-CF594 (clone RPA-2.10), and CD25-PE (clone M-A251), all from BD Biosciences.
For the second BLT study, unfrozen whole blood was shipped overnight for analysis. Blood was stained with LIVE/DEAD aqua dead cell stain and the same antibodies as described above, except that anti-CD2 was replaced with anti-mouse CD45-PE-CF594 (clone 30-F11). Cells and antibodies were incubated for 30 min at room temperature, and red blood cells were lysed for 5 min using ammonium-chloride-potassium (ACK) lysis buffer (Quality Biological, Gaithersburg, MD). Samples from both studies were assessed with a BD Biosciences LSRII flow cytometer equipped with 405-nm, 488-nm, 532-nm, and 640-nm lasers. Data were analyzed with FlowJo 9.9.6.
pMCT118 genotyping by PCR.
DNA was extracted from mouse peripheral blood and HTLV-1-infected primary CD4+ cells used for infection by using the DNeasy blood and tissue kit (Qiagen), according to the manufacturer’s instructions. PCR amplification was performed with 10 ng of DNA sample, 45 μl of Platinum Taq Hi-Fi SuperMix, and 0.2 μM concentrations of each primer (Fw, 5′-GAAACTGGCCTCCAAACACTGCCCGCCG-3′; Rv, 5′-GTCTTGTTGGAGATGCACGTGCCCCTTGC-3′) (71).
High-throughput sequencing.
To explore HTLV-1 clonality in humanized mice, we used an optimized HTS method to map proviral integration sites in the human genome and to simultaneously measure the abundance of the corresponding clones. The method includes several critical modifications in library preparation and data analysis, overcoming some of the limitations of previously published protocols (55). We examined the number of unique integration sites, corresponding to the number of independent HTLV-1-infected clones, and their abundance in PBMCs and spleen tissue isolated from HTLV-1-infected NSG-1d animals.
Immunohistochemistry.
A number of 5-μm-thick sections were prepared from paraffin-embedded tumor blocks. The immunohistochemical staining was performed with a Bond RX autostainer (Leica Biosystems, Vista, CA). Heat-induced epitope retrieval (HIER) EDTA 10 solution was used for anti-CD3, anti-CD4, and anti-CD8 antibodies, and HIER citrate 20 solution was used with cell adhesion molecule 1 (CADM1) (SynCAM). The primary antibodies used in this study included anti-CD3 (1:50 dilution, product no. CME324; Biocare Medical, Pachecho, CA), anti-CD4 (1:250 dilution, product no. ab133616; Abcam, Cambridge, MA), anti-CD8 (1:100 dilution, product no. ab101500; Abcam), and anti-CADM1 (1:2,000 dilution, product no. CM004-3; MBL International, Woburn, MA). All antibodies were incubated for 30 min and detected using the Bond Polymer Refine detection kit (Leica Biosystems), omitting the postprimary reagent. The kit uses the diaminobenzidine reaction to detect antibody labeling, with hematoxylin counterstaining. All immunohistochemical markers were optimized using positive and negative controls. Human tonsil tissue (formalin fixed and paraffin embedded) was used as a positive control for CD3, CD4, and CD8 staining. Mouse brain tissue was used as a positive control for SynCAM staining.
The stained slides were digitally scanned at ×20 magnification using Aperio AT2 whole-slide scanners. The images were viewed using Aperio ImageScope software, and the digital image analysis was performed using Aperio Image Toolbox (Leica Biosystems). The numbers of CD3+, CD4+, and CD8+ inflammatory cells were quantified in the spleen, liver, and lymph nodes. For scoring SynCAM, H-scores were measured in the spleen, liver, brain, and lymph nodes to calculate the percentage of positive pixels (data not shown).
ACKNOWLEDGMENTS
This work was supported with federal funds from the Intramural Program of the National Cancer Institute and the Office of AIDS Research, National Institutes of Health.
We thank David Ahern for editorial assistance.
We declare no competing interests.
REFERENCES
- 1.Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. 1980. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A 77:7415–7419. doi: 10.1073/pnas.77.12.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gallo RC. 2005. The discovery of the first human retrovirus: HTLV-1 and HTLV-2. Retrovirology 2:17. doi: 10.1186/1742-4690-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gallo RC. 2011. Research and discovery of the first human cancer virus, HTLV-1. Best Pract Res Clin Haematol 24:559–565. doi: 10.1016/j.beha.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Tsukasaki K, Hermine O, Bazarbachi A, Ratner L, Ramos JC, Harrington W, O’Mahony D, Janik JE, Bittencourt AL, Taylor GP, Yamaguchi K, Utsunomiya A, Tobinai K, Watanabe T. 2009. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: a proposal from an international consensus meeting. J Clin Oncol 27:453–459. doi: 10.1200/JCO.2008.18.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uchiyama T, Yodoi J, Sagawa K, Takatsuki K, Uchino H. 1977. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood 50:481–492. [PubMed] [Google Scholar]
- 6.Gessain A, Barin F, Vernant JC, Gout O, Maurs L, Calender A, de Thé G. 1985. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet 2:407–410. doi: 10.1016/s0140-6736(85)92734-5. [DOI] [PubMed] [Google Scholar]
- 7.Osame M, Usuku K, Izumo S, Ijichi N, Amitani H, Igata A, Matsumoto M, Tara M. 1986. HTLV-I associated myelopathy, a new clinical entity. Lancet 1:1031–1032. doi: 10.1016/s0140-6736(86)91298-5. [DOI] [PubMed] [Google Scholar]
- 8.Takatsuki K, Yamaguchi K, Kawano F, Hattori T, Nishimura H, Tsuda H, Sanada I, Nakada K, Itai Y. 1985. Clinical diversity in adult T-cell leukemia-lymphoma. Cancer Res 45(Suppl):4644s–4645s. [PubMed] [Google Scholar]
- 9.Kamoi K, Mochizuki M. 2012. HTLV-1 uveitis. Front Microbiol 3:270. doi: 10.3389/fmicb.2012.00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eguchi K, Matsuoka N, Ida H, Nakashima M, Sakai M, Sakito S, Kawakami A, Terada K, Shimada H, Kawabe Y. 1992. Primary Sjogren's syndrome with antibodies to HTLV-I: clinical and laboratory features. Ann Rheum Dis 51:769–776. doi: 10.1136/ard.51.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Terada K, Katamine S, Eguchi K, Moriuchi R, Kita M, Shimada H, Yamashita I, Iwata K, Tsuji Y, Nagataki S. 1994. Prevalence of serum and salivary antibodies to HTLV-1 in Sjogren's syndrome. Lancet 344:1116–1119. doi: 10.1016/s0140-6736(94)90630-0. [DOI] [PubMed] [Google Scholar]
- 12.Nishioka K, Maruyama I, Sato K, Kitajima I, Nakajima Y, Osame M. 1989. Chronic inflammatory arthropathy associated with HTLV-I. Lancet 1:441. doi: 10.1016/s0140-6736(89)90038-x. [DOI] [PubMed] [Google Scholar]
- 13.Morgan OS, Rodgers-Johnson P, Mora C, Char G. 1989. HTLV-1 and polymyositis in Jamaica. Lancet 2:1184–1187. doi: 10.1016/s0140-6736(89)91793-5. [DOI] [PubMed] [Google Scholar]
- 14.Jones KS, Petrow-Sadowski C, Huang YK, Bertolette DC, Ruscetti FW. 2008. Cell-free HTLV-1 infects dendritic cells leading to transmission and transformation of CD4+ T cells. Nat Med 14:429–436. doi: 10.1038/nm1745. [DOI] [PubMed] [Google Scholar]
- 15.de Castro-Amarante MF, Pise-Masison CA, McKinnon K, Washington Parks R, Galli V, Omsland M, Andresen V, Massoud R, Brunetto G, Caruso B, Venzon D, Jacobson S, Franchini G. 2015. Human T cell leukemia virus type 1 infection of the three monocyte subsets contributes to viral burden in humans. J Virol 90:2195–2207. doi: 10.1128/JVI.02735-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rizkallah G, Alais S, Futsch N, Tanaka Y, Journo C, Mahieux R, Dutartre H. 2017. Dendritic cell maturation, but not type I interferon exposure, restricts infection by HTLV-1, and viral transmission to T-cells. PLoS Pathog 13:e1006353. doi: 10.1371/journal.ppat.1006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alais S, Mahieux R, Dutartre H. 2015. Viral source-independent high susceptibility of dendritic cells to human T-cell leukemia virus type 1 infection compared to that of T lymphocytes. J Virol 89:10580–10590. doi: 10.1128/JVI.01799-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furtado MSBS, Andrade RG, Romanelli LCF, Ribeiro MA, Ribas JG, Torres EB, Barbosa-Stancioli EF, Proietti ABFC, Martins ML. 2012. Monitoring the HTLV-1 proviral load in the peripheral blood of asymptomatic carriers and patients with HTLV-associated myelopathy/tropical spastic paraparesis from a Brazilian cohort: ROC curve analysis to establish the threshold for risk disease. J Med Virol 84:664–671. doi: 10.1002/jmv.23227. [DOI] [PubMed] [Google Scholar]
- 19.Yamano Y, Nagai M, Brennan M, Mora CA, Soldan SS, Tomaru U, Takenouchi N, Izumo S, Osame M, Jacobson S. 2002. Correlation of human T-cell lymphotropic virus type 1 (HTLV-1) mRNA with proviral DNA load, virus-specific CD8+ T cells, and disease severity in HTLV-1-associated myelopathy (HAM/TSP). Blood 99:88–94. doi: 10.1182/blood.v99.1.88. [DOI] [PubMed] [Google Scholar]
- 20.Matsuzaki T, Nakagawa M, Nagai M, Usuku K, Higuchi I, Arimura K, Kubota H, Izumo S, Akiba S, Osame M. 2001. HTLV-I proviral load correlates with progression of motor disability in HAM/TSP: analysis of 239 HAM/TSP patients including 64 patients followed up for 10 years. J Neurovirol 7:228–234. [DOI] [PubMed] [Google Scholar]
- 21.Iwanaga M, Watanabe T, Utsunomiya A, Okayama A, Uchimaru K, Koh KR, Ogata M, Kikuchi H, Sagara Y, Uozumi K, Mochizuki M, Tsukasaki K, Saburi Y, Yamamura M, Tanaka J, Moriuchi Y, Hino S, Kamihira S, Yamaguchi K. 2010. Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: a nationwide prospective study in Japan. Blood 116:1211–1219. doi: 10.1182/blood-2009-12-257410. [DOI] [PubMed] [Google Scholar]
- 22.Murphy EL, Lee TH, Chafets D, Nass CC, Wang B, Loughlin K, Smith D. 2004. Higher human T lymphotropic virus (HTLV) provirus load is associated with HTLV-I versus HTLV-II, with HTLV-II subtype A versus B, and with male sex and a history of blood transfusion. J Infect Dis 190:504–510. doi: 10.1086/422398. [DOI] [PubMed] [Google Scholar]
- 23.Yakova M, Lézin A, Dantin F, Lagathu G, Olindo S, Jean-Baptiste G, Arfi S, Césaire R. 2005. Increased proviral load in HTLV-1-infected patients with rheumatoid arthritis or connective tissue disease. Retrovirology 2:4. doi: 10.1186/1742-4690-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brady J, Jeang KT, Duvall J, Khoury G. 1987. Identification of p40x-responsive regulatory sequences within the human T-cell leukemia virus type I long terminal repeat. J Virol 61:2175–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshida M, Inoue J, Fujisawa J, Seiki M. 1989. Molecular mechanisms of regulation of HTLV-1 gene expression and its association with leukemogenesis. Genome 31:662–667. doi: 10.1139/g89-121. [DOI] [PubMed] [Google Scholar]
- 26.Bex F, Gaynor RB. 1998. Regulation of gene expression by HTLV-I Tax protein. Methods 16:83–94. doi: 10.1006/meth.1998.0646. [DOI] [PubMed] [Google Scholar]
- 27.Albrecht B, Collins ND, Burniston MT, Nisbet JW, Ratner L, Green PL, Lairmore MD. 2000. Human T-lymphotropic virus type 1 open reading frame I p12I is required for efficient viral infectivity in primary lymphocytes. J Virol 74:9828–9835. doi: 10.1128/jvi.74.21.9828-9835.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartoe JT, Albrecht B, Collins ND, Robek MD, Ratner L, Green PL, Lairmore MD. 2000. Functional role of pX open reading frame II of human T-lymphotropic virus type 1 in maintenance of viral loads in vivo. J Virol 74:1094–1100. doi: 10.1128/jvi.74.3.1094-1100.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ciminale V, Zotti L, D'Agostino DM, Ferro T, Casareto L, Franchini G, Bernardi P, Chieco-Bianchi L. 1999. Mitochondrial targeting of the p13II protein coded by the x-II ORF of human T-cell leukemia/lymphotropic virus type I (HTLV-I). Oncogene 18:4505–4514. doi: 10.1038/sj.onc.1203047. [DOI] [PubMed] [Google Scholar]
- 30.Nicot C, Mulloy JC, Ferrari MG, Johnson JM, Fu K, Fukumoto R, Trovato R, Fullen J, Leonard WJ, Franchini G. 2001. HTLV-1 p12I protein enhances STAT5 activation and decreases the interleukin-2 requirement for proliferation of primary human peripheral blood mononuclear cells. Blood 98:823–829. doi: 10.1182/blood.v98.3.823. [DOI] [PubMed] [Google Scholar]
- 31.Landry S, Halin M, Vargas A, Lemasson I, Mesnard JM, Barbeau B. 2009. Upregulation of human T-cell leukemia virus type 1 antisense transcription by the viral tax protein. J Virol 83:2048–2054. doi: 10.1128/JVI.01264-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edwards D, Fenizia C, Gold H, de Castro-Amarante MF, Buchmann C, Pise-Masison CA, Franchini G. 2011. orf-I and orf-II-encoded proteins in HTLV-1 infection and persistence. Viruses 3:861–885. doi: 10.3390/v3060861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seiki M, Inoue J, Hidaka M, Yoshida M. 1988. Two cis-acting elements responsible for posttranscriptional trans-regulation of gene expression of human T-cell leukemia virus type I. Proc Natl Acad Sci U S A 85:7124–7128. doi: 10.1073/pnas.85.19.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukumoto R, Andresen V, Bialuk I, Cecchinato V, Walser JC, Valeri VW, Nauroth JM, Gessain A, Nicot C, Franchini G. 2009. In vivo genetic mutations define predominant functions of the human T-cell leukemia/lymphoma virus p12I protein. Blood 113:3726–3734. doi: 10.1182/blood-2008-04-146928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Migone TS, Lin JX, Cereseto A, Mulloy JC, O'Shea JJ, Franchini G, Leonard WJ. 1995. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science 269:79–81. doi: 10.1126/science.7604283. [DOI] [PubMed] [Google Scholar]
- 36.Johnson JM, Nicot C, Fullen J, Ciminale V, Casareto L, Mulloy JC, Jacobson S, Franchini G. 2001. Free major histocompatibility complex class I heavy chain is preferentially targeted for degradation by human T-cell leukemia/lymphotropic virus type 1 p12I protein. J Virol 75:6086–6094. doi: 10.1128/JVI.75.13.6086-6094.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mulloy JC, Crownley RW, Fullen J, Leonard WJ, Franchini G. 1996. The human T-cell leukemia/lymphotropic virus type 1 p12I proteins bind the interleukin-2 receptor β and γc chains and affects their expression on the cell surface. J Virol 70:3599–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takemoto S, Mulloy JC, Cereseto A, Migone TS, Patel BK, Matsuoka M, Yamaguchi K, Takatsuki K, Kamihira S, White JD, Leonard WJ, Waldmann T, Franchini G. 1997. Proliferation of adult T cell leukemia/lymphoma cells is associated with the constitutive activation of JAK/STAT proteins. Proc Natl Acad Sci U S A 94:13897–13902. doi: 10.1073/pnas.94.25.13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pise-Masison CA, de Castro-Amarante MF, Enose-Akahata Y, Buchmann RC, Fenizia C, Washington Parks R, Edwards D, Fiocchi M, Alcantara LC, Bialuk I, Graham J, Walser J-C, McKinnon K, Galvão-Castro B, Gessain A, Venzon D, Jacobson S, Franchini G. 2014. Co-dependence of HTLV-1 p12 and p8 functions in virus persistence. PLoS Pathog 10:e1004454. doi: 10.1371/journal.ppat.1004454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Prooyen N, Gold H, Andresen V, Schwartz O, Jones K, Ruscetti F, Lockett S, Gudla P, Venzon D, Franchini G. 2010. Human T-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc Natl Acad Sci U S A 107:20738–20743. doi: 10.1073/pnas.1009635107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fenizia C, Fiocchi M, Jones K, Parks RW, Ceribelli M, Chevalier SA, Edwards D, Ruscetti F, Pise-Masison CA, Franchini G. 2014. Human T-cell leukemia/lymphoma virus type 1 p30, but not p12/p8, counteracts toll-like receptor 3 (TLR3) and TLR4 signaling in human monocytes and dendritic cells. J Virol 88:393–402. doi: 10.1128/JVI.01788-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valeri VW, Hryniewicz A, Andresen V, Jones K, Fenizia C, Bialuk I, Chung HK, Fukumoto R, Parks RW, Ferrari MG, Nicot C, Cecchinato V, Ruscetti F, Franchini G. 2010. Requirement of the human T-cell leukemia virus p12 and p30 products for infectivity of human dendritic cells and macaques but not rabbits. Blood 116:3809–3817. doi: 10.1182/blood-2010-05-284141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Derse D, Mikovits J, Ruscetti F. 1997. X-I and X-II open reading frames of HTLV-I are not required for virus replication or for immortalization of primary T-cells in vitro. Virology 237:123–128. doi: 10.1006/viro.1997.8781. [DOI] [PubMed] [Google Scholar]
- 44.Collins ND, Newbound GC, Albrecht B, Beard JL, Ratner L, Lairmore MD. 1998. Selective ablation of human T-cell lymphotropic virus type 1 p12I reduces viral infectivity in vivo. Blood 91:4701–4707. [PubMed] [Google Scholar]
- 45.Walsh NC, Kenney LL, Jangalwe S, Aryee KE, Greiner DL, Brehm MA, Shultz LD. 2017. Humanized mouse models of clinical disease. Annu Rev Pathol 12:187–215. doi: 10.1146/annurev-pathol-052016-100332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tezuka K, Xun R, Tei M, Ueno T, Tanaka M, Takenouchi N, Fujisawa J. 2014. An animal model of adult T-cell leukemia: humanized mice with HTLV-1-specific immunity. Blood 123:346–355. doi: 10.1182/blood-2013-06-508861. [DOI] [PubMed] [Google Scholar]
- 47.Ginwala R, Caruso B, Khan ZK, Pattekar A, Chew GM, Corley MJ, Loonawat R, Jacobson S, Sreedhar S, Ndhlovu LC, Jain P. 2017. HTLV-1 infection and neuropathogenesis in the context of Rag1−/−γc−/− (RAG1-Hu) and BLT mice. J Neuroimmune Pharmacol 12:504–520. doi: 10.1007/s11481-017-9740-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huey DD, Niewiesk S. 2018. Production of humanized mice through stem cell transfer. Curr Protoc Mouse Biol 8:17–27. doi: 10.1002/cpmo.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith DJ, Lin LJ, Moon H, Pham AT, Wang X, Liu S, Ji S, Rezek V, Shimizu S, Ruiz M, Lam J, Janzen DM, Memarzadeh S, Kohn DB, Zack JA, Kitchen SG, An DS, Yang L. 2016. Propagating humanized BLT mice for the study of human immunology and immunotherapy. Stem Cells Dev 25:1863–1873. doi: 10.1089/scd.2016.0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, Othieno FA, Wege AK, Haase AT, Garcia JV. 2006. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med 12:1316–1322. doi: 10.1038/nm1431. [DOI] [PubMed] [Google Scholar]
- 51.Marsden MD, Kovochich M, Suree N, Shimizu S, Mehta R, Cortado R, Bristol G, An DS, Zack JA. 2012. HIV latency in the humanized BLT mouse. J Virol 86:339–347. doi: 10.1128/JVI.06366-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gonzalez L, Strbo N, Podack ER. 2013. Humanized mice: novel model for studying mechanisms of human immune-based therapies. Immunol Res 57:326–334. doi: 10.1007/s12026-013-8471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Percher F, Curis C, Pérès E, Artesi M, Rosewick N, Jeannin P, Gessain A, Gout O, Mahieux R, Ceccaldi PE, Van den Broeke A, Duc Dodon M, Afonso PV. 2017. HTLV-1-induced leukotriene B4 secretion by T cells promotes T cell recruitment and virus propagation. Nat Commun 8:15890. doi: 10.1038/ncomms15890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pérès E, Blin J, Ricci EP, Artesi M, Hahaut V, Van den Broeke A, Corbin A, Gazzolo L, Ratner L, Jalinot P, Duc Dodon M. 2018. PDZ domain-binding motif of Tax sustains T-cell proliferation in HTLV-1-infected humanized mice. PLoS Pathog 14:e1006933. doi: 10.1371/journal.ppat.1006933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Artesi M, Marcais A, Durkin K, Rosewick N, Hahaut V, Suarez F, Trinquand A, Lhermitte L, Asnafi V, Avettand-Fenoel V, Burny A, Georges M, Hermine O, Van den Broeke A. 2017. Monitoring molecular response in adult T-cell leukemia by high-throughput sequencing analysis of HTLV-1 clonality. Leukemia 31:2532–2535. doi: 10.1038/leu.2017.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosewick N, Durkin K, Artesi M, Marcais A, Hahaut V, Griebel P, Arsic N, Avettand-Fenoel V, Burny A, Charlier C, Hermine O, Georges M, Van den Broeke A. 2017. Cis-perturbation of cancer drivers by the HTLV-1/BLV proviruses is an early determinant of leukemogenesis. Nat Commun 8:15264. doi: 10.1038/ncomms15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dodon MD, Villaudy J, Gazzolo L, Haines R, Lairmore M. 2012. What we are learning on HTLV-1 pathogenesis from animal models. Front Microbiol 3:320. doi: 10.3389/fmicb.2012.00320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feuer G, Zack JA, Harrington WJ Jr, Valderama R, Rosenblatt JD, Wachsman W, Baird SM, Chen IS. 1993. Establishment of human T-cell leukemia virus type I T-cell lymphomas in severe combined immunodeficient mice. Blood 82:722–731. [PubMed] [Google Scholar]
- 59.Kondo A, Imada K, Hattori T, Yamabe H, Tanaka T, Miyasaka M, Okuma M, Uchiyama T. 1993. A model of in vivo cell proliferation of adult T-cell leukemia. Blood 82:2501–2509. [PubMed] [Google Scholar]
- 60.Imada K, Takaori-Kondo A, Uchiyama T. 1995. Analysis of in vivo cell proliferation of ATL using SCID mice. Rinsho Ketsueki 36:573–577. (In Japanese.) [PubMed] [Google Scholar]
- 61.Miyazato P, Yasunaga J, Taniguchi Y, Koyanagi Y, Mitsuya H, Matsuoka M. 2006. De novo human T-cell leukemia virus type 1 infection of human lymphocytes in NOD-SCID, common gamma-chain knockout mice. J Virol 80:10683–10691. doi: 10.1128/JVI.01009-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Banerjee P, Tripp A, Lairmore MD, Crawford L, Sieburg M, Ramos JC, Harrington W Jr, Beilke MA, Feuer G. 2010. Adult T-cell leukemia/lymphoma development in HTLV-1-infected humanized SCID mice. Blood 115:2640–2648. doi: 10.1182/blood-2009-10-246959. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63.Villaudy J, Wencker M, Gadot N, Gillet NA, Scoazec JY, Gazzolo L, Manz MG, Bangham CR, Dodon MD. 2011. HTLV-1 propels thymic human T cell development in “human immune system” Rag2−/− gamma c−/− mice. PLoS Pathog 7:e1002231. doi: 10.1371/journal.ppat.1002231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ju W, Zhang M, Petrus M, Maeda M, Pise-Masison CA, Waldmann TA. 2014. Combination of 9-aminoacridine with Campath-1H provides effective therapy for a murine model of adult T-cell leukemia. Retrovirology 11:43. doi: 10.1186/1742-4690-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mone A, Puhalla S, Whitman S, Baiocchi RA, Cruz J, Vukosavljevic T, Banks A, Eisenbeis CF, Byrd JC, Caligiuri MA, Porcu P. 2005. Durable hematologic complete response and suppression of HTLV-1 viral load following alemtuzumab in zidovudine/IFN-α-refractory adult T-cell leukemia. Blood 106:3380–3382. doi: 10.1182/blood-2005-01-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Phillips AA, Shapira I, Willim RD, Sanmugarajah J, Solomon WB, Horwitz SM, Savage DG, Bhagat G, Soff G, Zain JM, Alobeid B, Seshan VE, O'Connor OA. 2010. A critical analysis of prognostic factors in North American patients with human T-cell lymphotropic virus type-1-associated adult T-cell leukemia/lymphoma: a multicenter clinicopathologic experience and new prognostic score. Cancer 116:3438–3446. doi: 10.1002/cncr.25147. [DOI] [PubMed] [Google Scholar]
- 67.Tan C, Waldmann TA. 2002. Proteasome inhibitor PS-341, a potential therapeutic agent for adult T-cell leukemia. Cancer Res 62:1083–1086. [PubMed] [Google Scholar]
- 68.Zhang Z, Zhang M, Ravetch JV, Goldman C, Waldmann TA. 2003. Effective therapy for a murine model of adult T-cell leukemia with the humanized anti-CD2 monoclonal antibody, MEDI-507. Blood 102:284–288. doi: 10.1182/blood-2002-11-3601. [DOI] [PubMed] [Google Scholar]
- 69.Marsden MD, Zack JA. 2017. Humanized mouse models for human immunodeficiency virus infection. Annu Rev Virol 4:393–412. doi: 10.1146/annurev-virology-101416-041703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tanaka G, Okayama A, Watanabe T, Aizawa S, Stuver S, Mueller N, Hsieh CC, Tsubouchi H. 2005. The clonal expansion of human T lymphotropic virus type 1-infected T cells: a comparison between seroconverters and long-term carriers. J Infect Dis 191:1140–1147. doi: 10.1086/428625. [DOI] [PubMed] [Google Scholar]
- 71.Kasai K, Nakamura Y, White R. 1990. Amplification of a variable number of tandem repeats (VNTR) locus (pMCT118) by the polymerase chain reaction (PCR) and its application to forensic science. J Forensic Sci 35:1196–1200. [PubMed] [Google Scholar]