

Chandipura virus is a clinically important human pathogen of the Indian subcontinent. The rapidity of death associated with CHPV infection in addition to the absence of an effective vaccine or therapeutics results in poor clinical prognosis. The biology of the virus and its interaction with the host immune system, including the complement system, are understudied. Our investigation reveals the susceptibility of CHPV to fluid phase complement and also dissects the pathway involved and the mechanism of virus neutralization. Direct binding of C1q, an important upstream component of the classical pathway of complement to CHPV, and the strong dependency on C1q for virus neutralization highlight the significance of identifying such interactions to better understand CHPV pathogenesis and devise strategies to target this deadly pathogen.
KEYWORDS: aggregation, Chandipura virus, complement, complement C1q, virus neutralization
ABSTRACT
Among the innate immune sentinels, the complement system is a formidable first line of defense against pathogens, including viruses. Chandipura virus (CHPV), a neurotropic vesiculovirus of the family Rhabdoviridae, is a deadly human pathogen known to cause fatal encephalitis, especially among children. The nature of interaction and the effect of human complement on CHPV are unknown. Here, we report that CHPV is a potent activator of complement and, thus, is highly sensitive to complement proteins in normal human serum (NHS). Utilizing a panel of specific complement component depleted/reconstituted human serum, we have demonstrated that CHPV neutralization is C3, C4, and C1q dependent and independent of factor B, suggesting the importance of the classical pathway in limiting CHPV. Employing a range of biochemical approaches, we showed (i) a direct association of C1q to CHPV, (ii) deposition of complement proteins C3b, C4b, and C1q on CHPV, and (iii) virus aggregation. Depletion of C8, an important component of the pore-forming complex of complement, had no effect on CHPV, further supporting the finding that aggregation and not virolysis is the mechanism of virus neutralization. With no approved vaccines or treatment modalities in place against CHPV, insights into such interactions can be exploited to develop potent vaccines or therapeutics targeting CHPV.
IMPORTANCE Chandipura virus is a clinically important human pathogen of the Indian subcontinent. The rapidity of death associated with CHPV infection in addition to the absence of an effective vaccine or therapeutics results in poor clinical prognosis. The biology of the virus and its interaction with the host immune system, including the complement system, are understudied. Our investigation reveals the susceptibility of CHPV to fluid phase complement and also dissects the pathway involved and the mechanism of virus neutralization. Direct binding of C1q, an important upstream component of the classical pathway of complement to CHPV, and the strong dependency on C1q for virus neutralization highlight the significance of identifying such interactions to better understand CHPV pathogenesis and devise strategies to target this deadly pathogen.
INTRODUCTION
The complement system is one of the potent innate immune barriers faced by viruses during the course of infection. The complement cascade comprises a highly concerted group of proteins activated through three major pathways, namely, the classical pathway (CP), the lectin pathway (LP), and the alternative pathway (AP) (1). The primary trigger is a key factor determining the initiation of a specific pathway. The CP is initiated by an antigen-antibody immune complex; the LP by unique sugar moieties, such as mannose; and the AP by a range of pathogen-associated signatures (1, 2). Complement activity is not restricted to assembly of a lytic pore-forming unit called the membrane attack complex (MAC) but is diverse, spanning both the innate and the adaptive arms of the immune system (3–5). Complement-specific convertases mediate the proteolytic cleavage of complement proteins, resulting in the generation of bioactive components that mediate functions such as anaphylaxis, opsonization, sensitization, and stimulation of antigen-presenting cells and T and B lymphocytes (6). These interactions, besides facilitating rapid clearance of viral pathogens, are instrumental in generating long-term immunity (7, 8). The overall premise of this study is to decipher the effect of complement on Chandipura virus (CHPV), dissect the pathways involved, and understand the mechanisms of neutralization of CHPV by complement.
The involvement of either a single or multiple complement pathways in limiting pathogenesis varies among viruses (9). Viruses known to activate the AP include parainfluenza virus 5 (PIV5), Sindbis virus, and Epstein-Barr virus, among others (10–14). Specific signatures like sialic acid associated with viral glycoproteins have been shown to contribute to the degree of AP activation (10, 15). Recognition of unique carbohydrate moieties by mannose binding lectins (MBLs) on the surface of viruses such as influenza virus, herpes simplex virus 2, and hepatitis C virus results in activation of the LP (16–18). Classical pathway activation involves antigen-antibody immune complex engagement with C1q in viruses, including vesicular stomatitis virus (VSV), human immunodeficiency virus (HIV), and human T-cell lymphotropic virus (HTLV); however, an independent role for C1q has been observed in a few viruses, such as Zika virus (19–22). The role of multiple pathways in limiting virus infection has been demonstrated elegantly in the pandemic influenza A (H1N1) 2009 virus, wherein both the CP and AP were essential for maximal virus neutralization (23). The involvement of all three pathways in virus neutralization is not widely reported, but West Nile virus is a classic example (24). Thus, the complement system is capable of neutralizing a diverse range of viruses.
Rhabdoviruses, which include rabies virus, VSV, and CHPV, are potent animal and human pathogens. Chandipura virus is a vector-borne pathogen transmitted by sand flies (Phlebotomus sp.) and is widely distributed in many regions of the Indian subcontinent, causing sporadic outbreaks resulting in significant mortality ranging between 56% and 75% (25, 26). The swiftness of death in severe CHPV cases (24 to 48 h) (27) raises the most important question on the nature of the interaction of CHPV with one of the earliest immune barriers faced by the virus, the complement system. Insights gained so far into the rhabdovirus-complement interaction arise from the studies on VSV. Among the three pathways of complement, VSV was shown to preferentially activate the CP, resulting in virus neutralization (19, 28, 29). Marked differences have been observed among the same family of viruses with respect to the mechanism of complement-mediated neutralization. Mumps virus (MuV) and the closely related PIV5 both activate the AP, resulting in MuV lysis and PIV5 aggregation (11). Thus, it is clear that despite the close relatedness among viruses, major differences in the mechanism of complement activation and neutralization exist. Virolysis ensues upon exposure of VSV to human complement (30, 31). With both VSV and CHPV being related vesiculoviruses, it was intriguing to check if the CHPV-complement interaction mirrored that of VSV.
In this study, we have demonstrated that despite being a potent pathogen, CHPV is sensitive to serum complement. We have also shown that CHPV can activate the complement system, and the resulting neutralization is dependent on critical components like C3 and C4. Among the three pathways, CHPV preferentially activates the CP, and neutralization is highly dependent on C1q. Finally, we have also deciphered that the CHPV-complement interaction results in aggregation, rendering the virus inactive.
RESULTS
Activation of human complement by Chandipura virus results in virus neutralization.
To check if CHPV could activate complement, 2-fold dilutions of sucrose gradient-purified CHPV (2 to 0.18 μg) were incubated with normal human serum (NHS) for 15 min, and the reaction was stopped by adding sample loading buffer containing SDS and dithiothreitol (DTT) and boiled at 100°C for 5 min. Following Western blotting, C3a was detected with anti-human C3a polyclonal antibody. Enhancement of C3a levels corresponded with increasing concentrations of CHPV, saturating at higher CHPV concentrations (Fig. 1A). Activation was also found to be time dependent, with a gradual increase in the levels of C3a with increasing time. Serum incubated with virus had significantly higher concentrations of C3a than the serum-only controls, which had only background levels of C3a (Fig. 1B). Thus, CHPV was found to be a strong activator of complement.
FIG 1.
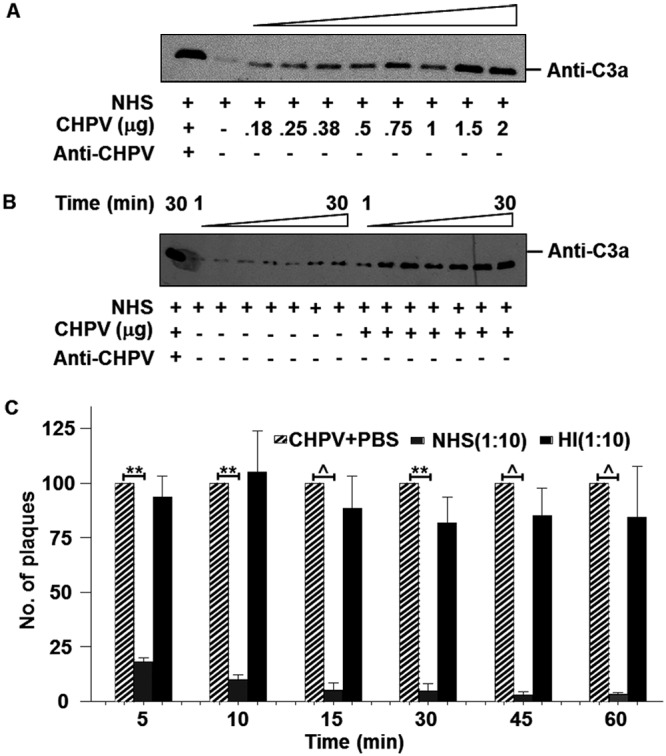
Chandipura virus is a potent activator of human complement, resulting in virus neutralization. (A) Various concentrations of CHPV as indicated were incubated with NHS (1:10 dilution) for 15 min followed by Western blotting to detect C3a, the activation product of C3. Positive control included NHS incubated with both CHPV and anti-CHPV antibody for 15 min, while the serum by itself was used to detect background levels of C3a. (B) Time course experiment was carried out by incubating CHPV with NHS for various time points as depicted followed by Western blotting to detect C3a. Serum-only samples incubated for the respective time points served as the control. (C) Effect of NHS on CHPV was determined by incubating 100 PFU of the virus with either NHS (gray bars) or heat-inactivated NHS (black bars) at a dilution of 1:10 for the respective time periods, and the remaining infectious virus was determined by plaque assays. Plaque reduction was compared against virus-only samples (hatched bars), with each bar representing 6 independent experiments. Differences in neutralization between CHPV+PBS and CHPV+NHS were found to be statistically significant with P values of ≤0.001 (**) and ≤0.0001 (^).
In order to test the effect of NHS on CHPV, pooled human serum was used in a plaque reduction (neutralization) assay. A set concentration of CHPV (100 PFU) was incubated with a 1:10 dilution of NHS for the specified time points, and the remaining virus was titrated out onto monolayers of Vero cells. Marked neutralization (>75%) was observed as early as 5 min, with more pronounced neutralization observed at the later time points (Fig. 1C). Heat-inactivated NHS (HI-NHS) was not capable of neutralizing the virus at all the time points tested. These data suggest that CHPV promotes complement activation and that heat labile factors in NHS, possibly complement, contribute significantly to CHPV neutralization.
C3 plays a pivotal role in CHPV neutralization.
Complement proteins are highly heat labile, and the added finding that exposure of NHS to CHPV resulted in C3a generation strengthened the hypothesis that the CHPV neutralizing factor in serum is complement. For further validation, a system comprised of C3-depleted and C3-reconstituted serum was employed. C3 depletion and reconstitution were checked using a standard hemolytic assay as described in Materials and Methods. The C3-depleted serum, compared with NHS, had limited or negligible hemolytic potential, which was reversed upon reconstitution with physiological concentrations of C3 (Fig. 2A). The neutralizing capacity of C3-depleted or -reconstituted NHS was tested against NHS at a dilution of 1:10. C3-depleted NHS had no effect on CHPV; however, upon reconstitution, marked neutralization was observed (Fig. 2B). Thus, CHPV neutralization is dependent on C3, which further validates the role of human complement in CHPV neutralization.
FIG 2.

Chandipura virus neutralization by NHS is dependent on C3. (A) Hemolytic assays were carried out with antibody-sensitized sheep erythrocytes (EA) to check reconstitution of C3 in C3-depleted serum which was compared against NHS. (B) Plaque reduction assays were carried out by incubating 100 PFU of CHPV with either PBS, NHS, C3-depleted serum (C3 dep.), or C3-depleted serum reconstituted with C3 (C3 dep.+C3). Each bar represents six independent experiments, and the differences between both the CHPV versus CHPV+NHS and C3 dep. versus C3 dep.+C3 were found to be statistically significant with P values of ≤0.001 (**) and ≤0.01 (^), respectively.
Neutralization of CHPV by human complement proceeds via the classical pathway.
Among the three major pathways of complement, the activation of CP and LP are dependent upon Ca2+, which can be blocked by the addition of EGTA, while EDTA is known to block all three pathways (32). In order to identify the pathway important for CHPV neutralization, 100 PFU of the virus was incubated with either phosphate-buffered saline (PBS), NHS, HI-NHS, 20 mM EDTA, or 15 mM EGTA containing 2 mM MgCl2 alone or NHS treated with EGTA-MgCl2 or EDTA. Only the NHS-treated sample showed significant virus neutralization (90%). Neither NHS-EDTA nor NHS-EGTA supported CHPV neutralization, suggesting that the Ca2+-dependent CP or LP is important for CHPV neutralization. The inhibitors by themselves had no effect on the virus (Fig. 3A).
FIG 3.

Neutralization of Chandipura virus by complement proceeds via the classical pathway. (A) To identify the pathway mediating CHPV neutralization, 100 PFU of virus was incubated with either NHS, HI-NHS, 20 mM EDTA, 15 mM EGTA+2 mM MgCl2, or NHS treated with EDTA or EGTA, and the remaining infectious titer was determined by plaque assays. Except the NHS-treated sample, none of the other treatments had any effect on the virus and, thus, were found to be statistically insignificant. (B) The total hemolytic activity of C4-depleted (C4 dep.) and -reconstituted serum (C4 dep.+C4) was tested against NHS using EA. (C) The significance of classical pathway in CHPV neutralization was tested by incubating CHPV (100 PFU) with either C4 dep. or C4 dep.+C4 with NHS serving as the control. The reduction in viral titer was determined by plaque assays. Statistical significance was calculated using values from six independent experiments, with ** denoting a P value of ≤0.005.
The functional relevance of the inability of EGTA-NHS to support CHPV neutralization and, thereby, the importance of the classical pathway in CHPV neutralization was tested using C4-depleted NHS. C4-depleted serum was incapable of causing hemolysis; however, upon reconstitution, the percent lysis mirrored that of the NHS (Fig. 3B). Plaque reduction assays carried out with C4-depleted and -reconstituted serum showed that CHPV was resistant to C4-depleted NHS, which was reversed upon reconstitution with C4. Neutralization of CHPV by C4-reconstituted NHS was comparable to that of NHS treatment, with >70% virus reduction (Fig. 3C). This clearly suggests a role for the classical pathway in CHPV neutralization.
To check if, in fact, the AP plays a less significant role in CHPV neutralization, serum depleted of a critical protein specific for the pathway, called factor B, was used. An AP-specific hemolytic assay with rabbit erythrocytes (RbEs) was carried out to check factor B depletion and reconstitution. Factor B reconstitution resulted in hemolysis similar to that of NHS, while the factor B-depleted serum had negligible hemolysis (Fig. 4A). Plaque reduction assays with factor B-depleted/reconstituted serum showed that depleted serum by itself was capable of neutralizing CHPV by greater than 80% (Fig. 4B). Reconstitution with factor B resulted in only a marginal increase in neutralization. These data clearly suggest that the CP or the LP is sufficient for CHPV neutralization and that the role of the AP is limited.
FIG 4.

Contribution of the alternative pathway of complement to Chandipura virus neutralization is limited. (A) Factor B reconstitution of the depleted serum was tested against NHS, with the depleted serum serving as the control using the standard hemolytic assay. (B) One hundred PFU of CHPV was incubated with either factor B-depleted (fB dep.) or -reconstituted serum (fB dep.+fB), and the CHPV+PBS or the CHPV+NHS served as the controls. Significance of the tests and NHS control was compared against the virus-only sample by taking the values from six independent experiments, with ** denoting a P value of ≤0.0001.
The classical pathway component C1q is essential for CHPV neutralization.
Since the AP did not contribute significantly to CHPV neutralization, we sought to further investigate the role of classical pathway components in neutralizing CHPV. A vital upstream component that required investigation was C1q, which has been shown to bind to immune complexes mediating CP activation. A C1q-specific enzyme-linked immunosorbent assay (ELISA) was carried out to check if C1q can bind directly to CHPV. A fixed concentration of gradient-purified CHPV (500 ng) was immobilized to Nunc-Maxisorp 96-well plates, blocked, and incubated with 2-fold dilutions of C1q, starting with 1,000 ng to 7.81 ng. Complement C1q binding to CHPV was found to be dose dependent, with maximum binding at 1 μg (absorbance at 405 nm [A405], 1.0), followed by a gradual decline in binding at lower concentrations (Fig. 5A). To further confirm C1q binding to CHPV, electron microscopy was carried out by adsorbing CHPV to Formvar and carbon-coated gold grids followed by incubation with purified C1q (3.6 μg). The bound C1q was detected with anti-C1q antibody and viral antigens with a mouse anti-CHPV antibody and with 12-nm and 6-nm Nanogold-labeled antibodies. Particles treated with C1q showed reactivity to anti-C1q antibodies (12 nm), suggesting that C1q can bind directly to the virion (Fig. 5B, bottom two panels). Anti-CHPV antibodies (6 nm) were found to be distributed throughout the virion, while the secondary control showed no reactivity (Fig. 5B, top panel), indicating specific binding of both anti-C1q and -CHPV antibodies.
FIG 5.

Classical pathway component C1q binds to Chandipura virus. (A) Gradient-purified CHPV (500 ng) was immobilized onto Nunc Maxisorp 96-well flat-bottom plates, and various concentrations of purified C1q were incubated with the bound virus followed by anti-C1q and HRP-conjugated secondary rabbit anti-goat antibody. (B) Sucrose gradient-purified CHPV was adsorbed onto Formvar and carbon-coated gold grids and treated with 180 ng of purified C1q followed by anti-C1q/CHPV primary antibodies and secondary gold conjugated antibodies (bottom two panels), while the top panel represents the secondary-only control. Samples were analyzed using a Jeol transmission electron microscope, and the closed triangles indicate anti-C1q antibody reactivity to bound C1q (12-nm Nanogold), while the arrows represent anti-mouse antibody (6-nm Nanogold) reacting to the bound anti-CHPV antibodies (bar represents 0.02 μm).
In order to check if C1q is important for CHPV neutralization, C1q-depleted or -reconstituted NHS was used. Hemolytic assays showed that the C1q-depleted serum could barely lyse EA, while the reconstituted serum had rates similar to that of NHS (Fig. 6A). Neutralization assays were carried out by incubating 100 PFU of CHPV with either PBS, NHS, or C1q-depleted or -reconstituted NHS. C1q-depleted serum had no effect on CHPV, with the number of plaques obtained similar to that of the virus-only control. Reconstitution with physiological concentrations of C1q resulted in >70% neutralization, and the levels were comparable to that of the NHS control. The very binding of C1q to CHPV suggests that upstream complement activation is possible without the need for immune-complex formation, unlike most CP activation. Augmentation of CHPV neutralization by C1q provides ample evidence that among the three complement pathways the CP is critical for CHPV neutralization compared with that of the LP or the AP.
FIG 6.

Chandipura virus neutralization by classical pathway of complement is dependent on C1q. (A) Hemolytic assay with sensitized sheep erythrocytes was employed to check reconstitution of C1q in the depleted serum against NHS. (B) One hundred PFU of the virus with either C1q-depleted (C1q dep.) or -reconstituted serum (C1q dep.+C1q) and the virus-only or the virus treated with NHS served as the controls. Remaining virus was detected by plaque assay. The data represent the average and standard deviation of values from six independent experiments, with ** denoting significance at a P value of ≤0.001.
Treatment of CHPV with NHS causes a differential shift in the mobility of virus particles through a sucrose gradient.
Having established a role for complement in CHPV neutralization, it was imperative to identify the mechanism of complement-mediated neutralization. Marked differences in mechanisms have been observed among viruses, which include virolysis, aggregation, and opsonization (33). Sucrose gradient ultracentrifugation was utilized to study the migration pattern of either the virus alone or virus incubated with NHS. Gradient-purified CHPV (50 μg) was incubated with either PBS or NHS (1:5 dilution) for 30 min at 37°C, and the samples were then layered on top of a 15% to 60% linear sucrose gradient and subjected to ultracentrifugation at 150,000 × g for 4 h at 4°C. Fractions of approximately 0.25 ml were collected by puncturing the bottom of the tube, and samples were subjected to SDS-PAGE and Western blotting. The blots were initially probed with anti-CHPV antibodies to detect the virus-containing fractions. Reactivity to CHPV antibodies was observed between the 26th to 32nd fractions from the bottom in the CHPV+PBS gradient (Fig. 7A). Interestingly, in the CHPV+NHS gradient, a significant shift in viral fractions (8 fractions) was observed, with the first reactive fraction from the bottom being the 17th and the last one being the 23rd (Fig. 7B). Purified CHPV (Fig. 7A and B, marked V) served as the positive control. Reactivity to CHPV antibodies was not observed in any other fractions, including the top of the gradient, suggesting that exposure to NHS did not result in virolysis. The shift in the migration pattern could be due to binding of complement proteins to the virus. Complement activation by many pathogens results in deposition of multiple complement proteins, most predominantly C3b. The peak fractions from the CHPV+NHS gradients (17–21) were further analyzed for the presence of complement proteins. All the peak fractions showed marked reactivity to C3 antibodies, suggesting association of C3 components with the virion (Fig. 7C). Since C1q was earlier shown to bind to CHPV (Fig. 5), we also tested if C1q is present in the viral fractions reactive to C3 antibodies. As can be seen in Fig. 7D, C1q could be detected in all the fractions between 17 and 21, suggesting comigration of C1q with CHPV.
FIG 7.

Differential shift in Chandipura virus fractions upon NHS treatment is due to association of complement components. Gradient-purified CHPV was incubated with PBS or NHS (1:5) and layered on top of a 15% to 60% linear sucrose gradient and subjected to ultracentrifugation. Western blot analysis was carried out on the fractions collected. Panel A represents the CHPV+PBS samples (fraction 25 to 32 shows reactivity to anti-CHPV antibodies), while blots in panel B represent the CHPV+NHS samples (fractions 17 to 24 are reactive to anti-CHPV antibodies). The letter V in every blot represents purified CHPV run as a control. The labels G, N, and M indicate CHPV proteins glycoprotein G, nucleocapsid, and matrix protein, respectively. The peak fractions were also probed to detect complement component C3 (C) and C1q (D) association. Purified proteins C3b and C1q were used as controls and are labeled thus in the blots.
To check the infectivity of the particles in the sucrose gradient, plaque assays were carried out on the peak fractions of CHPV+PBS and CHPV+NHS based on reactivity to anti-CHPV antibodies. Hardly any plaques could be visualized in the CHPV+NHS fractions even at the 10−2 dilution, while the PBS-treated fractions had an uncountable number of plaques at the 10−5 dilution (Fig. 8A). The viral titers of the CHPV+PBS samples were in the range of 1 × 107 to 108 PFU/ml (Fig. 8B). To further confirm the absence of infective particles in the CHPV+NHS samples, Vero cells infected with the respective samples were subjected to immunostaining followed by confocal analysis. Marked staining for viral antigens was observed only in the wells infected with PBS-treated CHPV, with hardly any signal in the NHS-treated samples (Fig. 8C). These observations clearly demonstrate that CHPV is susceptible to complement factors in NHS, rendering the virus noninfectious.
FIG 8.
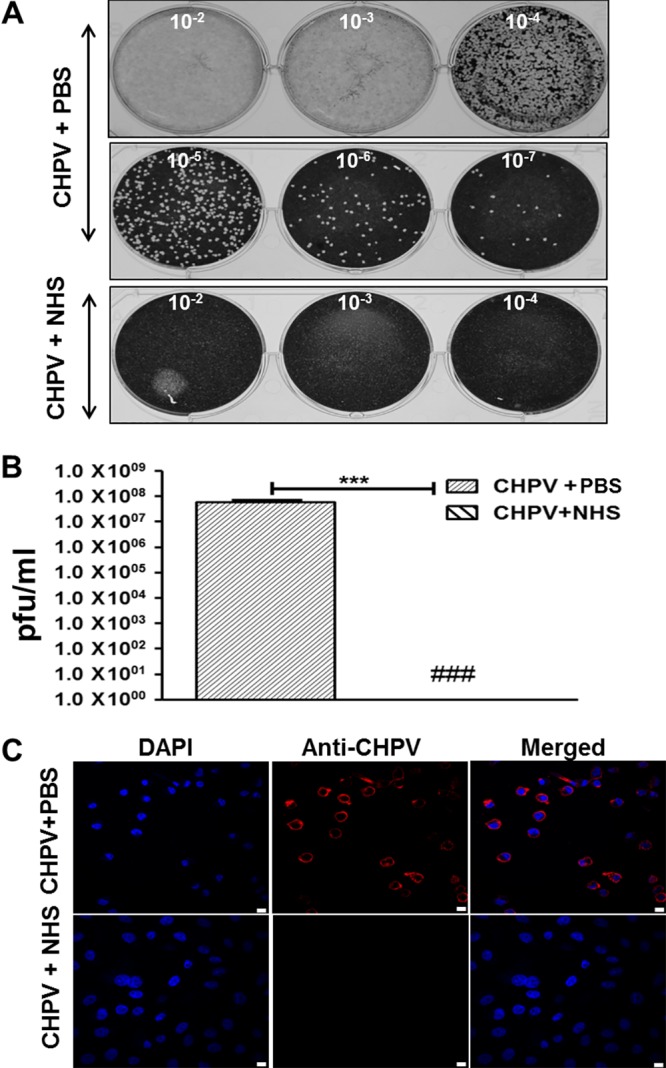
Peak fractions of CHPV treated with PBS and not NHS retain infectivity. Gradient-purified CHPV treated with either PBS or NHS was subjected to ultracentrifugation, and the peak fractions showing reactivity to anti-CHPV antibodies were used for plaque reduction and immunofluorescence assays. Photograph of plaques in monolayers of Vero cells in 6-well plates infected with either the peak fractions of PBS or NHS-treated CHPV is shown in (A). The respective dilution (10−2, 10−3, etc.) is marked within each well. Serum treatment resulted in marked reduction in the number of plaques compared to that of PBS treatment (A, bottom; B). The reduction in the number of plaques was statistically significant; ### denotes that no plaques were visualized, and *** denotes a P value of ≤0.001 (B). Immunofluorescence images showing the difference in infectivity of the peak gradient fractions of either PBS or NHS-treated samples. The scale bar in the images equals 10 μm.
To further confirm binding of complement proteins to CHPV, electron microscopy (EM) analysis was carried out by adsorbing 2 × 106 PFU of CHPV onto EM grids followed by incubation with NHS (1:10) and probing for C3, C4, and C1q deposition. From the EM images it may be inferred that complement components C3 (Fig. 9, top-middle and right), C4 (Fig. 9, center-middle and right), and C1q (Fig. 9, bottom-middle and right) are bound to CHPV upon exposure to NHS. No background staining was observed in the control samples stained with only the secondary antibodies. Taken together, these data suggest that interaction of CHPV with human complement results in a marked shift in migration through a sucrose gradient compared with that of the virus by itself. This may be due merely to the association of complement components or could be due to an additive effect of association followed by aggregation.
FIG 9.

Complement components C3, C4, and C1q bind to CHPV. Gradient-purified CHPV adsorbed to gold grids containing carbon, and Formvar was treated with NHS, blocked, and probed for C3, C4, and C1q deposition. The bound antibodies were detected with 12-nm gold-labeled secondary antibody (closed triangles). Anti-CHPV antibody followed by staining with 6-nm gold-labeled secondary antibody (arrows) was used to counterstain viral antigens. The images on the left (top, middle, and bottom) represent the secondary-only controls, while the two images to the right represent NHS-treated samples probed for deposition of C3 (top, right 2 panels), C4 (middle, right 2 panels), and C1q (bottom, right 2 panels). Samples were analyzed in a Jeol TEM with bar representing 0.02 μm.
Neutralization of CHPV by human complement is by aggregation.
Assays pertaining to complement deposition on CHPV by EM were earlier carried out by adsorbing the particles onto the grid, followed by NHS challenge (Fig. 9). In order to mimic the conditions in fluid phase, 2 × 106 PFU of gradient-purified CHPV and 1:5 dilutions of NHS were incubated for 0, 1, 3, 5, and 15 min at 37°C. After incubation, the mixture was loaded onto EM grids prechilled on ice and postadsorption was negatively stained with phosphotungstic acid (PTA) and analyzed using a Jeol TEM. Virus treated with NHS started clumping at an accelerated pace as time progressed, resulting in massive aggregates (Fig. 10A; note the scale bar). This phenomenon correlated with the neutralization data, wherein >80% neutralization was observed as early as 5 min (Fig. 1C). Particles treated with PBS for 15 min were found to be dispersed with no visible aggregation (Fig. 10A, top left). The aggregates, when probed with anti-CHPV and -C3 antibodies, showed positive reactivity to both, suggesting complement-mediated CHPV aggregation (data not shown).
FIG 10.

Neutralization of Chandipura virus by complement is by aggregation. Transmission electron microscopic analysis of the effect of complement on CHPV was carried out by incubating gradient-purified CHPV with NHS, followed by incubation at the indicated time points in minutes (A, top left corner of each image). Postincubation, the samples were layered on top of Formvar and carbon-coated gold grids, followed by negative staining. Note the dispersed particles upon PBS-only treatment (A, top extreme left panel); however, treatment with NHS resulted in aggregation starting as early as the zero time point (A, top middle panel) with massive aggregates forming thereafter. The bar lengths are variable and highlight the aggregation of CHPV over time. Virus treated with either PBS or NHS was subjected to centrifugation at 4,200 × g, and the pellet was examined for CHPV proteins by Western blotting (B). Note the intense signal in the NHS-treated sample compared with that of the PBS treated. G represents the surface glycoprotein of CHPV.
To confirm aggregation, PBS- or NHS-treated CHPV was incubated for 30 min at 37°C and subjected to centrifugation at 4,200 × g for 10 min. A prominent pellet could be visualized in the NHS-treated sample. Irrespective of whether a pellet was visible, the supernatant was removed and protein loading buffer (PLB)-containing DTT was added to the bottom of the tubes, followed by boiling, sonication, and Western blotting. Marked reactivity to CHPV antibodies was observed in the lanes containing NHS-treated CHPV, with very mild reactivity in the PBS-treated sample (Fig. 10B). Thus, exposure to complement resulted in virus aggregation which could pellet even at low centrifugal forces, while the majority of the PBS-treated particles remained in solution. This finding further supported the earlier observation of complement-mediated aggregation.
To test if the terminal complement components contribute to CHPV neutralization, we sought to address the effect of C8-deficient serum on CHPV. Formation of MAC results in virolysis, and C8 is highly essential for MAC assembly. When C8-deficient serum was reconstituted with physiological concentrations of C8, the serum could lyse EA similarly to NHS, while the depleted sera were incapable of causing hemolysis (Fig. 11A). CHPV treated with C8-depleted serum showed a significant reduction in the number of plaques comparable to that of NHS (>80%), and reconstitution did not lead to any further increase. These data further confirm that the terminal complement components do not contribute to CHPV neutralization and also that the inactivation of CHPV is not by virolysis.
FIG 11.

Chandipura virus neutralization is mediated by upstream and not downstream complement factors. (A) Complement component C8 reconstitution of the depleted serum was tested against NHS with the depleted serum serving as the control using the standard hemolytic assay. (B) The complement pathway culminates with the terminal pathway, of which C8 is a critical factor. One hundred PFU of the virus was incubated with either C8-depleted (C8 dep.) or -reconstituted serum (C8 dep.+C8), and CHPV+PBS or CHPV+NHS served as the controls. Significance of the tests and NHS control was compared against the virus-only sample by taking the values from six independent experiments, with ** denoting a P value of ≤0.001.
DISCUSSION
Among the multiple layers of immune resistance faced by viruses, the complement system acts as a potent front-line barrier. The nature of interaction of viruses with the complement system is so complex that major differences in mechanisms exist even among closely related viruses within the same family (11). Chandipura virus is a human pathogen of great clinical significance. How this virus interacts with the complement system had never been investigated before. Our major aim was to study the effect of human complement on CHPV in fluid phase, to dissect the complement pathways involved, and to understand the mechanism of virus neutralization. Both enveloped and nonenveloped viruses have been shown to activate complement to various degrees. Since the complement system lacks memory, the advent of a pathogen can trigger the complement cascade. Chandipura virus was found to be a potent activator of complement, as saturating levels of C3a were generated with minimal virus concentrations and time. Complement activation initiates the amplification loop, leading to complement deposition and virus neutralization in many cases. However, the outcome of complement activation by viruses does not always result in neutralization. A comparison of two paramyxoviruses, PIV5 and Nipah virus (NiV), showed that NiV was resistant to human complement even at a very high concentration, while PIV5 was readily neutralized (34). Chandipura virus was found to be highly sensitive to NHS, with neutralization observed as early as 5 min. Plaque reduction assays carried out with various concentrations of NHS showed readable plaques only at dilutions of 1:10 and above, with hardly any plaques observed at higher concentrations (data not shown). The inability of heat-inactivated serum to neutralize CHPV suggested that the neutralizing factor in NHS is heat labile. Complement proteins are highly heat labile, and the finding that CHPV neutralization is dependent on C3 further confirmed complement as the neutralizing factor.
Complement-mediated neutralization of viruses has been found to be dependent on either single or multiple pathways. Neutralization among the paramyxoviruses PIV5 and mumps virus is largely dependent on the AP, while neutralization of human parainfluenza virus 2 (HPIV2) is dependent on the CP and that of viruses such as West Nile virus has shown dependency on all three pathways (11, 24, 35). Among the rhabdoviruses, the prototypic VSV has been shown to activate complement with resultant neutralization dependent on the CP. The progression of complement pathways is largely dependent on the metal ions Ca2+ and Mg2+. While EDTA can block all three pathways, EGTA, being a calciumion chelating agent, can effectively block both the CP and the LP (32). Interestingly, NHS treated with 15 mM EGTA and 2 mM MgCl2, despite possessing an intact AP, was unable to neutralize CHPV, suggesting that the CP/LP and not the AP is important for CHPV neutralization. Complement component C4 is essential for the progression of both CP and LP, while factor B is a critical component of the AP. The dependency on C4 and not on factor B for CHPV neutralization further confirmed the role of CP or LP in limiting CHPV. Although in the case of CP a role for antibodies can be defined, the overall involvement of viral factors in determining specificity warrants further investigation.
Neutralization of VSV is largely via the CP and is either dependent or independent of antibodies (19, 28, 29). Direct binding of C1q without the involvement of IgM has not often been reported, with a few exceptions, as in dengue virus (NS1) and more recently in Zika virus (NS1 and E protein) (22, 36). Given the sporadic outbreaks, we hypothesized that the overall anti-CHPV antibody pool in the general population would be less and that the probable mechanism is direct binding of C1q to CHPV. Binding studies with immobilized CHPV (by ELISA) and the electron microscopy experiment results supported our hypothesis. Antibodies purified from NHS of local volunteers did not bind to CHPV, were unable to neutralize CHPV, and could not enhance the neutralizing potential of complement (data not shown). This suggested a direct role for C1q in CHPV neutralization. C1q has been shown to have differential effects on many viruses. For example, direct binding of C1q to murine leukemia virus (MuLV) activated the CP, leading to virolysis without the involvement of antibodies (37). In contrast, viruses have also been shown to utilize the initial C1q binding to activate complement and use the resultant opsonization as a tool to evade neutralization, e.g., HIV-1 (20). Although C1q could directly bind to CHPV, the binding did not affect the infectivity of the virus (data not shown). Neutralization of CHPV by C1q-reconstituted serum supports a role for downstream complement components C3 and C4, after the initial C1q interaction. These observations provide ample evidence that CHPV neutralization by complement occurs predominantly via the CP and is largely dependent on C1q.
Complement induces virus neutralization by multiple mechanisms like virolysis, aggregation, and opsonization (33). Virolysis is effected by the assembly of terminal complement components C5b-9, called the membrane attack complex, resulting in disassembly of the virus, e.g., Zika virus and MuLV (22, 37). Complement activation could result in deposition of complement components like C3b and C4b, resulting in further amplification of the complement pathways and virus neutralization, but differences do exist, as seen with NiV, wherein only limited complement deposition was observed (34). Immunoblots of sucrose gradient fractions of CHPV+NHS showed a marked shift in the migration of virus to the denser layers compared with that of the PBS-treated virus, suggesting comigration of complement factors due to complement deposition. Western blotting of the peak fractions and electron microscopy further confirmed the association of complement components C3b and C1q with the virion. The eight-fraction shift in the migration of the CHPV+NHS sample compared with CHPV+PBS alluded to substantial aggregation. The differential shift could be due to covalent binding of complement proteins like C3b or to complement-induced virus aggregation. Of much significance is the finding that the peak fractions of CHPV treated with NHS were unable to infect cells, confirming virus inactivation. Complement deposition experiments employing electron microscopy were carried out by adsorbing CHPV followed by NHS treatment. Such a treatment does not mimic the properties of particles in solution. Incubation of CHPV with NHS in solution showed marked aggregation. It had been demonstrated earlier that exposure to human complement results in gradual aggregation of PIV5; however, this process was more rapid in the case of CHPV (11). The viral factor inducing aggregation and the underlying mechanism need further investigation.
As mentioned earlier, the assembly of C5b-9 (MAC) on the pathogen leads to lysis. The formation of C5 convertase initiates the first step in the terminal phase of the complement cascade, facilitating the proteolytic cleavage of C5 into C5b and C5a. C5b then associates with the other components to form MAC, which includes another key component called C8. Depletion of C8 essentially checks MAC progression, and utilization of C8-depleted serum would provide functional insights into mechanisms like virolysis and aggregation. Treatment of CHPV with C8-depleted serum was in itself sufficient to neutralize the virus by >80%, which suggested that the terminal pathway has a limited role in CHPV neutralization. Further evidence for the absence of virolysis comes from the observation that the top-most fractions of the CHPV+NHS gradients were not reactive to anti-CHPV antibodies, which would have been the reverse in the case of virolysis. This observation is in contrast to the prototypic VSV, for which marked lysis was observed upon exposure to NHS (31). What causes this difference even among related viruses like CHPV and VSV with just a single surface protein, namely, the G protein, is not clear. Such a difference was observed earlier only in the case of the paramyxoviruses PIV5 and mumps virus, which are both known to have two major surface glycoproteins, namely, the fusion protein and hemagglutinin-neuraminidase (11). As CHPV is an enveloped virus, the contribution of the acquired cellular factors cannot be totally eliminated, but the roles of specific viral signatures in determining the mechanism of neutralization cannot be ruled out either. Taken together, the results of our study demonstrate that the human complement system can act as a potent barrier against CHPV infection.
Chandipura virus is an emerging pathogen. A recent report on the screening of sand flies in the Gujarat area of India showed unprecedented levels of CHPV in male sandflies (38). Although we have demonstrated that CHPV is sensitive to serum complement, it is also known to cause considerable mortality. Therefore, the most intriguing question is how the virus disseminates to distal sites, overcoming complement. The levels of complement at local sites are significantly lower than that in the serum; thus, our observation of complement binding to CHPV may have a different outcome at local sites of infection compared with that of the serum. Many RNA viruses have been shown to possess ingenious strategies to evade complement, and whether CHPV also has a certain mechanism to overcome complement needs further investigation. Since no approved therapeutics or vaccine is in place against CHPV, understanding these basic interactions is imperative to developing strategies to target this virus effectively.
MATERIALS AND METHODS
Ethics statement.
Animal experiments were performed after obtaining approval from the Institutional Animal Ethics Committee (IAEC) of the Rajiv Gandhi Centre for Biotechnology, Thiruvananthapuram (RGCB), India. Chandipura virus usage was approved by the Institutional Biosafety Committee (IBSC), RGCB.
Cells and viruses.
Monolayer cultures of Vero cells were grown in Eagle’s minimal essential medium (MEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), penicillin and streptomycin (1,000 U/ml), and l-glutamine (200 mM) and maintained in a humidified CO2 incubator at 5% CO2 at 37°C. Chandipura virus National Institute of Virology (NIV) number (65351.4)/Mk No(M-1461) was kindly provided by the director of the National Institute of Virology, Pune, India. The virus is a designated biosafety level 2 (BSL2) pathogen, and all cultures and experiments involving CHPV were carried out in a biosafety class 2, type B2 cabinet (Esco, Singapore). Viral RNA was isolated from CHPV-infected Vero cells, and cDNA was synthesized. Chandipura virus-specific genes N, M, and G were amplified and subjected to partial sequencing in a 3730xl DNA analyzer (Applied Biosystems, CA). The sequences were found to be 99.9% identical to that of CHPV isolate CIN 0451 (GenBank accession number NC_020805).
Chandipura virus was cultured in Vero cells, and the culture supernatant was collected 24 h postinfection and clarified by centrifugation at 3,200 × g for 15 min at room temperature. The harvested virus was stored with 0.75% bovine serum albumin in small aliquots at –80°C. The concentration of CHPV was determined by centrifuging the virus culture supernatant in an Optima XPN-100 ultracentrifuge (Beckman Coulter) at 115,000 × g for 1 h in a Ti 50.2 rotor; the pellet obtained was further purified by centrifugation on a 15% to 60% sucrose gradient (150,000 × g, 10 h, SW41 rotor). The virus band obtained at the interphase of 15% and 60% was collected, pelleted, and resuspended in MEM containing protease inhibitors and stored at –80°C.
Complement reagents.
Normal human serum (NHS) collected from healthy volunteers was pooled and used as a source of complement for the assays. After letting the blood clot for 1 h at 37°C, the clot was retracted and the serum was separated, aliquoted, and stored at –80°C. Complement-inactivated serum was prepared by incubating NHS at 56°C for 30 min. In order to block complement pathways, NHS was treated with 15 mM EGTA and 2 mM MgCl2 specifically for the classical and MBL pathways and with 20 mM EDTA to block all three complement pathways. To dissect the pathway of complement important for CHPV neutralization, C1q-, C3-, C4-, and C8-depleted sera (Complement Technologies, TX) were reconstituted with physiological concentrations of purified human C1q (180 μg/ml), C3 (1.2 mg/ml), C4 (640 μg/ml), and C8 (80 μg/ml) (Complement Technologies) and used in CHPV neutralization assays.
Hemolytic assays.
Complement-depleted serum and complement-depleted serum reconstituted with the specific depleted proteins were compared with NHS by measuring the total hemolytic complement activity (CH50) for classical pathways as described earlier (11). Normal human serum (NHS) or complement component-depleted NHS or the respective reconstituted NHS diluted 2-fold from a starting dilution of 1:50 was mixed with 5 μl of antibody-sensitized sheep erythrocytes (EA; 1 × 109 cells/ml), and the volume was made up to 250 μl with dextrose-gelatin-Veronal buffered saline containing 0.5 mM MgCl2 and 0.15 mM CaCl2 supplemented with 2.5% dextrose (DGVB++; Complement Technologies, TX) and incubated at 37°C for 30 min with vortexing at regular intervals. For the alternative pathway, total hemolytic complement activity (AH50) was measured by an alternative pathway-specific hemolytic assay (modified from reference 39). Briefly, 2-fold-diluted NHS or reconstituted serum was mixed with 10 μl of rabbit erythrocytes (RbE; 1 × 108 cells/ml), and the volume was made up to 300 μl with AP buffer (gelatin Veronal buffer without calcium or magnesium containing 5 mM EGTA and 5 mM MgCl2). The mixture was incubated at 37°C for 30 min with intermittent vortexing. For both AP and CP hemolytic assays, the reactions were stopped by placing samples on ice, immediately followed by centrifugation at 4°C for 5 min at 1,400 × g, and hemolysis in 200 μl of the supernatant was measured by reading the absorbance at 405 nm for CP and at 412 nm for AP using a multimode plate reader (Tecan, Switzerland). Erythrocytes (EA or RbE) treated with water or incubated with buffer only served as positive and negative controls, respectively, in both assays.
Virus neutralization assays.
Complement-dependent neutralization assays were carried out as described previously with slight modifications (11). One hundred PFUs of CHPV was incubated with NHS, heat-inactivated serum (HI), or complement-depleted (C3, C4, C1q, factor B, or C8) or -reconstituted serum at a dilution of 1:10. Virus incubated with PBS only served as the control. After incubation at 37°C for variable times (time course assay) or 30 min (for all other experiments), the viral titers were determined by plaque assay. Reduction in the number of plaques from the input virus (100 PFU) represented virus neutralization. Individual data points represented an average and standard deviation from six independent experiments, and the Student’s t test was used for statistical analysis.
Complement activation assays.
Complement activation by CHPV, both concentration and time dependent, was monitored by determining the level of C3a generated by Western blotting. In order to determine the effect of virus concentration on complement activation, various concentrations of sucrose gradient-purified CHPV (0.18, 0.25, 0.38, 0.5, 0.75, 1.0, 1.5, and 2 μg) were incubated with NHS (1:10 dilution) for 15 min. For the time course assay, 1 μg of CHPV was incubated with NHS (1:10) for an increasing period of time starting from 1 min to 30 min. The reactions were stopped by adding SDS sample loading buffer containing DTT and boiling at 100°C for 5 min. Following SDS-PAGE and Western blotting, the C3a generated was detected with goat anti-human C3a antibody (Complement Technologies, TX). The positive control included NHS incubated with both CHPV and anti-CHPV antibodies. Background levels of C3a were determined from the serum-only controls.
ELISA.
Equal concentrations of sucrose gradient-purified CHPV were added to MaxiSorp 96-well ELISA plates (Nunc) and incubated at 4°C overnight. Following the coating step, the wells were blocked with 200 μl of PBS containing 2% skimmed milk for 2 h at room temperature (RT). Two-fold dilutions of purified C1q in PBS (500 to 0.18 ng) were added to the wells and incubated at 37°C for 1 h, and wells were washed five times with PBS containing 0.2% Tween before incubation for 1 h with goat anti-human C1q antibody (Complement Technologies, TX) followed by rabbit anti-goat secondary antibody (Bio-Rad Laboratories, CA). The horseradish peroxidase (HRP) substrate 2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid) (ABTS; Roche, Switzerland) was added to each well and incubated in the dark for 20 min, and absorbance was determined at 405 nm on a Spark 10M multimode plate reader (Tecan, Switzerland). The data represented are the averages and standard deviations from three independent experiments.
Electron microscopy.
C3, C4, and C1q deposition on the virus particles was analyzed by incubating 10 μl of 3 × 108 PFU/ml purified virus particles on Formvar carbon film on 200-mesh gold grids (Electron Microscopy Sciences, PA) at RT for 1 h. Adsorbed virus was treated with 10 μl of a 1:10 dilution of NHS and probed with goat anti-human C3, C4, and C1q antibodies at a dilution of 1:10. Mouse polyclonal antibody against CHPV at a 1:10 dilution was used to probe for viral antigens. Bound antibody was then detected with 12- nm colloidal gold anti-goat and 6-nm colloidal gold anti-mouse antibodies (Jackson ImmunoResearch Laboratories, PA) at a 1:20 dilution. Particles were then negative stained with 2% phosphotungstic acid (PTA; pH 6.6) and analyzed under a JEM 1011 transmission electron microscope (Jeol, MA).
To study the effects of complement under physiological conditions, purified CHPV particles were mixed with NHS at a dilution of 1:5 and incubated for various time points (0, 3, 5, and 15 min) at 37°C, followed by adsorption of the samples on Formvar and carbon-coated 200-mesh gold grids. After adsorption for 5 min on ice, grids were washed with PBS and negatively stained with 2% PTA, followed by transmission electron microscopy.
Ultracentrifugation and Western blotting.
Chandipura virus (sucrose gradient purified; 10 μl of 3 × 108 PFU/ml) was mixed with a 1:5 dilution of NHS and incubated at 37°C for 30 min. The reaction mixture was layered on top of a 15% to 60% preequilibrated linear sucrose gradient in water. The samples were centrifuged in an Optima XPN-100 model ultracentrifuge (Beckman Coulter, CA) at 150,000 × g for 4 h at 4°C in a SW41 rotor. After centrifugation, 0.25-ml fractions were collected by puncturing the bottom of the tube, and the fractions were analyzed by SDS-PAGE followed by Western blotting as described previously (11) using mouse anti-CHPV antibodies. Alternatively, blots were probed with goat anti-human C3 (1:2,500), goat anti-human C1q, and goat anti-human C4 (1:1,000) from Complement Technologies to detect the associated complement proteins. Blots were visualized using West-Pico chemiluminescent substrate (Pierce, IL) followed by exposure to X-ray film.
The peak viral fractions obtained after ultracentrifugation were also used in plaque reduction and immunofluorescence assays. Plaque reduction assays were performed on these fractions by carrying out 10-fold dilutions in series. The latter part of the plaque assay was carried out as described earlier.
Benchtop centrifugation and immunofluorescence assays.
Five microliters of gradient-purified CHPV (0.7 mg/ml) was incubated with a 1:5 dilution of NHS or PBS in a total volume of 20 μl for 15 min at 37°C. The samples were centrifuged at 4,200 × g for 5 min at 4°C in an Eppendorf benchtop centrifuge. After centrifugation, the supernatant was removed and PLB containing DTT was added to the tubes, vortexed, and boiled at 95°C for 5 min. The samples were then analyzed by Western blotting to detect CHPV antigens using in-house-generated mouse anti-CHPV polyclonal antibodies.
Immunofluorescence assays were carried out by infecting monolayers of Vero cells in chamber slides with 0.1 ml of the peak viral fractions. After 12 h, the cells were fixed with 3.7% paraformaldehyde and probed with anti-CHPV antibody (1:400 dilution), followed by goat anti-mouse Alexa Flour 633 antibody (Invitrogen, NY). The cells were mounted with ProLong Gold antifade mountant with DAPI (Invitrogen), and images were acquired using a Nikon A1Rsi laser scanning confocal microscope (Japan) and processed using NIS Elements software.
Polyclonal antibody generation.
Wild-type BALB/c male mice were bred and housed in the pathogen-free barrier-maintained animal facility of RGCB. For generating anti-CHPV hyperimmune sera, approximately 1,000 PFU of UV-inactivated CHPV diluted in 250 μl PBS mixed with an equal volume of Freund’s complete adjuvant was injected via the intraperitoneal route followed by three booster doses in Freund’s incomplete adjuvant (Sigma-Aldrich, CA). Seroconversion was validated by performing ELISA, Western blotting, and immunofluorescence using the sera from the immunized mice. Preimmune sera were used as the negative control in all the assays. Only the immune sera were found to react with the CHPV antigens in both Western blots and infected Vero cells. ELISA performed by coating gradient-purified CHPV showed that the immune sera had a significant antibody titer.
ACKNOWLEDGMENTS
We thank M. Radhakrishna Pillai, Director, RGCB for his constant support. D.T. Mourya, Director, NIV, and A. Basu are duly acknowledged for providing Chandipura virus. We thank Jackson James, Arumugam Rajavelu, and K. R. Mahendran for critically reviewing the manuscript and for their valuable comments. We also acknowledge the RGCB Transmission Electron Microscopy and Image Processing Core and the Animal Research Facility and their respective personnel Gopi Krishnan and V. Jiji and Vishnu Sunil Kumar for services rendered.
This work was supported by Department of Science and Technology-Science and Engineering Research Board (DST-SERB) grant SERB-ECR/2015/000261, Department of Biotechnology-Ramalingaswami Fellowship BT/RLF/Re-entry/29/2012, a Ministry of Science and Technology grant (to J.B.J.), and a KSCSTE and UGC, India, fellowship (to U.K.).
We declare no conflicts of interest.
REFERENCES
- 1.Walport MJ. 2001. Complement. N Engl J Med 344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 2.Wallis R. 2007. Interactions between mannose-binding lectin and MASPs during complement activation by the lectin pathway. Immunobiology 212:289–299. doi: 10.1016/j.imbio.2006.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carroll MC. 2004. The complement system in regulation of adaptive immunity. Nat Immunol 5:981–986. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- 4.Gasque P. 2004. Complement: a unique innate immune sensor for danger signals. Mol Immunol 41:1089–1098. doi: 10.1016/j.molimm.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 5.Roozendaal R, Carroll MC. 2006. Emerging patterns in complement-mediated pathogen recognition. Cell 125:29–32. doi: 10.1016/j.cell.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 6.Kemper C, Atkinson JP. 2007. T-cell regulation: with complements from innate immunity. Nat Rev Immunol 7:9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]
- 7.Kopf M, Abel B, Gallimore A, Carroll M, Bachmann MF. 2002. Complement component C3 promotes T-cell priming and lung migration to control acute influenza virus infection. Nat Med 8:373–378. doi: 10.1038/nm0402-373. [DOI] [PubMed] [Google Scholar]
- 8.Carroll MC, Isenman DE. 2012. Regulation of humoral immunity by complement. Immunity 37:199–207. doi: 10.1016/j.immuni.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoermer KA, Morrison TE. 2011. Complement and viral pathogenesis. Virology 411:362–373. doi: 10.1016/j.virol.2010.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mc Sharry JJ, Pickering RJ, Caliguiri LA. 1981. Activation of the alternative complement pathway by enveloped viruses containing limited amounts of sialic acid. Virology 114:507–515. doi: 10.1016/0042-6822(81)90230-0. [DOI] [PubMed] [Google Scholar]
- 11.Johnson JB, Capraro GA, Parks GD. 2008. Differential mechanisms of complement-mediated neutralization of the closely related paramyxoviruses simian virus 5 and mumps virus. Virology 376:112–123. doi: 10.1016/j.virol.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirsch RL, Winkelstein JA, Griffin DE. 1980. The role of complement in viral infections. III. Activation of the classical and alternative complement pathways by Sindbis virus. J Immunol 124:2507–2510. [PubMed] [Google Scholar]
- 13.Mold C, Bradt BM, Nemerow GR, Cooper NR. 1988. Activation of the alternative complement pathway by EBV and the viral envelope glycoprotein, gp350. J Immunol 140:3867–3874. [PubMed] [Google Scholar]
- 14.McConnell I, Klein G, Lint TF, Lachmann PJ. 1978. Activation of the alternative complement pathway by human B cell lymphoma lines is associated with Epstein‐Barr virus transformation of the cells. Eur J Immunol 8:453–458. doi: 10.1002/eji.1830080702. [DOI] [PubMed] [Google Scholar]
- 15.Hirsch RL, Winkelstein JA, Griffin DE. 1981. Host modification of Sindbis virus sialic acid content influences alternative pathway activation and virus clearance. J Immunol 127:1740–1743. [PubMed] [Google Scholar]
- 16.Hartshorn KL, Sastry K, White MR, Anders EM, Super M, Ezekowitz RA, Tauber AI. 1993. Human mannose-binding protein functions as an opsonin for influenza A viruses. J Clin Invest 91:1414–1420. doi: 10.1172/JCI116345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gadjeva M, Paludan SR, Thiel S, Slavov V, Ruseva M, Eriksson K, Lowhagen GB, Shi L, Takahashi K, Ezekowitz A, Jensenius JC. 2004. Mannan-binding lectin modulates the response to HSV-2 infection. Clin Exp Immunol 138:304–311. doi: 10.1111/j.1365-2249.2004.02616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishii Y, Shimomura H, Itoh M, Miyake M, Ikeda F, Miyaike J, Fujioka SI, Iwasaki Y, Tsuji H, Tsuji T. 2001. Cold activation of serum complement in patients with chronic hepatitis C: study on activating pathway and involvement of IgG. Acta Med Okayama 55:229–236. [DOI] [PubMed] [Google Scholar]
- 19.Beebe DP, Cooper NR. 1981. Neutralization of vesicular stomatitis virus (VSV) by human complement requires a natural IgM antibody present in human serum. J Immunol 126:1562–1568. [PubMed] [Google Scholar]
- 20.Ebenbichler CF, Thielens NM, Vornhagen R, Marschang P, Arlaud GJ, Dierich MP. 1991. Human immunodeficiency virus type 1 activates the classical pathway of complement by direct C1 binding through specific sites in the transmembrane glycoprotein gp41. J Exp Med 174:1417–1424. doi: 10.1084/jem.174.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikeda F, Haraguchi Y, Jinno A, Iino Y, Morishita Y, Shiraki H, Hoshino H. 1998. Human complement component C1q inhibits the infectivity of cell-free HTLV-I. J Immunol 161:5712–5719. [PubMed] [Google Scholar]
- 22.Schiela B, Bernklau S, Malekshahi Z, Deutschmann D, Koske I, Banki Z, Thielens NM, Würzner R, Speth C, Weiss G, Stiasny K, Steinmann E, Stoiber H. 2018. Active human complement reduces the Zika virus load via formation of the membrane-attack complex. Front Immunol 9:2177. doi: 10.3389/fimmu.2018.02177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rattan A, Pawar SD, Nawadkar R, Kulkarni N, Lal G, Mullick J, Sahu A. 2017. Synergy between the classical and alternative pathways of complement is essential for conferring effective protection against the pandemic influenza A (H1N1) 2009 virus infection. PLoS Pathog 13:e1006248. doi: 10.1371/journal.ppat.1006248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehlhop E, Diamond MS. 2006. Protective immune responses against West Nile virus are primed by distinct complement activation pathways. J Exp Med 203:1371–1381. doi: 10.1084/jem.20052388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhatt PN, Rodrigues FM. 1967. Chandipura: a new arbovirus isolated in India from patients with febrile illness. Indian J Med Res 55:1295–1305. [PubMed] [Google Scholar]
- 26.Sudeep AB, Bondre VP, Gurav YK, Gokhale MD, Sapkal GN, Mavale MS, George RP, Mishra AC. 2014. Isolation of Chandipura virus (Vesiculovirus: Rhabdoviridae) from Sergentomia sp. of sand flies from Nagpur, Maharashtra, India. Ind J Med Res 139:769–772. [PMC free article] [PubMed] [Google Scholar]
- 27.Sapkal GN, Sawant PM, Mourya DT. 2018. Chandipura viral encephalitis: a brief review. Open Virol J 12:44–51. doi: 10.2174/1874357901812010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leddy JP, Simons RL, Douglas RG. 1977. Effect of selective complement deficiency on the rate of neutralization of enveloped viruses by human sera. J Immunol 118:28–34. [PubMed] [Google Scholar]
- 29.Mills BJ, Beebe DP, Cooper NR. 1979. Antibody-independent neutralization of vesicular stomatitis virus by human complement. II. Formation of VSV-lipoprotein complexes in human serum and complement-dependent viral lysis. J Immunol 123:2518–2524. [PubMed] [Google Scholar]
- 30.Mills BJ, Cooper NR. 1978. Antibody-independent neutralization of vesicular stomatitis virus by human complement: I. Complement requirements. J Immunol 121:1549–1557. [PubMed] [Google Scholar]
- 31.Johnson JB, Lyles DS, Alexander MA, Parks GD. 2012. Virion associated complement regulator CD55 is more potent than CD46 in mediating resistance of mumps virus and vesicular stomatitis virus to neutralization. J Virol 86:9929–9940. doi: 10.1128/JVI.01154-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Des Prez RM, Bryan CS, Hawiger J, Colley DG. 1975. Function of the classical and alternate pathways of human complement in serum treated with ethylene glycol tetraacetic acid and MgClII-ethylene glycol tetraacetic acid. Infect Immun 11:1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernet J, Mullick J, Singh AK, Sahu A. 2003. Viral mimicry of the complement system. J Biosci 28:249–264. doi: 10.1007/BF02970145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson JB, Borisevich V, Rockx B, Parks GD. 2015. A novel factor I activity in Nipah virus inhibits human complement pathways through cleavage of C3b. J Virol 89:989–998. doi: 10.1128/JVI.02427-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vasantha S, Coelingh KL, Murphy BR, Dourmashkin RR, Hammer CH, Frank MM, Fries LF. 1988. Interactions of a non-neutralizing IgM antibody and complement in parainfluenza virus neutralization. Virology 167:433–441. [PubMed] [Google Scholar]
- 36.Douradinha B, McBurney SP, Soares de Melo KM, Smith AP, Krishna NK, Barratt-Boyes SM, Evans JD, Nascimento EJ, Marques ET Jr. 2014. C1q binding to dengue virus decreases levels of infection and inflammatory molecules transcription in THP-1 cells. Virus Res 179:231–234. doi: 10.1016/j.virusres.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bartholomew RM, Esser AF, Müller-Eberhard HJ. 1978. Lysis of oncornaviruses by human serum. Isolation of the viral complement (C1) receptor and identification as p15E. J Exp Med 147:844–853. doi: 10.1084/jem.147.3.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Damle RG, Sankararaman V, Bhide VS, Jadhav VK, Walimbe AM, Gokhale MD. 2018. Molecular evidence of Chandipura virus from Sergentomyia species of sand flies in Gujarat. Jpn J Infect Dis 71:247–249. doi: 10.7883/yoken.JJID.2017.397. [DOI] [PubMed] [Google Scholar]
- 39.Morgan BP. 2000. Measurement of complement hemolytic activity, generation of complement depleted sera, and production of hemolytic intermediates. Methods Mol Biol 150:61–71. [DOI] [PubMed] [Google Scholar]