

Human T-cell leukemia virus type 1 (HTLV-1) causes a variety of diseases, ranging from a fatal form of leukemia to immune-mediated inflammatory diseases. These diseases occur rarely, arising from one or a small subset of virally infected cells infrequently evolving into a pathogenic state. Thus, the process of HTLV-1 cell-to-cell transmission within the host helps influence the probability of disease development. HTLV-1 primarily infects T cells and initially spreads within this cell population when virally infected T cells dock to uninfected target T cells and then transfer HTLV-1 virus particles to the target cells. Here we found that the viral protein HTLV-1 bZIP factor (HBZ) promotes infectivity. HBZ accomplishes this task by increasing the surface abundance of a cellular adhesion protein known as intercellular adhesion molecule 1 (ICAM-1), which helps initiate and stabilize contact (docking) between infected and target T cells. These results define a novel and unexpected function of HBZ, diverging from its defined functions in cellular survival and proliferation.
KEYWORDS: Cell-to cell infection, HBZ, HTLV-1, ICAM-1, retrovirus
ABSTRACT
Human T-cell leukemia virus type 1 (HTLV-1) causes multiple pathological effects, ranging from a form of leukemia to a spectrum of inflammation-mediated diseases. These diseases arise from one or several infected CD4+ T cells among thousands acquiring proliferation and survival advantages and ultimately becoming pathogenic. Given the low incidence of HTLV-1-associated diseases among carriers, such cellular evolutionary processes appear to occur rarely. Therefore, infectious spread of HTLV-1 within the T-cell population may be one underlying factor influencing disease development. Free HTLV-1 virions are poorly infectious, so infection of T cells relies on direct contact between infected and target cells. Following contact, virions pass to target cells through a virological synapse or cellular conduits or are transferred to target cells within an extracellular matrix. Lymphocyte functioning antigen 1 (LFA-1) on the surface of the target cell engaging with its ligand, ICAM-1, on the surface of the infected cell (effector cell) initiates and stabilizes cell-cell contact for infection. We found that stable expression of an HTLV-1 accessory protein, HTLV-1 bZIP factor (HBZ), in Jurkat T cells increases homotypic aggregation. This phenotype was attributed to elevated ICAM-1 expression in the presence of HBZ. Using a single-cycle replication-dependent luciferase assay, we found that HBZ expression in Jurkat cells (used as effector cells) increases HTLV-1 infection. Despite this effect, HBZ could not replace the critical infection-related functions of the HTLV-1 regulatory protein Tax. However, in HTLV-1-infected T cells, knockdown of HBZ expression did lead to a decrease in infection efficiency. These overall results suggest that HBZ contributes to HTLV-1 infectivity.
IMPORTANCE Human T-cell leukemia virus type 1 (HTLV-1) causes a variety of diseases, ranging from a fatal form of leukemia to immune-mediated inflammatory diseases. These diseases occur rarely, arising from one or a small subset of virally infected cells infrequently evolving into a pathogenic state. Thus, the process of HTLV-1 cell-to-cell transmission within the host helps influence the probability of disease development. HTLV-1 primarily infects T cells and initially spreads within this cell population when virally infected T cells dock to uninfected target T cells and then transfer HTLV-1 virus particles to the target cells. Here we found that the viral protein HTLV-1 bZIP factor (HBZ) promotes infectivity. HBZ accomplishes this task by increasing the surface abundance of a cellular adhesion protein known as intercellular adhesion molecule 1 (ICAM-1), which helps initiate and stabilize contact (docking) between infected and target T cells. These results define a novel and unexpected function of HBZ, diverging from its defined functions in cellular survival and proliferation.
INTRODUCTION
Globally, approximately 10 million people are estimated to be infected with a complex retrovirus known as human T-cell leukemia virus type 1 (HTLV-1) (1). De novo viral transmission occurs through contact with body fluids containing infected cells, such as blood, semen, and breast milk. Following transmission, the virus can infect a variety of cell types; however, in vivo, the vast majority of infected cells are CD4+ T cells (2, 3). In line with this pattern, pathological effects associated with HTLV-1 are often linked to the expansion of specific infected CD4+ T-cell clones (defined by the chromosomal integration site of the provirus). Such expansions are associated mainly with two distinct diseases caused by HTLV-1: a fatal malignancy known as adult T-cell leukemia (ATL; monoclonal expansion) and a progressive neurodegenerative disorder known as HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP; oligoclonal expansion) (4, 5). In addition, HTLV-1 infection is associated with a variety of other immune-inflammatory disorders. Interestingly, these diseases develop in only a small portion (∼5%) of the HTLV-1-infected population (6), suggesting that the chance of a newly infected T cell acquiring the survivability, proliferative, and pathogenic properties to ultimately elicit disease is extremely low. This premise is supported by estimates that healthy HTLV-1 carriers harbor tens of thousands of infected T-cell clones (7). One factor that is believed to contribute to the potential for an infected T cell to ultimately cause disease (especially HAM/TSP) is the chromosomal integration site of the provirus affecting levels of viral gene transcription (7, 8). Considering that HTLV-1 integration is a near-random event (9), infectious spread of HTLV-1 in the T-cell population appears to be an important determinant of disease development.
As infection of T cells by free HTLV-1 virions is extremely inefficient (10–12), T-cell infection occurs through mechanisms involving contact between an effector cell (infected with HTLV-1) and a target cell. In the context of T-cell-to-T-cell infection, initiation and stabilization of effector cell-target cell contact is mediated by intercellular adhesion molecule 1 (ICAM-1) on the surface of the effector T cell engaging with lymphocyte functioning antigen 1 (LFA-1) on the target cell (13, 14). Once these contacts are established, viral particles can be transferred to the target cell by one of three known mechanisms that potentially serve to circumvent immune surveillance (15). One mechanism involves polarization of the effector cell and transfer of viral particles through a specialized microenvironment known as a viral synapse that forms at the cell-cell interphase (16). Another mechanism involves transfer of viral particles to the target cell as part of an extracellular matrix known as a viral biofilm (17). A third mechanism involves the transfer of viral particles through cellular conduits extending from the effector to the target cell (18).
Among the nonstructural HTLV-1 proteins, Tax and p8 have been shown to regulate events involved in cell-to-cell infection. While p8 functions in conduit formation (18), Tax appears to have a more central and multifaceted role in viral infection. It increases the expression of ICAM-1, which is important for effector cell-target cell interactions (19). It also assists in polarizing the microtubule-organizing center (MTOC) in the infected cell toward the contacted target cell (14), which appears to be essential to achieving a functional virological synapse (13, 16). In addition, it may activate expression of certain cellular genes involved in forming the viral biofilm (17). Finally, it activates expression of Fascin and Gem, both of which regulate actin dynamics to promote HTLV-1 infection (20, 21).
In this study, we provide evidence that in addition to Tax and p8, HTLV-1 basic leucine zipper protein (HBZ) participates in HTLV-1 infection. HBZ is known to localize to the nucleus, where it affects gene expression through interactions with a variety of transcriptional regulators (22). Using Jurkat T cells, we have established clonal cell lines that express HBZ or carry the empty expression vector. Interestingly, the HBZ-expressing cells display increased homotypic aggregation compared to the empty-vector cells. We attributed this phenotype to an increase in ICAM-1 expression and found that ICAM-1 expression is also elevated by HBZ expression in SupT1 and HeLa cells. As effector cells in single-cycle replication-dependent luciferase assays, the HBZ-expressing Jurkat clones exhibited a higher proficiency for infecting target cells than the empty-vector clones. Consistent with this observation, knocking down HBZ expression in HTLV-1-infected SLB-1 cells reduced the infection efficiency of these cells. Finally, while HBZ increased the efficiency of infection, it was incapable of replacing the essential functions of Tax in this process. These overall results support a novel role for HBZ in the infectious spread of HTLV-1.
RESULTS
HBZ promotes homotypic aggregation through a mechanism involving LFA-1.
To better understand the molecular function of HBZ in T cells, we established clonal cell lines from Jurkat T cells that constitutively express HBZ and control clonal cell lines containing the empty pcDNA3.1 expression vector. To compare quantities of the HBZ protein in these cell lines to those in HTLV-1-infected cell lines, we performed a Western blot analysis of HBZ that was precipitated from cell lysates through its high-affinity interaction with the KIX domain of the cellular coactivator CBP (23). Overall, the abundance of HBZ in the clones was higher than in the infected cells (Fig. 1A). In culture, the HBZ clones displayed marked homotypic aggregation, which was not apparent with the empty-vector clones but is a typical phenotype of HTLV-1-infected T-cell lines (Fig. 1B and C).
FIG 1.

HBZ expression in Jurkat cells stimulates homotypic aggregation. (A) Western blot (WB) detection of HBZ expression in Jurkat clonal cell lines and HTLV-1-infected T-cell lines. HBZ was affinity purified from whole-cell extracts using the CH1-KIX domain of CBP fused to GST. The upper panel shows the membrane probed with an HBZ antibody. The lanes shown are from the same membrane/same scanned image. The lower panel shows the same membrane stained with Ponceau S. An asterisk denotes a nonspecific band. (B) Homotypic aggregation of the indicated T-cell lines. The cells indicated were plated at 5 × 105 cells/ml, at which time cellular aggregates were completely disrupted. Cells were cultured for 2 h to allow aggregates to reform and then were photographed. (C) Homotypic aggregation of the indicated empty-vector and HBZ-expressing Jurkat clones. The cells indicated were plated at 1 × 106 cells/ml, at which time cellular aggregates were completely disrupted. Cells were cultured for 3 h to allow aggregates to reform and then were photographed.
Given that integrins are major effectors of T-cell adhesion (24), we tested whether they played a role in the aggregation of the HBZ clones. Integrins contain a variety of divalent metal ion-binding sites that can affect their ligand interactions (25). We found that chelation of divalent metal ions in the culture medium with EDTA, but not EGTA (which favors Ca2+), blocked aggregation of both the HBZ clones and the HTLV-1-infected T-cell lines (Fig. 2A). In complementary experiments, the HBZ clones and HTLV-1-infected cells failed to form clusters in medium lacking divalent metal ions but underwent aggregation following the addition of Mg2+ but not Ca2+ (Fig. 2B). Together, these observations implied that an integrin was involved in the homotypic aggregation displayed by the HBZ clones. More specifically, they indicate the involvement of LFA-1, which is a heterodimer composed of the β2 (CD18) and αL (CD11a) integrins. Indeed, while Ca2+ increases the affinity of some integrin complexes for their ligands, it inhibits adhesion by LFA-1 (25).
FIG 2.

Mg2+ induces homotypic aggregation of HBZ-expressing Jurkat clones. (A) Effects of divalent metal ion chelators on homotypic aggregation. The cells indicated were plated at 5 × 105 cells/ml in the absence or present of the indicated chelators, at which time cellular aggregates were completely disrupted. Cells were cultured for 6 h to allow to aggregates to reform and were then photographed. (B) Effects of Mg2+ and Ca2+ on homotypic aggregation. The cells indicated were plated at 5 × 105 cells/ml in HEPES-buffered Tyrode’s solution without Mg2+ or Ca2+ (unsupplemented) or containing 1 mM Mg2+ or Ca2+ as indicated. Cells were cultured for 6 h and then photographed.
To identify the specific integrin modulating the phenotype, we used a series of antibodies that block integrin interactions. Among those that we tested, only antibodies against β2 (CD18) and αL (CD11a) integrins were found to inhibit aggregation of the HBZ clones (Fig. 3A). Indeed, as shown in Fig. 3A, antibodies against αM and LFA-3, which were among several of the known blocking antibodies we tested, did not affect aggregation of the HBZ clones. In HTLV-1-infected T cells (SLB-1 cells), the antibody against αL also inhibited cell aggregation (Fig. 3B, left panels). Similarly, the antibody against β2 reduced aggregation of another HTLV-1-infected T-cell line (ATL-2s) (Fig. 3B, right panels). In parallel with the divalent metal ion analysis, these results suggested that cell adhesion through LFA-1 controlled the homotypic aggregation of the HBZ clones.
FIG 3.

Antibodies against LFA-1 block homotypic aggregation of HBZ-expressing Jurkat clones. Effects of integrin-blocking antibodies on homotypic aggregation are shown. Empty-vector and HBZ-expressing Jurkat clones (1 × 106 cells/ml) (A) or HTLV-1-infected SLB-1 and ATL-2s cells (2.5 × 105 cells/ml and 4.5 × 105 cells/ml, respectively) (B) were plated in the presence of the indicated antibodies, at which time cellular aggregates were completely disrupted. The Jurkat clones were cultured for 4 h, and the SLB-1 and ATL-2s cells were cultured for 6 h and 2 h, respectively, to allow aggregates to reform prior to being photographed.
HBZ upregulates the LFA-1 ligand ICAM-1 to promote homotypic aggregation.
Given the observed effect of the LFA-1-blocking antibodies, we were interested in testing whether HBZ increases expression of the ITGB2 and/or ITGAL genes that encode β2 and αL, respectively. Such a mechanism would coincide with the general role of HBZ in regulating gene expression through its interactions with cellular transcriptional regulators in the nucleus (22). Unexpectedly, we did not observe a significant difference in ITGB2 or ITGAL mRNA levels between the Jurkat HBZ and empty-vector clones (Fig. 4A). Furthermore, there was no difference in the cell surface abundance of these LFA-1 subunits (Fig. 4B). We also analyzed the cell surface abundance of LFA-1 in its open, high-affinity conformation, which is known to augment cell adhesion, using an antibody that specifically binds the open conformation of the β2 subunit (26). Despite two of three empty-vector clones displaying higher levels of the activated form of LFA-1 (Fig. 4C), the average geometric mean intensity of the three empty-vector clones was not significantly greater than that of the HBZ clones in this and replicate experiments (data not shown). These results show that HBZ does not affect expression of LFA-1 or inside-out signaling that modulates transition to the high-affinity conformation of LFA-1.
FIG 4.

HBZ does not affect LFA-1 expression or activation. (A) LFA-1 mRNA levels in empty-vector and HBZ-expressing Jurkat clones. The graph on the left shows qRT-PCR results for the ITGAL gene, which expresses the αL subunit of LFA-1. The graph on the right shows qRT-PCR results for the ITGB2 gene, which expresses the β2 subunit of LFA-1. Data are the average of results of three independent experiments and were normalized to values for the empty-vector clone C4 (set to 1). Error bars show standard deviations. (B) Flow cytometry analysis of LFA-1 on empty-vector and HBZ-expressing Jurkat clones. Histograms in the left panel show relative cell surface levels of αL, and histograms in the right panel show relative cell surface levels of β2 (empty-vector cells, yellow; HBZ-expressing cells, blue; secondary antibody probing clone G8, gray). (C) Flow cytometry analysis of activated LFA-1 on empty-vector and HBZ-expressing Jurkat clones. The histograms show the relative cell surface levels of the activated conformation of the β2 subunit of LFA-1 (empty-vector cells, yellow; HBZ-expressing cells, blue; secondary antibody probing clone G8, gray).
These results led us to examine whether HBZ influenced expression of an LFA-1 ligand, ICAM-1. In first comparing gene expression between the sets of Jurkat clones, we found that the ICAM1 mRNA levels were higher in the HBZ clones than in the empty-vector clones (Fig. 5A). Moreover, ICAM-1 cell surface abundance was higher for the HBZ clones (Fig. 5B). We additionally examined ICAM-1 expression in other cell lines that had been modified to stably express HBZ. Using another T-cell line, SupT1, we established cells that stably express HBZ or carry the empty expression vector (Fig. 5C). As with the Jurkat clones, we observed higher ICAM-1 expression in the SupT1 cells expressing HBZ (Fig. 5D). This trend extended to HeLa cells (Fig. 6A).
FIG 5.

HBZ upregulates ICAM-1 expression. (A) ICAM1 mRNA levels in empty-vector and HBZ-expressing Jurkat clones. The graph shows qRT-PCR data averaged from seven independent experiments, with data normalized to values for empty-vector clone C4 (set to 1). Error bars show standard deviations. Significance was determined by a one-way ANOVA comparing the clones (P < 0.001), followed by a Tukey honestly significant difference (HSD) test; double asterisks denote a P value of <0.01 as the highest P value among pairwise comparisons between empty-vector and HBZ clones. (B) Flow cytometry analysis of ICAM-1 on empty-vector and HBZ-expressing Jurkat clonal cells. Histograms on the left show relative cell surface levels of ICAM-1 (empty-vector clones, yellow; HBZ-expressing clones, blue; secondary antibody probing clone F10, gray). The bar graph on the right shows the geometric mean fluorescence intensities (Geo-MFI) averaged for the three empty-vector clones versus the three HBZ-expressing clones. Data represent one of three independent experiments. Error bars show standard deviations. Significance was determined by a two-tailed Student's t test (**, P < 0.01). (C) Western blot analysis of whole-cell extracts prepared from SupT1 cells stably transduced with an HBZ expression vector or the empty vector and, for comparison, selected Jurkat clones. Membranes were probed with an antibody against Myc to detect the C-terminal epitope tag on HBZ and then stripped and probed for actin. (D) Flow cytometry analysis of ICAM-1 on empty-vector and HBZ-expressing SupT1 cells. Histograms show the relative cell surface levels of ICAM-1 (empty-vector cells, yellow; HBZ-expressing cells, blue; secondary antibody probing empty-vector cells, gray).
FIG 6.

Among the ICAM family members, HBZ-mediated activation is restricted to ICAM-1 and does not depend on JunD. (A) ICAM1 mRNA levels in empty-vector and HBZ-expressing HeLa clones. The graph shows qRT-PCR data averaged from three independent experiments, with data normalized to values for the empty-vector clone (set to 1). Mutations in the activation domain, leucine zipper domain, and the start codon of HBZ are indicated as mutAD, mutZIP, and mutATG, respectively; wt, wild type. Error bars show standard deviations. Significance was determined by a two-tailed Student's t test (***, P < 0.001). Western blot analysis was performed using whole-cell extracts prepared from HeLa clones. Membranes were probed with an antibody against Myc to detect the C-terminal epitope tag on HBZ and actin. (B) JUND and ICAM1 mRNA levels in an HBZ wild-type-expressing HeLa clone following siRNA-mediated knockdown of JUND. The graph shows qRT-PCR data averaged from three independent experiments for ICAM1 and two experiments for JUND, with data normalized to values for the control siRNA (set to 1). (C) ICAM2, ICAM3, ICAM4, and ICAM5 mRNA levels in empty-vector and HBZ-expressing Jurkat clones. The graph shows qRT-PCR data averaged from three independent experiments, with data normalized to values for empty-vector clone C2 (set to 1). Error bars show standard deviations.
We also analyzed ICAM1 mRNA levels in HeLa clonal cell lines expressing each of the following HBZ mutants: HBZ mutAD contains LL→AA substitutions in the two LXXLL motifs in the N-terminal activation domain (AD), which disables binding to the cellular coactivator CBP/p300 (27); HBZ mutZIP contains L→C substitutions in the second and fourth heptad repeat of the leucine zipper, which disables binding to cellular bZIP factors (28); and HBZ mutATG contains an A→T substitution in the start codon that prevents translation of the transcript. We found that cells expressing HBZ mutAD contained levels of ICAM1 mRNA comparable to those of cells expressing wild-type HBZ, while cells expressing HBZ mutZIP or HBZ mutATG contained significantly lower levels of the transcript (Fig. 6A). These results imply that HBZ regulates ICAM-1 expression through an interaction with a cellular bZIP factor.
Among the cellular bZIP factors bound by HBZ, JunD stood out as possibly being involved in HBZ-mediated activation of ICAM-1 expression. JunD is a component of the transcription factor AP-1, which has been shown to directly regulate ICAM1 transcription (29), and importantly, heterodimers composed of HBZ and JunD have been shown to activate transcription from AP-1 sites and through DNA-bound Sp1 (30–33). Indeed, in addition to harboring AP-1-binding sites, the ICAM-1 promoter contains an Sp1 binding site (29). To test whether JunD played a role in the activation of ICAM1 transcription, we transfected the HBZ-expressing HeLa cells with small interfering RNA (siRNA) targeting JunD or, separately, with control siRNA. An analysis of mRNA from the transfected cells indicated the efficient knockdown of JunD; however, this effect was not accompanied by a significant reduction in ICAM1 mRNA (Fig. 6B). This pattern suggests that JunD is not involved in the activation of ICAM-1 expression by HBZ. Finally, it is important to note that HBZ did not affect mRNA levels of the other ICAM family members (Fig. 6C). Overall, these results indicated that the enhanced homotypic aggregation caused by HBZ involves increased expression of the LFA-1 ligand ICAM-1.
As previously reported (34), ICAM1 mRNA levels were found to be elevated in HTLV-1-infected T cells, which we confirmed (Fig. 7A). This effect was reported to involve activation of ICAM1 transcription by Tax (19, 35). Given our findings, we compared levels of ICAM-1 activation elicited by Tax versus HBZ. For this analysis, we used the clonal empty-vector and HBZ-expressing HeLa cell lines and HeLa cells transiently transfected with a Tax expression vector (pSG-Tax) or the corresponding empty vector (pSG5). An analysis of both ICAM1 mRNA levels and the cell surface abundance of ICAM-1 showed that each viral protein produced a similar increase in ICAM-1 expression, with Tax causing a slightly greater increase in activation (Fig. 7B and C).
FIG 7.

Levels of ICAM1 activation by HBZ and Tax are similar. (A) ICAM1 mRNA levels in the indicated T-cell lines. The graph shows qRT-PCR results averaged from two independent experiments in which values were normalized to those for the resting CD4 T cells (set to 1). Numbers above the bars indicate fold differences relative to the resting CD4 T cells. TL-Om1 cells (dark gray bar) express only HBZ, as the proviral sense strand-encoding genes are turned off. (B) ICAM1 mRNA levels from the empty-vector cells, HBZ-expressing HeLa clone, and Tax-transfected HeLa cells. The graph shows qRT-PCR data averaged from three independent experiments, with data normalized to values from cells carrying the empty vector (pcDNA or pSG5; set to 1). Error bars show standard deviations. Western blot analysis was performed using whole-cell extracts prepared from the empty-vector cells, HBZ-expressing HeLa clone, and Tax-transfected HeLa cells. Membranes were probed with an antibody against His to detect the C-terminal epitope tag on HBZ and Tax and then stripped and probed for actin. (C) Flow cytometry analysis of ICAM-1 on the empty-vector cells, HBZ-expressing HeLa clone, and Tax-transfected HeLa cells. Histograms on the left show relative cell surface levels of ICAM-1 on the HBZ-expressing clone, and histograms in the middle show relative cell surface levels of ICAM-1 on Tax-transfected cells (empty-vector cells, yellow; HBZ- and Tax-expressing cells, blue; isotype control antibody probing the HBZ- and Tax-expressing cells, gray). The bar graph on the right shows the fold change in the geometric mean fluorescence intensities (Geo-MFI) between empty-vector and HBZ- or Tax-expressing cells.
To test whether ICAM-1 contributed to homotypic aggregation of the HBZ clones, we first tested whether an ICAM-1-blocking antibody disrupted cell-cell interactions in the culture. Like the LFA-1 antibodies, the ICAM-1-blocking antibody inhibited formation of aggregates by the HBZ clones, although not to the same extent as the LFA-1 antibodies (Fig. 8A, upper panels). This difference may reflect a variation in the blocking capabilities between the LFA-1 and ICAM-1 antibodies. Indeed, despite the known function of ICAM-1 in modulating adhesion of HTLV-1-infected T cells (34), the ICAM-1 antibody we used also appeared to be less effective than the LFA-1 antibodies in disrupting SLB-1 cell aggregates (Fig. 8A, lower panels). To further analyze ICAM-1-mediated aggregation, we tested whether an LFA-1 peptide also inhibited homotypic aggregation of the HBZ clones. The blocking peptide used consisted of amino acids 466 to 491 of the αL subunit (GenBank accession no. AAC31672), which was previously reported to block ICAM-1, thereby disrupting T-cell aggregation (36). Like the LFA-1- and ICAM-1-blocking antibodies, the αL peptide disrupted homotypic aggregation of the HBZ clones and the HTLV-1-infected cells (Fig. 8B).
FIG 8.

Blocking adhesion through ICAM-1 disrupts homotypic aggregation of HBZ-expressing Jurkat clones. (A) Effects of an ICAM-1-blocking antibody on homotypic aggregation. Upper panels, HBZ-expressing Jurkat clonal cells were plated at 5 × 105 cells/ml and cultured for 6 h prior to being photographed. Lower panels, HTLV-1-infected SLB-1 cells were plated at 1 × 106 cells/ml and cultured for 8.5 h prior to being photographed. At time zero, cellular aggregates were completely disrupted, and the antibodies indicated were added. The graph shows relative areas of aggregates averaged from three independent experiments, with the average aggregate area from the antibody-treated cells normalized to the average aggregate area from untreated cells (set to 100). The asterisk denotes a P value of <0.05 for a two-tailed Student's t test. Aggregate areas were determined using ImageJ. (B) Effects of an αL peptide on homotypic aggregation. HBZ-expressing Jurkat cells (upper panels) or HTLV-1-infected SLB-1 cells (lower panels) were plated at 5 × 105 cells/ml in the presence of the αL peptide or the DMSO vehicle, at which time cellular aggregates were completely disrupted. Cells were cultured for 6 h to allow aggregates to reform and were then photographed.
HBZ enhances the efficiency of HTLV-1 infection.
These findings led to the hypothesis that HBZ participates in the HTLV-1 infection process, as infection between T cells occurs through direct cell-cell contact that involves ICAM-1 on the surface of the infected cell engaging with LFA-1 on the target cell (13). This hypothesis was supported by findings that the HBZ-expressing Jurkat clones were able to incorporate normal Jurkat cells (defined as not having been transfected/transduced) into the cellular aggregates (Fig. 9A), showing that HBZ-expressing cells efficiently adhere to cells lacking the viral protein. To compare the infection efficiencies of the HBZ-expressing and empty-vector clones, we used the single-cycle replication-dependent luciferase assay developed by Mazurov et al. (37). In these assays, we cotransfected the Jurkat clones with the pCMV-HT1Δenv packaging vector that is defective for Env expression, the pCRU5H-inLuc reporter plasmid, and either the CMV-ENV expression vector or a negative-control CMV-ENVΔPvuII in which the env gene contains an early nonsense mutation (38). Mazurov et al. provided a detailed illustration of this system in the original study in which this system was developed and used (37). Based on this system, the transfected Jurkat clones produce HTLV-1 virus-like particles containing a luciferase trans-gene, defining these cells in the assay as effector cells. Infection of target cells with virus-like particles leads to luciferase expression in the target cells. In initial experiments, using normal Jurkat cells as the target cells, we observed significantly higher luciferase activity produced from cocultures with effector cells that were the HBZ-expressing clones than from those with effector cells that were the empty-vector clones (Fig. 9B). These results indicated that HBZ increases the efficiency of HTLV-1 infection.
FIG 9.

HBZ increases the infection efficiency of Jurkat cells when used as effector cells in infection assays. (A) Jurkat cells expressing HBZ form aggregates with HBZ-negative Jurkat cells. Jurkat cells stably expressing HBZ (6.7 × 105 cells/ml) were stained with Hoechst 33342 and combined with an equal quantity of unstained normal Jurkat cells. Cellular aggregates were then completely disrupted, and cells were cocultured for 2 h and then photographed. (B) As effector cells, the HBZ-expressing Jurkat clones produce higher levels of infection than the empty-vector clones. Empty-vector and HBZ-expressing clones were cotransfected with pCMV-HT1Δenv, pCRU5H-inLuc, and either CMV-ENV (light bars) or CMV-ENVΔPvuII (dark bars) and cocultured with normal Jurkat cells. Luciferase assays were performed 48 h later. The graph shows infection as a measure of relative luminescence units (RLUs) normalized to the highest value in each experiment (set to 100). The effector cells used are indicated. Data were averaged from five independent experiments. Error bars show standard deviations. Significance was determined by a one-way ANOVA comparing the clones (P < 0.001), followed by a Tukey’s HSD test; an asterisk denotes a P value of <0.05 as the highest P value among pairwise comparisons between empty-vector and HBZ clones. The Western blot shows Tax expression in the indicated Jurkat clones cotransfected with pCMV-HT1Δenv, pCRU5H-inLuc, and CMV-ENV. The membrane was stripped and reprobed with an antibody against actin. (C) As effector cells, the Jurkat clones infect the intended target cells. HBZ-expressing cells (light bars; Jurkat clone F10) and empty-vector cells (dark bars; Jurkat clone C4) were transfected with pCMV-HT1Δenv, pCRU5H-inLuc, and either CMV-ENV or CMV-ENVΔPvuII and cocultured with normal CHO or CHO-LFA-1 cells for 1.5 to 2 h. Effector cells were then removed, and luciferase assays were performed on the CHO-LFA-1 cells 48 h later. The graph shows infection as a measure of relative luminescence units (RLUs) produced by the indicated effector cells with data averaged from five replicates per treatment from a single experiment. The graph is representative of three independent experiments. The horizontal dashed line shows the averaged background values (effector cells cotransfected with CMV-ENVΔPvuII). Error bars show standard deviations. Significance was determined by a two-tailed Student's t test (*, P < 0.05). The Western blot shows αL and β2 expression in CHO-LFA-1 cells. The membrane was probed for each LFA-1 subunit and actin. (D) Blocking ICAM-1 reduces the level of infection. HBZ-expressing Jurkat cells were cotransfected with pCMV-HT1Δenv, pCRU5H-inLuc, and CMV-ENV and cocultured with normal Jurkat cells in the absence or presence of an ICAM-1-blocking antibody. Luciferase assays were performed 48 h later. The graph shows data from five independent experiments, with infection shown as a measure of relative luminescence units (RLUs): luminescence from the antibody-treated coculture was normalized to luminescence from the untreated cocultures (set to 100) in each experiment. ***, P < 0.001 (for a two-tailed Student's t test).
While we showed that the HBZ clones formed clusters with parental Jurkat cells, it was possible that the efficient infection in cocultures containing the HBZ clones was due to preferential infection of other HBZ clones rather than the intended normal Jurkat cells. Therefore, assays were performed using adherent CHO cells stably expressing LFA-1 as the target cells (Fig. 9C, right panel). In these assays, the transfected Jurkat clones were washed out of the wells following coculture, leaving only the adherent CHO-LFA-1 cells. Therefore, luciferase activity corresponded specifically to infection of the CHO-LFA-1 cells. Results from these assays showed that the CHO-LFA-1 cells exhibited significantly higher luciferase activity when cocultured with effector HBZ clones than when cocultured with the effector empty-vector clones (Fig. 9C). These observations verify that the HBZ clones exhibit an increased infection efficiency toward the intended target cells.
We were also interested in confirming the positive role of ICAM-1 in HTLV-1 infection. For this purpose, we used the single-cycle replication-dependent luciferase assay to examine the effect of an ICAM-1-blocking antibody on infection. In these experiments, HBZ-expressing Jurkat cells were used as effector cells and normal Jurkat cells were used as target cells. We found that the addition of the blocking antibody to cocultures caused a significant reduction in luciferase activity (Fig. 9D). This observation provides evidence that ICAM-1 expression contributes to the efficiency of HTLV-1 infection.
HBZ does not functionally replace Tax in modulating HTLV-1 infection.
T-cell-to-T-cell infection of HTLV-1 was previously shown to be modulated by the viral protein Tax (14, 20, 37). Given the substantial increase in infection efficiency in Jurkat cells expressing HBZ, we tested whether HBZ could replace Tax specifically with respect to its infection-related functions. To test this hypothesis, we used the single-cycle replication-dependent luciferase assay, in which pCMV-HT1Δenv was replaced with the pCMVHT1M-Tax9Q packaging vector that expresses Env but not Tax (37). To compare infection efficiencies in the absence and presence of Tax, cells were also cotransfected with pSG5 or pSG-Tax, respectively. We found that independent of the presence of HBZ, luciferase activity did not exceed the background signal in the absence of Tax (Fig. 10), suggesting that HBZ is unable to replace essential functions of Tax in mediating infection.
FIG 10.
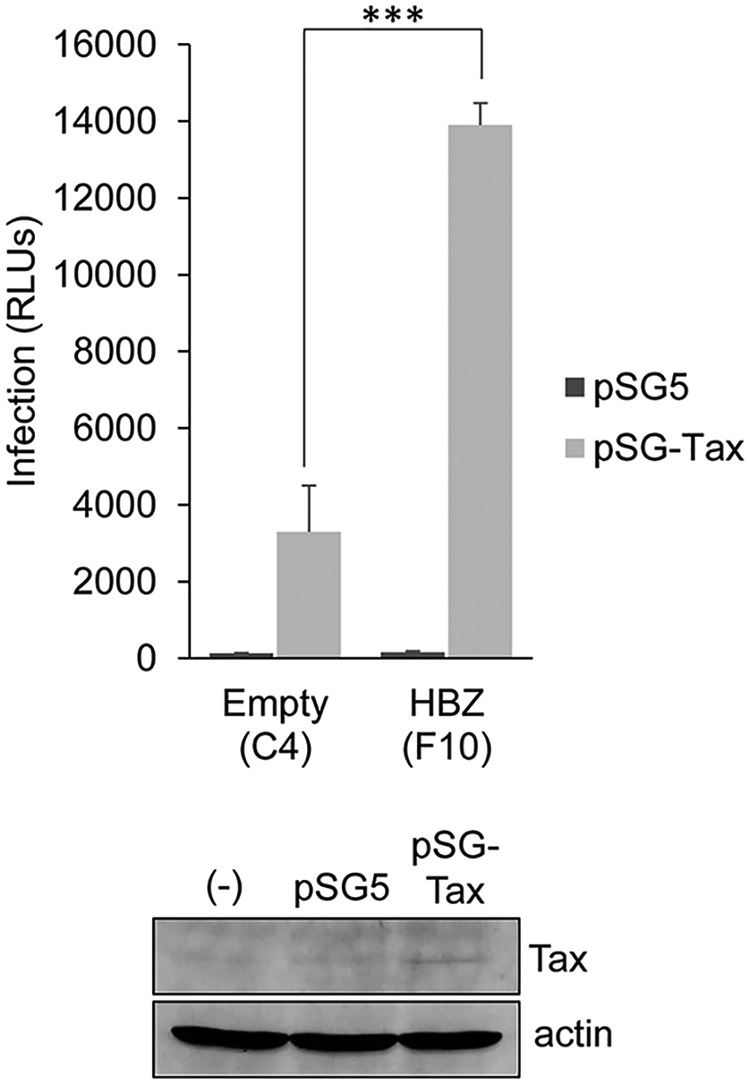
HBZ does not replace Tax during the infection process. The HBZ-expressing clone F10 and the empty-vector clone C4 were transfected with pCMVHT1M-Tax9Q, pCRU5H-inLuc, and either pSG-Tax (light bars) or pSG5 (dark bars) and cocultured with CHO-LFA-1 cells for 1.5 to 2 h. Effector cells were then removed, and luciferase assays were performed on the CHO-LFA-1 cells 48 h later. The graph shows infection as a measure of relative luminescence units (RLUs) produced by the indicated effector cells, with data averaged from three replicates per treatment from a single experiment. The graph is representative of three independent experiments. Error bars show standard deviations. Significance was determined by a two-tailed Student's t test (***, P < 0.001). The Western blot shows Tax expression in the F10 Jurkat clone cotransfected with pCMVHT1M-Tax9Q, pCRU5H-inLuc, and pSG5 (lane pSG5) or pSG-Tax (lane pSG-Tax). (-), untransfected Jurkat cells.
Low expression of HBZ is sufficient to enhance the efficiency of infection.
The Jurkat clones used in initial single-cycle replication-dependent luciferase assays expressed higher levels of HBZ than chronically infected HTLV-1-infected T-cell lines (see Fig. 1A), raising the possibility that the increased infection efficiency of the Jurkat clones requires abnormally high expression of HBZ. Therefore, we tested a Jurkat clone that expresses a low level of HBZ (Fig. 11A). This clone was established by electroporation, while the clones with high HBZ expression were established using a cationic lipid transfection reagent. Like the other HBZ-expressing clones, this clone displayed the aggregation phenotype (Fig. 11B). Interestingly, the cell surface abundance of ICAM-1 on the clone with low HBZ expression was elevated to a level similar to that on clones with high HBZ expression (Fig. 11C). Furthermore, in single-cycle replication-dependent luciferase assays, the clone with low HBZ expression exhibited an infection efficiency similar to that of the clones with high HBZ expression (Fig. 11D). These observations suggest that lower and potentially more physiologically relevant levels of HBZ are sufficient to enhance infection.
FIG 11.

Jurkat cells with low HBZ expression maintain the ability to increase infection efficiency. (A) HBZ expression in Jurkat clone G6 is lower than in the other clones. Whole-cell extracts from the indicated Jurkat clonal cell lines were analyzed by Western blotting using an antibody against the C-terminal 6×His epitope tag (His) on HBZ. The membrane was stripped and reprobed with an antibody against actin. (B) Homotypic aggregation of the G6 clone. Cells were plated at 1 × 106 cells/ml, at which time cellular aggregates were completely disrupted. Cells were cultured for 3 h to allow aggregates to reform and then were photographed. (C) Cell surface abundance of ICAM-1 is elevated in Jurkat cells with low HBZ expression. Histograms show the relative cell surface abundance of ICAM-1 on the Jurkat empty-vector clones (yellow; C2, C4, and C5) and HBZ-expressing clones (blue; F10 and G6). Histograms for clones F10 and G6 are indicated by arrows. The gray histogram represents clone C2 cells probed with secondary antibody. (D) As effector cells, Jurkat cells with low HBZ expression produce higher levels of infection than the empty-vector clones. The indicated HBZ-expressing (light bars) and empty-vector (dark bars) Jurkat clones were transfected with pCMVHT1M-Tax9Q, pCRU5H-inLuc, and pSG-Tax. Clone F10 was separately cotransfected with pSG5 in place of pSG-Tax [F10 (-)]. Effector cells were cocultured with CHO-LFA-1 cells for 1.5 to 2 h and removed, and luciferase assays were performed on the CHO-LFA-1 cells 48 h later. The graph shows infection as a measure of relative luminescence units (RLUs) produced by the indicated effector cells, with data averaged from at least three replicates per treatment from a single experiment. The graph is representative of the results of two independent experiments. Error bars show standard deviations. Significance was determined by a two-tailed Student's t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
HTLV-1-infected T cells with reduced HBZ expression display decreased infection efficiency.
Finally, we were interested in testing whether HBZ contributes to infection in the context of naturally infected T cells, as the single-cycle replication-dependent luciferase assay might not fully recapitulate the precise infection functions modulated through the HTLV-1 provirus. We established a clonal cell line expressing a short hairpin RNA (shRNA) vector targeting the dominant splice 1 isoform of HBZ and a clonal line containing the empty expression vector (39). These cells were produced from the HTLV-1-infected T-cell line SLB-1, which exhibits relatively higher expression of HBZ than most of the other HTLV-1-infected T-cell lines that we have tested (23). Cells expressing the shRNA targeting HBZ were confirmed to have lower quantities of HBZ mRNA and protein than cells with the empty vector (Fig. 12A and C). In contrast, in both SLB-1 cell lines, HTLV-1 Env and Tax expression levels were relatively similar (Fig. 12B). To verify that these cells were deficient for HBZ-mediated functions, we examined BDNF mRNA levels; BDNF transcription is upregulated by HBZ (23). In the three separate RNA specimen sets prepared from the clones, BDNF mRNA levels were lower in the HBZ knockdown clones than in the empty-vector clones (Fig. 12C, lower panels), which is indicative of effects caused by knockdown of HBZ. In examining ICAM-1 expression, we observed lower levels of the transcript and the cell surface protein with the HBZ knockdown cells (Fig. 12C and D). To test the infection efficiency of these lines, we established Jurkat cells stably transfected with pminLUC-viral cyclic AMP response element (CRE) (40), which contains the luciferase reporter gene regulated by three tandem copies of the viral CRE found proximal to the transcription start site in the HTLV-1 promoter. Therefore, following infection with HTLV-1, these cells will express the viral trans-activator protein Tax, which will then activate transcription of the luciferase gene from the viral CREs (17). In infection assays using the SLB-1 clones as effector cells and the Jurkat pminLUC-viral CRE cells as target cells, significantly lower luciferase activity was obtained from cocultures containing the HBZ knockdown cells than from the empty-vector cells (Fig. 12E). This observation supports a legitimate role for HBZ in facilitating HTLV-1 infection.
FIG 12.

Knockdown of HBZ in HTLV-1-infected T cells reduces infection efficiency. (A) An shRNA targeting the HBZ S1 transcript reduces HBZ expression in SLB-1 cells (HTLV-1 positive). Clonal cell lines were established from SLB-1 cells stably transfected with a vector expressing an shRNA targeting HBZ S1 (shHBZ) or with the empty vector. Chromatin fractions from the indicated cell lines were analyzed by Western blotting using an antibody against the bZIP domain of HBZ (upper panel). The membrane was stained with Ponceau S prior to being blocked and probed (lower panel). (B) Env expression is unaffected by the shRNA targeting the HBZ. Whole-cell extracts from the indicated cell lines were analyzed by Western blotting using an antibody against Env. The membrane was stripped and reprobed with an antibody against actin. (C) Knockdown of HBZ leads to a reduction in the level of ICAM1 and BDNF mRNA. ICAM1, HBZ S1, and BDNF transcript levels were compared between SLB-1 shHBZ and empty-vector cell lines. The graph shows qRT-PCR data averaged from three independent experiments, with data normalized to values for the empty-vector clone (set to 100). Error bars show standard deviations. Significance was determined by a two-tailed Student's t test (**, P < 0.01). The gel shows RT-PCR analysis of BDNF-IV mRNA levels from the three experimental sample sets. (D) Knockdown of HBZ leads to a reduction in cell surface abundance of ICAM-1. Cell surface abundance of ICAM-1 was analyzed by flow cytometry on empty-vector (yellow histogram) and shHBZ (blue histogram) SLB-1 cells. The gray histogram represents shHBZ cells probed with secondary antibody. (E) Knockdown of HBZ leads to a reduction in infection efficiency. The graph shows infection as a measure of relative luminescence units (RLUs) produced by the indicated effector cells cocultured with Jurkat pminLUC-viral CRE cells. Data are averaged from six replicates per treatment from a single experiment. The graph is representative of three independent experiments. Error bars show standard deviations. Significance was determined by a two-tailed Student's t test (***, P < 0.001).
DISCUSSION
In this study, we provide evidence that the HTLV-1 accessory protein HBZ contributes to viral infection. For an HTLV-1-infected T cell to effectively transfer viral particles to a target T cell, it must first establish stable contact with target cells. This cell-cell interaction is initiated and stabilized primarily by ICAM-1 on the surface of the infected cell engaging with its ligand, LFA-1, on the surface of the target cell (13, 16). Our compiled results show that HBZ upregulates ICAM-1 expression, thereby enhancing cell-to-cell adhesion and, in turn, HTLV-1 infection. This function of HBZ is surprising, considering that the viral protein Tax serves as a key modulator of infection and the functions of HBZ often oppose those of Tax (41). While HBZ mirrors Tax in promoting infection, in single-cycle replication-dependent luciferase assays, it cannot replace Tax during the infection process. Indeed, Tax has multiple, diverse functions associated with HTLV-1 infection, including augmenting infected cell-target cell interactions (19, 42), modulating formation of the virological synapse and cytoskeletal remodeling (14, 20, 21), and potentially activating the expression of components of HTLV-1 biofilm-like structures (17). In contrast, our current data indicate that the role of HBZ in HTLV-1 infection is centered on its ability to augment effector cell-target cell interactions. While we show that this role involves upregulation of ICAM-1 expression, we cannot rule out the possibility that other cellular proteins regulated by HBZ contribute to the increased infection efficiency.
Our observations suggest that activation of ICAM-1 expression by HBZ occurs through an indirect mechanism. We found that HBZ upregulates ICAM1 expression in cells established to stably express HBZ, suggesting that long-term expression of the viral protein is required for this phenomenon. This observation is consistent with knockdown of JunD failing to affect ICAM1 mRNA levels in HBZ-expressing cells. Therefore, HBZ/JunD heterodimers, which have been shown to function as transcriptional activators (30–33), do not appear to regulate transcription from AP-1 sites or the Sp1 site in the ICAM1 promoter. Further supporting this premise, an in silico analysis of HBZ-binding sites from published chromatin immunoprecipitation sequencing (ChIP-seq) data (43) did not reveal any peaks of HBZ enrichment within the ICAM1 promoter or in proximity to the gene in ATL cells (data not shown). However, an in silico analysis of other ChIP-seq data from the same study showed overlapping peaks of BATF3 and IRF4 enrichment within the ICAM1 promoter (data not shown). Interestingly, in this previous study, HBZ was shown to associate with a superenhancer to drive BATF3 expression. Therefore, the requirement for long-term expression of HBZ to upregulate ICAM1 may relate to HBZ first activating the expression of BATF3, which then forms a complex with IRF4 to regulate ICAM1 transcription. Further work is needed to test this hypothesis.
In contrast to our results, a previous in vitro analysis did not reveal a role for HBZ in infection (44). The discrepancy between studies may be due to different approaches taken in each analysis. In the previous study, irradiated 729 B cells stably transfected with a wild-type HTLV-1 molecular clone or an HBZ-defective molecular clone were used as effector cells, and infection was measured by immortalization of cocultured peripheral blood mononuclear cells (PBMCs). While differences in the effector cells used in each study may have influenced the results, we believe the main distinction lies in the methods used to measure infection. Compared to the single-cycle replication-dependent luciferase assay and use of the Jurkat pminLUC-viral CRE reporter target cells, immortalization of PBMCs may be a less-specific measure of infectivity, as it involves long-term coculture and is affected by other factors, such as cell proliferation and survival. Interestingly, in this previous report, the same effector cells were also used to examine infection with a rabbit model, and rabbits inoculated with 729 cells containing an HBZ-defective molecular clone produced lower proviral loads than rabbits inoculated with cells carrying the wild-type HTLV-1 molecular clone. Interestingly, infection through a molecular clone expressing HBZ deleted of the leucine zipper also resulted in a lower proviral load. This observation is consistent with our findings that HBZ mutZIP fails to augment ICAM-1 expression (Fig. 4E) and consistent with the seminal role of ICAM-1 in cell-cell-mediated infection of HTLV-1. Therefore, while the results from the rabbit infection were speculated to involve an HBZ-mediated survival effect, they may have alternatively, or in part, arisen from the contribution of HBZ to HTLV-1 infectivity. This possibility coincides with results from a separate animal study in which macaques were inoculated with 729 B cells containing the same HBZ-defective molecular clone used in the rabbit studies (45). This HTLV-1 molecular clone contains an early nonsense mutation in the hbz gene that prevents HBZ translation. Strikingly, during the course of the infection in the macaques, the hbz mutation reverted to the wild-type sequence, which would reestablish expression of HBZ. As with results from the rabbit infection, data from the macaque infection are consistent with a function of HBZ related to HTLV-1 infectivity.
While our analysis showed that HBZ does not replace Tax during the infection process, it does effect a dramatic increase in the efficiency of infection, suggesting that HBZ contributes to the infectious spread of HTLV-1 within the T-cell population in HTLV-1 carriers. Current data suggest that a high frequency of infectious spread of HTLV-1 occurs immediately following the initial transmission of the virus (the acute stage of infection) and then declines significantly, but is not eliminated, once the adaptive immune response is mounted against the virus (the persistent stage of infection) (46–48). Given our results, we expect that HBZ participates in the initial, rapid infectious spread of the virus.
In addition, HBZ might play an especially important role in the ongoing low-level infection occurring during the persistent stage of infection. At this point, HTLV-1-infected T cells expressing Tax are undergoing elimination by the cytotoxic T-cell response due to the high immunogenicity of Tax (49, 50). However, in some infected T cells, expression of Tax becomes repressed due to actions of a variety of trans-acting factors that target the 5′ long terminal repeat (LTR) promoter and through epigenetic modifications within this region of the provirus (51). Even though these cells typically maintain expression of HBZ from the 3′ LTR promoter (52, 53), they are likely to avoid immune-mediated elimination, as the immunogenic response to HBZ is poor (54, 55). Interestingly, Tax repression has been found to be reversible, and consequently, Tax has been found to be transiently present in infected cells (56, 57). This phenomenon is currently implicated in promoting the survival and proliferation of infected cells, while limiting their exposure to immune surveillance. Given that Tax activates the transcription of the provirus and cellular genes, its transient expression might also lead to the accumulation of viral particles and some host factors that contribute to infection, such as components involved in formation of the viral biofilm. It is therefore possible that once Tax repression is reestablished, these cells will remain poised to infect, and HBZ serves to complete the infection process by promoting interactions with target T cells.
The scenario described above does not account for Tax-dependent formation of the virological synapse that is established upon contact of an infected T cell with a target cell. However, it is possible that this structure is dispensable during certain infection events, as transfer of the viral biofilm appears capable of occurring outside the region of contact between the effector and target cell (17). Alternatively, overexpression of ICAM-1 by HBZ may in itself be sufficient to trigger formation of the virological synapse. Indeed, polarization of the MTOC in the infected cell toward the target cell is a key process that marks formation of the virological synapse (16). This event has been found to be controlled by Tax and cell surface engagement of ICAM-1 (13, 14). Considering that Tax activates expression of ICAM-1, it is possible that the role of Tax in MTOC polarization is exclusively to increase the cell surface abundance of ICAM-1, and a subsequent signal through engagement of ICAM-1 modulates MTOC polarization. In support of this premise, Tax mutants that are defective for activation of the CREB and NF-κB pathways are unable to assist in MTOC polarization (58), which coincides with the involvement of potentially both pathways in activating ICAM1 transcription (29). Furthermore, signaling through RhoA and ERK have been reported to be required for MTOC polarization in response to ICAM-1 engagement in HTLV-1-infected cells (29); these are both pathways that, in various cell lines, are activated by ICAM-1 engagement (59). Considering these observations in conjunction with data from our current study and previous results showing that ICAM-1 is upregulated in HTLV-1-infected T cells (34), it would be interesting to analyze signaling through ICAM-1 in HTLV-1-infected T cells and how it relates to the mechanism of viral infection.
MATERIALS AND METHODS
Cell culture and generation of cell lines.
CD4+ lymphocytes, Jurkat, CEM, and HUT-78 cells, and HTLV-1-infected cells (MT-2, C10/MJ, SLB-1, C8166/45, and ATL-2s) were cultured in Iscove's modified Dulbecco medium (IMDM) supplemented with 10% fetal bovine serum (FBS) or 12.5% FetalPlex serum (Gemini Bio-Products) and 2 mM l-glutamine, 100 U/ml penicillin, and 50 μg/ml streptomycin. To obtain CD4+ lymphocytes, human buffy coat (purchased from ZenBio) was centrifuged over Ficoll-Paque Plus medium (GE Healthcare) according to the manufacturer’s instructions. The PBMC fraction was then processed using the CD4+ T-cell isolation kit (Miltenyi Biotec) according to the manufacturer’s instructions. A fraction of the isolated CD4+ lymphocytes was activated in culture wells coated with anti-CD3 and anti-CD28 antibodies. Jurkat pminLUC-viral CRE cells and Jurkat clonal cell lines were supplemented with 1.5 mg/ml G418. SLB-1 clonal cell lines were supplemented with 1 mg/ml G418. HTLV-1 infected cells (1185, TL-Om1, and SP), SupT1-HBZ, and SupT1–empty-vector cells were cultured in RPMI medium supplemented with 10% FBS or 12.5% FetalPlex serum and 2 mM l-glutamine, 100 U/ml penicillin, 50 μg/ml streptomycin, and 0.5 μg/ml puromycin. 1185 and SP cells were cultured in the presence of interleukin 2 (IL-2). HeLa clonal cell lines (28), HEK293T cells, HeLa cells, CHO cells, and CHO-LFA-1 clones were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS or 10% FetalPlex serum, 2 mM l-glutamine, 100 U/ml penicillin, and 50 μg/ml streptomycin. HeLa and CHO clones were supplemented with 0.5 mg/ml G418. The Jurkat clone with low HBZ expression has been described previously (60). Clones with high HBZ expression were established by transfecting Jurkat cells with pcDNA-HBZ-SP1-Myc (61) using Lipofectamine LTX with Plus reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. SLB-1 clones were established by electroporating SLB-1 cells with pRNAT/U6.2 or pRNAT/U6.2shHBZ-V4 (39). Cells (3 × 106) were exposed to a single exponential decay pulse of 250 V/950 μF in 0.4-cm cuvettes containing 10 μg plasmid DNA in 300 μl RPMI–10 mM dextrose–0.1 mM dithiothreitol. Jurkat pminLuc-viral CRE cells were established by using the same procedure, but with the pminLUC-viral CRE plasmid (40). Jurkat and SLB-1 clones were established from single cells as described previously (60). CHO-LFA-1 clones were established by electroporating CHO cells with pAPRM8.LFA-1alpha and pcDNA3.1-CD18 (Addgene plasmids 8630 and 8640, respectively; gifts from T. Springer) (62, 63), as described previously (64), and isolating/expanding cell colonies that survived 0.5 mg/ml G418 selection. SupT1-HBZ and SupT1–empty-vector cells were established by lentivirus transduction as follows. HEK293T cells (5 × 106) were plated on 10-cm dishes, cultured overnight, and then transfected with 39 μg pUMVC, 9.1 μg pHCMV-G, and 49 μg pQCXIP (ClonTech; empty vector) or pQC-HBZ-IP using CaCl2 (pQC-HBZ-IP was established by PCR amplifying the HBZ sequence from pcDNA-HBZ-SP1-Myc and inserting it into the EcoRI site in pQCXIP). The culture medium was replaced 24 h posttransfection with 6 ml supplemented RPMI medium, and the cells were cultured for another 24 h. The culture medium (viral supernatant) was then passed through a 0.4-μm polyethersulfone filter; the transfected HEK293T cells were maintained in culture by adding back 6 ml fresh supplemented RPMI medium. Pelleted SupT1 cells (3 × 106) were resuspended in the filtered viral supernatant with 12 μg/ml Polybrene and cultured overnight. The transduction process was repeated on the same SupT1 cells using the second batch of viral supernatant from the transfected HEK293T cells. Transduced cells were selected with 0.5 μg/ml of puromycin.
Protein fractionation and purification and Western blotting.
Whole-cell extracts were prepared as described previously (65). HBZ was purified from whole-cell extracts through its high-affinity interaction with the KIX domain of CBP as described before (23). However, glutathione S-transferase (GST)-CH1-KIX was used in place of GST-KIX, as the smaller GST-KIX fusion polypeptide obscured detection of HBZ-Myc-6×His expressed by the Jurkat clones. Chromatin fractions (HBZ is enriched in this fraction) were prepared as described previously (66). Western blotting was performed as described previously (65). The antibodies used were as follows: anti-HBZ serum provided by J. M. Mesnard (67); anti-actin clone C4 and anti-Myc clone 4A6 purchased from Millipore-Sigma; anti-6×His (Abcam; ab9108); anti-αL clone MHM24 and anti-B2 clone purchased from the University of Iowa Developmental Studies Hybridoma Bank; and anti-HTLV-1 Env (NIH AIDS Reagent Program; no. 1578).
Cell aggregation assays.
Cells were normalized to 5 × 105 cells/ml and cultured overnight in unsupplemented IMDM. Cells were then collected by centrifugation, suspended in unsupplemented IMDM to 2.5 × 105 to 1 × 106 cells/ml, and transferred to a 96-well plate (200 μl/well). Cells were pipetted up and down to fully dissociate aggregates; elimination of cellular aggregates was verified by light microscopy. Plates were incubated at 37°C for 2 to 8.5 h before photographing the cells with a T-mounted camera on an Olympus CK2 inverted microscope. For some experiments, 5 mM EDTA or EGTA, 5 μg/well of a specific blocking antibody, or 0.25 mM an LFA-1 peptide was added at time zero. Blocking antibodies used in the experiments shown were as follows: anti-αM (M1/70; EMD Millipore), anti-αL (TS1/22; University of Iowa Developmental Studies Hybridoma Bank), anti-β2 (TS1/18; Thermo Fisher Scientific), anti-LFA-3 (TS2/9; Thermo Fisher Scientific), and anti-ICAM-1 (P2A4; EMD Millipore). Antibody stocks did not contain sodium azide. The blocking peptide, GVDVDQDGETELLLIGAPLFYGEQRG (amino acids 466 to 491 of the sequence deposited under GenBank accession no. AAC31672), was reported to disrupt ICAM-1-dependent cell aggregation (36). It was synthesized and purified by GenWay Biotech, reconstituted in dimethyl sulfoxide (DMSO), and supplemented with ammonium hydroxide to adjust the pH to approximately 7. In coculture assays, Jurkat clones (2 × 106 cells) were stained with 0.25 μg/ml Hoechst 33342 (Thermo Fisher Scientific) in 3 ml of unsupplemented IMDM for 20 min, washed three times in unsupplemented IMDM, and then suspended in 3 ml of unsupplemented IMDM. Stained cells were combined with an equal volume/concentration of unstained normal Jurkat cells and cocultured for 2 h. The cells were then photographed with a Nikon DS-Fi3 camera mounted on a Nikon Eclipse E600 fluorescence microscope.
siRNA-mediated knockdown of JunD.
The siGENOME human Jun D (3727) SMART pool M-003900-05-0005 was used to knock down JunD, while the siGENOME nontargeting siRNA pool 1 D-001206-13-05 was used as a control (Dharmacon). Cells were seeded to reach ∼50% confluence on the day of transfection. Cells were transfected with 50 nM siRNA using DharmaFECT 1 siRNA transfection reagent (Dharmacon) according to the manufacturer's instructions. RNA was extracted 48 h after transfection.
RNA extraction, cDNA synthesis, and quantitative reverse transcriptase PCR (qRT-PCR).
RNA was extracted from cells using TRIzol reagent (Thermo Fisher Scientific), and cDNA was synthesized using a RevertAid kit (Thermo Fisher Scientific) as described by the manufacturer. Random hexamer primers were used for all reactions except those analyzing hbz mRNA. Oligo(dT) primers were used for hbz cDNA synthesis. The HBZ-S1 and UBE2D2 PCR primers have been described previously (28). The other PCR primers used in the data presented were as follows: ICAM1-F, 5′-GCTATTCAAACTGCCCTGATG; ICAM1-R, 5′-AGCGTAGGGTAAGGTTCTTG; ITGAL-F, 5′-CACAGGAAGCCTCTATCAGT; ITGAL-R, 5′-GTCTGTTGCCAAGGTCATTC; ITGB2-F, 5′-CATTCTCCTGCTGGTCAT; ITGB2-R, 5′-CTTCACCAAGTGCTCCTAAC. Other primers used in the study are available upon request. Real-time PCR was performed, and relative mRNA levels were determined as described previously (28). Serial dilutions of an appropriate experimental sample were used to generate standard curves for all primer sets included on a PCR plate. From the compilation of all the standard curves for all primers and all PCR plates (analyses), the amplification efficiencies ranged from 80 to 120%, with correlation coefficients ranging from 0.98 to 1.0.
Transfection and infection assays.
Single-cycle replication-dependent luciferase assays were used to quantify cell contact-dependent infection (37). Electroporation was done as described above. Cells were electroporated with 7.1 μg of pCMVHT1M-ΔEnv or pCMVHT1M-Tax9Q (37), 13.2 μg of pCRU5HT1-inLuc (37) and 1.8 μg of CMV-ENV or CMV-ENVΔPvuII (38) (used with pCMVHT1M-ΔEnv) or pSG5 or pSG-Tax (68) (used with pCMVHT1M-Tax9Q) per electroporation cuvette. At 48 h postelectroporation, cells were washed in supplemented IMDM and cocultured with target cells. In experiments using Jurkat cells as target cells, 1 × 106 of both effector and target cells were combined in 5 ml of supplemented IMDM and cocultured for 48 h. For ICAM-1 antibody blocking tests, cells were cocultured in 24-well plates (1 ml/well), and cocultures were supplemented with 20 μg of anti-ICAM-1 antibody (RR1/1; Thermo Fisher Scientific). In experiments using CHO-LFA-1 cells as target cells, 8 × 104 CHO-LFA-1 cells were plated per well in 24-well plates, cultured for 24 h, and then exposed to 5 × 105 effector cells/well for 1.5 to 2 h. The wells were then washed four times with phosphate-buffered saline (PBS) to remove effector cells, and the remaining CHO-LFA-1 cells were cultured in supplemented DMEM for an additional 48 h. In experiments using the SLB-1 clones to infect Jurkat pminLUC-viral CRE cells, 2.5 × 105 effector and target cells were cocultured in 1 ml/well in a 24-well plate for 48 h. Cells were lysed with 100 μl of cell culture lysis reagent or passive lysis buffer (Promega), samples were normalized according to total protein, and luciferase activity was measured using the luciferase assay system (Promega) and a GloMax 20/20 luminometer (Promega). HeLa cells (4 × 105) were transfected with 10 μg of plasmid (10 μg of pSG5 or 5 μg of pSG-Tax-His [69] and 5 μg of pSG5) using calcium phosphate. Cells were collected 24 h after transfection for RNA analysis and whole-cell extract preparation and at 48 h for flow cytometry analysis.
Flow cytometry.
Cells were normalized to 5 × 105 cells/ml and cultured for 24 h prior to analysis. A total of 5 × 105 cells/labeling reaction were collected by centrifugation at 800 × g for 3 min at 4°C, washed once in 2 ml of cold PBS–0.2% bovine serum albumin (BSA) (fluorescence activated cell sorting [FACS] buffer), and recentrifuged, and cell pellets were then suspended in 50 μl of cold FACS buffer to which 2 μg of primary antibody was added. Antibodies used were as follows: anti-αL (TS1/22; University of Iowa Developmental Studies Hybridoma Bank), anti-β2 (TS1/18; Thermo Fisher Scientific), anti-activated β2 (MEM-148; Thermo Fisher Scientific), and anti-ICAM-1 (P2A4; Millipore Corporation). Cells were labeled on ice for 1 h and then washed with 2 ml of FACS buffer. Following centrifugation, cell pellets were suspended in 50 μl of FACS buffer to which 0.25 μg of fluorescein isothiocyanate (FITC) goat anti-mouse Ig (Southern Biotech) was added. Cells were labeled on ice for 30 m, washed with 2 ml of FACS buffer, suspended in 500 μl FACS buffer, and analyzed using a BD LSR II flow cytometer (BD Biosciences). The resulting data were analyzed using Flowing Software 2.5.1 (developed by P. Terho, University of Turku; http://flowingsoftware.btk.fi/index.php?page=1).
Statistical analysis.
Two-tailed Student’s t tests were used for two-group comparisons. A one-way analysis of variance (ANOVA) was used to compare the repeated measures for experiments examining the six Jurkat clones. When statistical significance (P < 0.05) was identified, means were compared using a Tukey post hoc test using JMP Pro 14 (SAS Institute, Inc.).
ACKNOWLEDGMENTS
We thank J. M. Mesnard for the anti-HBZ rabbit serum, M. Matsuoka for the ATL-2s and TL-Om1 cell lines, P. Green for the pRNAT/U6.2shHBZ-V4 plasmid, D. Mazurov for the single-cycle replication-dependent luciferase assay plasmids, and C. Pique for the CMV-ENV and CMV-ENVΔPvuII plasmids.
Funding for this study was provided by the National Institute of Allergy and Infectious Diseases (NIH/NIAID grant AI133412) to I.L.
The funding agency had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Gessain A, Cassar O. 2012. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol 3:388. doi: 10.3389/fmicb.2012.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richardson JH, Edwards AJ, Cruickshank JK, Rudge P, Dalgleish AG. 1990. In vivo cellular tropism of human T-cell leukemia virus type 1. J Virol 64:5682–5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koyanagi Y, Itoyama Y, Nakamura N, Takamatsu K, Kira J, Iwamasa T, Goto I, Yamamoto N. 1993. In vivo infection of human T-cell leukemia virus type I in non-T cells. Virology 196:25–33. doi: 10.1006/viro.1993.1451. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida M, Seiki M, Yamaguchi K, Takatsuki K. 1984. Monoclonal integration of human T-cell leukemia provirus in all primary tumors of adult T-cell leukemia suggests causative role of human T-cell leukemia virus in the disease. Proc Natl Acad Sci U S A 81:2534–2537. doi: 10.1073/pnas.81.8.2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wattel E, Vartanian JP, Pannetier C, Wain-Hobson S. 1995. Clonal expansion of human T-cell leukemia virus type I-infected cells in asymptomatic and symptomatic carriers without malignancy. J Virol 69:2863–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Panfil AR, Martinez MP, Ratner L, Green PL. 2016. Human T-cell leukemia virus-associated malignancy. Curr Opin Virol 20:40–46. doi: 10.1016/j.coviro.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bangham CR, Cook LB, Melamed A. 2014. HTLV-1 clonality in adult T-cell leukaemia and non-malignant HTLV-1 infection. Semin Cancer Biol 26:89–98. doi: 10.1016/j.semcancer.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meekings KN, Leipzig J, Bushman FD, Taylor GP, Bangham CR. 2008. HTLV-1 integration into transcriptionally active genomic regions is associated with proviral expression and with HAM/TSP. PLoS Pathog 4:e1000027. doi: 10.1371/journal.ppat.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doi K, Wu X, Taniguchi Y, Yasunaga J-i, Satou Y, Okayama A, Nosaka K, Matsuoka M. 2005. Preferential selection of human T-cell leukemia virus type I provirus integration sites in leukemic versus carrier states. Blood 106:1048–1053. doi: 10.1182/blood-2004-11-4350. [DOI] [PubMed] [Google Scholar]
- 10.Pique C, Jones KS. 2012. Pathways of cell-cell transmission of HTLV-1. Front Microbiol 3:378. doi: 10.3389/fmicb.2012.00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alais S, Mahieux R, Dutartre H. 2015. Viral source-independent high susceptibility of dendritic cells to human T-cell leukemia virus type 1 infection compared to that of T lymphocytes. J Virol 89:10580–10590. doi: 10.1128/JVI.01799-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meissner ME, Mendonca LM, Zhang W, Mansky LM. 2017. Polymorphic nature of human T-cell leukemia virus type 1 particle cores as revealed through characterization of a chronically infected cell line. J Virol 91:e00369-17. doi: 10.1128/JVI.00369-17.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnard AL, Igakura T, Tanaka Y, Taylor GP, Bangham CR. 2005. Engagement of specific T-cell surface molecules regulates cytoskeletal polarization in HTLV-1-infected lymphocytes. Blood 106:988–995. doi: 10.1182/blood-2004-07-2850. [DOI] [PubMed] [Google Scholar]
- 14.Nejmeddine M, Barnard AL, Tanaka Y, Taylor GP, Bangham CR. 2005. Human T-lymphotropic virus, type 1, Tax protein triggers microtubule reorientation in the virological synapse. J Biol Chem 280:29653–29660. doi: 10.1074/jbc.M502639200. [DOI] [PubMed] [Google Scholar]
- 15.Gross C, Thoma-Kress AK. 2016. Molecular mechanisms of HTLV-1 cell-to-cell transmission. Viruses 8:74. doi: 10.3390/v8030074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, Tanaka Y, Osame M, Bangham CR. 2003. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299:1713–1716. doi: 10.1126/science.1080115. [DOI] [PubMed] [Google Scholar]
- 17.Pais-Correia AM, Sachse M, Guadagnini S, Robbiati V, Lasserre R, Gessain A, Gout O, Alcover A, Thoulouze MI. 2010. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat Med 16:83–89. doi: 10.1038/nm.2065. [DOI] [PubMed] [Google Scholar]
- 18.Van Prooyen N, Andresen V, Gold H, Bialuk I, Pise-Masison C, Franchini G. 2010. Hijacking the T-cell communication network by the human T-cell leukemia/lymphoma virus type 1 (HTLV-1) p12 and p8 proteins. Mol Aspects Med 31:333–343. doi: 10.1016/j.mam.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka Y, Fukudome K, Hayashi M, Takagi S, Yoshie O. 1995. Induction of ICAM-1 and LFA-3 by Tax1 of human T-cell leukemia virus type 1 and mechanism of down-regulation of ICAM-1 or LFA-1 in adult-T-cell-leukemia cell lines. Int J Cancer 60:554–561. doi: 10.1002/ijc.2910600421. [DOI] [PubMed] [Google Scholar]
- 20.Gross C, Wiesmann V, Millen S, Kalmer M, Wittenberg T, Gettemans J, Thoma-Kress AK. 2016. The Tax-inducible actin-bundling protein fascin is crucial for release and cell-to-cell transmission of human T-cell leukemia virus type 1 (HTLV-1). PLoS Pathog 12:e1005916. doi: 10.1371/journal.ppat.1005916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chevalier SA, Turpin J, Cachat A, Afonso PV, Gessain A, Brady JN, Pise-Masison CA, Mahieux R. 2014. Gem-induced cytoskeleton remodeling increases cellular migration of HTLV-1-infected cells, formation of infected-to-target T-cell conjugates and viral transmission. PLoS Pathog 10:e1003917. doi: 10.1371/journal.ppat.1003917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma G, Yasunaga J, Matsuoka M. 2016. Multifaceted functions and roles of HBZ in HTLV-1 pathogenesis. Retrovirology 13:16. doi: 10.1186/s12977-016-0249-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Polakowski N, Terol M, Hoang K, Nash I, Laverdure S, Gazon H, Belrose G, Mesnard JM, Cesaire R, Peloponese JM, Lemasson I. 2014. HBZ stimulates brain-derived neurotrophic factor/TrkB autocrine/paracrine signaling to promote survival of human T-cell leukemia virus type 1-Infected T cells. J Virol 88:13482–13494. doi: 10.1128/JVI.02285-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertoni A, Alabiso O, Galetto AS, Baldanzi G. 2018. Integrins in T cell physiology. Int J Mol Sci 19:E485. doi: 10.3390/ijms19020485.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stuiver I, O'Toole TE. 1995. Regulation of integrin function and cellular adhesion. Stem Cells 13:250–262. doi: 10.1002/stem.5530130306. [DOI] [PubMed] [Google Scholar]
- 26.Drbal K, Angelisova P, Cerny J, Hilgert I, Horejsi V. 2001. A novel anti-CD18 mAb recognizes an activation-related epitope and induces a high-affinity conformation in leukocyte integrins. Immunobiology 203:687–698. doi: 10.1016/S0171-2985(01)80017-6. [DOI] [PubMed] [Google Scholar]
- 27.Clerc I, Polakowski N, Andre-Arpin C, Cook P, Barbeau B, Mesnard JM, Lemasson I. 2008. An interaction between the human T cell leukemia virus type 1 basic leucine zipper factor (HBZ) and the KIX domain of p300/CBP contributes to the down-regulation of Tax-dependent viral transcription by HBZ. J Biol Chem 283:23903–23913. doi: 10.1074/jbc.M803116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polakowski N, Gregory H, Mesnard JM, Lemasson I. 2010. Expression of a protein involved in bone resorption, Dkk1, is activated by HTLV-1 bZIP factor through its activation domain. Retrovirology 7:61. doi: 10.1186/1742-4690-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roebuck KA, Finnegan A. 1999. Regulation of intercellular adhesion molecule-1 (CD54) gene expression. J Leukoc Biol 66:876–888. doi: 10.1002/jlb.66.6.876. [DOI] [PubMed] [Google Scholar]
- 30.Thebault S, Basbous J, Hivin P, Devaux C, Mesnard JM. 2004. HBZ interacts with JunD and stimulates its transcriptional activity. FEBS Lett 562:165–170. doi: 10.1016/S0014-5793(04)00225-X. [DOI] [PubMed] [Google Scholar]
- 31.Kuhlmann AS, Villaudy J, Gazzolo L, Castellazzi M, Mesnard JM, Duc Dodon M. 2007. HTLV-1 HBZ cooperates with JunD to enhance transcription of the human telomerase reverse transcriptase gene (hTERT). Retrovirology 4:92. doi: 10.1186/1742-4690-4-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borowiak M, Kuhlmann AS, Girard S, Gazzolo L, Mesnard JM, Jalinot P, Dodon MD. 2013. HTLV-1 bZIP factor impedes the menin tumor suppressor and upregulates JunD-mediated transcription of the hTERT gene. Carcinogenesis 34:2664–2672. doi: 10.1093/carcin/bgt221. [DOI] [PubMed] [Google Scholar]
- 33.Gazon H, Lemasson I, Polakowski N, Cesaire R, Matsuoka M, Barbeau B, Mesnard JM, Peloponese JM Jr. 2012. Human T-cell leukemia virus type 1 (HTLV-1) bZIP factor requires cellular transcription factor JunD to upregulate HTLV-1 antisense transcription from the 3′ long terminal repeat. J Virol 86:9070–9078. doi: 10.1128/JVI.00661-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukudome K, Furuse M, Fukuhara N, Orita S, Imai T, Takagi S, Nagira M, Hinuma Y, Yoshie O. 1992. Strong induction of ICAM-1 in human T cells transformed by human T-cell-leukemia virus type 1 and depression of ICAM-1 or LFA-1 in adult T-cell-leukemia-derived cell lines. Int J Cancer 52:418–427. doi: 10.1002/ijc.2910520316. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka Y, Hayashi M, Takagi S, Yoshie O. 1996. Differential transactivation of the intercellular adhesion molecule 1 gene promoter by Tax1 and Tax2 of human T-cell leukemia viruses. J Virol 70:8508–8517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tibbetts SA, Seetharama Jois D, Siahaan TJ, Benedict SH, Chan MA. 2000. Linear and cyclic LFA-1 and ICAM-1 peptides inhibit T cell adhesion and function. Peptides 21:1161–1167. doi: 10.1016/S0196-9781(00)00255-2. [DOI] [PubMed] [Google Scholar]
- 37.Mazurov D, Ilinskaya A, Heidecker G, Lloyd P, Derse D. 2010. Quantitative comparison of HTLV-1 and HIV-1 cell-to-cell infection with new replication dependent vectors. PLoS Pathog 6:e1000788. doi: 10.1371/journal.ppat.1000788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delamarre L, Rosenberg AR, Pique C, Pham D, Dokhelar MC. 1997. A novel human T-leukemia virus type 1 cell-to-cell transmission assay permits definition of SU glycoprotein amino acids important for infectivity. J Virol 71:259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnold J, Zimmerman B, Li M, Lairmore MD, Green PL. 2008. Human T-cell leukemia virus type-1 antisense-encoded gene, Hbz, promotes T-lymphocyte proliferation. Blood 112:3788–3797. doi: 10.1182/blood-2008-04-154286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giebler HA, Loring JE, Van Orden K, Colgin MA, Garrus JE, Escudero KW, Brauweiler A, Nyborg JK. 1997. Anchoring of CREB binding protein to the human T-cell leukemia virus type 1 promoter: a molecular mechanism of Tax transactivation. Mol Cell Biol 17:5156–5164. doi: 10.1128/mcb.17.9.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuoka M, Yasunaga J. 2013. Human T-cell leukemia virus type 1: replication, proliferation and propagation by Tax and HTLV-1 bZIP factor. Curr Opin Virol 3:684–691. doi: 10.1016/j.coviro.2013.08.010. [DOI] [PubMed] [Google Scholar]
- 42.Hieshima K, Nagakubo D, Nakayama T, Shirakawa AK, Jin Z, Yoshie O. 2008. Tax-inducible production of CC chemokine ligand 22 by human T cell leukemia virus type 1 (HTLV-1)-infected T cells promotes preferential transmission of HTLV-1 to CCR4-expressing CD4+ T cells. J Immunol 180:931–939. doi: 10.4049/jimmunol.180.2.931. [DOI] [PubMed] [Google Scholar]
- 43.Nakagawa M, Shaffer AL III, Ceribelli M, Zhang M, Wright G, Huang DW, Xiao W, Powell J, Petrus MN, Yang Y, Phelan JD, Kohlhammer H, Dubois SP, Yoo HM, Bachy E, Webster DE, Yang Y, Xu W, Yu X, Zhao H, Bryant BR, Shimono J, Ishio T, Maeda M, Green PL, Waldmann TA, Staudt LM. 2018. Targeting the HTLV-I-regulated BATF3/IRF4 transcriptional network in adult T cell leukemia/lymphoma. Cancer Cell 34:286–297. doi: 10.1016/j.ccell.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arnold J, Yamamoto B, Li M, Phipps AJ, Younis I, Lairmore MD, Green PL. 2006. Enhancement of infectivity and persistence in vivo by HBZ, a natural antisense coded protein of HTLV-1. Blood 107:3976–3982. doi: 10.1182/blood-2005-11-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valeri VW, Hryniewicz A, Andresen V, Jones K, Fenizia C, Bialuk I, Chung HK, Fukumoto R, Parks RW, Ferrari MG, Nicot C, Cecchinato V, Ruscetti F, Franchini G. 2010. Requirement of the human T-cell leukemia virus p12 and p30 products for infectivity of human dendritic cells and macaques but not rabbits. Blood 116:3809–3817. doi: 10.1182/blood-2010-05-284141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gillet NA, Cook L, Laydon DJ, Hlela C, Verdonck K, Alvarez C, Gotuzzo E, Clark D, Farre L, Bittencourt A, Asquith B, Taylor GP, Bangham CR. 2013. Strongyloidiasis and infective dermatitis alter human T lymphotropic virus-1 clonality in vivo. PLoS Pathog 9:e1003263. doi: 10.1371/journal.ppat.1003263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cook LB, Melamed A, Demontis MA, Laydon DJ, Fox JM, Tosswill JH, de Freitas D, Price AD, Medcalf JF, Martin F, Neuberger JM, Bangham CR, Taylor GP. 2016. Rapid dissemination of human T-lymphotropic virus type 1 during primary infection in transplant recipients. Retrovirology 13:3. doi: 10.1186/s12977-015-0236-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bangham CRM, Matsuoka M. 2017. Human T-cell leukaemia virus type 1: parasitism and pathogenesis. Philos Trans R Soc Lond B Biol Sci 372:20160272. doi: 10.1098/rstb.2016.0272.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kannagi M, Harada S, Maruyama I, Inoko H, Igarashi H, Kuwashima G, Sato S, Morita M, Kidokoro M, Sugimoto M, Funahashi S-i, Osame M, Shida H, Honjo T. 1991. Predominant recognition of human T-cell leukemia virus type I (HTLV-I) pX gene products by human CD8+ cytotoxic T cells directed against HTLV-I-infected cells. Int Immunol 3:761–767. doi: 10.1093/intimm/3.8.761. [DOI] [PubMed] [Google Scholar]
- 50.Goon PK, Bangham CR. 2004. Interference with immune function by HTLV-1. Clin Exp Immunol 137:234–236. doi: 10.1111/j.1365-2249.2004.02524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Polakowski N, Lemasson I. 2010. Regulation of HTLV-1 transcription by viral and cellular proteins, p 129–169. In Lever AML, Jeang KT (ed), Recent advances in human retroviruses: principles of replication and pathogenesis advances in retroviral research. World Scientific Publishing Co, Singapore. [Google Scholar]
- 52.Satou Y, Yasunaga J, Yoshida M, Matsuoka M. 2006. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci U S A 103:720–725. doi: 10.1073/pnas.0507631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Usui T, Yanagihara K, Tsukasaki K, Murata K, Hasegawa H, Yamada Y, Kamihira S. 2008. Characteristic expression of HTLV-1 basic zipper factor (HBZ) transcripts in HTLV-1 provirus-positive cells. Retrovirology 5:34. doi: 10.1186/1742-4690-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suemori K, Fujiwara H, Ochi T, Ogawa T, Matsuoka M, Matsumoto T, Mesnard JM, Yasukawa M. 2009. HBZ is an immunogenic protein, but not a target antigen for human T-cell leukemia virus type 1-specific cytotoxic T lymphocytes. J Gen Virol 90:1806–1811. doi: 10.1099/vir.0.010199-0.. [DOI] [PubMed] [Google Scholar]
- 55.Rowan AG, Suemori K, Fujiwara H, Yasukawa M, Tanaka Y, Taylor GP, Bangham CR. 2014. Cytotoxic T lymphocyte lysis of HTLV-1 infected cells is limited by weak HBZ protein expression, but non-specifically enhanced on induction of Tax expression. Retrovirology 11:116. doi: 10.1186/s12977-014-0116-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Billman MR, Rueda D, Bangham C. 2017. Single-cell heterogeneity and cell-cycle-related viral gene bursts in the human leukaemia virus HTLV-1. Wellcome Open Res 2:87. doi: 10.12688/wellcomeopenres.12469.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mahgoub M, Yasunaga JI, Iwami S, Nakaoka S, Koizumi Y, Shimura K, Matsuoka M. 2018. Sporadic on/off switching of HTLV-1 Tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc Natl Acad Sci U S A 115:E1269–E1278. doi: 10.1073/pnas.1715724115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nejmeddine M, Negi VS, Mukherjee S, Tanaka Y, Orth K, Taylor GP, Bangham CR. 2009. HTLV-1-Tax and ICAM-1 act on T-cell signal pathways to polarize the microtubule-organizing center at the virological synapse. Blood 114:1016–1025. doi: 10.1182/blood-2008-03-136770. [DOI] [PubMed] [Google Scholar]
- 59.Hubbard AK, Rothlein R. 2000. Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radic Biol Med 28:1379–1386. doi: 10.1016/S0891-5849(00)00223-9. [DOI] [PubMed] [Google Scholar]
- 60.Wurm T, Wright DG, Polakowski N, Mesnard JM, Lemasson I. 2012. The HTLV-1-encoded protein HBZ directly inhibits the acetyl transferase activity of p300/CBP. Nucleic Acids Res 40:5910–5925. doi: 10.1093/nar/gks244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hivin P, Basbous J, Raymond F, Henaff D, Arpin-Andre C, Robert-Hebmann V, Barbeau B, Mesnard JM. 2007. The HBZ-SP1 isoform of human T-cell leukemia virus type I represses JunB activity by sequestration into nuclear bodies. Retrovirology 4:14. doi: 10.1186/1742-4690-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Larson RS, Corbi AL, Berman L, Springer T. 1989. Primary structure of the leukocyte function-associated molecule-1 alpha subunit: an integrin with an embedded domain defining a protein superfamily. J Cell Biol 108:703–712. doi: 10.1083/jcb.108.2.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kishimoto TK, O'Connor K, Lee A, Roberts TM, Springer TA. 1987. Cloning of the beta subunit of the leukocyte adhesion proteins: homology to an extracellular matrix receptor defines a novel supergene family. Cell 48:681–690. doi: 10.1016/0092-8674(87)90246-7. [DOI] [PubMed] [Google Scholar]
- 64.van den Hoff MJ, Christoffels VM, Labruyere WT, Moorman AF, Lamers WH. 1995. Electrotransfection with “intracellular” buffer. Methods Mol Biol 48:185–197. doi: 10.1385/0-89603-304-X:185. [DOI] [PubMed] [Google Scholar]
- 65.Lemasson I, Nyborg JK. 2001. Human T-cell leukemia virus type I Tax repression of p73beta is mediated through competition for the C/H1 domain of CBP. J Biol Chem 276:15720–15727. doi: 10.1074/jbc.M100131200. [DOI] [PubMed] [Google Scholar]
- 66.Winter HY, Marriott SJ. 2007. Human T-cell leukemia virus type 1 Tax enhances serum response factor DNA binding and alters site selection. J Virol 81:6089–6098. doi: 10.1128/JVI.02179-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, Mesnard JM. 2002. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol 76:12813–12822. doi: 10.1128/jvi.76.24.12813-12822.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rousset R, Desbois C, Bantignies F, Jalinot P. 1996. Effects on NF-kappa B1/p105 processing of the interaction between the HTLV-1 transactivator Tax and the proteasome. Nature 381:328–331. doi: 10.1038/381328a0. [DOI] [PubMed] [Google Scholar]
- 69.Laverdure S, Polakowski N, Hoang K, Lemasson I. 2016. Permissive sense and antisense transcription from the 5′ and 3′ long terminal repeats of human T-cell leukemia virus type 1. J Virol 90:3600–3611. doi: 10.1128/JVI.02634-15. [DOI] [PMC free article] [PubMed] [Google Scholar]