

SUMMARY
Carbohydrate response element binding protein (ChREBP) is a key transcriptional regulator of de novo lipogenesis (DNL) in response to carbohydrates and in hepatic steatosis. As such, mechanisms underlying nutrient modulation of ChREBP are under active investigation. Here, we identify Host Cell Factor 1 (HCF-1) as a previously unknown ChREBP-interacting protein that is enriched in liver biopsies of nonalcoholic steatohepatitis (NASH) patients. Biochemical and genetic studies show HCF-1 is O-GlcNAcylated in response to glucose as a prerequisite for its binding to ChREBP and subsequent recruitment of OGT, ChREBP O-GlcNAcylation and activation. The HCF-1:ChREBP complex resides at lipogenic gene promoters, where HCF-1 regulates H3K4 trimethylation to prime recruitment of the Jumonji C domain-containing histone demethylase PHF2 for epigenetic activation of these promoters. Overall, these findings define HCF-1’s interaction with ChREBP as a previously unappreciated mechanism whereby glucose signals are both relayed to ChREBP and transmitted to epigenetic regulation of lipogenic genes.
Graphical Abstract
eTOC
Lane, Choi et al. identify HCF-1 as a ChREBP-modulatory protein relevant for de novo lipogenesis. Glucose stimulation triggers HCF-1 O-GlcNAcylation, priming its association with ChREBP and OGT recruitment for ChREBP O-GlcNAcylation and increased ChREBP transcriptional activity. The HCF-1:ChREBP complex imparts glucose-dependent epigenetic regulation to lipogenic promoters by recruiting epigenetic modifiers.
INTRODUCTION
Metabolic adaptation to fed and fasted states is intricately controlled by nutrient and hormonal signals. In the fed state, the liver stores glucose as glycogen, and once glycogen stores are replete, it converts excess glucose to fat through de novo lipogenesis (DNL), a process regulated by insulin and glucose-derived metabolic signals (Abdul-Wahed et al., 2017; Sanders and Griffin, 2016; Uyeda and Repa, 2006; Yecies et al., 2011). While storage of excess glucose as lipids is an important component of metabolic adaptation, excessive DNL is associated with pathologies such as nonalcoholic fatty liver disease (NAFLD), where up to 30% of hepatic fat accumulation can be linked to increased hepatic DNL (Softic et al., 2016).
Glucose metabolism provides both biosynthetic precursors and regulatory signals for DNL, and evidence indicates that the Carbohydrate Response Element Binding Protein (ChREBP) is an important transcription factor and effector of glucose-derived signals in this process (Abdul-Wahed et al., 2017; Agius, 2016a; Alves-Bezerra and Cohen, 2017; Uyeda and Repa, 2006). Notably, polymorphisms in ChREBP are linked to increased plasma triglycerides (Kooner et al., 2008), and increased ChREBP protein levels are associated with human NAFLD (Benhamed et al., 2012). Glucose activates ChREBP through multiple mechanisms (reviewed in (Abdul-Wahed et al., 2017)). For example, several glucose-derived metabolites can activate ChREBP, including G6P (glucose-6-phosphate), F2-6BP (fructose-2,6-bisphosphate) and X5P (xyulose-5-phosphate) (Arden et al., 2012; Dentin et al., 2012; Iizuka et al., 2013). There is some evidence that X5P can trigger dephosphorylation and nuclear translocation of ChREBP through activation of protein phosphatase 2A (PP2A) (Kabashima et al., 2003). However, ChREBP phospho-mutants remain glucose responsive (Tsatsos and Towle, 2006), and a pool of ChREBP resides in the nucleus even in the absence of glucose stimulation (Kim et al., 2016), indicating the existence of additional glucose-dependent regulatory mechanisms. Other glucose-derived metabolic signals in the form of acetyl-CoA and UDP-N-acetylglucosamine (UDP-GlcNAc) can activate ChREBP through acetylation and O-GlcNAcylation, respectively (Bricambert et al., 2010; Guinez et al., 2011; Sakiyama et al., 2010; Yang and Qian, 2017). Specifically, increased glucose metabolism can augment UDP-GlcNAc pools through the hexosamine biosynthesis pathway (HBP), and O-GlcNAcylation of ChREBP increases its DNA binding affinity, transcriptional activity, and protein stability (Guinez et al., 2011; Sakiyama et al., 2010; Yang et al., 2017). However, the molecular details underlying the glucose-dependent activation of ChREBP, including specific mechanisms controlling its O-GlcNAcylation are not fully understood.
The myriad of glucose-dependent signals that converge on ChREBP reflect a complex network of mechanisms regulating gene expression in response to glucose and carbohydrate surplus. In the liver, these signals are controlled by the glucose phosphorylating enzyme glucokinase (GK, Hexokinase IV), the product of the maturity onset diabetes of the young type 2 (MODY2) gene (Matschinsky, 2009; Nissim et al., 2012). In the fed state, hepatic GK regulates glucose utilization and storage by stimulating glycogen and DNL while suppressing hepatic glucose production (Dentin et al., 2004; Velho et al., 1996). Genetic evidence also indicates that GK is required for activation of ChREBP by glucose and carbohydrate feeding (Dentin et al., 2004). Hepatic GK activity is regulated by a number of binding partners including GKRP (glucokinase regulatory protein), PFK2/FBPase2 (6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase) and BAD (BCL-2-associated Agonist of Cell Death) (reviewed in (Agius, 2016b)). These endogenous modulators of GK could be components of additional signaling pathways that exert physiologic control over ChREBP activation in response to lipogenic signals. BAD activates GK when phosphorylated at serine 155 within its BH3 (BCL2 homology 3) domain downstream of the insulin signaling pathway, independent of its capacity to bind other BCL-2 family proteins (Agius, 2016b; Danial et al., 2003; Gimenez-Cassina and Danial, 2015; Giménez-Cassina et al., 2014). This involves direct binding of the BAD BH3 helix near the active site of the enzyme (Szlyk et al., 2014). Activation of GK by phosphorylated BAD stimulates hepatic glucose utilization while suppressing gluconeogenesis (Giménez-Cassina et al., 2014). However, whether BAD phosphorylation is relevant to glucose stimulation of DNL is not known and could not be predicted given the multitude of hormonal and nutrient signals that can regulate hepatic GK, ChREBP activity, and lipid synthesis. Here, we show that BAD phosphorylation is necessary for glucose stimulation of DNL and utilize the BAD mutant hepatocyte system as a discovery platform to gain new molecular insights into how ChREBP is normally activated in response to glucose.
Our unbiased proteomics analyses comparing ChREBP-containing nuclear complexes in control versus BAD-deficient hepatocytes, where glucose stimulation of ChREBP is impaired, revealed Host Cell Factor 1 (HCF-1) as a previously unknown ChREBP binding protein. A combination of biochemical, genetic, and functional studies pinpoint distinct steps at which HCF-1 modulates activation of ChREBP and the lipogenic program by glucose. In particular, the formation of the HCF-1:ChREBP complex is first primed by HCF-1 glycosylation in response to glucose stimulation or carbohydrate surplus and is followed by HCF-1-dependent recruitment of OGT to ChREBP and ChREBP O-GlcNAcylation. HCF-1 is also required for glucose-stimulated increase in activating histone marks and epigenetic control of ChREBP target promoters. These data reveal shared control of ChREBP O-GlcNAcylation/activation and epigenetic modulation of lipogenic gene promoters via HCF-1.
RESULTS
ChREBP-dependent de novo lipogenesis is regulated by BAD phosphorylation
To initially assess glucose modulation of DNL following alterations in BAD, we quantified incorporation of glucose carbons into lipid fractions of wild type (WT) and Bad −/− primary hepatocytes following incubation with low (5 mM) versus high (25 mM) glucose concentrations. Compared to WT hepatocytes, the contribution of glucose to DNL was significantly lower in Bad −/− hepatocytes (Figure 1A). This defect was fully reversed by genetic reconstitution with the phosphomimic BAD S155D variant capable of activating GK but not the phospho-deficient BAD AAA variant, harboring triple Ala mutations within the BAD BH3 domain (L151A, S155A, and D156A) that blunt its GK-activating capacity (Danial et al., 2008; Giménez-Cassina et al., 2014) (Figures 1A and S1A). The BAD S155D and AAA mutants are particularly informative in this setting as they are deficient in binding to BCL-2 family proteins known to interact with the BAD BH3 domain (Danial et al., 2008; Giménez-Cassina et al., 2014). This enabled assessment of the GK-activating property of BAD without the confounding effects of other BAD interacting proteins. In WT hepatocytes, BAD S155D did not lead to additional augmentation of lipogenesis beyond the level seen in GFP-expressing controls (Figure 1A), indicating that, at least in this acute setting, phospho-BAD does not lead to hyper-stimulation of the lipogenic program by glucose.
Figure 1. Altered ChREBP-dependent de novo lipogenesis following BAD modifications.

(A) Incorporation of glucose into lipid fractions of WT and Bad −/− hepatocytes reconstituted with the indicated adenoviruses and treated with 1μCi of U14C-glucose for 4 hrs. Data are presented as percent increase in label incorporation from 5 to 25 mM glucose (glc) treatment (n=3-4).
(B) Relative expression of lipogenic genes in WT and Bad −/− primary hepatocytes reconstituted with the indicated adenoviruses and treated with 5 or 25 mM glucose for 20 hrs (n=5).
(C) ChoRE luciferase reporter activity in WT and Bad −/− primary hepatocytes treated as in (B). Data are represented as percent increase in reporter activity upon glucose stimulation (n=4).
(D) Relative occupancy of ChREBP at L-Pk and Acc promoters in WT and Bad −/− primary hepatocytes reconstituted with the indicated adenoviruses and treated with 5 or 25 mM glucose for 4 hrs (n=7).
(E) Relative occupancy of ChREBP at the promoters of L-Pk and Acc in livers of WT and Bad −/− mice reconstituted with the indicated adenoviruses one week prior to being fasted for 24 hrs and refed a HCD for 18 hrs (n=3-4).
(F) DNL assays in WT and Bad −/− hepatocytes co-infected with the indicated adenoviruses and assessed as in (A) (n=4).
Error bars in A-F are means ± SEM, *p < 0.05; **p < 0.01; ***p < 0.001; n.s., nonsignificant; one way ANOVA (A, C, F) or two way ANOVA (B, D, E).
See also Figures S1, S2 and Table S1.
Gene abbreviations in (B): L-PK, liver pyruvate kinase; Acly, ATP citrate lyase; Acc1, acetyl-CoA carboxylase 1; Fas, fatty acid synthase; Elovl6, Elovl fatty acid elongase 6; Scd-1, stearoyl-CoA desaturase; Chrebp, carbohydrate response element binding protein; Srebp-1c, sterol regulatory element-binding protein-1C; Pdhk, pyruvate dehydrogenase kinase
Hepatic lipogenesis is substantially regulated at the transcriptional level by the sterol regulatory element-binding protein-1C (SREBP-1C) and ChREBP transcription factors, which have overlapping target genes and mediate the input from insulin and glucose, respectively (Abdul-Wahed et al., 2017; Agius, 2016b; Herman et al., 2012; Uyeda and Repa, 2006; Wang et al., 2015; Yecies et al., 2011). Because our primary focus was on regulation of lipogenesis by glucose, we performed all subsequent measurements in hepatocytes in response to glucose alone without insulin stimulation (Petrie et al., 2013). As expected, substantial increase in the mRNA abundance of lipogenic genes was observed upon glucose stimulation of WT hepatocytes (Figures 1B and S1B). Chrebpα mRNA levels were not substantially regulated by glucose but those of the Chrebpβ isoform were induced (Figure 1B), which is consistent with previous reports (Herman et al., 2012; Stamatikos et al., 2016). In BAD-deficient hepatocytes, glucose induction of mRNAs for lipogenic genes was significantly blunted (Figure 1B). However, the expression of other genes whose transcription is not glucose responsive, such as Chrebpα, Srebp-1C and pyruvate dehydrogenase kinase (Pdhk) (Stamatikos et al., 2016; Zhang et al., 2015), was comparable to WT hepatocytes stimulated with glucose (Figure 1B). As in lipogenesis assays (Figure 1A), BAD S155D but not AAA restored glucose induction of lipogenic gene transcripts in Bad −/− hepatocytes (Figure 1B). Importantly, BAD S155D does not rescue lipogenic gene expression in Bad −/− hepatocytes subjected to Gk knockdown (Figure S1C). Likewise, BAD S155D does not rescue lipogenic gene expression in GK-deficient hepatocytes, which similar to Bad −/− hepatocytes display a defect in glucose stimulation of DNL gene expression (Dentin et al., 2004) (Figure S1D). Collectively, these observations, together with the deficiency of the non-GK-activating BAD AAA mutant in stimulating DNL in response to glucose, indicate that the effect of phospho-BAD requires an intact GK. BAD-dependent changes in lipogenic gene expression are associated with altered ChREBP activity as evident from two complementary functional readouts; luciferase reporters containing the Carbohydrate Response Elements (ChoREs) of L-Pk and Acc genes and chromatin immunoprecipitation (ChIP) assays examining promoter occupancy of ChREBP at target genes. Glucose induction of ChoRE reporters and recruitment of ChREBP to endogenous promoters of L-Pk and Acc genes were significantly lower in Bad −/− compared to WT primary hepatocytes but were restored to WT levels following genetic rescue with the GK-activating BAD S155D but not the BAD AAA mutant (Figures 1C–D).
As an in vivo correlate to our findings in hepatocyte cultures, we also examined hepatic ChREBP activity and the lipogenic gene program in liver biopsies from WT and Bad −/− mice subjected to short term (18 hr) high-carbohydrate diet (HCD) feeding, a treatment known to activate hepatic ChREBP (Agius, 2016a). ChREBP binding to L-Pk and Acc gene promoters as well as transcriptional induction of lipogenic genes in response to HCD were significantly attenuated in Bad −/− liver (Figures 1E and S1E). Hepatic reconstitution of Bad −/− mice with BAD S155D or AAA corroborated our findings in primary hepatocytes that full induction of ChREBP activity and lipogenic gene expression require BAD phosphorylation (Figures 1E, S1E–F). Notably, BAD S155 phosphorylation is normally induced in the liver upon HCD feeding or in primary hepatocytes following glucose stimulation (Figures S1G–H). Thus, BAD phosphorylation is a downstream effector of lipogenic signals that converge on ChREBP activation.
To establish a cause-and-effect relationship between phospho-BAD and ChREBP activity, we tested the effects of BAD S155D on DNL in the context of ChREBP deficiency. Chrebp knockdown in WT primary hepatocytes led to diminished DNL and transcriptional induction of lipogenic genes in response to glucose consistent with previous reports (Dentin et al., 2006), and completely ablated the ability of BAD S155D to restore lipogenesis in Bad −/− hepatocytes (Figures 1F, S2A–B). In Bad −/− hepatocytes, ChREBP depletion had no additional effect on reducing DNL (Figure 1F). Taken together, these data indicate that the effect of phospho-BAD on glucose stimulation of lipogenesis is mediated by ChREBP. Thus, although BAD is one of many GK-modulatory mechanisms in the liver, its modification is sufficient to alter glucose stimulation of ChREBP activity and DNL.
Identification of HCF-1 as a ChREBP binding and regulatory protein
We next took advantage of the WT and Bad −/− hepatocytes, in which glucose induction of ChREBP is preserved or blunted, as an unbiased discovery platform to learn about new biochemical mechanisms that can contribute to glucose regulation of ChREBP. To this end, we initially examined changes in ChREBP-containing protein complexes in glucose-stimulated WT hepatocytes compared to BAD-deficient counterparts using immunoprecipitation (IP) coupled to mass spectrometry (MS). ChREBP was immunoprecipitated from the nuclear fractions of primary WT and Bad −/− hepatocytes expressing Flag-tagged ChREBP and cultured in 25 mM glucose. Among proteins captured by the anti-Flag antibody in WT hepatocytes, were known ChREBP binding proteins such as Max-like protein X (Mlx), an obligate partner of ChREBP (Ma et al., 2006) (Figures S3A–C). We then prioritized candidates that were ≥ 3 fold more abundant in ChREBP IPs in WT compared to Bad −/− hepatocytes, and focused on proteins with direct or indirect functional annotations in gene transcription and glucose signaling by gene ontology, STRING, and DAVID bioinformatic tools. Using this filtering approach, the only protein with known roles in both transcription and glucose signalling was Host Cell Factor (HCF)-1, which was enriched by 3.5 fold in ChREBP IPs of WT compared to Bad −/− samples (Figures S3B and D). HCF-1 is a transcriptional co-regulator whose interaction with ChREBP or role in DNL has not been previously reported. HCF-1 is proteolytically processed by N-acetylglucosamine (GlcNAc) transferase (OGT) at multiple cleavage sites, which produces N- and C-terminal fragments that remain non-covalently bound, forming a conformation that is important for HCF-1 function (Capotosti et al., 2011; Daou et al., 2011; Janetzko et al., 2016; Lazarus et al., 2013).
We next validated the association of endogenous ChREBP and HCF-1 in WT hepatocytes and confirmed that it was diminished in the absence of BAD (Figure 2A). These experiments were performed in the presence of the proteasome inhibitor MG132 to normalize ChREBP protein stability (Guinez et al., 2011), because endogenous ChREBP levels were found to be lower in Bad −/− hepatocytes and liver compared to WT controls (see below). Notably, the association of HCF-1 and ChREBP in WT hepatocytes and liver samples is induced in response to glucose stimulation or HCD, respectively (Figures 2A and S3E). To our knowledge, this is the first example of an HCF-1-containing protein complex that is induced by nutrient stimulation. In comparison, the HCF-1:PGC1α complex is enriched only at low/basal glucose concentrations or in the fasted state in hepatocytes and liver (Ruan et al., 2012), and other HCF-1-containing complexes are not known to be nutrient responsive (Mazars et al., 2010; Thomas et al., 2016; Tyagi et al., 2007).
Figure 2. HCF-1 interacts with ChREBP and is required for its O-GlcNAcylation.

(A) Representative western blots (top) and quantification by densitometry of n=4 experiments (bottom) of ChREBP association with HCF-1 and OGT in WT and Bad −/− primary hepatocytes cultured in 5 or 25 mM glucose for 6 hrs in the presence of 20 μM MG132.
(B) Representative western blots (top) and quantification of n=3 experiments (bottom) of ChREBP association with HCF-1 and OGT following Hcf-1 knockdown in WT hepatocytes cultured in 25 mM glucose for 6 hrs in the presence of 20 μM MG132.
(C) Representative western blots (top) and quantification of n=3 experiments (bottom) of O-GlcNAcylated ChREBP (ChREBPOG) in wheat germ agglutinin (WGA) pull down (PD) assays in WT hepatocytes subjected to Hcf-1 knockdown and cultured as in (A).
(D) Representative western blots (left) and quantification of n=3 experiments (right) of ChREBP protein levels in WT hepatocytes subjected to Hcf-1 knockdown and cultured in 25 mM glucose for 6 hrs in the absence or presence of 20 μM MG132.
(E) Representative western blots (top) and quantification of n=3 experiments (bottom) of ChREBPOG in primary WT and Bad −/− hepatocytes reconstituted with the indicated BAD mutants, subjected to Hcf-1 knockdown, and cultured in 25 mM glucose for 6 hrs in the presence or absence of 20 μm MG132.
Error bars are means ± SEM, *p < 0.05; **p < 0.01; ***p < 0.001; n.s., nonsignificant; two way ANOVA (A, C, D, E) or one way ANOVA (B).
See also Figures S3 and S4.
HCF-1 is first and foremost known as a transcriptional co-regulator, however, in limited settings, it may facilitate protein Ser/Thr O-GlcNAcylation by recruiting OGT to specific partner proteins (Han et al., 2017; Ruan et al., 2012). This adaptor-like function of HCF-1 has been demonstrated in the context of two interacting partners, PGC1α and NRF1 (Han et al., 2017; Ruan et al., 2012), and remains to be further studied. In addition to HCF-1, OGT was also present in ChREBP immunoprecipitates in a glucose- and HCD-responsive manner (Figures 2A and S3E). Notably, depletion of HCF-1 in WT hepatocytes led to diminished capture of OGT in ChREBP immunoprecipitates (Figure 2B), indicating that HCF-1 is required for recruitment of OGT to ChREBP. Consistent with these observations, HCF-1 depletion also led to decreased ChREBP O-GlcNAcylation (ChREBPOG) in response to 25 mM glucose (Figures 2C, S4A–B). Reduced ChREBP O-GlcNAcylation in this setting was commensurate with lower ChREBP protein levels (Figures 2D and S4B), consistent with reports that ChREBP stability is regulated by O-GlcNAcylation (Guinez et al., 2011; Sakiyama et al., 2010). In conditions where glucose stimulation of ChREBP activity is blunted as in the absence of GK or BAD, the interaction between HCF-1 and OGT with ChREBP is significantly diminished in response to glucose stimulation or HCD (Figures 2A, S3E, S4C–D). This parallels a corresponding decrease in ChREBPOG and ChREBP protein levels in Bad −/− hepatocytes, which were rescued with BAD S155D but not BAD AAA (Figures 2E, lanes 1-4, and S4E–F). Notably, BAD S155D was ineffective in the context of Hcf-1 knockdown (Figures 2E, lane 3 vs 7 and S4F), further corroborating the necessity of HCF-1 for ChREBP O-GlcNAcylation. Collectively, our findings indicate that HCF-1 is a ChREBP binding partner that is required for recruitment of OGT to ChREBP and ChREBP O-GlcNAcylation in a glucose- and HCD-sensitive protein complex.
HCF-1 O-GlcNAcylation is induced by lipogenic signals and selectively regulates its interaction with ChREBP
OGT modifies HCF-1 both through proteolytic cleavage, which proceeds via an intermediary non-canonical glutamate side chain glycosylation, and through canonical O-GlcNAcylation of Ser/Thr residues (Janetzko et al., 2016). However, whether HCF-1 glycosylation is regulated by nutrient stimulation and dietary treatment has not been previously examined. This is a relevant question because factors beyond UDP-GIcNAc levels can regulate protein O-GlcNAcylation as evident from preferential O-GlcNAcylation of some OGT substrates under conditions where UDP-GlcNAc levels are reduced (Ruan et al., 2012; Taylor et al., 2009; Yang and Qian, 2017). Therefore, HCF-1 O-GlcNAcylation may not necessarily be responsive to nutrient fluctuations. However, we found that HCF-1 O-GlcNAcylation is significantly increased in primary hepatocytes in response to glucose stimulation and in liver derived from HCD-treated mice (Figures 3A and S5A). Moreover, HCF-1 O-GlcNAcylation in response to glucose or HCD is blunted in BAD-deficient hepatocytes or in hepatocytes subjected to acute knockdown of Gk or Bad (Figures 3A–B and S5A). Notably, this defect can be rescued by BAD S155D in a GK-dependent manner (Figures S5B–C). Collectively, these data further corroborate the sensitivity of HCF-1 glycosylation to changes in glucose metabolism.
Figure 3. HCF-1 O-GlcNAcylation selectively regulates its association with ChREBP.

(A) Representative western blots (left) and quantification of n=3 experiments (right) of O-GlcNAcylated HCF-1 (HCF-1OG) in WT and Bad −/− primary hepatocytes cultured in 5 or 25 mM glucose for 6 hrs in the presence of 20 μM MG132.
(B) Representative western blots (left) and quantification of n=3 experiments (right) of HCF-1OG in WT primary hepatocytes subjected to Bad or Gk knockdown and cultured in 25 mM glucose for 6 hrs in the presence of 20 μM MG132.
(C-D) Schematic of the experimental system to generate glycosylated or non-glycosylated recombinant HCF-1 and test its interaction with ChREBP (C). Representative western blots (left) and quantification of n=3 experiments (right) showing the interaction between recombinant HCF-1 generated as in (C) and ChREBP (D). rOGT, recombinant OGT
(E-F) Interaction of Flag-tagged ChREBP (E) compared with Flag-tagged PGC1α (F) with differentially glycosylated HCF-1 in 293T cells transfected with OGT WT or D554N.
Error bars in A, B, D-F are means ± SEM, *p < 0.05; **p < 0.01; ***p < 0.001; n.s., nonsignificant; two way ANOVA (A, D) or one way ANOVA (B, E, F).
See also Figure S5.
To date, no functional roles have been ascribed to HCF-1 Ser/Thr O-GlcNAcylation, motivating us to investigate whether this modification is required for HCF-1 binding to ChREBP. To this end, we incubated in vitro transcribed and translated HCF-1 with increasing amounts of recombinant WT OGT or the OGT D554N mutant previously shown to be selectively impaired in canonical Ser/Thr O-GlcNAcylation but not HCF-1 proteolytic cleavage (Janetzko et al., 2016) (Figures 3C–D and S5D). This enabled generation of two HCF-1 variants that underwent proteolytic processing (Figure 3D, input), but differed in O-GlcNAcylation (Figure 3D, WGA PD). The proteolytic processing of HCF-1 by the OGT D554N mutant was more efficient compared to WT OGT, consistent with published characterization of this mutant (Janetzko et al., 2016). We then examined the capacity of these differentially O-GlcNAcylated HCF-1 proteins to interact with ChREBP that was immunoprecipitated from primary hepatocytes, and found that HCF-1 processed by the OGT D554N mutant is significantly impaired in ChREBP binding compared with HCF-1 processed by WT OGT (Figure 3D, ChREBP IP). Collectively, these data indicate that HCF-1 glycosylation is required for its binding to ChREBP and is stimulated by lipogenic signals, providing a biochemical explanation for the observed enrichment of the ChREBP:HCF-1 complex in response to glucose stimulation or HCD treatment (Figures 2A and S3E).
To determine whether HCF-1 glycosylation is generally relevant for its binding interactions or is a specific requirement for its association with ChREBP, we also examined the effect of HCF-1 O-GlcNAcylation on its capacity to bind PGC1α. To this end, we expressed Flag-tagged PGC1α or ChREBP and either WT or D554N OGT in 293T cells. As with the above in vitro system, HCF-1 was comparably cleaved by WT and D554N OGT but differentially glycosylated in 293T cells (Figures 3E–F). In comparison to the in vitro system where HCF-1 glycosylation was undetectable in the presence of OGT D554N (Figure 3D), basal HCF-1 glycosylation could be detected in 293T cells expressing this mutant likely due to endogenous OGT activity (Figures 3E–F). Nonetheless, expression of WT OGT produced a larger increase in HCF-1 glycosylation and, unlike the D554N OGT mutant, led to higher capture of HCF-1 in Flag-ChREBP immunoprecipitates (Figure 3E). In contrast, HCF-1 was equally captured in Flag-PGC1α immunoprecipitates from cells expressing WT or D554N mutant OGT (Figure 3F). These data indicate that, unlike its interaction with ChREBP, HCF-1 interaction with PGC1α is insensitive to the HCF-1 glycosylation status. These findings establish a specific biochemical function of HCF-1 Ser/Thr O-GlcNAcylation by demonstrating its selective requirement for ChREBP binding, and reveal HCF-1 glycosylation as a molecular mechanism dictating the specificity of HCF-1 and ChREBP interaction.
Regulation of ChREBP by HCF-1 is sensitive to changes in the hexosamine biosynthesis pathway
Our findings suggest that lipogenic signals or alterations in glucose metabolism regulate HCF-1-dependent recruitment of OGT to ChREBP, thereby modulating its glycosylation. Beyond OGT recruitment, it is possible that changes in the OGT substrate, UDP-GlcNAc, also regulate this process. However, individual OGT substrates have differential sensitivity to fluctuations in UDP-GlcNAc levels (Shen et al., 2012). Thus, the extent to which HCF-1 O-GlcNAcylation and its binding to ChREBP, which requires HCF-1 to be glycosylated (Figures 3D–E), might be sensitive to changes in HBP and UDP-GlcNAc levels is not evident. To address this question, we utilized Bad −/− hepatocytes as a model system where HBP metabolites were found to be lower in response to glucose stimulation commensurate with diminished HCF-1 glycosylation and association with ChREBP without global diminution in protein O-GlcNAcylation (Figure 4A, Table S2 and data not shown). HBP metabolites were rescued by genetic reconstitution of Bad −/− hepatocytes with BAD S155D but not BAD AAA in keeping with phospho-BAD-dependent modulation of GK activity and hepatic glucose metabolism (Giménez-Cassina et al., 2014) (Figure 4A). We reasoned that if lower UDP-GlcNAc levels contribute to diminished HCF-1 glycosylation in this setting, then supplementation with HBP metabolites should rescue HCF-1 O-GlcNAcylation. To test this possibility, we used GlcNAc or glucosamine (GlcN), which bypasses the rate-limiting reaction catalysed by glutamine-fructose-6-phosphate aminotransferase (GFAT) to enter the HBP upon direct phosphorylation (Guinez et al., 2011), and found that both were sufficient to restore HCF-1 O-GlcNAcylation in Bad −/− hepatocytes (Figures 4B and S6A). Remarkably, HBP metabolite supplementation was also sufficient to fully rescue the interaction between ChREBP with HCF-1 and OGT in this setting (Figures 4C and S6B). This is in keeping with the finding that HCF-1 glycosylation regulates its binding to ChREBP (Figures 3D–E). Metabolite rescue of this HCF-1- and OGT-containing ChREBP complex was accompanied by restoration of ChREBP O-GlcNAcylation, ChREBP protein levels and target gene activation in response to glucose stimulation (Figures S6C–E). These results are significant because they indicate that HCF-1 O-GlcNAcylation and the attendant HCF-1:ChREBP complex formation are sensitive to glucose modulation of UDP-GlcNAc pools.
Figure 4. Regulation of the HCF-1:ChREBP complex by hexosamine pathway metabolites.

(A) LC-MS quantification of N-acetyglucosamine-6-phosphate and UDP-GlcNAc in WT and Bad −/− primary hepatocytes reconstituted with the indicated adenoviruses and cultured in 25 mM glucose for 5 hrs (n=4-8).
(B) Representative western blots (top) and quantification of n=3 experiments (bottom) of HCF-1OG in WT and Bad −/− primary hepatocytes cultured in 25 mM glucose (glc) and 20 μM MG132 in the absence or presence of 20 mM GlcNAc or 5 mM GlcN for 6 hrs.
(C) Representative western blots (left) and quantification of n=3 experiments (right) of ChREBP association with HCF-1 and OGT in WT and Bad −/− primary hepatocytes cultured in the absence or presence of GlcNAc as in (B).
(D) DNL assays in WT and Bad −/− hepatocytes cultured in the absence or presence of 20 mM GlcNAc and assessed as in Figure 1A (n=4).
Error bars are means ± SEM, *p < 0.05; **p < 0.01; ***p < 0.001; n.s., non-significant; two way ANOVA.
We next examined the extent to which the lipogenic program is reinstated by GlcNAc supplementation of Bad −/− hepatocytes beyond restoration of ChREBP transcriptional activity. We found that GlcNAc treatment led to significant rescue of the biochemical defect in lipogenesis (Figure 4D). While significant, the rescue of lipogenesis was partial despite full restoration of ChREBP O-GlcNAcylation and lipogenic gene expression (Figures 4D, S6C and E). This may reflect the requirement of multiple branch points of glucose metabolism downstream of GK for induction of DNL beyond glucose stimulation of ChREBP activity. These include malonyl-CoA produced downstream of the glycolytic and oxidative arms of glucose metabolism, as well as NADPH generated through the pentose phosphate pathway, which are not expected to be restored by GlcNAc treatment (Alves-Bezerra and Cohen, 2017; Sanders and Griffin, 2016).
ChREBP-dependent binding of HCF-1 to lipogenic gene promoters is required for the recruitment of the epigenetic activator PHF2
The finding that HCF-1 is required for ChREBP O-GlcNAcylation warranted more detailed examination of its role in DNL. In WT primary hepatocytes, glucose incorporation into lipids and lipogenic gene expression were significantly impaired in response to glucose following Hcf-1 knockdown (Figures 5A–B). ChIP assays further indicated that, similar to ChREBP, HCF-1 is normally recruited to lipogenic gene promoters in response to glucose stimulation (Figure 5C). HCF-1 does not contain a DNA binding domain, however, its recruitment to lipogenic gene promoters is mediated by ChREBP as evident from diminution of the HCF-1 ChIP signal in WT hepatocytes subjected to Chrebp knockdown (Figure 5D). Similarly, in Bad −/− hepatocytes, where the HCF-1:ChREBP complex is at low abundance, HCF-1 recruitment to lipogenic gene promoters is significantly blunted (Figure 5C).
Figure 5. HCF-1 binds to the promoters of lipogenic genes in a ChREBP dependent manner and is required for the recruitment of PHF2.

(A) DNL assays in primary hepatocytes subjected to Hcf-1 knockdown and assessed as in Figure 1A (n=3).
(B) Relative expression of Acc in primary hepatocytes subjected to Hcf-1 knockdown and treated with 5 or 25 mM glucose for 20 hrs (n=3).
(C) Relative occupancy of ChREBP and HCF-1 at the Acc promoter in WT and Bad −/− primary hepatocytes treated with 5 or 25 mM glucose for 4 hrs (n=9 experimental repeats in hepatocytes derived from 3 mice per genotype).
(D) Relative occupancy of HCF-1 at the Acc promoter in primary hepatocytes subjected to Chrebp knockdown and treated with 5 or 25 mM glucose for 4 hrs (n=6 experimental repeats in hepatocytes derived from 3 mice per genotype).
(E) Representative western blots (left) and quantification of n=3 experiments (right) of HCF-1 association with ChREBP and PHF2 in primary hepatocytes cultured in 5 or 25 mM glucose for 6 hrs in the presence of 20 μM MG132.
(F) Representative western blots (left) and quantification of n=5 experiments (right) of ChREBP association with PHF2 in primary hepatocytes subjected to Hcf-1 knockdown and cultured in 25 mM glucose for 6 hrs in the presence of 20 μM MG132.
(G) Relative occupancy of PHF2 at the Acc promoter in primary hepatocytes subjected to Hcf-1 knockdown and treated with 25 mM glucose for 4 hrs (n=6 experimental repeats in hepatocytes derived from 3 mice per genotype).
(H) Relative H3K4me3 occupancy at the Acc promoter in primary hepatocytes subjected to Hcf-1 knockdown and cultured in 5 or 25 mM glucose for 4 hrs (n=4).
Error bars are means ± SEM, *p < 0.05; **p < 0.01; ***p < 0.001; n.s., nonsignificant; two way ANOVA (B-D, G and H) or Student’s t test (A, E-F)
See also Figure S7.
ChREBP was recently shown to recruit the epigenetic co-activator Plant Homeodomain Finger 2 (PHF2) also known as KDM7C, which in turn demethylates inhibitory H3K9me2 marks at the promoters of ChREBP target genes to enhance transcription (Bricambert et al., 2018). This prompted examination whether PHF2 is a component of the ChREBP:HCF-1 promoter complex. HCF-1 immunoprecipitation assays indicated that, in addition to ChREBP, PHF2 interacts with HCF-1 in primary hepatocytes and this association is enriched in response to glucose stimulation (Figure 5E). The interaction between ChREBP and PHF2 is sensitive to GlcNAc levels as evident from the observation that this complex is reduced in Bad −/− hepatocytes treated with 25 mM glucose and is rescued upon GlcNAc supplementation similar to ChREBP and HCF-1 interaction (Figure S7). Remarkably, Hcf-1 knockdown in WT hepatocytes leads to diminished association between ChREBP and PHF2 and inhibits the recruitment of PHF2 to lipogenic gene promoters (Figures 5F–G). This indicates that PHF2 recruitment to ChREBP-containing promoter complexes is mediated by HCF-1 and, together with the data in Figure 5D, suggests a previously unappreciated tiered promoter recruitment process in that ChREBP recruits HCF-1 to lipogenic promoters, which in turn recruits PHF2.
PHF2 binds H3K4me3 histone tails and H3K4me3 is a key histone mark priming PHF2 recruitment to transcriptionally active promoters (Bricambert et al., 2018). The finding that HCF-1 is required for the binding of PHF2 to lipogenic gene promoters warranted examination of whether HCF-1 modulates H3K4me3 at these promoters, thereby facilitating PHF2 recruitment. We directly examined this possibility using H3K4me3 ChIP assays comparing hepatocytes subjected to control versus Hcf-1 knockdown. In contrast to control hepatocytes where glucose stimulation augments H3K4me3 at lipogenic gene promoters, this histone modification was significantly lower in HCF-1 depleted hepatocytes (Figure 5H). Thus, the necessity of HCF-1 for PHF2 recruitment to lipogenic gene promoters is consistent with HCF-1-dependent modulation of H3K4 trimethylation. Overall, these findings suggest that HCF-1 modulation of lipogenic gene transcription is linked to epigenetic regulation of ChREBP target promoters.
HCF-1 levels in NASH liver biopsies
Under normal physiological conditions, human hepatic DNL is kept at low levels (Sanders and Griffin, 2016). However, excessive DNL can contribute to hepatic steatosis in response to high carbohydrate overload and in NAFLD (Softic et al., 2016). The relevance of ChREBP in fatty liver disease is highlighted by studies showing that ChREBP overexpression in mice leads to hepatic steatosis and that ChREBP proteins levels are elevated in the livers of humans with NAFLD (Benhamed et al., 2012; Dentin et al., 2006; Hurtado del Pozo et al., 2011). Identification of HCF-1 as a ChREBP binding protein required for lipogenic gene expression prompted assessment of HCF-1 in a pathologic setting of excessive hepatic DNL as in human patients with fatty liver. ChREBP and PHF2 protein levels were higher in NASH (nonalcoholic steatohepatitis) patients compared to healthy liver donors consistent with previous reports (Figure 6 and Table S3) (Bricambert et al., 2018). Remarkably, HCF-1 levels were significantly enriched in NASH liver biopsies along with BAD phosphorylation on S118 (equivalent to S155 in the mouse BAD sequence) (Figure 6). These data, in conjunction with our findings that HCF-1 regulates ChREBP, suggest that the HCF-1-ChREBP biochemical axis, including its regulation by phosho-BAD, may be relevant in the pathophysiology of fatty liver disease. This possibility awaits future investigation of the consequences of hepatic HCF-1 alterations in mouse models of fatty liver.
Figure 6. Increased HCF-1 levels in livers of human NASH patients.

Immunoblot analysis (top) and quantification (bottom) of the indicated proteins in liver biopsies from healthy subjects and NASH patients (n=7).
Error bars are means ± SEM, *p < 0.05; **p < 0.01; n.s., nonsignificant; Student’s t test.
See also Table S3.
DISCUSSION
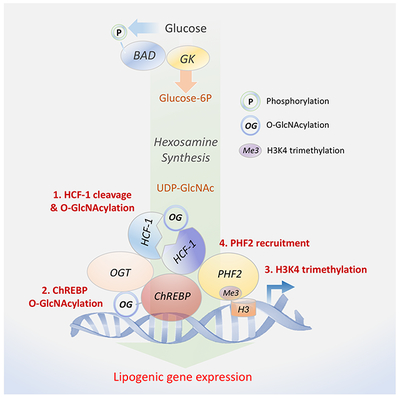
Our studies uncover an intricate interplay between lipogenic signals and transcriptional activation of hepatic DNL via stepwise assembly of an active promoter complex containing ChREBP and HCF-1, a transcriptional co-regulator we identified as a ChREBP binding protein. Specifically, we provide multiple lines of biochemical evidence that HCF-1 O-GlcNAcylation in response to glucose or HCD first recruits OGT to ChREBP, leading to ChREBP O-GlcNAcylation and activation (Figure 7). In turn, HCF-1 is recruited to lipogenic gene promoters in a ChREBP-dependent manner, where it is required for H3K4 trimethylation and subsequent recruitment of the epigenetic activator PHF2 (Figure 7).
Figure 7. Proposed biochemical model for HCF-1 modulation of de novo lipogenesis.

Increased hepatic glucose metabolism through GK promotes DNL by both providing biosynthetic precursors such as malonyl-CoA and by stimulating lipogenic gene expression via a glucose-sensitive protein complex containing HCF-1, OGT and ChREBP. Full induction of the ChREBP transcriptional program by glucose requires HCF-1 O-GlcNAcylation (1), mediating its interaction with ChREBP and subsequent recruitment of OGT to ChREBP (2). This augments ChREBP O-GlcNAcylation and activity. Through its association with ChREBP, HCF-1 is recruited to lipogenic gene promoters in response to glucose, where it mediates an increase in H3K4me3 histone marks (3), thereby recruiting the epigenetic activator PHF2 for enhanced transcription (4).
The discovery of HCF-1 as a regulator of ChREBP and DNL was facilitated by informative gain- and loss-of-function genetic approaches we undertook to modulate GK and the attendant glucose signals that regulate ChREBP. In addition to manipulating GK directly, we took advantage of the necessity of BAD phosphorylation for GK-mediated activation of ChREBP to effectively create a genetic system with “tuneable” glucose signaling for altering ChREBP activity. This not only provided a well-controlled platform for unbiased proteomics discovery of HCF-1 in complex with ChREBP, but also enabled interrogation of the mechanisms whereby glucose regulates the assembly and function of this complex.
Numerous post-translational mechanisms modulate ChREBP activity in response to lipogenic signals (Abdul-Wahed et al., 2017; Agius, 2016a). Among these, O-GlcNAcylation regulates ChREBP DNA binding and transcriptional activity, as well as protein stability (Guinez et al., 2011; Sakiyama et al., 2010; Yang and Qian, 2017). However, a specific mechanism for regulation of ChREBP O-GlcNAcylation has not been previously reported. Protein O-GlcNAcylation is the outcome of the net balance between OGT and O-GlcNAcase (OGA) activities, which transfer and remove the sugar modification, respectively. Remarkably, OGT and OGA are each encoded by a single gene, yet the pair regulates O-GlcNAcylation of a multitude of substrates. Several scenarios have been proposed as to how a single OGT gene can specifically O-GlcNAcylate hundreds of substrates. These include specific subcellular localization of different OGT splice variants, the capacity of OGT tetratricopeptide repeats (TPRs) to control the access of specific substrates to the active site, differential sensitivity of OGT substrates to intracellular UDP-GlcNAc concentrations, and adaptor proteins that recruit the enzyme to distinct substrates (Yang and Qian, 2017). We provide evidence that HCF-1 is required for OGT recruitment to ChREBP. Moreover, this process is glucose-dependent as evident from loss- and gain-of-function approaches we undertook to modulate GK activity and examine its effect on ChREBP association with HCF1 and OGT.
Although HCF-1 is primarily known as a transcriptional co-regulator rather than an adaptor for OGT, it can also recruit OGT and regulate the glycosylation of two other transcription factors, NRF1 and PGC1α (Lin et al., 2002; Ruan et al., 2012). However, unlike the HCF-1:ChREBP complex, HCF-1 association with PGC1α and PGC1α O-GlcNAcylation are diminished in response to glucose stimulation or in the fed state (Ruan et al., 2012), indicating additional mechanisms are at play to regulate the specificity of HCF-1 binding interactions and its effect on glycosylation of select partner proteins. Our studies have uncovered such a mechanism and demonstrate that HCF-1 O-GlcNAcylation itself, which we show is triggered by glucose stimulation or HCD treatment, is selectively required for its binding to ChREBP but dispensable for its interaction with PGC1α. To our knowledge, this is also the first evidence assigning a specific biochemical function to HCF-1 O-GlcNAcylation. Moreover, the observation that GlcNAc or GlcN supplementation is sufficient to restore HCF-1 O-GlcNAcylation, HCF-1:ChREBP complex formation and ChREBP O-GlcNAcylation under conditions where HBP pathway activity is decreased without global diminution in protein O-GlcNAcylation, suggests that the HCF-1-ChREBP biochemical axis may be particularly sensitive to fluctuations in UDP-GlcNAc levels. As such, lipogenic signals may be required to secure a sufficient pool of UDP-GlcNAc for HCF-1 glycosylation and its regulated binding to ChREBP. From a conceptual standpoint, this mode of regulation is intriguing because it provides an explanation for regulated enrichment of the HCF:ChREBP complex in response to lipogenic signals.
Our findings also point to more complex effects of HCF-1 on lipogenic gene expression beyond regulation of ChREBP glycosylation. These include HCF-1-dependent H3K4 trimethylation and the recruitment of the epigenetic activator PHF2 to these promoters in a glucose-dependent manner. While PHF2 was recently shown to be required for transcriptional activation of ChREBP target promoters (Bricambert et al., 2018), a specific mechanism for its binding to ChREBP or recruitment to these promoters was not known. As such, our findings that HCF-1 is required for PHF2 recruitment to ChREBP target promoters in response to glucose stimulation add significant new molecular details on nutrient regulation of epigenetic events at ChREBP target promoters. As a transcriptional co-regulator, HCF-1 can modulate the activity of epigenetic modifiers such as histone methyl transferases (Deplus et al., 2013; Tyagi et al., 2007; Wysocka et al., 2003; Zhou et al., 2013). While our Hcf-1 knockdown experiments clearly show its requirement for H3K4 trimethylation in response to glucose stimulation, the identity of the specific histone methyl transferase in charge of this modification at the lipogenic gene promoters awaits further studies. Regardless, our findings indicate that ChREBP and PHF2 are part of a larger nutrient sensitive promoter complex that contains HCF-1 as a key co-regulator and prerequisite for upregulation of activating histone marks at ChREBP target promoters.
The role of HCF-1 in hepatic metabolism is not fully understood and is likely to be context-dependent. Its capacity to interact with and regulate PGC1α, a chief transcriptional regulator of fatty acid oxidation and gluconeogenesis, would be consistent with its involvement in hepatic fasting responses such as increased glucose production (Lin et al., 2002; Ruan et al., 2012). On the other hand, we show HCF-1 is also required for transcriptional regulation of DNL through its binding and regulation of ChREBP in response to glucose stimulation or carbohydrate feeding. Within this context, nutrient-induced HCF-1 O-GlcNAcylation as a specific determinant of its binding to ChREBP but not PGC1α is an attractive mechanism that could underlie the toggling of HCF-1’s transcriptional regulatory roles between these two distinct nutritional states. The capacity to regulate both fatty acid oxidation and synthesis argues for a potentially broader role of HCF-1 in hepatic substrate metabolism and warrants future investigation of its significance in systemic physiology and in NAFLD.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nika N. Danial (nika_danial@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Bad −/− and conditional Gk mice (Gklox/lox) have been previously described (Giménez-Cassina et al., 2014; Postic et al., 1999). Unless otherwise indicated, mice received a standard chow diet and were housed in a barrier facility with 12 hr light and dark cycles. For high carbohydrate diet experiments, mice were fasted for 24 hrs and refed a high carbohydrate diet (70% sucrose) (Envigo, TD.150694) for 18 hrs.
All animal procedures were approved by the Institutional Animal Care and Use Committee at the Dana-Farber Cancer Institute.
Human Liver Tissue
Frozen human liver biopsies classified as normal (n = 7) or fatty liver (NASH) (n = 7) were provided by the Liver Tissue Cell Distribution System (LTCDS, Minneapolis, Minnesota), which was funded by the National Institutes of Health Contract # HSN276201200017C (Table S3). This study used collected specimens that had been de-identified by the LTCDS and were exempted by the Dana-Farber Cancer Institute Institutional Review Board (IRB) because it was determined that the project activities do not meet the definition of human subjects research set forth at 45 CFR 46.102 (protocol # 16-324).
Hepatocyte Isolation and Culture Conditions
Primary hepatocyte isolation and cultures were carried out as previously described with a two-step digestion process (Matsumoto et al., 2007). Briefly, livers from 8-12 week old male mice were drained of blood by perfusion via vena cava with 42°C perfusion buffer (0.4 g/L KCl, 1.0 g/L Dextrose, 1.8 g/L NaHCO3, 0.2 g/L EDTA ) for 3 min. Connective tissue within the liver was digested by perfusion of 42°C liver digest media (0.4 g/L KCl, 1.0 g/L Dextrose, 1.8 g/L NaHCO3, 0.5 g/L CaCl2, 10 g/L BSA, 30 mg/L Collagenase) for 10 min. The liver was mechanically dissociated in plating media (DMEM: 10% FBS, 2 mM Sodium Pyruvate, 2% Pen/Strep, 1 μM Dexamethasone, 0.1 μM Insulin) at 4°C, strained through 70 micron cell strainer, and hepatocytes were collected by centrifugation at 50 g for 3 min. Hepatocytes were further isolated from other cells on a percoll gradient with centrifugation at 650 RPM for 5 min. The cell pellet was washed and resuspended in plating media, and cells were plated in plating media at a density of 8x105 cells/well in 6 well plates or 4x106 cells/10 cm dish unless otherwise indicated and cultured with 5% CO2 at 37°C. Cells were starved in serum-free M199 media supplemented with 0.2% BSA (2% Pen/Strep, 2 mM Sodium pyruvate) for 12-16 hrs prior to experimental treatments.
For glucosamine (GlcN) and N-acetylglucosamine (GlcNAc) treatments, primary hepatocytes were treated with 5 mM glucosamine hydrochloride (Sigma) or 20 mM N-acetylglucosamine (Sigma) in high glucose M199 serum free media at pH 7.4 for the indicated length of time.
For MG132 treatment, primary hepatocytes treated with the indicated adenoviruses and/or siRNAs were cultured in low (5 mM) or high (25 mM) glucose for 6 hrs. After 3.5 hrs, the cells were treated with MG132 to a final concentration of 20 μM for the remaining 2.5 hrs of the 6 hr treatment.
METHOD DETAILS
Adenovirus Production and Viral Transduction
Bad shRNA, Gk shRNA or scrambled shRNA and recombinant GFP, BAD S155D, and BAD AAA adenoviruses were described previously (Bain et al., 2004; Giménez-Cassina et al., 2014). Adenoviruses carrying FLAG-ChREBP were a kind gift of Dr. Donald Scott (Mount Sinai) (Metukuri et al., 2012). Adenoviruses containing the Chrebp shRNA were created using the following sequence : gatccGTGTTGGCAATGCTGACATGttcaagagaCATGTCAGCATTGCCAACAttttttggag against ChREBP from Dentin et al. (Dentin et al., 2012). The shRNAs were ligated into pSIREN-DNR-DsRed express plasmid at BamH1 and EcoRI RE sites. The construct was excised at Sall and Spe1 sites to shuttle the hU6 promoter with the Chrebp shRNA sequence and ligated into pAdTrack-promoterless at the Sall and EcoRV RE sites. The adenovirus was generated using pAdEasy system (Stratagene). All virus amplification, purification, titration and verification were done using the services of ViraQuest Inc.
Adenoviral transduction of hepatocyte cultures was carried out during plating at a viral dose of 3 pfu/cell for 2-4 hrs followed by 2-4 hr recovery in maintenance media (DMEM: 8 mM Glucose, 1 nM Insulin, 0.1 M Dexamethasone, 2 mM Sodium Pyruvate, 2% Pen/Strep, 10% FBS) before overnight serum starvation in M199 media (0.2% BSA) unless otherwise noted. For knockdown experiments, hepatocytes were cultured for an additional 24 hrs in maintenance media before serum starvation in M199 media.
For hepatic reconstitution assays, mice were injected with a viral dose of 3x108 pfu/g of body weight via tail vein, and livers were harvested 1 week after injection.
Primary Hepatocyte siRNA Transfection
Reverse siRNA transfection was performed with Lipofectamine RNAi Max (Life Technologies) per manufacturer’s protocol. In brief, 150 pmol of control or Hcf-1 RNA duplex and 15 μL Lipofectamine RNAi max were gently mixed in 1 mL Opti-MEM and incubated for 10 min at RT before adding to freshly plated hepatocytes. For experiments that combined Hcf-1 siRNA treatment and adenoviral expression of BAD variants, cells treated with Hcf-1 siRNA, were left to recover for 2 hrs in maintenance media (DMEM: 8 mM Glucose, 1 nM Insulin, 0.1 M Dexamethasone, 2 mM Sodium Pyruvate, 2% Pen/Strep, 10% FBS) prior to infection with adenoviruses. Cells were then harvested for analysis after 30 hrs.
De Novo Lipogenesis Assay
Hepatocytes were plated at 106 cells per well in 6 well plates. After overnight serum starvation, hepatocytes were pretreated in 0.8 mL serum free M199 media containing 5 or 25 mM glucose for 30 min and then spiked with 1 μCi of U-14C-labeled glucose (American Radiolabeled Chemicals) for 4 hrs. Lipids were extracted in methanol with a modified Folch method. Briefly, cells were washed with PBS and then harvested in 200 μL of 0.5% Triton in PBS, and 500 μL of 2:1 chloroform:methanol was added to 150 μL before vortexing at high speed for 20-30 sec and incubating on ice for 1 hr. The remaining protein lysate was used for protein quantification and normalization. The samples were vortexed, and 125 μL of dH2O was added before vortexing again and centrifuging for 15 min at 1000 RPM at 4°C. The bottom phase was transferred to a large 20 mL glass scintillation vial containing 3 mL of scintillation fluid. Incorporation of 14C carbons into the lipid fraction was measured using a Tri-Carb 2900 TR liquid scintillation counter (Packard), and all reads were normalized to protein content.
RNA Preparation and Quantitative Real-Time PCR
mRNA was isolated from hepatocytes plated at a density of 8x105 cells/well in 6 well plates using 0.5 mL of Trizol (Life Technologies) per well, and 1 μg of RNA was used to generate cDNA by the Superscript III reverse transcriptase reagents (Life Technologies). cDNA was diluted to 200 μL (1:10 dilution), and qPCR was performed using an Applied Biosystems 7900HT Fast Real-Time PCR instrument with Syber Green reagents (Life Technologies) to a final volume of 10 μL in 384 well qPCR plates with 3 μL of diluted cDNA (1.5 μL for cyclophilin D control primers) and 300 μM primers for the indicated genes. Gene expression levels were normalized to Cyclophilin D (CycD) using the 2−ΔΔCt method and are presented as relative transcript levels. The sequence of primers is available in Table S1.
Dual Luciferase Assay
Hepatocytes were transfected with 3 μg/mL of Acc (O’Callaghan et al., 2001) or L-Pk (Lou et al., 1999) ChoRE firefly luciferase reporters (kind gift from Dr. Howard Towle) and 10 ng/mL of CMV Renilla luciferase control plasmids (Thermo Fisher Scientific) using the Targefect-hepatocyte (Targeting Systems) transfection reagent. Briefly, transfection complexes were formed in 1 mL serum free high glucose DMEM without antibiotics by first adding 3 μg of DNA followed by 7.5 μL of targefect reagent and 10 μL of virofect reagent per well followed by a 20 min incubation at 37°C. The cells were washed in antibiotic free medi a and 1 mL of the transfection complex was added per well in 6 well plate. Hepatocytes were cultured in maintenance media without antibiotics (DMEM: 8 mM Glucose, 1 nM Insulin, 0.1 M Dexamethasone, 2 mM Sodium Pyruvate, 10% FBS) for 4 hrs for recovery and then infected with 2 pfu/cell of the indicated adenoviruses for 2 hrs before overnight culture in serum free (0.2% BSA) M199 media containing 5 mM glucose. Hepatocytes were then treated for an additional 20 hrs in M199 media containing 5 or 25 mM glucose. ChREBP transcriptional activity was measured by performing Promega dual luciferase assay. Briefly, cells were lysed in 500 μL of 1X passive lysis buffer (Promega), and 20 μL of lysate for each condition was transferred to a 96 well white plate. The samples were analyzed using a Berthold Technologies Mithras LB 940 instrument to inject 100 μL of LAR II reagent, measure firefly luciferase activity followed by 100 μL of Stop & Glo reagent. Renilla luciferase activity for each well was then measured individually.
Chromatin Immunoprecipitation
After overnight serum starvation, primary hepatocytes were treated with 5 or 25 mM glucose for 4 hrs and washed with PBS before incubation in 1% formaldehyde (in PBS) for 10 min at 37°C. Cells were washed once in 0.125 M glycine in PBS and collected in 1 mL of PBS before centrifugation at 2000 g for 5 min at 4°C. The pell et was resuspended in 500 μL of SBAR (50 mM HEPES pH 7.4, 140 mM NaCl, 1 mM EDTA, 1% Triton, 0.1% NaDOC, 0.1% SDS, 0.5 mM PMSF, Complete™ EDTA-free Protease Inhibitor Cocktail), and passed through an insulin syringe several times.
Cells in SBAR buffer were sonicated in a Biorupter sonicator at middle intensity 3 times for 5 min with a 30 sec ON 30 sec OFF cycle at 4°C. Lysates were centrifuged for 20 min and the supernatant was transferred to a new tube. DNA concentration was measured and the equivalent of 75 μg of DNA was used per ChIP in a total volume of 200 μL of SBAR (10 μL was kept for input). Samples were incubated with 1 μg of the indicated antibody at 4°C overnight, and 10 μL of dynabeads Protein A (Invitrogen) pre-blocked with salmon sperm DNA were added to the sample and rotated for 1.5 hr at 4°C. Consecutive washes were performed in the following buffers: Buffer I (1% Triton, 0.1% NaDOC, 150 mM NaCl, 10 mM Tris.Cl pH 8), Buffer II (1% NP-40, 1% NaDOC, 150 mM KCL, 10 mM TrisCl), Buffer III (0.5% Triton, 0.1% NaDOC, 500 mM NaCl, 10 mM TrisCl), Buffer IV ( 0.5% NP-40, 0.5% NaDOC, 250 mM LiCl, 20 mM TrisCl, 1 mM EDTA), Buffer V (0.1% NP-40, 150 mM NaCl, 20 mM TrisCl, 1 mM EDTA), and TE for 5 min each. 100 μL of 10% Chelex resin was added to all samples to recover eluates, and 5 μL of 2 mg/mL proteinase K was added to the eluates (20 mg/mL for inputs) followed by 30 min incubation at 55°C. The digestion was then stopped by boiling the samples for 10 min. qPCR was performed using 3 μL per replica for eluates and 1 μL per replica for input. A standard curve was generated using dilutions of a pooled input sample, and ChIP samples were normalized to their respective inputs. Primers used are available in Table S1.
For liver chromatin immunoprecipitation assays, approximately 80% of the liver sample was homogenized in 5 mL of SHB (15 mM HEPES pH 7.9, 15 mM KCl, 1 mM EDTA, 50 μM spermine, 50 μM spermidine, 1.9 M sucrose, 0.5 mM PMSF, Complete™ EDTA-free Protease Inhibitor Cocktail) using a low speed motor-driven pestle 3 times. The liver homogenate was layered on top of 7.5 mL of cold SHB in 13.5 mL Beckman Ultra-Clear tubes and centrifuged at 25,000 RPM for 1 hr at 4°C. The pellet was gently resuspended in 400 μL of SucC buffer (15 mM HEPES pH 7.9, 60 mM KCL, 15 mM NaCl, 15 μM β-ME, 0.2 mM MgCl2, 0.34 M sucrose, 0.5 mM PMSF, Complete™ EDTA-free Protease Inhibitor Cocktail), and transferred to a new Eppendorf tube. Formaldehyde was added to a final concentration of 1%, and the tubes were incubated for 13 min at 37°C to crosslink the DNA and protein. Crosslinking was stopped by adding glycine to a final concentration of 0.125 M followed by a 5 min incubation on ice. The homogenate was transferred to a second cushion of SucC buffer with 0.9 M sucrose in a 15 mL centrifuge tube and spun at 4,000 RPM for 15 min at 4°C before resuspending the pellet in 400 μL of SBAR buffer.
Co-Immunoprecipitation and Western Blotting
For immunoprecipitation assays (IPs), cells were lysed in buffer containing (20 mM Tris/HCl pH 7.5, 137 mM NaCl, 5% glycerol, 2 mM EDTA, 2 mM Na3VO4, Complete™ EDTA-free Protease Inhibitor Cocktail, phosphatase inhibitor cocktail 3, 2 μM Thiamet-G, 10 μM PUGNAc, 1 μM TSA, 0.15% Triton-X100). Cells were mechanically disrupted by passing through an insulin syringe and rotating for 1.5 hr at 4°C, pelleted at maximum speed in a desktop centrifuge, and the supernatant was transferred to a new tube. Immunoprecipitation was carried out with 3 mg of protein lysates and 3 μg of ChREBP antibody (Novus Biologicals, lots Q1-Q3) rotating overnight at 4°C. After overnight incubation with the antibody, 50 μL of protein G agarose beads (Sigma-Aldrich) was added to each sample followed by rotation for an additional 2 hrs at 4°C. The IPs were washed once in lysis buffer, and proteins were eluted in 2X LDS sample buffer (Life Technologies) with a 10 min boil.
For western blotting without immunoprecipitation, cells were lysed in RIPA buffer (10 mM Tris/HCl, 150 mM NaCl, 1% Triton X-100 [v/v], 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 10 μM PUGNAc , and 2 μM Thiamet-G , and protease inhibitors; pH 7.4), and the protein amount was quantified by BCA assay. For western blots, 10-30 μg of protein lysate was loaded.
Samples were prepared for western blotting using the NuPAGE 4X sample buffer and NuPAGE gel systems (Life Technologies). Proteins were transferred onto PVDF membrane in Towbin buffer (3.03 g/L Tris Base, 7.57 g/L Glycine, 20% methanol), and blotted with the indicated antibodies. The ImageJ software was used for quantification of band intensities.
Wheat Germ Agglutinin (WGA) Pull Down
WGA pull down assays were performed as previously described (Guinez et al., 2011). In brief, primary hepatocytes subjected to the indicated genetic manipulations and cell culture conditions were treated with MG132 (20 μM) and lysed in RIPA lysis buffer (10 mM Tris/HCl, 150 mM NaCl, 1% Triton X-100 [v/v], 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 10 μM PUGNAc, 2 μM Thiamet-G , and protease inhibitors; pH 7.4) supplemented with or without 0.5 M of GlcNAc for 20 min on ice. Samples were centrifuged at 16,400 RPM for 20 min, and 1.5 mg of protein lysate was incubated with 50 μL of WGA beads (VWR) for 2 hrs at 4°C.
Beads were centrifuged briefly at 2,000 RPM and subsequently washed 3 times with the following buffers: I) RIPA buffer; II) 1:1 mixture of RIPA and RIPA containing 500 mM NaCl; III) TNE (10 mM Tris/HCl, 150 mM NaCl, and 1 mM EDTA; pH 7.4). Samples were boiled in 50 μL of 2X LDS sample buffer (Life Technologies) and subjected to SDS-PAGE followed by western blotting with the indicated antibodies.
In Vitro Cleavage and O-GlcNAcylation of HCF1 and Pull Down Assays
HCF-1 was in vitro transcribed and translated using the S6 promoter of the pCMV-Sport6 HCF-1 plasmid and the TnT® Coupled Wheat Germ Extract System per manufacturer’s instructions (Promega, L4130).
His-OGT WT or OGT D554N proteins were purified from E. Coli as previously described (Janetzko et al., 2016), and used to process the in vitro translated HCF-1. Briefly, 5 μl of the in vitro synthesized HCF-1 protein was incubated with 35 μl of in vitro cleavage and O-GlcNAcylation assay buffer (20 mM Tris/HCl pH 7.5, 120 mM NaCl, 2 mM MgCl, 2 mM UDP-GlcNAc, 1mM fresh DTT) containing 1 or 2 μg of His-OGT WT or OGT D554N for 3 hrs at 37°C.
For interaction assays, endogenous ChREBP was bound to protein G agarose beads as described in the immunoprecipitation method above. 30 μl of pre-washed, ChREBP bound beads was incubated with 40 μl of the in vitro processed HCF-1 protein that was diluted in 110 μl of Co-IP lysis buffer (20 mM Tris/HCl pH 7.5, 137 mM NaCl, 5% glycerol, 2 mM EDTA, 2 mM Na3VO4, Complete™ EDTA-free Protease Inhibitor Cocktail, Phosphatase inhibitor cocktail 3, 2 μM Thiamet-G, 10 μM PUGNAc, 1 μM TSA, 0.15% Triton-X100). After 1 hr rotation at 4°C, the complex was washed 2 times with lysis buffer, eluted in 50 μl of 2X LDS sample buffer (Life Technologies) and boiled for 15 min for western blot analysis.
Expression of OGT WT and D554N for HCF-1 Interaction Studies in HEK293T Cells
For mammalian expression of OGT, full-length human OGT cDNA (NM_181673.2) was amplified by PCR and inserted by isothermal assembly between the BamHI and Xhol sites in plasmid pENTR1A no ccDB (w48-1) (Gibson et al., 2009). The D554N OGT mutant was generated using the Q5 site-directed mutagenesis kit according to the manufacturer’s protocols (New England Biolabs). WT and D554N OGT were inserted into the pLenti PGK Neo DEST (w531-1) vector using LR Clonase II enzyme mix (Thermo Fisher Scientific). All constructs were verified by Sanger sequencing of the complete OGT insert before use. pENTR1A no ccDB (w48-1) and pLenti PGK Neo DEST (w531-1) were gifts from Drs. Eric Campeau and Paul Kaufman (Addgene plasmid # 17398 and 19067, respectively).
Transfection of 293T cells was carried out using Lipofectamine 2000™ according to the manufacturer’s instructions. In brief, one day after plating 293T cells in a 10 cm dish, 4.0 μg of the DNA mixture was incubated with 13.5 μl of Lipofectamine 2000™ in 1 ml of Opti-MEM™ for 10 min at RT. Cell media was replaced with 5 ml of pre-warmed Opti-MEM™ and the DNA mixture was added. After a 5 hr incubation, cells were incubated with newly replaced DMEM media for an additional 24 hrs and subjected to WGA pull down or Co-IP experiments. For ChREBP interaction studies, the DNA mixture for transfection included the following expression vectors: 1.5 μg HCF-1, 0.5 μg ChREBP, 0.5 μg pCDN3.1, and 1.5 μg OGT WT or D554N. For PGC1α interaction studies, the DNA mixture for transfection included the following expression vectors: 1.5 μg HCF-1, 1 μg PGC1α (Fan et al., 2004), and 1.5 μg OGT WT or D554N.
Hexosamine Pathway Metabolite Measurement
Primary hepatocytes infected with the indicated adenoviruses were serum starved overnight and subsequently treated with 25 mM glucose for 5 hrs. For each measurement, extracts from three 10 cm dishes were used. Cells were quickly washed twice on ice with cold 150 mM ammonium formate at pH 7.4. Plates were then placed on dry ice and scraped in 230 μL of 50% MeOH/50% HPLC H2O solution and transferred to a pre-chilled tube with a metal bead. Tubes were washed with an additional 150 μL of 50% MeOH/ 50% HPLC H2O solution and vortexed for 10 sec. 220 μL of ice cold acetonitrile was added and the tubes were vortexed again for an additional 10 sec. Thawed samples were placed in pre-chilled bead beater racks and beaten for 2-5 min at 30 Hz. 600 μL of dichloromethane and 300 μL of ice cold H2O were added and tubes were vortexed for 15 sec prior to 10 min incubation on ice. The samples were spun down for 5 min at 4,000 RPM at 1°C and the upper aqueous phase was transferred to a new tube on ice. The samples were spun 1 min at 13,000 RPM to remove any remaining contaminants and supernatants were transferred to a new tube on dry ice. The aqueous phase was dried using a chilled speedvac and samples were stored at −80°C u ntil ready for LC-MS/MS data collection.
For targeted analysis and semi-quantitative concentration determination of the hexosamine metabolites, 5 uL of each sample was re-suspended in 50 μL of water and injected in an Agilent 6430 Triple Quadrupole (QQQ)-LC-MS/MS. Chromatography was performed using a 1290 Infinity ultra-performance LC system (Agilent Technologies) consisting of vacuum degasser, autosampler and a binary pump. The mass spectrometer was equipped with an electrospray ionization (ESI) source and samples were analyzed in negative mode. Multiple reaction monitoring (MRM) transitions were optimized on standards for each quantitated metabolite. MRM transitions and metabolite retention times are shown in Table S2. Gas temperature and flow were set at 350°C and 10 L/min respectively, nebulizer pressure was set at 50 psi and capillary voltage was set at ±4000V.
Chromatographic separation was performed on a Unison UK-Amino column 3 μm, 2.0x150mm (Imtakt Corp) maintained at 55°C. The chromatograph ic gradient started at 90% mobile phase B (acetonitrile) with a 2 min hold followed with a 18 min gradient to 100% mobile phase A (200 mM ammonium acetate in H2O) and then held at 100% A for 10 min at a flow rate of 0.6 mL/min. This was followed by a 6 min re-equilibration at 100% mobile phase B before the next injection. Relative concentrations were determined from external calibration curves prepared in H2O. Additional corrections for potential sample ion suppression were not made. Data were analyzed using MassHunter Quant (Agilent Technologies).
Nuclear Flag-ChREBP IP and LC-MS Analysis
Primary hepatocytes were rinsed once with cold PBS and once with cold H2O before being scraped in 400 μL of hypotonic lysis buffer (40 mM Tris PH 7.4, 10 mM NaCl, 1 mM EDTA, 2 mM Na3VO4, Complete™ EDTA-free Protease Inhibitor Cocktail). Cells were incubated on ice for 20 min and CHAPS was added to a final concentration of 0.5%. Cells were passed up and down in an insulin syringe 4-5 times, and nuclei were pelleted by centrifugation at 1,600 g for 7 min. The nuclei were washed 3 times in hypotonic wash buffer (hypotonic lysis buffer containing 0.5% CHAPS) and resuspended in 150 μL of hypertonic lysis buffer (40 mM Tris PH 7.4, 420 mM NaCl, 1 mM EDTA, 0.5 mM DTT, 2 mM Na3VO4, 0.5 mM PMSF, Complete™ EDTA-free Protease Inhibitor Cocktail, 0.5% CHAPS) using a hand held pestle homogenizer. Nuclei were incubated on ice for an additional 30 min with periodic vortexing, spun at 10,000 g for 30 min. The nuclear fraction (supernatant) was then transferred to a new tube.
For Co-IP experiments, nuclear fractions were concentrated using Amicon Ultra centrifugal filter units with a 3K cut off, and samples were diluted such that the final buffer concentrations was the same as Co-IP lysis buffer (20 mM Tris/HCL pH 7.4, 137 mM NaCl, 5% glycerol, 2 mM EDTA, 2 mM Na3VO4, Complete™ EDTA-free Protease Inhibitor Cocktail, 0.5% CHAPS). Anti-FLAG® M2 magnetic beads (Sigma-Aldrich) were washed 3 times in Co-IP lysis buffer and the equivalent of 20 μL of initial slurry was added to 3.5 mg of nuclear lysate. Samples were rotated for 8 hrs at 4°C and washed 3 times in Co-IP lysis buffer. After the 3rd wash, proteins were eluted by resuspending the beads in 50 μL of Co-IP buffer containing 15 μg of FLAG peptides followed by incubation on a vortex platform for 30 min at 4°C. The eluate was transferred to a centrifugal filter prewashed with Co-IP buffer and the elution step was repeated. Eluates were then pooled in a single tube, TCA precipitated, and submitted for proteomic analysis at the Taplin Mass Spectrometry Facility at Harvard Medical School.
For proteomics analysis, the TCA-precipitated samples were reduced in a solution of 1 mM DTT and 50 mM ammonium bicarbonate for 30 min at 60°C. The samples were then cooled to room temperature (RT) and iodacetamide was added to a concentration of 5 mM for 15 min in the dark at RT. DTT was then added to a 5 mM concentration to quench the reaction. Sequence grade trypsin was added at a concentration of 5 ng/ μL followed by overnight incubation at 37°C. The samples were then desalted by an in house-made desalting column, dried in a speed vac, and stored at 4°C until analysis.
On the day of analysis, the samples were reconstituted in 5-10 μl of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid). A nano-scale reverse-phase HPLC capillary column was created by packing 2.6 μm C18 spherical silica beads into a fused silica capillary (100 μm inner diameter x ~30 cm length) with a flame-drawn tip (Shevchenko et al., 1996). After equilibrating the column, each sample was loaded via a Famos auto sampler (LC Packings) onto the column. A gradient was formed and peptides were eluted with increasing concentrations of solvent B (97.5% acetonitrile, 0.1% formic acid).
As peptides eluted they were subjected to electrospray ionization and entered into an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific). Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences (and hence protein identity) were determined by matching protein databases with the acquired fragmentation pattern by the software program, Sequest (Thermo Fisher Scientific) (Eng et al., 1994). All databases include a reversed version of all the sequences and the data was filtered to between a one and two percent peptide false discovery rate (FDR).
The spectral counts for the identified proteins in Flag-ChREBP immunoprecipitates were normalized to ChREBP spectral counts in each IP sample. Proteins that were enriched 3-fold or more in WT compared to Bad −/− samples were prioritized for analysis using online databases, including DAVID, STRING, Panther, UniProt, and PubMed literature search. Proteins known to interact with other transcription factors were highlighted by STRING analysis. Proteins with direct or indirect functional annotations in metabolism and transcription were identified by gene ontology in DAVID and Panther, and proteins with indirect annotations in metabolism and transcription were identified by UniProt and PubMed literature searches. Proteins with exclusively mitochondrial or endoplasmic reticulum localization were excluded from the list.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean ± SEM of the indicated number of independent hepatocyte isolations or mice per genotype in the figure legends. Statistical significance among the groups was tested with unpaired or paired Student’s t test and ANOVA with Tukey’s post-hoc multiple comparison test when appropriate using the GraphPad Prism software. Differences were considered significant at p < 0.05. Unless otherwise indicated, n denotes the number of independent hepatocyte isolations or mice.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ChREBP | Novus Biologicals | NB400135 lots Q1-Q3 |
| Rabbit polyclonal anti-HCF-1 | Bethyl laboratories | A301-400A |
| Rabbit monoclonal anti-OGT | Cell Signaling | 24083 |
| Rabbit monoclonal anti-PHF2 | Cell Signaling | 3497S |
| Rabbit polyclonal anti-GCK | Santa Cruz | sc7908 |
| Rabbit monoclonal anti-BAD | Abcam | ab32445 |
| Rabbit polyclonal anti-phospho BAD (S155) | Cell Signaling | 9297 |
| Mouse monoclonal anti-Actin | Sigma-Aldrich | A5316 |
| Rabbit IgG Isotype Control | Invitrogen | 31235 |
| Mouse monoclonal anti FLAG M2- Peroxidase | Sigma-Aldrich | A8592 |
| Mouse Anti-rabbit IgG (Conformation Specific) | Cell Signaling | 5127S |
| Goat anti-rabbit IgG HRP | Jackson ImmunoResearch Laboratories | 111-035-003 |
| Goat anti-mouse IgG HRP | Jackson ImmunoResearch Laboratories | 115-035-003 |
| Bacterial and Virus Strains | ||
| BAD SD Adenovirus | Gimenez-Cassina et al, 2014 | PMID: 24506868 |
| BAD AAA Adenovirus | Gimenez-Cassina et al, 2014 | PMID: 24506868 |
| Gk shRNA Adenovirus | Gift of Dr. Christopher Newgard (Duke University) Bain et al., 2004 | PMID: 15331526 |
| Bad shRNA Adenovirus | Gimenez-Cassina et al, 2014 | PMID: 24506868 |
| Chrebp shRNA Adenovirus | Designed from sequence in Dentin et al, 2006 | PMID: 16873678 |
| Ctrl shRNA Adenovirus | Gimenez-Cassina et al, 2014 | PMID: 24506868 |
| FLAG-ChREBP Adenovirus | Gift from Dr. Donald Scott (Mount Sinai) (Metukuri et al, 2012) | PMID: 22586588 |
| Biological Samples | ||
| Human Liver Tissues Healthy and NASH Patients | Liver Tissue Cell Distribution System, Minneapolis, Minnesota | NIH Contract # HHSN276201200017C |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phosphatase Inhibitor Cocktail 2 aqueous solution | Sigma-Aldrich | P5726 |
| Protein Phosphatase Inhibitor Cocktail 3 | Sigma-Aldrich | P0044 |
| Phosstop, Phosphatase Inhibitor | Sigma-Aldrich | 4906837001 |
| Complete™, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | 11873580001 |
| PuGNAC | Sigma-Aldrich | A7229 |
| Thiamet G | Sigma-Aldrich | SML 0244 |
| Sodium Orthovanadate | Sigma-Aldrich | S6508 |
| Western Lightning Plus-ECL | Thermo Fisher Scientific | 509049325 |
| SuperSignal West Femto | Thermo Fisher Scientific | PI34095 |
| NuPAGE LDS Sample Buffer (4X) | Life Technologies | NP0008 |
| FLAG Peptide | Sigma-Aldrich | F3290 |
| UDP-GlcNAC | Sigma-Aldrich | |
| N-Acetyl-D-glucosamine | Sigma-Aldrich | A8625-5G |
| Proteinase K | Life technologies | 25530015 |
| CHELEX 100 200-400 MB | Bio-Rad Laboratories | 1421253 |
| Formaldehyde (37% solution) | Santa Cruz | sc-203049 |
| Collagenase | Sigma-Aldrich | C5138 |
| Insulin | Sigma-Aldrich | I6634 |
| Dexamethasone | Sigma-Aldrich | D-2915 |
| Percoll | Sigma-Aldrich | P4937 |
| MG132 | Sigma-Aldrich | C2211 |
| BSA Fraction IV Fatty acid free | Fischer Scientific | NC9227912 |
| Glucosamine hydrochloride | Sigma-Aldrich | G4875 |
| Ammonium Formate | Sigma-Aldrich | 17843 |
| Acetonitrile | Sigma-Aldrich | AX0145-1 |
| HPLC grade water | Sigma-Aldrich | WX0008 |
| Ammonium Acetate | Sigma-Aldrich | 5330040050 |
| U14C labeled glucose | American Radiolabled Chemical | ARC-122 |
| Critical Commercial Assays | ||
| Dual luciferase reagent | Promega | E1910 |
| Targefect hepatocyte | Targeting systems | HEP-01 |
| Trizol | Life Technologies | 15596026 |
| Syber Green Master Mix | Life Technologies | |
| Superscript III 1st Strand Synthesis | Life Technologies | 18080051 |
| Lipofectamine RNAiMax | Life Technologies | 13778150 |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | E0554S |
| Gateway™ LR Clonase™ II Enzyme mix | Thermo Fischer Scientific | 11791100 |
| TnT® Coupled Wheat Germ Extract System | Promega | L4130 |
| Deposited Data | ||
| Unprocessed gel images presented in this manuscript | http://dx.doi.org/10.17632/by6b3tn4g2.1 | |
| Experimental Models: Cell Lines | ||
| HEK293T Cells | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6: WT | ||
| C57BL/6-KO (BAD) | Gimenez-cassina et al, 2014 | PMID: 24506868 |
| GKlox/lox | Gift from Dr. Mark Magnuson (Vanderbilt university): Postic et al, 1999 | PMID: 9867845 |
| C57BL/6-alb-cre mice | Jackson Laboratory | 003574 |
| Oligonucleotides | ||
| Ctrl siRNA: siGenome Non-targeting siRNA #1 | Dharmacon | D-001210 |
| Hcf1 siRNA-1 sense sequence: GGA GCU UAU AGU GGU GUU U | Dharmacon | N/A |
| Hcf1 siRNA-2 sense sequence: AGA ACA ACA UUC CGA GGU A | Dharmacon | N/A |
| Primers for qPCR: See Table S1 for sequence | IDT | N/A |
| Recombinant DNA | ||
| L-Pk ChORE firefly luciferase reporter | Gift from Dr. Howard Towle: Lou D-Q et al, 1999 | PMID: 10497199 |
| Acc ChORE firefly luciferase reporter | Gift from Dr. Howard Towle: O’Callaghan BL et al, 2001 | PMID: 11340083 |
| pCMV-Green Renilla Luciferase control plasmid: | Thermo Fisher Scientific | PI16153 |
| pCMV10 3X FLAG-ChREBP | Gift from Dr. Mark Herman (Duke University) | N/A |
| pCMV-SPORT6 HCF-1 Plasmid | Harvard PlasmID | MmCD00320375 |
| His-OGT WT | Janetzko et al, 2016 | PMID: 27618188 |
| His-OGT D554N | Janetzko et al, 2016 | PMID: 27618188 |
| OGT WT | This Paper | N/A |
| OGT D554N | This Paper | N/A |
| PCDNA3.1 FLAG-PGC1α | Gift from Dr. Pere Puigserver: Fan et al, 2004 | PMID: 14744933 |
| pCDNA3.1 | Invitrogen | V790-20 |
| Software and Algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | N/A |
| Prism | Graph Pad | N/A |
| SDS 2.4 (for Q-PCR) | Applied Biosystems | N/A |
| MassHunter Quant | Agilent Technologies | N/A |
| Sequest | Thermo Fischer Scientific | N/A |
| Other | ||
| Serum | Gemini | 100-106 lot A16E00E |
| High Carbohydrate Diet (70% sucrose) | Envigo | TD.150694 |
| WGA Beads | Vector Laboratories | AL-1023 |
| Anti-FLAG® M2 Magnetic Beads | Sigma-Aldrich | M8823 |
| Protein G Agarose | Sigma-Aldrich Aldrich | 11719416001 |
| DynaBeads Protein A | Thermo Fisher Scientific | 10001D |
| Amicon Ultra Centrifugal Filters | Millipore | UFC500324 |
Highlights.
HCF-1 binds ChREBP and is required for glucose stimulation of de novo lipogenesis
HCF-1 recruits OGT to ChREBP, stimulating ChREBP O-GlcNAcylation and activation
Nutrient induction of HCF-1 O-GlcNAcylation is required for its binding to ChREBP
HCF-1 recruits epigenetic activators to promoters of lipogenic genes
Acknowledgments
Taplin Mass Spectrometry Facility at Harvard Medical School under the direction of Ross Tomaino and Steven Gygi for proteomic analysis of FLAG-ChREBP IPs; Daina Avizonis and Gaëlle Bridon for metabolite measurements at the Rosalind and Morris Goodman Cancer Research Centre Metabolomics Core Facility supported by the Canada Foundation for Innovation, The Dr. John R. and Clara M. Fraser Memorial Trust, the Terry Fox Foundation (TFF Oncometabolism Team Grant 1048 in partnership with the Foundation du Cancer du Sein du Quebec), and McGill University; The Liver Tissue Cell Distribution System, Minneapolis, Minnesota funded by NIH Contract #HHSN276201200017C for providing normal and NASH human liver samples; Mark Magnuson, Christopher Newgard, Pere Puigserver, Mark Herman, Donald Scott and Howard Towle for reagents; Pere Puigserver, Suzanne Gaudet, Loren Walensky, and Mark Herman for helpful discussions. E.A.L was supported by an NIH F31 Ruth-Kirschstein Predoctoral Fellowship. This work was supported by the U.S. NIH grants R01GM094263 (S.W.) R01DK078081 (N.N.D) and Claudia Adams Barr Award in Innovative Basic Cancer Research (N.N.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
REFERENCES
- Abdul-Wahed A, Guilmeau S, and Postic C (2017). Sweet Sixteenth for ChREBP: Established Roles and Future Goals. Cell Metab 26, 324–341. [DOI] [PubMed] [Google Scholar]
- Agius L (2016a). Dietary carbohydrate and control of hepatic gene expression: mechanistic links from ATP and phosphate ester homeostasis to the carbohydrate-response element-binding protein. Proc Nutr Soc 75, 10–18. [DOI] [PubMed] [Google Scholar]
- Agius L (2016b). Hormonal and Metabolite Regulation of Hepatic Glucokinase. Annu Rev Nutr 36, 389–415. [DOI] [PubMed] [Google Scholar]
- Alves-Bezerra M, and Cohen DE (2017). Triglyceride Metabolism in the Liver. Compr Physiol 8, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden C, Tudhope S, Petrie J, Al-Oanzi Z, Cullen K, Lange A, Towle H, and Agius L (2012). Fructose 2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. The Biochemical journal 443, 111–123. [DOI] [PubMed] [Google Scholar]
- Bain JR, Schisler JC, Takeuchi K, Newgard CB, and Becker TC (2004). An adenovirus vector for efficient RNA interference-mediated suppression of target genes in insulinoma cells and pancreatic islets of langerhans. Diabetes 53, 2190–2194. [DOI] [PubMed] [Google Scholar]
- Benhamed F, Denechaud P-DD, Lemoine M, Robichon C, Moldes M, Bertrand-Michel J, Ratziu V, Serfaty L, Housset C, Capeau J, et al. (2012). The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. The Journal of clinical investigation 122, 2176–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricambert J, Alves-Guerra MC, Esteves P, Prip-Buus C, Bertrand-Michel J, Guillou H, Chang CJ, Vander Wal MN, Canonne-Hergaux F, Mathurin P, et al. (2018). The histone demethylase Phf2 acts as a molecular checkpoint to prevent NAFLD progression during obesity. Nat Commun 9, 2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricambert J, Miranda J, Benhamed F, Girard J, Postic C, and Dentin R (2010). Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J Clin Invest 120, 4316–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capotosti F, Guernier S, Lammers F, Waridel P, Cai Y, Jin J, Conaway JW, Conaway RC, and Herr W (2011). O-GlcNAc transferase catalyzes site-specific proteolysis of HCF-1. Cell 144, 376–388. [DOI] [PubMed] [Google Scholar]
- Danial N, Walensky L, Zhang C-Y, Choi C, Fisher J, Molina A, Datta S, Pitter K, Bird G, Wikstrom J, et al. (2008). Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nature medicine 14, 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, et al. (2003). BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 424, 952–956. [DOI] [PubMed] [Google Scholar]
- Daou S, Mashtalir N, Hammond-Martel I, Pak H, Yu H, Sui G, Vogel JL, Kristie TM, and Affar el B (2011). Crosstalk between O-GlcNAcylation and proteolytic cleavage regulates the host cell factor-1 maturation pathway. Proc Natl Acad Sci U S A 108, 2747–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JR, Girard J, and Postic C (2006). Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 55, 2159–2170. [DOI] [PubMed] [Google Scholar]
- Dentin R, Pegorier J-P, Benhamed F, Foufelle F, Ferre P, Fauveau V, Magnuson M, Girard J, and Postic C (2004). Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. The Journal of biological chemistry 279, 20314–20326. [DOI] [PubMed] [Google Scholar]
- Dentin R, Tomas-Cobos L, Foufelle F, Leopold J, Girard J, Postic C, and Ferre P (2012). Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. Journal of hepatology 56, 199–209. [DOI] [PubMed] [Google Scholar]
- Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E, et al. (2013). TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET 1/COMPASS. EMBO J 32, 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Fan M, Rhee J, St-Pierre J, Handschin C, Puigserver P, Lin J, Jaeger S, Erdjument-Bromage H, Tempst P, and Spiegelman BM (2004). Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: modulation by p38 MAPK. Genes Dev 18, 278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Gimenez-Cassina A, and Danial NN (2015). Regulation of mitochondrial nutrient and energy metabolism by BCL-2 family proteins. Trends Endocrinol Metab 26, 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giménez-Cassina A, Garcia-Haro L, Choi CS, Osundiji MA, Lane EA, Huang H, Yildirim MA, Szlyk B, Fisher JK, Polak K, et al. (2014). Regulation of hepatic energy metabolism and gluconeogenesis by BAD. Cell metabolism 19, 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinez C, Filhoulaud G, Rayah-Benhamed F, Marmier S, Dubuquoy C, Dentin R, Moldes M, Burnol AF, Yang X, Lefebvre T, et al. (2011). O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 60, 1399–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JW, Valdez JL, Ho DV, Lee CS, Kim HM, Wang X, Huang L, and Chan JY (2017). Nuclear factor-erythroid-2 related transcription factor-1 (Nrf1) is regulated by O-GlcNAc transferase. Free Radic Biol Med 110, 196–205. [DOI] [PubMed] [Google Scholar]
- Herman MA, Peroni OD, Villoria J, Schon MR, Abumrad NA, Bluher M, Klein S, and Kahn BB (2012). A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 484, 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado del Pozo C, Vesperinas-Garcia G, Rubio MA, Corripio-Sanchez R, Torres-Garcia AJ, Obregon MJ, and Calvo RM (2011). ChREBP expression in the liver, adipose tissue and differentiated preadipocytes in human obesity. Biochim Biophys Acta 1811, 1194–1200. [DOI] [PubMed] [Google Scholar]
- Iizuka K, Wu W, Horikawa Y, and Takeda J (2013). Role of glucose-6-phosphate and xylulose-5-phosphate in the regulation of glucose-stimulated gene expression in the pancreatic beta cell line, INS-1E. Endocr J 60, 473–482. [PubMed] [Google Scholar]
- Janetzko J, Trauger SA, Lazarus MB, and Walker S (2016). How the glycosyltransferase OGT catalyzes amide bond cleavage. Nat Chem Biol 12, 899–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashima T, Kawaguchi T, Wadzinski BE, and Uyeda K (2003). Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc Natl Acad Sci U S A 100, 5107–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Krawczyk SA, Doridot L, Fowler AJ, Wang JX, Trauger SA, Noh HL, Kang HJ, Meissen JK, Blatnik M, et al. (2016). ChREBP regulates fructose-induced glucose production independently of insulin signaling. J Clin Invest 126, 4372–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooner J, Chambers J, Aguilar-Salinas C, Hinds D, Hyde C, Warnes G, Gomez Perez F, Frazer K, Elliott P, Scott J, et al. (2008). Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nature genetics 40, 149–151. [DOI] [PubMed] [Google Scholar]
- Lazarus MB, Jiang J, Kapuria V, Bhuiyan T, Janetzko J, Zandberg WF, Vocadlo DJ, Herr W, and Walker S (2013). HCF-1 is cleaved in the active site of O-GlcNAc transferase. Science 342, 1235–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Puigserver P, Donovan J, Tarr P, and Spiegelman BM (2002). Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta ), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem 277, 1645–1648. [DOI] [PubMed] [Google Scholar]
- Lou DQ, Tannour M, Selig L, Thomas D, Kahn A, and Vasseur-Cognet M (1999). Chicken ovalbumin upstream promoter-transcription factor II, a new partner of the glucose response element of the L-type pyruvate kinase gene, acts as an inhibitor of the glucose response. J Biol Chem 274, 28385–28394. [DOI] [PubMed] [Google Scholar]
- Ma L, Robinson LN, and Towle HC (2006). ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J Biol Chem 281, 28721–28730. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM (2009). Assessing the potential of glucokinase activators in diabetes therapy. Nat Rev Drug Discov 8, 399–416. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Pocai A, Rossetti L, Depinho RA, and Accili D (2007). Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab 6, 208–216. [DOI] [PubMed] [Google Scholar]
- Mazars R, Gonzalez-de-Peredo A, Cayrol C, Lavigne AC, Vogel JL, Ortega N, Lacroix C, Gautier V, Huet G, Ray A, et al. (2010). The THAP-zinc finger protein THAP1 associates with coactivator HCF-1 and O-GlcNAc transferase: a link between DYT6 and DYT3 dystonias. J Biol Chem 285, 13364–13371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metukuri MR, Zhang P, Basantani MK, Chin C, Stamateris RE, Alonso LC, Takane KK, Gramignoli R, Strom SC, O’Doherty RM, et al. (2012). ChREBP mediates glucose-stimulated pancreatic beta-cell proliferation. Diabetes 61, 2004–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissim I, Horyn O, Nissim I, Daikhin Y, Wehrli SL, Yudkoff M, and Matschinsky FM (2012). Effects of a glucokinase activator on hepatic intermediary metabolism: study with 13C-isotopomer-based metabolomics. Biochem J 444, 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan BL, Koo SH, Wu Y, Freake HC, and Towle HC (2001). Glucose regulation of the acetyl-CoA carboxylase promoter PI in rat hepatocytes. J Biol Chem 276, 16033–16039. [DOI] [PubMed] [Google Scholar]
- Petrie JL, Al-Oanzi ZH, Arden C, Tudhope SJ, Mann J, Kieswich J, Yaqoob MM, Towle HC, and Agius L (2013). Glucose induces protein targeting to glycogen in hepatocytes by fructose 2,6-bisphosphate-mediated recruitment of MondoA to the promoter. Mol Cell Biol 33, 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, and Magnuson MA (1999). Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274, 305–315. [DOI] [PubMed] [Google Scholar]
- Ruan HB, Han X, Li MD, Singh JP, Qian K, Azarhoush S, Zhao L, Bennett AM, Samuel VT, Wu J, et al. (2012). O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab 16, 226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakiyama H, Fujiwara N, Noguchi T, Eguchi H, Yoshihara D, Uyeda K, and Suzuki K (2010). The role of O-linked GlcNAc modification on the glucose response of ChREBP. Biochem Biophys Res Commun 402, 784–789. [DOI] [PubMed] [Google Scholar]
- Sanders FW, and Griffin JL (2016). De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose. Biol Rev Camb Philos Soc 91, 452–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen DL, Gloster TM, Yuzwa SA, and Vocadlo DJ (2012). Insights into O-linked N-acetylglucosamine ([0–9]O-GlcNAc) processing and dynamics through kinetic analysis of O-GlcNAc transferase and O-GlcNAcase activity on protein substrates. J Biol Chem 287, 15395–15408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, and Mann M (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68, 850–858. [DOI] [PubMed] [Google Scholar]
- Softic S, Cohen DE, and Kahn CR (2016). Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig Dis Sci 61, 1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatikos AD, da Silva RP, Lewis JT, Douglas DN, Kneteman NM, Jacobs RL, and Paton CM (2016). Tissue Specific Effects of Dietary Carbohydrates and Obesity on ChREBPalpha and ChREBPbeta Expression. Lipids 51, 95–104. [DOI] [PubMed] [Google Scholar]
- Szlyk B, Braun CR, Ljubicic S, Patton E, Bird GH, Osundiji MA, Matschinsky FM, Walensky LD, and Danial NN (2014). A phospho-BAD BH3 helix activates glucokinase by a mechanism distinct from that of allosteric activators. Nat Struct Mol Biol 21, 36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RP, Geisler TS, Chambers JH, and McClain DA (2009). Up-regulation of O-GlcNAc transferase with glucose deprivation in HepG2 cells is mediated by decreased hexosamine pathway flux. J Biol Chem 284, 3425–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LR, Foshage AM, Weissmiller AM, Popay TM, Grieb BC, Qualls SJ, Ng V, Carboneau B, Lorey S, Eischen CM, et al. (2016). Interaction of MYC with host cell factor-1 is mediated by the evolutionarily conserved Myc box IV motif. Oncogene 35, 3613–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsatsos N, and Towle H (2006). Glucose activation of ChREBP in hepatocytes occurs via a two-step mechanism. Biochemical and biophysical research communications 340, 449–456. [DOI] [PubMed] [Google Scholar]
- Tyagi S, Chabes AL, Wysocka J, and Herr W (2007). E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol Cell 27, 107–119. [DOI] [PubMed] [Google Scholar]
- Uyeda K, and Repa J (2006). Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell metabolism 4, 107–110. [DOI] [PubMed] [Google Scholar]
- Velho G, Petersen KF, Perseghin G, Hwang JH, Rothman DL, Pueyo ME, Cline GW, Froguel P, and Shulman GI (1996). Impaired hepatic glycogen synthesis in glucokinase-deficient (MODY-2) subjects. J Clin Invest 98, 1755–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Viscarra J, Kim SJ, and Sul HS (2015). Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol 16, 678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocka J, Myers MP, Laherty CD, Eisenman RN, and Herr W (2003). Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev 17, 896–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AQ, Li D, Chi L, and Ye XS (2017). Validation, Identification, and Biological Consequences of the Site-specific O-GlcNAcylation Dynamics of Carbohydrate-responsive Element-binding Protein (ChREBP). Mol Cell Proteomics 16, 1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, and Qian K (2017). Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 18, 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yecies JL, Zhang HH, Menon S, Liu S, Yecies D, Lipovsky AI, Gorgun C, Kwiatkowski DJ, Hotamisligil GS, Lee CH, et al. (2011). Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab 14, 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Kumar A, Katz LS, Li L, Paulynice M, Herman MA, and Scott DK (2015). Induction of the ChREBPbeta Isoform Is Essential for Glucose-Stimulated beta-Cell Proliferation. Diabetes 64, 4158–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Wang Z, Yuan X, Zhou C, Liu L, Wan X, Zhang F, Ding X, Wang C, Xiong S, et al. (2013). Mixed lineage leukemia 5 (MLL5) protein regulates cell cycle progression and E2F1-responsive gene expression via association with host cell factor-1 (HCF-1). J Biol Chem 288, 17532–17543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.