

Abstract
Classification of variants of unknown significance is a challenging technical problem in clinical genetics. As up to one third of disease-causing mutations are thought to affect pre-mRNA splicing, it is important to accurately classify splicing mutations in patient sequencing data. Several consortia and healthcare systems have conducted large-scale patient sequencing studies, which discover novel variants faster than they can be classified. Here we compare the advantages and limitations of several high-throughput splicing assays aimed at mitigating this bottleneck, and describe a dataset of ~5000 variants that we analyzed using our Massively Parallel Splicing Assay (MaPSy). The Critical Assessment of Genome Interpretation group (CAGI) organized a challenge, in which participants submitted machine learning models to predict the splicing effects of variants in this dataset. We discuss the winning submission of the challenge (MMSplice) which outperformed existing software. Finally, we highlight methods to overcome the limitations of MaPSy and similar assays, such as tissue-specific splicing, the effect of surrounding sequence context, classifying intronic variants, synthesizing large exons, and amplifying complex libraries of minigene species. Further development of these assays will greatly benefit the field of clinical genetics, which lack high-throughput methods for variant interpretation.
Keywords: Splicing, precision medicine, disease, variant, assay, high-throughput
Introduction:
The cost of next generation sequencing (NGS) has fallen several thousand-fold in the last ten years, which has allowed for whole-genome sequencing and whole-exome sequencing to become common approaches in personal genomics and clinical medicine. A typical exome sequencing study will reveal thousands of variants of unknown significance (Telenti et al., 2016). The effects of these coding variants on protein function are particularly difficult to interpret, as individual functional assays do not exist for most proteins. However, variants in splice-regulatory elements typically result in deleterious phenotypes. Splicing mutations are not only harmful, but they are also prevalent. In fact, it has been predicted that one-third of all disease-causing variants confer some degree of aberrant splicing (Lim, Ferraris, Filloux, Raphael, & Fairbrother, 2011). The effect of variants on splicing is measurable through the use of minigene assays (Cooper, 2005). Because splicing mutations are deleterious, prevalent, and measurable, splicing minigene assays are a valuable method for interpreting the pathogenicity of variants discovered.
As thousands of variants are discovered in sequencing studies, the challenge for precision medicine lies in the ability to classify variants at the same rate they are discovered. Recently, our group developed a Massively Parallel Splicing Assay (MaPSy) to screen ~5,000 disease-causing exonic mutations for splicing defects. Using highly stringent criteria, this study showed that 10% of exonic mutations altered splicing (Soemedi, Cygan, Rhine, Wang, et al., 2017). The ability to evaluate variants for defective splicing is beginning to emerge as an achievable goal with the advent of massively-parallel reporter assays (MPRAs) and high-throughput screens (Adamson, Zhan, & Graveley, 2018; Ke et al., 2018; Soemedi, Cygan, Rhine, Wang, et al., 2017). Computational methods aimed at leveraging MPRAs and high-throughput assay data have led to improved predictive models for classifying splicing variants that have not been empirically verified (Bretschneider, Gandhi, Deshwar, Zuberi, & Frey, 2018; Desmet et al., 2009; Fairbrother, Yeh, Sharp, & Burge, 2002; Mort et al., 2014). The Critical Assessment of Genome Interpretation (CAGI) recognized the need for a community effort in advancing the computational methods in predicting the impacts of genomic variation and devised a prediction challenge. In the challenge, participants were asked to identify variants causing splicing defects and estimate the severity of each defect. A variety of different machine learning approaches were submitted, and the top performer, a program called MMSplice, was recently described in a publication (Jun Cheng et al., 2018). Here we outline methods, challenges, and future directions for this hybrid experimental/computational approach. We focus in particular on the role of MPRAs in functional genomics and clinical medicine. In addition to applications of this technology to consortia sequencing science and discussing drug screening technologies, the effect of sequence context on splicing in MPRAs and technical issues relating to oligonucleotide synthesis are discussed.
CAGI and the MaPSy dataset challenge:
Increasingly sophisticated predictive models have been developed to estimate the effect of variants on splicing. Many of these models are trained on data from various implementations of MPRAs (FAS-ESS, ESRseq scores, and HAL) (Ke et al., 2011; Rosenberg, Patwardhan, Shendure, & Seelig, 2015; Wang et al., 2004). However, these models lack training on large datasets describing the effect of single nucleotide variants on the process of splicing, hindering their ability to produce reliable splicing predictions for variant interpretation. The development of the MaSPy has now offered the splicing and machine learnings fields with a rich training dataset describing the effect of ~5,000 single nucleotide variants on splicing. Recognizing the need for improved prediction models for variant interpretation, CAGI devised a competition where multiple machine learning teams were challenged to construct splicing variant predictive models to aid in the variant interpretation demands facing precision medicine. The following section describes the MaPSy training and test sets provided to CAGI, the challenge posed to the machine learning teams, and the resulting machine learning splicing model which outperformed the competing CAGI teams.
Massively Parallel Splicing Assay (MaPSy) Experiment:
The challenge was prepared from a splicing analysis of publicly available disease-causing variants. Nonsynonymous mutations classified as disease-causing (DM) were downloaded from Human Genome Mutation Database (Stenson et al., 2009). Mutations were mapped to internal exons of 100 nucleotides or less in length and selected for those that fit into 170 nucleotide genomic windows. The genomic window included 15 nucleotides of downstream intronic sequence and at least 55 nucleotides of upstream intronic sequence (n = 4,964). The mutant and wildtype versions of the 170-mer genomic fragments were flanked with 15-mer common primers and synthesized as a 200-mer oligo library (Figure 1A).
Figure 1: MaPSy challenge.
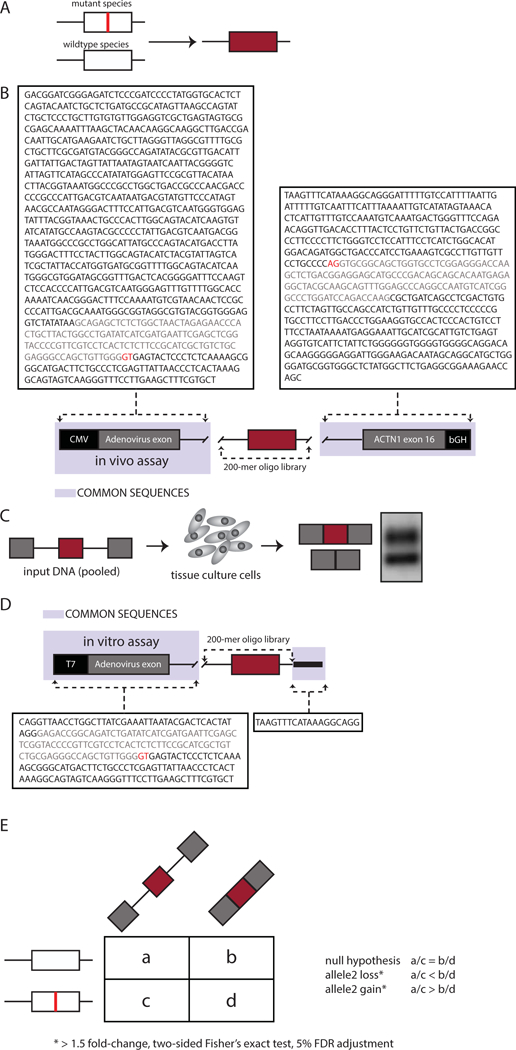
A. Mutant and wildtype versions of 170-mer genomic fragments flanked by 15-mer common. B. The in vivo splicing reporter consists of the Cytomegalovirus (CMV) promoter and Adenovirus (pHMS81) exon with part of its downstream intron at the 5’ end, followed by the 200-mer oligo library, and exon16 of ACTN1 with part of intron15 and bGH PolyA signal sequence at the 3’ end. C. The in vivo reporters were transfected in hek293 cells. D. The in vitro reporter includes a T7 promoter and Adenovirus (pHMS81) exon. E. Contingency tables were created for each mutant/wildtype pair and include the counts obtained from deep sequencing of the input pool as well as the output-spliced fractions to assess defects in splicing.
An additional 797 mutations were mapped to exons greater than 100 nucleotides. Each of these longer variant exons were “cut” in a way to 1) preserve the 5’ and 3’ splice site signals and 2) a middle portion of the exon was removed to decrease the size of the exon to 100 nucleotides or less to meet oligonucleotide synthesis size restrictions.
A three exon in vivo splicing reporter was constructed to include a Cytomegalovirus (CMV) promoter and a common first exon, followed by the 200-mer oligo library, and a common downstream exon (Figure 1B). The resulting in vivo reporters were transfected to human embryonic kidney HEK293T cells. RNA was extracted 24 hours post transfection (Figure 1C). Input reporters and spliced species were sequenced by Illumina HiSeq2500.
The in vitro splicing reporter includes a T7 promoter and a common first exon, followed by the oligo library (Figure 1D). In vitro reporters were obtained via in vitro transcription using T7 RNA Polymerase. The resulting RNA was gel purified and used for splicing reactions in 40% HeLa-S3 nuclear extract for 80 min at 30°C. Pools of input and spliced RNAs were converted to cDNA and prepped into an Illumina library for deep sequencing.
A contingency table was created for each mutant/wild-type pair and includes the counts obtained from deep sequencing of the input pool as well as the output-spliced fractions (Figure 1E). To determine pairs with significant allelic skew we required at least 1.5-fold change and a two-sided Fisher’s exact test adjusted with 5% false discovery rate (FDR). The following formula was used to calculate allelic skew: , where is the count of reads in the spliced fraction for the mutant, is the count of reads in the input for the mutant, is the count of reads in the spliced fraction for the wildtype, and is the count of reads in the input for the wildtype.
Prediction Challenge:
Two sets of variants that were tested by MaPSy were provided to CAGI, the training set and the test set. The training set included all 4,964 published variants (Soemedi, Cygan, Rhine, Wang, et al., 2017). The test set includes all 797 mutant/wildtype pairs of variants that fall within exons that needed to by ‘cut’ to fit in the oligonucleotide library. The sequence for both constructs including exon/intron boundaries needed to evaluate allelic ratio, as well as the counts for the input for both mutant and wildtype species for both panels were provided.
CAGI participants were asked to submit predictions on the subset of variants in the test set that passed our threshold as disruptors of splicing both in vitro and in vivo and therefore were categorized as exonic splicing mutations. Participants provided the probability that each variant is an exonic splicing mutation and which allele from each pair spliced better. In addition, given the input read counts, the participants predicted the log2 allelic skew ratio for in vivo and in vitro panels for each pair in the test set.
Prediction Winner: Modular Modeling of Splicing
The winning prediction model, modular modeling of splicing (MMSplice), trained a set of neural network modules separately for exons, 3’ and 5’ splice-sites, and intronic sequences. This method of building modules for individual splicing-relevant sequence regions allowed the group to leverage multiple datasets to predict percent spliced-in (psi) values, splicing efficiency, and pathogenicity. The resulting program, MMSplice, was shown to outperform previous highly predictive models on predicting the effect genetic variants have on splicing (Jun Cheng et al., 2018).
High-throughput methods in splicing
The success of the challenge prompted an examination of the potential for this technology in precision medicine. Building from advancements in solid-phase oligonucleotide synthesis technologies, massively-parallel reporter assays (MPRAs) have become an increasingly attractive approach for the study of alternative splicing (Park, Pan, Zhang, Lin, & Xing, 2018). Extensive libraries of sequence variants constructed into minigene reporters, can be screened in parallel for functional impacts on splicing. The study of wildtype and variant exons in minigene cassettes allows for direct assessment of sequence contributions to splicing outcomes (Singh & Cooper, 2006). Such MPRAs have been used to analyze the ability of sequence variants in degenerate or mutationally saturated libraries to influence 5’ and 3’ splice site selection (Rosenberg et al., 2015; Wong, Kinney, & Krainer, 2018) and exon definition (Ke et al., 2018). Recently, an MPRA that measured mutations within the primate lineage helped identify a mathematical equation to calculate the magnitude of splicing disruption caused by a novel exonic mutation. In this equation, exonic mutations have a maximal impact in exons with an intermediate degree of splicing (Baeza-Centurion, Minana, Schmiedel, Valcarcel, & Lehner, 2019). In other words, less efficient splicing substrates are more prone to splicing defects. MaPSy, another MPRA, provides a direct measure of splicing disruption caused by exonic mutations. Mutations from thousands of different exons can be assayed in parallel, offering both insights into the determinants of splicing aberrations and a potential high-throughput technology for the classification of disease variants (Soemedi, Cygan, Rhine, Wang, et al., 2017).
MPRAs are not the only approach for testing the effects of variation on splicing. The CRISPR-Cas9 system has also been used to screen thousands of mutations in parallel within endogenous genomic loci (CRISPR-arrays). In CRISPR-arrays, pools of guide RNAs are used to introduce numerous mutations at one locus. For example, a 6-bp region of BRCA1 exon 18 was replaced with all possible hexamers. The utility of CRISPR-arrays is widely applicable in functional genomics. They have identified novel regulatory elements, pathogenic variants, and quantified effects such as nonsense-mediated decay (Canver et al., 2017; Findlay, Boyle, Hause, Klein, & Shendure, 2014; Sanjana, 2017).
CRISPR-arrays have some unique advantages. By editing endogenous genes, they capture the physiologic context of the cell. All relevant cis-elements or secondary structural components are preserved. They are also unconstrained by size limitations of solid-state oligonucleotide synthesis. Therefore, any full-length exon may be screened by this method. It is also more technically straightforward to construct a pool of guide RNAs than a pool of minigene species, which may require PCR and other molecular biology techniques to assemble. Despite these advantages, there are some important drawbacks to consider. Haplotype cells lines have been required in order to achieve efficient multiplex gene editing with CRISPR-Cas9. CRISPR-array throughput can test variants to saturation, but only within a small window. In other words, CRISPR screening is limited to one exon per experiment, and also requires sufficient gene expression for downstream analysis. Lastly, genes considered essential for cell survival may pose additional limitations (Findlay et al., 2014). Splicing mutations in essential genes may have lethal effects, because the only copy of the gene is mutated in these assays.
MPRAs have several advantages over CRISPR-arrays. Because they utilize minigenes, MPRAs are not dependent on endogenous gene expression. MPRAs tend to represent a pure measure of splicing effects. Many clinical whole exome sequencing datasets are being generated, which return large numbers of variants for interpretation. MPRAs that leverage minigenes are better suited for studying the functional consequences of these variants at the scale and widespread genomic distribution of variants returned by exome sequencing. For example, MPRAs can assay many or potentially all variants of interest from a whole exome sequencing study instead of being restricted to one gene or exon (Adamson et al., 2018; Soemedi, Cygan, Rhine, Wang, et al., 2017). Therefore, MPRAs are the method of choice to analyze variants from consortia sequencing because of these unique advantages over other methods, like CRISPR-arrays.
There are still several challenges that limit the potential of MPRAs. First, splicing is a tissue-specific process. For example, the brain has the highest degree of exon skipping, and splicing in the liver is almost entirely limited to cryptic alternative splicing events (alternative 5’ and 3’ splice sites) (Yeo, Holste, Kreiman, & Burge, 2004). MPRAs only report splicing outcomes in one tissue type, and cannot be extrapolated to other tissue types. However, we can identify similar splicing events across tissues from RNA-seq studies in multiple tissues, such as the GTEx consortium, in to determine the potential effect of a variant across tissue types (Consortium et al., 2017). In addition, MPRAs rely on artificial minigene constructs that lack valuable surrounding endogenous sequence context that may impact splicing. Exons that are tested in these assays are typically flanked by common exons to all species in a minigene library as opposed to the exons from the endogenous transcripts. Sequence context from the whole pre-mRNA transcript affects the order of intron removal and can lead to alternative splicing events not captured in mingenes (Kim et al., 2017). Moreover, as splicing is a co-transcriptional process, chromatin binding state, absent in minigenes, has also been shown to affect splicing (Jaganathan et al., 2019). Lastly, large datasets containing genetic variants for screening by MPRAs often lack corresponding phenotypic or other relevant patient information and thus limit the use of MPRAs in returning informative variant discoveries.
New scientific and healthcare initiatives: Geisinger Health System and Simons Foundation of Autism Research Initiative
Many datasets reporting disease-causing or disease-associated variants, such as the Human Gene Mutation Database (Stenson et al., 2009) and ClinVar (Landrum et al., 2016), have limited information on clinical phenotypes and lack methods in contacting and/or requesting biospecimens from patients. Fortunately, new scientific and healthcare initiatives have recognized the need in accurately identifying and interpreting genomic findings that will prove relevant to clinical efforts and precision medicine. Such relationships provide a direct means for validation of functional genomic approaches, return incidental findings to patients, and further analyze the relationship between variants and patient phenotypic characteristics.
A prominent example of this type of integrated dataset can be found in the DiscovEHR cohort (Dewey et al., 2016). Through a partnership with the Regeneron Genetics Center, Geisinger has created the DiscovEHR cohort (Dewey et al., 2016). The DiscovEHR cohort is a large population of patients from the Geisinger healthcare system who have had exome sequencing added to their electronic health care records to pair genotype with phenotype in a single dataset. This patient cohort currently includes 92,805 participants drawn entirely from participants in the MyCode® Community Health Initiative (Carey et al., 2016). MyCode participants are consented for collection of biospecimens to be used in conjunction with all the data from their electronic health record (EHR). The participants in MyCode have an average of 14 years of medical records that can be linked with their exome sequencing results, including; clinical notes, lab values, ICD10 Codes, medications, and imaging studies. This combination of genotypic and detailed phenotypic information provides for an extremely rich dataset for genomic discovery. MyCode participants are also consented for recontact for additional research which allows for additional clinical evaluation with more targeted phenotyping to supplement the rich dataset that already exists in the EHR. DiscovEHR has already been proven to be a tremendous resource for genomic discovery (Abul-Husn et al., 2018; Gusarova et al., 2018; Verma et al., 2019).
An integrated dataset such as the DiscovEHR cohort is a suitable platform for the aggregation of data from additional functional genomic experiments like high-throughput splicing assays. By comparing the comprehensive profiles of patients with splicing variants to matched controls, overrepresented phenotypes can be discovered that are representative of known gene effects and perhaps even discover phenotypes related to these variants that have not previously been described. For example, splicing defects could be a tissue-specific phenomenon, which could alter the presentation of particular genetic disorders. The interactive nature of the healthcare system allows researchers to recontact and assess patients for phenotypic features that may not be in their medical record through additional clinical evaluation, laboratory testing, or imaging studies. The sheer size of the DiscovEHR cohort which accounts for a large amount of rare variation allows for a more complete analysis of the phenotypic consequences of rare variants and the power of this resource increases as it continues to grow (Mirshahi et al., 2018).
In contrast to initiatives identifying variants across individuals sampled from a population, additional initiatives are taking a disease-centric approach to identify variants relating to a single disease. For example, the Simons Foundation of Autism Research Initiative (SFARI) was launched in 2003 to fund innovative research to understand the etiology of autism spectrum disorders (ASD). SFARI Simons Simplex Collection (SSC) has performed whole exome sequencing on families with one ASD affected child, and unaffected parents and siblings (quad families) to identify inherited and de novo variants. In combination with genomic data, SSC has collected extensive phenotypic data (i.e. IQ, cognitive, developmental, behavioral, etc.) and biospecimens (i.e. blood samples/cell lines) for each participant. This wealth of data offers a unique advantage to researchers attempting to decipher the phenotypic and genetic heterogeneity that characterizes ASD. More specifically, we can leverage this data by identifying potentially deleterious variants and ASD risk gene through the use of MaPSy, validate splicing defects using the relevant biospecimens, and even analyze phenotypic attributes that may have arisen due to defective splicing.
Technical Challenges: Designing and Utilizing Complex Libraries to Assay Splicing
The design of libraries for use in MaPSy assays is constrained by several technical challenges. Most notably, only mutations in exons of fewer than 100 nt can be included as a consequence of current limitations in oligonucleotide synthesis technology. As the median length of internal exons is approximately 130 nucleotides, more than half of all human exons are excluded from MaPSy splicing characterization. Advances in solid-phase oligonucleotide synthesis will continue to expand the window size for sequence design moving forward. Currently, exons greater than 100 nucleotides can be truncated to preserve 5’ and 3’ splice site signals and proximal regions, to approximate the effect of potential splice variants.
MPRAs and intronic variants
An additional technical challenge with MaPSy, and MPRAs using minigene approaches, lies in the analysis of intronic variants. As introns are excised, identifying variants in introns are lost during splicing and cDNA generated from mutant and wildtype alleles become indistinguishable. Recently Adamson et al. devised a barcoding strategy using an eight-nucleotide barcode that, after subcloning into a reporter plasmid, designated a particular variant at the end of a transcript. These extra steps can potentially limit library complexity and random octamers may themselves affect gene expression. To circumvent this issue in MaPSy, a one-step barcoding strategy has been developed to allow for the identification of the mutant and wildtype exons from the spliced product. In our method, a barcode was added to every intronic mutant species as a unique marker. Each oligonucleotide library species consisted of a mutant or wildtype intronic sequence and 26 nucleotides of the endogenous exon. The last 6 nucleotides of the endogenous exons were used to design each barcode (Figure 2A). All possible variants within the barcode window were submitted to the Spliceman prediction software, and the three variants least likely to disrupt splicing were selected as barcodes for each mutant species (Lim & Fairbrother, 2012). Therefore, each barcode consisted of unique point variants for intronic mutant identification, and each intronic mutant species was tested in triplicate using three unique barcodes.
Figure 2: New MaPSy barcoding strategy.
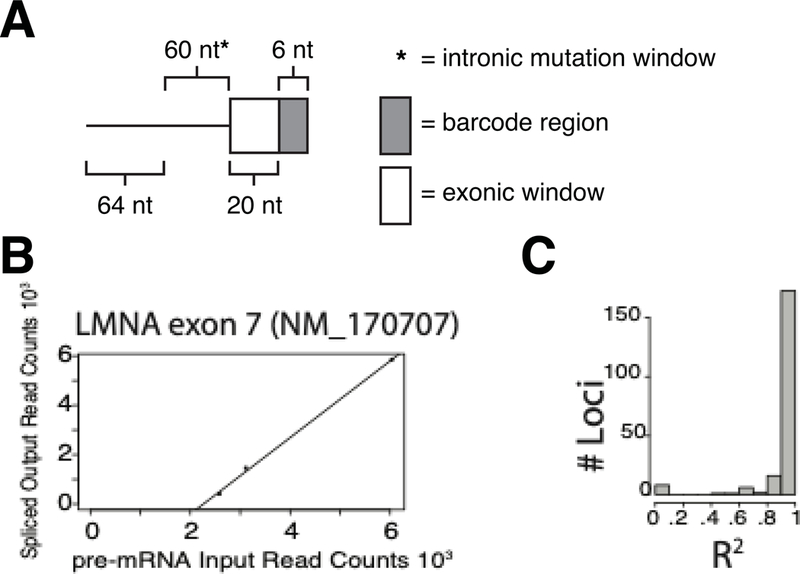
A. The schematic of the barcoding strategy shows that the last 6 nt of the region containing endogenous sequence (150 nt) was used to design barcodes. Mutations tested by the assay fell within 60 nt upstream of the 3’ss. B. Preliminary Data demonstrates correlation between three barcoded triplicates for LMNA exon 7. C. Distribution of R squared values across barcoded triplicates of 208 other exons demonstrates selected barcodes do not alter splicing.
To evaluate the effect of the barcodes on splicing, the counts for each intronic mutant barcoded triplicate in the unspliced input vs. the spliced output were plotted to test for a correlation. Presumably if the representation of each barcoded species in the unspliced input and spliced output are similar, we can be confident the barcodes are likely not affecting splicing. Of the 208 triplicates which had at least 10 reads each, 175 (84%) of these were highly correlated (r2 >0.9), suggesting that the barcoding strategy was effective (Figure 2B,C). This result suggests the observed allelic imbalances in splicing are more likely to be caused by the mutant being tested, and not a result of the barcode. This new approach will allow for the expansion of analysis into intronic variants.
Challenges with complex library amplifications
A related technical issue arises from difficulties in maintaining initial oligonucleotide library complexity during amplification. Library synthesis provides a highly complex pool of oligonucleotides, each at sub pmol quantities. In order to apply library contents to MPRAs, amplification through polymerase chain reaction (PCR) is necessary to acquire experimentally tractable quantities of DNA. However, amplification may alter the overall composition of the library, changing both the overall content and the ratio of constituents possessed there within. Such changes can be the result of either PCR drift or PCR selection (Polz & Cavanaugh, 1998). PCR drift is a bias that is assumed to be the result of stochastic variation in early cycles of amplification, and is not reproducible in replicate PCR amplifications. Alternatively, PCR selection operates on mechanisms which inherently favor amplification of particular templates relative to others. Factors such as the GC content and relative structure of oligonucleotides may dictate their representation in an amplified library. PCR of complex libraries also holds the heightened potential to generate artifacts in the form of chimeras and heteroduplexes, which can quickly change the compositional landscape of a library (Qiu et al., 2001). In order to circumvent these issues, amplification can most effectively be achieved through multiple rounds of fewer amplification cycles (5–10) followed by size selected purification of PCR products. However, even with optimized protocols, population dynamics are observed to shift between rounds of amplification (Figure 3A) suggesting that PCR selection continues to restrict the maximum complexity that can be achieved in an applied library. Fortunately, within the context of paired oligonucleotides (wildtype and variant), both species seem to behave similarly during amplification (Figure 3B). Overall representation, including observed dropout, is typically conserved in final datasets between compared oligonucleotides, minimizing the number of unproductive reads. In moving forward, the use of low pass sequencing allows for optimization of amplification protocols that place an emphasis on retaining both complexity of oligonucleotide content and uniformity of representation there within.
Figure 3: Amplification of complex oligonucleotide libraries.
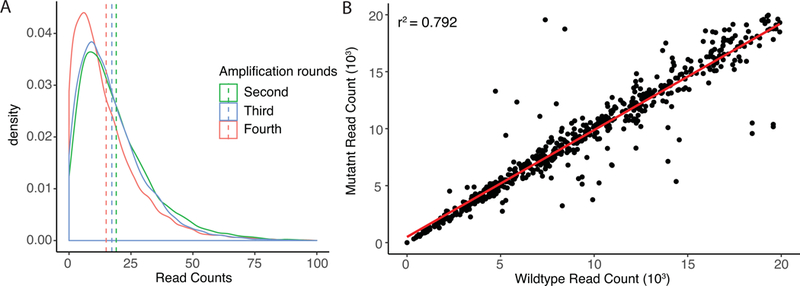
A. Density of oligonucleotide library substrate read counts between successive rounds of amplification (second, third, and fourth). Initial library contained 7520 oligonucleotide species generated using Agilent solid-phase oligonucleotide synthesis technologies. Next generation sequencing performed using Illumina MiSeq. Dashed lines represent mean substrate read counts. B. Relationship of paired mutant and wildtype oligonucleotide substrate read counts after initial amplification of an oligonucleotide library containing 1,504 substrates. Deep sequencing was performed using Illumina HiSeq 3000 (2×150).
The effect of flanking sequence context on variant perturbation in MaPSy
Another limitation lies in the accuracy of MRPAs in representing physiological outcomes. Previous reports have suggested that surrounding sequence context is an important determinant in splicing outcome (Kim et al., 2017). Our original MaPSy tested ~5,000 disease-causing exonic variants in a three exon minigene where each variant was flanked by an upstream adenovirus exon and a downstream ACTN1 exon (Figure 1). To determine the potential contribution of sequence context to splicing outcome, we re-implemented MaPSy to assess potential splicing defects in 748 alleles caused by de novo variants reported in the SSC using three slightly different three exon minigene reporters. Instead of using the adenovirus upstream exon as described in (Soemedi, Cygan, Rhine, Wang, et al., 2017), three exons representing a range of 5’ss strengths, determined by MaxEntScan (Yeo & Burge, 2004), were synthesized into three separate in vivo minigene reporters and assayed in parallel. Each reporter contained either the VCP exon 15, EMC7 exon 3, or VCP exon 10, a 230-mer genomic fragment containing either the mutant or wild type (reference) sequence, and a downstream ACTN4 exon (Supp. Figure S1). This resulted in each de novo variant being assayed in triplicate under three separate upstream exonic sequence contexts. Deep sequencing of input libraries and output spliced fractions were used to determine the allelic ratio of mutant/wild type pairs (M/W splice ratio) as described previously (cite Nat gen paper) (Figure 4A, Supp. Table S1). Despite the differences in the sensitivity of different reporter constructs, general agreements were observed between the relative allelic imbalances (i.e. M/W splice ratios) in all three assay runs (Figure 4B). Although sequence context does impose an effect on splicing outcome, as described previously (Kim et al., 2017), the validation rate of the original MaPSy (~83%) (Soemedi, Cygan, Rhine, Wang, et al., 2017) and the general agreement between the variants allelic imbalance given the three new minigene constructs, suggests that the MaPSy assay offers a reliable method for prioritizing variants by their ability to affect splicing.
Figure 4: Context dependence in MaPSy.
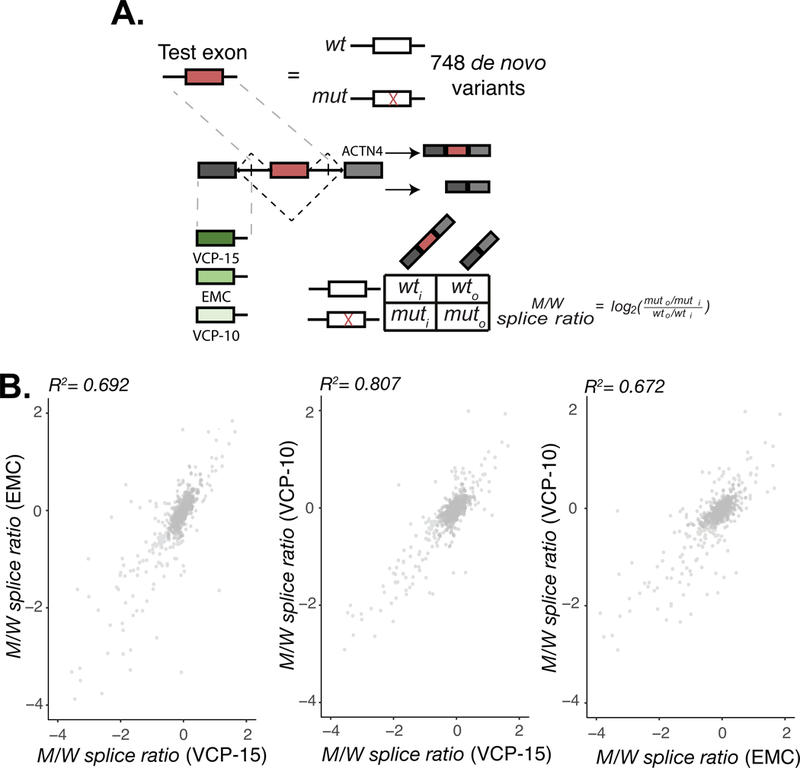
A. 748 de novo mutant exons and their corresponding wildtype counterparts were incorporated into three different three exon in vivo constructs. Both the unspliced input and spliced output library were deep sequenced to establish allelic imbalance between mutant and wildtype species. B. Comparison of individual allelic ratios of variants in the reporter constructs.
Future Potential
In summary, MPRAs show great promise for future efforts in precision medicine and drug discovery. Variants identified in consortia sequencing and integrated genetic datasets such as the Geisinger MyCode program, are well suited for MPRAs. MPRAs can help interpret incidental findings in clinical sequencing studies and guide clinical decisions. Moreover, the relatively low cost of deep sequencing has and will continue to produce large datasets containing novel variants. MPRAs are currently the ideal method for interpreting the functional consequences of novel variants, as they keep pace with discovery, and in the case of MaPSy, accurately assess a variant’s effect on splicing with a ~83% validation rate (Soemedi, Cygan, Rhine, Wang, et al., 2017). In addition to the functional interpretation of variants, the data generated from MPRAs have also been used to train predictive models for the effects of novel variants on splicing (J. Cheng et al., 2019; Ke et al., 2011; Rosenberg et al., 2015; Wang et al., 2004), offering additional tools for variant interpretation. A recent analysis evaluated the predictive power of three splicing variant prediction programs (SPANR (Xiong et al., 2015), ESRseq scores (Ke et al., 2011), and Hexplorer (Erkelenz et al., 2014)) and found that the model trained on a minigene screening of all possible hexamers, ESRseq, was the most predictive in nature (Soukarieh et al., 2016). Even more recently, the predictive ability of MMSplice, the splicing prediction model trained on the single nucleotide variant MaPSy data and additional splicing MPRA implementations, was shown to outperform multiple splicing prediction programs (J. Cheng et al., 2019). These analyses further highlight the utility of MRPAs in not only assessing functionally a variant’s effect on splicing but also in the construction of predictive models. In addition to MPRA-trained prediction models, a recent splicing model was trained on primary sequence alone and produced reliable splicing predictions (Jaganathan et al., 2019). The advantage of this model is that it can identify long-range sequence features that are not captured in the minigenes used in MPRAs. In time, it is likely that multiple algorithms will be combined when making classifications to improve the interpretation of variants. In general, the advantage of computational methods is their ability to assess more variants than can be assayed in a single MPRA and will help improve the novel variant classification problem facing clinical sequencing studies.
In addition to variant interpretation, MPRAs can identify the effects of drugs on splicing. Many drugs, such as amiloride, demonstrate widespread, but tolerable effects on splicing (Chang et al., 2011; Soemedi, Cygan, Rhine, Glidden, et al., 2017). These drugs can be screened against a library of variants in order to determine their personalized effects on patients with rare diseases (Soemedi, Vega, Belmont, Ramachandran, & Fairbrother, 2014). For example, a drug may be found to exacerbate a splicing defect in a patient. The patient’s physician could be informed of the adverse event, and a safer drug may be prescribed instead. Conversely, some splicing defects may be rescued by a drug. In this case, follow-up studies may be indicated, which might lead to the discovery of novel therapeutics for diseases that are too rare to justify the expense of other methods such as high-throughput screening.
Although there are some limitations to the scope and scale of MPRAs, viable strategies are being developed to circumvent them. Solid-phase oligonucleotide synthesis technology currently limits the length of the species to be tested in parallel to a few hundred base pairs. For splicing, this limits the number of full-length exons that can be tested to less than half of all human exons. This challenge can be addressed by designing chimeric exons that contain only one of the splice sites of larger exons. Intronic variants are also more challenging to test, because the fully-spliced species of the mutant and wild-type are degenerate. We have discussed barcoding methods that help identify degenerate species after splicing. The Vex-seq library design utilized one of these methods to test intronic variants (Adamson et al., 2018).
The winners of this CAGI challenge, who developed the MMSplice prediction software, can accurately predict the splicing outcomes of novel variants. The CAGI dataset we have generated represents the importance and future promise of variant interpretation algorithms. Similar datasets are likely to be generated from future clinical sequencing studies. Platforms such as MMSplice will help classify novel variants from these studies. Such classifications will help both for returning incidental findings to patients, and for determining the safety and efficacy of drugs for patients with rare variants.
Supplementary Material
Acknowledgments:
The work was funded by NIH R01 GM127472.
The CAGI experiment coordination is supported by NIH U41 HG007346 and the CAGI conference by NIH R13 HG006650.
We would like to acknowledge Dr. Julien Gagneur and Jun Cheng for their text recommendations. We would also like to acknowledge CAGI for providing a platform to critically assess predictive models and their application to larger datasets.
Grant Numbers
NIH R01 GM127472, NIH U41 HG007346, NIH R13 HG006650
References:
- Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, . . . Dewey FE (2018). A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N Engl J Med, 378(12), 1096–1106. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/29562163. doi: 10.1056/NEJMoa1712191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson SI, Zhan L, & Graveley BR (2018). Vex-seq: high-throughput identification of the impact of genetic variation on pre-mRNA splicing efficiency. Genome Biol, 19(1), 71 Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/29859120. doi: 10.1186/s13059-018-1437-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeza-Centurion P, Minana B, Schmiedel JM, Valcarcel J, & Lehner B (2019). Combinatorial Genetics Reveals a Scaling Law for the Effects of Mutations on Splicing. Cell, 176(3), 549–563 e523. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/30661752. doi: 10.1016/j.cell.2018.12.010 [DOI] [PubMed] [Google Scholar]
- Bretschneider H, Gandhi S, Deshwar AG, Zuberi K, & Frey BJ (2018). COSSMO: predicting competitive alternative splice site selection using deep learning. Bioinformatics, 34(13), i429–i437. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/29949959. doi: 10.1093/bioinformatics/bty244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver MC, Lessard S, Pinello L, Wu Y, Ilboudo Y, Stern EN, . . . Orkin SH (2017). Variant-aware saturating mutagenesis using multiple Cas9 nucleases identifies regulatory elements at trait-associated loci. Nat Genet, 49(4), 625–634. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/28218758. doi: 10.1038/ng.3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey DJ, Fetterolf SN, Davis FD, Faucett WA, Kirchner HL, Mirshahi U, . . . Ledbetter DH (2016). The Geisinger MyCode community health initiative: an electronic health record-linked biobank for precision medicine research. Genet Med, 18(9), 906–913. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/26866580. doi: 10.1038/gim.2015.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JG, Yang DM, Chang WH, Chow LP, Chan WL, Lin HH, . . . Yang WK (2011). Small molecule amiloride modulates oncogenic RNA alternative splicing to devitalize human cancer cells. PLoS One, 6(6), e18643. doi: 10.1371/journal.pone.0018643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Nguyen TYD, Cygan KJ, Celik MH, Fairbrother W, Avsec Z, & Gagneur J (2018). Modular modeling improves the predictions of genetic variant effects on splicing. bioRxiv. doi: 10.1101/438986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Nguyen TYD, Cygan KJ, Celik MH, Fairbrother WG, Avsec Z, & Gagneur J (2019). MMSplice: modular modeling improves the predictions of genetic variant effects on splicing. Genome Biol, 20(1), 48 Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/30823901. doi: 10.1186/s13059-019-1653-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GT, Laboratory DA, Coordinating Center -Analysis Working, G., Statistical Methods groups-Analysis Working, G., Enhancing G. g., Fund NIHC, . . . Montgomery SB (2017). Genetic effects on gene expression across human tissues. Nature, 550(7675), 204–213. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/29022597. doi: 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TA (2005). Use of minigene systems to dissect alternative splicing elements. Methods, 37(4), 331–340. doi: 10.1016/j.ymeth.2005.07.015 [DOI] [PubMed] [Google Scholar]
- Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, & Beroud C (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res, 37(9), e67 Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/19339519. doi: 10.1093/nar/gkp215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey FE, Murray MF, Overton JD, Habegger L, Leader JB, Fetterolf SN, . . . Carey DJ (2016). Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science, 354(6319). Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/28008009. doi: 10.1126/science.aaf6814 [DOI] [PubMed] [Google Scholar]
- Erkelenz S, Theiss S, Otte M, Widera M, Peter JO, & Schaal H (2014). Genomic HEXploring allows landscaping of novel potential splicing regulatory elements. Nucleic Acids Res, 42(16), 10681–10697. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/25147205. doi: 10.1093/nar/gku736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother WG, Yeh RF, Sharp PA, & Burge CB (2002). Predictive identification of exonic splicing enhancers in human genes. Science, 297(5583), 1007–1013. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/12114529. doi: 10.1126/science.1073774 [DOI] [PubMed] [Google Scholar]
- Fairbrother WG, Yeo GW, Yeh R, Goldstein P, Mawson M, Sharp PA, & Burge CB (2004). RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res, 32(Web Server issue), W187–190. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/15215377. doi: 10.1093/nar/gkh393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay GM, Boyle EA, Hause RJ, Klein JC, & Shendure J (2014). Saturation editing of genomic regions by multiplex homology-directed repair. Nature, 513(7516), 120–123. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/25141179. doi: 10.1038/nature13695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusarova V, O’Dushlaine C, Teslovich TM, Benotti PN, Mirshahi T, Gottesman O, . . . Gromada J (2018). Genetic inactivation of ANGPTL4 improves glucose homeostasis and is associated with reduced risk of diabetes. Nat Commun, 9(1), 2252 Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/29899519. doi: 10.1038/s41467-018-04611-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, . . . Farh KK (2019). Predicting Splicing from Primary Sequence with Deep Learning. Cell, 176(3), 535–548 e524. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/30661751. doi: 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- Ke S, Anquetil V, Zamalloa JR, Maity A, Yang A, Arias MA, . . . Chasin LA (2018). Saturation mutagenesis reveals manifold determinants of exon definition. Genome Res, 28(1), 11–24. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/29242188. doi: 10.1101/gr.219683.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Shang S, Kalachikov SM, Morozova I, Yu L, Russo JJ, . . . Chasin LA (2011). Quantitative evaluation of all hexamers as exonic splicing elements. Genome Res, 21(8), 1360–1374. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/21659425. doi: 10.1101/gr.119628.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SW, Taggart AJ, Heintzelman C, Cygan KJ, Hull CG, Wang J, . . . Fairbrother WG (2017). Widespread intra-dependencies in the removal of introns from human transcripts. Nucleic Acids Res, 45(16), 9503–9513. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/28934498. doi: 10.1093/nar/gkx661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, . . . Maglott DR (2016). ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res, 44(D1), D862–868. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/26582918. doi: 10.1093/nar/gkv1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KH, & Fairbrother WG (2012). Spliceman--a computational web server that predicts sequence variations in pre-mRNA splicing. Bioinformatics, 28(7), 1031–1032. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/22328782. doi: 10.1093/bioinformatics/bts074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KH, Ferraris L, Filloux ME, Raphael BJ, & Fairbrother WG (2011). Using positional distribution to identify splicing elements and predict pre-mRNA processing defects in human genes. Proc Natl Acad Sci U S A, 108(27), 11093–11098. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/21685335. doi: 10.1073/pnas.1101135108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirshahi UL, Luo JZ, Manickam K, Wardeh AH, Mirshahi T, Murray MF, & Carey DJ (2018). Trajectory of exonic variant discovery in a large clinical population: implications for variant curation. Genet Med. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/30449888. doi: 10.1038/s41436-018-0353-5 [DOI] [PubMed] [Google Scholar]
- Mort M, Sterne-Weiler T, Li B, Ball EV, Cooper DN, Radivojac P, . . . Mooney SD (2014). MutPred Splice: machine learning-based prediction of exonic variants that disrupt splicing. Genome Biol, 15(1), R19 Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/24451234. doi: 10.1186/gb-2014-15-1-r19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Pan Z, Zhang Z, Lin L, & Xing Y (2018). The Expanding Landscape of Alternative Splicing Variation in Human Populations. Am J Hum Genet, 102(1), 11–26. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/29304370. doi: 10.1016/j.ajhg.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polz MF, & Cavanaugh CM (1998). Bias in template-to-product ratios in multitemplate PCR. Appl Environ Microbiol, 64(10), 3724–3730. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/9758791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Wu L, Huang H, McDonel PE, Palumbo AV, Tiedje JM, & Zhou J (2001). Evaluation of PCR-generated chimeras, mutations, and heteroduplexes with 16S rRNA gene-based cloning. Appl Environ Microbiol, 67(2), 880–887. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/11157258. doi: 10.1128/AEM.67.2.880-887.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Patwardhan RP, Shendure J, & Seelig G (2015). Learning the sequence determinants of alternative splicing from millions of random sequences. Cell, 163(3), 698–711. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/26496609. doi: 10.1016/j.cell.2015.09.054 [DOI] [PubMed] [Google Scholar]
- Sanjana NE (2017). Genome-scale CRISPR pooled screens. Anal Biochem, 532, 95–99. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/27261176. doi: 10.1016/j.ab.2016.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, & Cooper TA (2006). Minigene reporter for identification and analysis of cis elements and trans factors affecting pre-mRNA splicing. Biotechniques, 41(2), 177–181. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/16925019. doi: 10.2144/000112208 [DOI] [PubMed] [Google Scholar]
- Soemedi R, Cygan KJ, Rhine CL, Glidden DT, Taggart AJ, Lin CL, . . . Fairbrother WG (2017). The effects of structure on pre-mRNA processing and stability. Methods, 125, 36–44. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/28595983. doi: 10.1016/j.ymeth.2017.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soemedi R, Cygan KJ, Rhine CL, Wang J, Bulacan C, Yang J, . . . Fairbrother WG (2017). Pathogenic variants that alter protein code often disrupt splicing. Nat Genet, 49(6), 848–855. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/28416821. doi: 10.1038/ng.3837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soemedi R, Vega H, Belmont JM, Ramachandran S, & Fairbrother WG (2014). Genetic variation and RNA binding proteins: tools and techniques to detect functional polymorphisms. Adv Exp Med Biol, 825, 227–266. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/25201108. doi: 10.1007/978-1-4939-1221-6_7 [DOI] [PubMed] [Google Scholar]
- Soukarieh O, Gaildrat P, Hamieh M, Drouet A, Baert-Desurmont S, Frebourg T, . . . Martins A (2016). Exonic Splicing Mutations Are More Prevalent than Currently Estimated and Can Be Predicted by Using In Silico Tools. PLoS Genet, 12(1), e1005756. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/26761715. doi: 10.1371/journal.pgen.1005756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, & Cooper DN (2009). The Human Gene Mutation Database: 2008 update. Genome Med, 1(1), 13 Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/19348700. doi: 10.1186/gm13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenti A, Pierce LC, Biggs WH, di Iulio J, Wong EH, Fabani MM, . . . Venter JC (2016). Deep sequencing of 10,000 human genomes. Proc Natl Acad Sci U S A, 113(42), 11901–11906. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/27702888. doi: 10.1073/pnas.1613365113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A, Bang L, Miller JE, Zhang Y, Lee MTM, Zhang Y, . . . Discov EHRC (2019). Human-Disease Phenotype Map Derived from PheWAS across 38,682 Individuals. Am J Hum Genet, 104(1), 55–64. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/30598166. doi: 10.1016/j.ajhg.2018.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, & Burge CB (2004). Systematic identification and analysis of exonic splicing silencers. Cell, 119(6), 831–845. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/15607979. doi: 10.1016/j.cell.2004.11.010 [DOI] [PubMed] [Google Scholar]
- Wong MS, Kinney JB, & Krainer AR (2018). Quantitative Activity Profile and Context Dependence of All Human 5’ Splice Sites. Mol Cell, 71(6), 1012–1026 e1013. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/30174293. doi: 10.1016/j.molcel.2018.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, . . . Frey BJ (2015). RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science, 347(6218), 1254806. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/25525159. doi: 10.1126/science.1254806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, & Burge CB (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol, 11(2–3), 377–394. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/15285897. doi: 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
- Yeo G, Holste D, Kreiman G, & Burge CB (2004). Variation in alternative splicing across human tissues. Genome Biol, 5(10), R74 Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/15461793. doi: 10.1186/gb-2004-5-10-r74 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.