

Abstract
Recent applications of gene panel sequencing analysis have significantly helped with identifying genetic causes for inherited diseases. However, large amounts of candidate variants remain a major challenge for prioritizing, often requiring arbitrary cutoffs in multiple steps. In addition, existing tools often prioritize a list of promising candidates that require much manual work to evaluate. To this end, we designed an automated, basically cutoff-free scoring scheme named Context and Hereditary Event based Scoring Scheme (CHESS), that scores all possible inheritance events in each gene, by taking into consideration phenotypes, genotypes, and how the manual prioritization works. We applied CHESS to the Critical Assessment of Genome Interpretation 5 intellectual disability panel challenge, to assign clinical phenotypes to patients based on gene panel sequencing data. Through this blind testing, CHESS proved to be a leading and useful tool for genetic diagnosis in a research setting. Further analyses showed that precise phenotype terms played an important role in variant prioritization and that multiple etiologies may exist for some patients. CHESS also successfully identified many of the causal, putative and contributing variants. In the postchallenge analysis, we showed that our best submission performed slightly better than the predictions made by a state-of-the-art tool. We believe that CHESS can provide aid to this and many other diagnostic scenarios.
Keywords: gene panel sequencing, genetic diagnosis, intellectual disability, variant prioritization
1 |. INTRODUCTION
Technology advances in recent years have made next-generation sequencing more and more affordable (Hayden, 2014). While whole-exome sequencing and whole-genome sequencing are in their infancy in clinical practice, gene panel sequencing has already been widely used in clinical laboratories for diagnosing diseases and identifying genetic causes, owing to the relatively lower cost, higher sequencing depth, and the disease-specific focus (Christensen, Dukhovny, Siebert, & Green, 2015; Prokop et al., 2018; Sun et al., 2015). Multiple studies have highlighted the accuracy, reproducibility, and general usefulness of gene-panel-based genetic testing for both rare and complex diseases (Dohrn et al., 2017; Manchanda & Gaba, 2018; Schwarze, Buchanan, Taylor, & Words-worth, 2018; Sturm et al., 2018; Trump et al., 2016; Turnbull et al., 2018). On the flip side, gene panel based procedures have shortcomings—they do not provide information for genes outside of the target set, alternative exons, and deep intron and intergenic information, making it important to define their scale of usage (Bodian, Kothiyal, & Hauser, 2018; Xue, Ankala, Wilcox, & Hegde, 2015). Nevertheless, gene panels should be designed according to the research or application objectives.
Upon obtaining variants through a gene panel sequencing, the next question would be to identify causal, pathogenic variants relevant to the phenotype(s) of interest. Many methods have been developed for evaluating the pathogenicity of variants, such as SIFT (Ng & Henikoff, 2006), PolyPhen (Adzhubei et al., 2010), CADD (Kircher et al., 2014), and REVEL (Ioannidis et al., 2016). There are also multiple methods developed for evaluating the association of the genes (that the variants are located in) with the disease phenotypes, such as PHIVE (Robinson et al., 2014), Phevor (Singleton et al., 2014), and Phenolyzer (Yang, Robinson, & Wang, 2015). Pipeline-type tools, such as the Exomiser (Robinson et al., 2014), have integrated both types of methods, to produce prioritized candidate causal variants. Such methods and tools can be used in gene panel sequencing analyses, as well as in exome/genome sequencing analyses.
Gene-panel-based genetic testing has been applied to the research and clinical diagnosis of many diseases, including neurode-velopmental diseases (Dohrn et al., 2017; Trump et al., 2016). Many neurodevelopmental disorders have profound heritability, highly heterogeneous genetic causes, and significant comorbidities (Blacher & Kasari, 2016; Brainstorm et al., 2018; Lo-Castro & Curatolo, 2014). For instance, researchers have found that intellectual disability and epilepsy can have larger than 70% co-occurrence rates with autism spectrum disorders in some population (Mpaka et al., 2016). With prior knowledge of such disorders, the Padua Diagnostic Laboratory designed a gene panel primarily for studying the genetic mechanisms underlying the comorbidity of intellectual and autism spectrum disorders. The gene panel covered exons and exon-adjacent regions of 74 genes, known associated with seven major types of traits—the important questions to answer were whether this gene panel can help diagnose patients, and what tools could help uncover causal variants from the sequencing data generated by this gene panel. The Critical Assessment of Genome Interpretation (CAGI) organization provides a “blind test” platform for assessing computational tools designed for interpreting variants in the genome, through community experiments and competitions. To this end, the Padua Diagnostic Laboratory provided the 74-gene panel sequencing results of 150 cases with neurodevelopmental disorders, as the data for the CAGI5 intellectual disability gene panel challenge. The challenge was to use the precalled genetic variants to predict the disease terms (among the seven traits) of cases as well as to prioritize the true genetic causes (causal variants). The submissions were then assessed by a designated independent assessor, primarily on how well the predicted disease terms aligned with the actual records. The predicted variants were also shared back with the data provider.
Given a large number of cases and disease terms to test, we used a fully-automated scoring scheme named Context and Hereditary Event based Scoring Scheme (CHESS). We designed the original form of CHESS for analyzing whole-exome sequencing family data. This CAGI challenge provided a chance for unbiasedly assessing this scoring scheme, so we made several adjustments to CHESS suitable for the data type of this challenge—gene panel sequencing data without family information, and generated three submissions named “stringent,” “medium stringent,” and “less stringent.” After the challenge, the assessor reported that the CHESS “less stringent” submission showed the best overall performance in predicting disease terms among all submissions by participants of this challenge. In this manuscript, we presented the CHESS scoring scheme and the predictions of disease terms in details. We also discussed postchallenge analyses based on the latest released causal, putative, and contributing variants by the data provider, as well as a comparison with predictions by a state-of-the-art tool for genetic diagnosis.
2 |. METHOD
2.1 |. Data preprocess
The precalled variants generated from the gene panel sequencing data were provided by the data provider. We then annotated the variants with VEP (McLaren et al., 2016), gnomAD v2.0.2 (Lek et al.,2016), and the precalculated REVEL scores (Ioannidis et al., 2016). REVEL was used for this challenge mainly because REVEL showed better performance than most other individual and ensemble methods for predicting rare variant pathogenicity (Ioannidis et al., 2016; Li et al., 2018). Variants mapped to multiple precalculated REVEL positions were scored as the largest REVEL score among the positions. Severest annotation by VEP (across all possible transcripts) was used for each variant. Only variants with putatively protein-altering annotation were used in the following analysis. We also excluded common variants--variants with minor allele frequency (MAF) ≧ 5%--from our analysis, since the seven diseases of interest were relatively rare genetic diseases. After these preprocess steps, a data matrix (see an example in the Supporting Information Data) containing variant locations and the above annotation information was generated for each sample, and was then used as the input data for the CHESS scoring step.
2.2 |. The scoring scheme of CHESS and settings for this challenge
The scoring scheme of CHESS contains two steps. As shown in Figure 1, the first step is variant scoring. The score of each variant is based on three major components—variant quality, predicted deleteriousness, and phenotype match. The calculation is designed as basically cutoff free, and will prioritize variants if any of the three parts stand out. Specifically, the equation is as follows:
FIGURE 1.
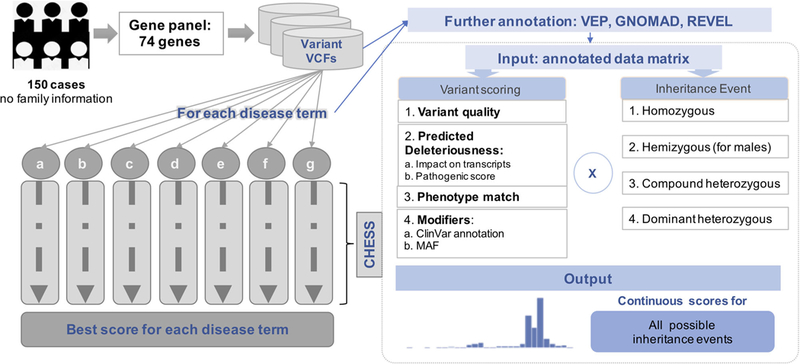
The workflow for predictions made by CHESS. The left half of this figure shows the scheme of the solution to the CAGI intellectual disability gene panel challenge. The right half of this figure shows the process of input data preparation and the CHESS scoring steps (the box by dotted line). CAGI, Critical Assessment of Genome Interpretation; CHESS, Context and Hereditary Event based Scoring Scheme
where for this challenge:
-
a)
The variant quality “qual” was defined as GQ value transformed to the scale of 0 to 1
-
b)
The predicted deleteriousness “del” was defined as a sum of variant impact annotation (.5 for frameshift, and .25 for missense and inframe InDel) and REVEL score (transformed to 0–.5); notethat the parameters for different types of variants were set based on best knowledge and specifically for this challenge, and they may subject to changes if applied to different scenarios
-
c)
The phenotype match score “pheno” was calculated using Phenolyzer (Yang et al., 2015) for each gene with each disease term (also 0–1)
-
d)
For convenience, the modifiers “m1,” “m2,” and “m3” were all set to the rarity ((.01-MAF)/.05).
The second step of CHESS is to integrate variant scores with all possible inheritance events. For each possible inheritance event (dominant or recessive events, on autosomal chromosomes or the X chromosome), a final score is calculated based on score(s) of the variant(s) involved in this inheritance event.
2.3 |. Adjustments and the three submissions
The following items were the special adjustments on the CHESS scoring scheme, made for generating our three submissions for the CAGI intellectual disability gene panel challenge:
-
(1)
Since family information was not available, the estimated “de novo dominant” events could have a high false positive rate (FPR). The scores for such events were divided by two as an adjustment.
-
(2)
For the same reason, the scores for estimated “compound heterozygous” events were calculated as the sum of scores of the two involved variants divided by 3 (or 2, in “less stringent”).
-
(3)
Special adjustments for the “stringent” submission: (a) variants called in more than 30% of the samples were excluded from the analysis; (b) only scores ≧.5 were reported; (c) only the score of the top event (if >.5) for each disease was reported as the final score.
-
(4)
Special adjustments for the “medium stringent” submission: (a) variants called in more than 30% of the samples were excluded from the analysis; (b) only the score of the top event for each disease was reported as the final score.
-
(5)
Special adjustments for the “less stringent” submission: (a) variants called in more than 50% of the samples were excluded from the analysis; (b) top two events for each disease were reported, and the final score was set as the mean of the two (in rare cases if the two top scored events come from the same gene, the larger score was used)
Based on the CHESS scoring scheme and all the adjustments discussed above, we calculated the final score for each of the seven disease terms for each case. We then used the final score of a disease term in each case as the predicted probability of the case having this disease. The variants involved in the top event(s) used for each final score were defined as the prioritized candidate variants. Please note that these adjustments were specifically made for this challenge, and should be evaluated case-by-case if the CHESS scoring scheme will be applied to other circumstances (see “CHESS for general usage” in the Supporting Information Data).
2.4 |. Predictions made by the state-of-the-art method Exomiser
Since the Exomiser (Robinson et al., 2014) took HPO IDs, instead of disease terms, as the input, we first selected a single obviously closest HPO ID for each disease term based on our best knowledge. With each HPO ID, we then ran the Exomiser (Version 11.0.0) on the variants of each case using default settings. Following a similar strategy as “less stringent”, we took the average score of the top two variants (across all the inheritance events used in default settings) of the Exomiser as the predicted probability score and used these two variants as the Exomiser “Top 2” prioritized candidate variants.
2.5 |. Scripts and packages
In-house scripts for CHESS were written in Perl. Most postchallenge analyses were performed using R. Receiver operating characteristic (ROC) curves were plotted using roc() from the R package pROC (Robin et al., 2011), and area under the ROC curve (AUCs) were calculated using auc() from the same R package.
3 |. RESULTS
3.1 |. Overview of the predictions in seven diseases
We used a scoring scheme named CHESS for predicting disease terms and prioritizing candidate variants for the CAGI5 intellectual disability gene panel challenge (see Section 2; Figure 1). Among the three submissions we made for this challenge, the third one (“less stringent”) had the best overall performance, when evaluated by AUC across all the seven disease terms—intellectual disability, autism spectrum disorder, epilepsy, microcephaly, macrocephaly, hypotonia, and ataxic gait (Figure 2; AUCLessStringent = .58). According to the assessor, our “less stringent” submission was also the best among all submissions received for this challenge based on the same measure, followed very closely by the second best, which was by another group (see the assessment of the CAGI5 intellectual disability gene panel challenge published in the same special issue). However, all the submissions were actually only slightly, though significantly (according to the assessor), better than a random guess, highlighting the need to analyze the details and perhaps also a demand for better-geared tools.
FIGURE 2.
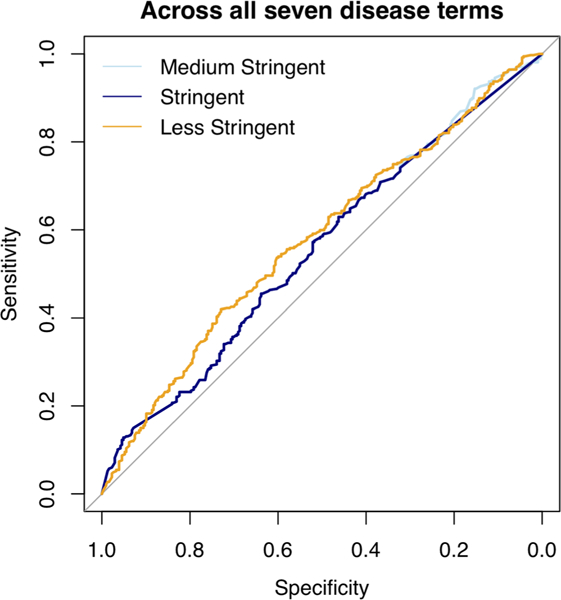
The ROC curves of the three submissions. The light blue line represents CHESS “Medium Stringent”; the dark blue line represents CHESS “stringent”; the orange line represents CHESS “less stringent”. CHESS, Context and Hereditary Event based Scoring Scheme; ROC, receiver operating characteristic
When breaking down into individual disease terms, “less stringent” predictions showed better performance in intellectual disability, macrocephaly, and microcephaly among the seven. The performance was not correlated with the affected rate of each disease in these patients. For all disease terms except “ataxia gait,” “less stringent” performed significantly better than random (1,000 times randomization; T test p value ≦.05). However, the actual AUC for each term was only slightly larger than the 1,000 times randomization average (Figure 3). Surprisingly, despite that this gene panel was originally designed for both intellectual disability and autism spectrum disorder, and that there was high comorbidity (68.9%) of the two diseases in this dataset, “less stringent” gave a much worse performance for autism spectrum disorder than for intellectual disability. We believe this was partially due to the complex nature of autism spectrum disorder—while intellectual disability was more of a type of rare inherited diseases.
FIGURE 3.
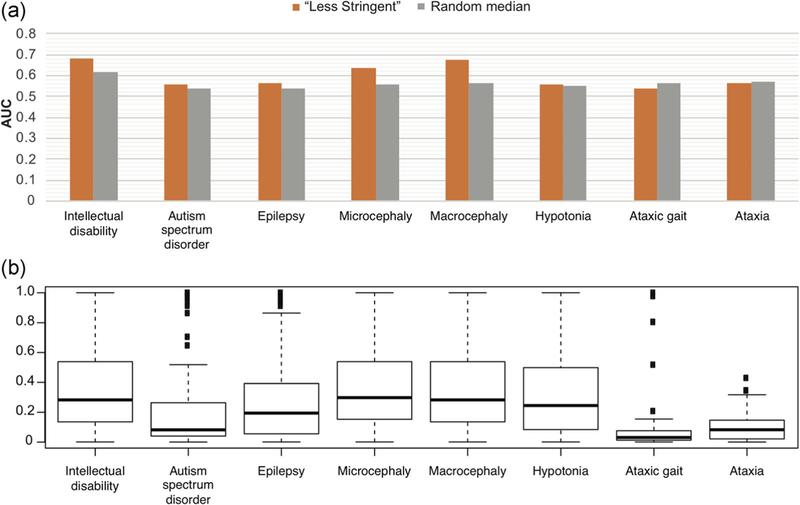
(a) Comparisons of prediction performances (quantified by AUCs) of CHESS “less stringent” versus “random median.” “Random median” refers to the median value of 1,000 times randomizations. The randomization was done by shuffling of case IDs. Performances for all seven disease terms as well as the term “ataxia” are shown here. (b) The distribution of phenotype match scores of all the genes in the gene panel, for each of the seven disease terms as well as the additional term “ataxia.” AUCs, areas under the ROC curve; CHESS, Context and Hereditary Event based Scoring Scheme
The complexity of the diseases and the amount of knowledge available for each disease can be partially reflected by the range of gene phenotype scores automatically generated by Phenolyzer (Yang et al., 2015) for each disease term, as shown in Figure 3. We observed a moderate positive correlation between the performance AUCs and the median gene phenotype scores (Pearson correlation coefficient = .78, p value = .04). Since we used the disease terms, instead of fixed HPO terms, to generate the gene phenotype scores, the choice of words could potentially affect the outcome. For instance, in the submission, we used the term “ataxia gait” provided by the data provider, while most genes in the panel showed very low phenotype scores relating to this term, and the prediction performance was worse than random. After the challenge, we tried the term “ataxia,” which led to a slightly higher overall gene phenotype scores and slightly better performance (Figure 3). This was not unexpected as the gene panel was not primarily designed for ataxia gait or ataxia.
3.2 |. Categorization of cases
We compared the predicted disease terms and the actual disease terms in details. Here we set the FPR for each disease term at .3, and defined that a disease term was predicted for a case if the prediction score was higher than the score at this FPR. For intellectual disability, the FPR was effectively .5, since there were only four cases without this disease. Of the 150 cases, 106 have been predicted with at least one disease term. We then categorized each case into the following three types: (a) fully predicted, where all the actual disease terms for the case were predicted; (b) partially predicted, where at least one actual term was predicted and at least one actual term was missed; (c) fully missed, where none of the actual terms was predicted. In short, we identified 60, 46, and 44 cases as “fully predicted”, “partially predicted”, and “fully missed”, respectively. Many of the “fully predicted” cases have been predicted with terms in addition to their actual disease terms. On the other hand, 65% of the “fully predicted” cases had no information for at least three of the seven disease terms, indicating a lack of complete medical information for such cases.
All the 46 “partially predicted” cases were correctly predicted with intellectual disability. For 36 of the “partially predicted” cases, autism spectrum disorder was missed by our prediction. Among the 36, nine were actually predicted with a relatively high score for autism spectrum disorder—only smaller than the cutoff by less than .05. Similarly, among the 46 “partially predicted” cases, 14 of 17 epilepsy cases, 9 of 10 microcephaly cases, 12 of 13 hypotonia cases, and all 3 ataxic gait cases were missed, while only 1 of 4 macrocephaly cases was missed. On the other hand, the predictions correctly marked most cases that did not have a certain disease term —there were few overpredictions. Overall, we observed a relatively poor recall in the “partially predicted” cases at the cutoff we chose (FPR at .3).
Surprisingly, the “fully missed” cases turned out to be cases with no predicted disease terms at all. The prediction scores of the “fully missed” cases for almost every disease term were much lower than the cutoffs. In fact, for all disease terms except ataxic gait, “fully missed” cases showed a lower affected rate compared to all other cases (odds ratios = .94, .85, .86, .24, .63, and .70 for intellectual disability, autism spectrum disorder, epilepsy, micro-cephaly, macrocephaly, and hypotonia, respectively). This indicated that our predictions did capture some true signal useful for separating different types of cases. The true genetic causes of these cases either failed to be prioritized by our predictions or did not exist in the genomic regions covered by this gene panel.
3.3 |. Postchallenge analyses: identification of causative variants
After the challenge was completed, the data provider released “causative variants,” “putative variants,” and “contributing factors” for 24, 16, and 12 cases, respectively. According to their definition, “causative variants” were identified through segregation analysis and genotype-phenotype correlation; “putative variants” were rare or novel variants, predicted as pathogenic, while without available segregation analysis; “contributing factors” were rare or novel variants, predicted as pathogenic, in relevant genes, while shared with healthy parents. Interestingly, there were no two cases that shared the same “causative variants,” “putative variants” or “contributing factors.”
In our best submission--“less stringent,” top two potential inheritance events for each disease term for each case were prioritized, which corresponded to two to four variants per case per disease term. In the same cases, the prioritized variants were often the same or similar across different disease terms—only 16% cases had more than four total prioritized variants. We successfully identified “causal variants” for 11 cases (45.8%; note that in one case one of the two “causal variants” was missed), “putative variants” for 9 cases (56.3%), and “contributing factors” for 9 cases (75.0%; one case with one of the two “contributing factors” missed). Given that our predictions were made without access to the gender or family information, we consider “less stringent” did well in prioritizing candidate variants.
Interestingly, of the 29 unique cases that “less stringent” predicted correct variants for, the disease terms of ten cases were “fully missed” by “less stringent” (FPR at .3), as shown in Table 1. This indicated that (a) “less stringent” have the power to identify the “correct” candidate variants, but the connections between variants and specific disease terms have not been well established; (b) there may be other disease-causing or disease-contributing variants that are missed by both “less stringent” and the date provider, or are not included in this gene panel analysis.
TABLE 1.
The categories of cases that are predicted with “causal variants,” “putative variants,” “contributing factors”
| No. of cases | Predicted with |
||
|---|---|---|---|
| “Causal variants” |
“Putative variants” |
“Contributing factors” |
|
| Terms “fully predicted” | 7 | 4 | 0 |
| Terms “partially predicted” | 1 | 3 | 4 |
| Terms “fully missed” | 3 | 2 | 5 |
3.4 |. Postchallenge analyses: comparison with the state-of-the-art tool Exomiser
We further evaluated our predictions by comparing them with the predictions by the state-of-the-art tool Exomiser (Robinson et al., 2014). The Exomiser (Version 11.0.0) was applied to each case for each disease term following default settings, and the results were collected using a similar strategy as CHESS “less stringent” (see Section 2). In this analysis, the top two variants, as well as their scores predicted by the Exomiser, were used for each disease term for each case, so we termed this application of the Exomiser as Exomiser “Top 2”. For five out of the seven disease terms, the AUCs of CHESS “less stringent” were better than those of Exomiser “Top 2” (Figure 4; see also Figure S1 for comparison with Exomiser “Top 1”). When pooling predictions for all seven disease terms together, Exomiser “Top 2” performed slightly better (AUC .62 Vs. .58). This indicated that Exomiser “Top 2” and CHESS “less stringent” could capture information in ways appropriate for different types of diseases.
FIGURE 4.
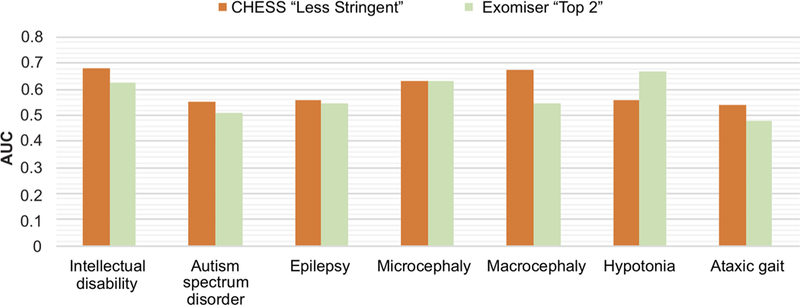
Comparisons of prediction performances (quantified by AUCs) of CHESS “less stringent” versus Exomiser “Top 2”. Performances for all seven disease terms are shown here. AUCs, areas under the ROC curve; CHESS, Context and Hereditary Event based Scoring Scheme
As discussed previously, for each case with each disease term, Exomiser “Top 2” prioritized the top two variants, while CHESS “less stringent” prioritized variants in the top two events (usually 2–4 variants). We found that the prioritized variants of Exomiser tended to be more varied among different disease terms, than those by CHESS “less stringent”—Exomiser “Top 2” prioritized a median of seven unique variants for each case across all disease terms, while CHESS “less stringent” only prioritized a median of three unique variants for each case across all disease terms. However, Exomiser “Top 2” hit fewer cases with “correct variants” than CHESS “less stringent” (25 unique cases compared with 29), indicating that Exomiser “Top 2” variants may have a higher FPR. Nevertheless, the overlap between the 25 and 29 cases were only 17 cases, meaning that both Exomiser “Top 2” and CHESS “Less Stringent” could provide novel “correct variants” for several cases that the other missed (Table 2).
TABLE 2.
The numbers of cases predicted with “correct variants” by CHESS “less stringent” and Exomiser “Top 2”
| No. of cases predicted with |
||||
|---|---|---|---|---|
| “Causal variants” |
“Putative variants” |
“Contributing factors” |
Any of the three types of variants |
|
| Exomiser “Top 2” | 13 | 7 | 6 | 25 |
| CHESS “Less Stringent” | 11 | 9 | 9 | 29 |
| Overlap | 8 | 5 | 4 | 17 |
Abbreviation: CHESS, Context and Hereditary Event based Scoring Scheme.
Taken together, we consider CHESS “less stringent” performed slightly better than Exomiser “Top 2” in predicting five of the seven disease terms, and in prioritizing candidate variants. However, each of the two could provide some unique useful information that the other missed. We cautiously suggest that this conclusion may only apply to certain disease terms, as the scale of this analysis was very specific. In addition, since both CHESS and Exomiser used comprehensive phenotype-matching based gene scores to help prioritize variants, detailed clinical phenotype data (in addition to the disease terms) could potentially improve the outcome of each method.
4 |. DISCUSSION
We presented the application of CHESS in the CAGI5 intellectual disability gene penal challenge, a fully automated and cutoff-free inheritance event-based scoring scheme for prioritizing candidate causal variants. The “less stringent” submission of CHESS performed slightly better than other submissions in predicting disease terms of patients in this challenge, and also slightly out-performed Exomiser “Top 2” in five of the seven disease terms. Overall, the prediction AUC for each disease was not very high, though significantly higher than random. CHESS “less stringent” successfully prioritized “causal,” “putative” or “contributing” variants for 29 cases out of the 50 cases provided with such variants by the data provider, with relatively fewer “false positives” compared with Exomiser “Top 2”.
Here are several interesting observations that could be useful for future investigation and perhaps future CAGI challenge designs. First, CHESS “less stringent” was the only one among our three submissions that used the top two events instead of trying to find one best event, yet it performed the best. This indicated that multiple, instead of single, etiologies may exist in at least some of the patients. The diseases of interest might have a large spectrum across the traditional concepts of “rare” and “complex” diseases. Second, we showed in the analyses that using slightly different terms may generate different results. This might suggest that current methods to associate genes with phenotypes were not perfect, but also highlighted the importance of standardizing disease terms and categorization. Third, it was tricky to try to set an FPR for intellectual disability, since there were only four true negative cases. We consider that more balanced numbers of positive and negative cases could provide more power to the overall assessment of performance.
There were two obvious pitfalls in our submissions. One was that we assumed these diseases should more or less follow the patterns of rare inherited diseases, so that we predicted disease terms fully based on the most possible genetic variants. The overall low performances might be partially owing to that the assumption was not entirely true. The other pitfall was that in our method we interrogated each disease term one by one, as if they were independent diseases. However, several of the diseases had high comorbidities, as previously reported (Blacher & Kasari, 2016; Lo-Castro & Curatolo, 2014; Mpaka et al., 2016). In fact, the gene panel used in this CAGI challenge was originally designed to study the genetics underlying the comorbidities of intellectual disability and autism spectrum disorders. When dealing with this challenge, we did not have a good genetic model to follow, but we hope that with such data researchers can find better models to use in the future.
We consider it possible that the patients could have genetic causes outside of the 74 genes of this gene panel. After all, there have been at least hundreds of genes reported associated with intellectual disability (Wieczorek, 2018). With technology advancement leading to lower sequencing price, whole exome sequencing could be appropriate for identifying other genetic causes. Also, whole genome sequencing might help find causal structural variants for some cases, as reported by others that structural genomic variations were also among the important causes of intellectual disability (Vissers, Gilissen, & Veltman, 2016).
All in all, the CAGI5 intellectual disability gene panel challenge has provided a great opportunity for assessing tools as well as investigating the genetic causes of neurodevelopmental diseases. We consider that CHESS could provide useful results when analyzing genetic testing data such as in this CAGI challenge. CHESS was mainly designed for rare inherited diseases and could be applied to both gene panel and whole exome data. In this specific analysis, CHESS was performed using input data based on VEP annotation, gnomAD MAF, and REVEL scores. Nevertheless, researchers can use the CHESS framework based on annotation generated based on other appropriate tools as well. We hope that this analysis would help inform future research, and hope that CAGI will continue to contribute to the genome interpretation area, both in research and in clinical applications.
Supplementary Material
ACKNOWLEDGMENT
The author would like to thank Dr Steven Brenner for encouraging the development of a cutoff-free method, and Drs Zhiqiang Hu and Yaqiong Wang for inspiring discussions about and suggestions on the CHESS scoring scheme. The development of CHESS and the submissions for the CAGI challenge were done when the author was part of the Brenner Lab. The author would also like to thank all other members of the Brenner Lab for their encouragement and support. The CAGI experiment coordination is supported by NIH U41 HG007346 and the CAGI conference by NIH R13 HG006650. The efforts for developing CHESS and generating the prediction submissions were supported by a gift from the Jeffrey Modell Foundation and the California Initiative to Advance Precision Medicine, and the efforts for postchallenge analyses and drafting the manuscript were supported by the Shanghai Municipal Science and Technology Major Project (No.2018SHZDZX01) and ZHANGJIANG LAB, as well as the National Natural Science Foundation of China (61772368, 61572363, 91530321).
In addition, the author would like to acknowledge the Genome Aggregation Database (gnomAD) and the groups that provided exome and genome variant data to this resource. A full list of contributing groups can be found at http://gnomad.broadinstitute. org/about.
Funding information
Jeffrey Modell Foundation, Grant/Award Number: gift; National Institutes of Health, Grant/Award Numbers: NIH R13 HG006650, NIH U41 HG007346; National Natural Science Foundation of China, Grant/Award Numbers: 91530321, 61572363, 61772368; ZHANGJIANG Lab, Shanghai; Shanghai Municipal Science and Technology Major Project, Grant/Award Number: No.2018SHZDZX01; California Initiative for Advancing Precision Medicine
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
DATA AVAILABILITY
The scripts used for the CAGI challenge are provided in the Supporting Information Data.
CONFLICT OF INTERESTS
The author declares that there is no conflict of interests.
The author is no longer affiliated with 3, but the submission for the CAGI challenge was completed while affiliated with 3. The author completed the postchallenge analysis and wrote the manuscript, while hosted by 1&2
REFERENCES
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P,… Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacher J, & Kasari C (2016). The intersection of autism spectrum disorder and intellectual disability. Journal of Intellectual Disability Research, 60(5), 399–400. 10.1111/jir.12294 [DOI] [PubMed] [Google Scholar]
- Bodian DL, Kothiyal P, & Hauser NS (2018). Pitfalls of clinical exome and gene panel testing: Alternative transcripts. Genetics in Medicine, 21, 1240–1245. 10.1038/s41436-018-0319-7 [DOI] [PubMed] [Google Scholar]
- Brainstorm C, Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, … Murray R (2018). Analysis of shared heritability in common disorders of the brain. Science, 360(6395), eaap8757. 10.1126/science.aap8757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen KD, Dukhovny D, Siebert U, & Green RC (2015). Assessing the costs and cost-effectiveness of genomic sequencing. Journal of Personalized Medicine, 5(4), 470–486. 10.3390/jpm5040470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohrn MF, Glockle N, Mulahasanovic L, Heller C, Mohr J, Bauer C, . Biskup S. (2017). Frequent genes in rare diseases: Panel-based next generation sequencing to disclose causal mutations in hereditary neuropathies. Journal of Neurochemistry, 143(5), 507–522. 10.1111/jnc.14217 [DOI] [PubMed] [Google Scholar]
- Hayden EC (2014). Technology: The $1,000 genome. Nature, 507(7492), 294–295. 10.1038/507294a [DOI] [PubMed] [Google Scholar]
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, … Sieh W (2016). REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. American Journal of Human Genetics, 99(4), 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’roak BJ, Cooper GM, & Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, … MacArthur DG (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao T, Zhang Y, Zhang K, Shi L, Chen Y, … Sun, Z. (2018). Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Research, 46(15), 7793–7804. 10.1093/nar/gky678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo-Castro A, & Curatolo P (2014). Epilepsy associated with autism and attention deficit hyperactivity disorder: Is there a genetic link? Brain and Development, 36(3), 185–193. 10.1016/j.braindev.2013.04.013 [DOI] [PubMed] [Google Scholar]
- Manchanda R, & Gaba F (2018). Population based testing for primary prevention: A systematic review. Cancers (Basel), 10(11), E424 10.3390/cancers10110424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A,… Cunningham F (2016). The ensembl variant effect predictor. Genome Biology, 17(1), 122 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mpaka DM, Okitundu DL, Ndjukendi AO, N’Situ AM, Kinsala SY, Mukau JE.,… Steyaert J. (2016). Prevalence and comorbidities of autism among children referred to the outpatient clinics for neurodevelopmental disorders. The Pan African Medical Journal, 25, 82 10.11604/pamj.2016.25.82.4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, & Henikoff S (2006). Predicting the effects of amino acid substitutions on protein function. Annual Review of Genomics and Human Genetics, 7, 61–80. [DOI] [PubMed] [Google Scholar]
- Prokop JW, May T, Strong K, Bilinovich SM, Bupp C, Rajasekaran S, . Lazar J. (2018). Genome sequencing in the clinic: The past, present, and future of genomic medicine. Physiological Genomics, 50(8), 563–579. 10.1152/physiolgenomics.00046.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, & Muller M (2011). pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics, 12, 77 10.1186/1471-2105-12-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Kohler S, Oellrich A, Wang K, Mungall CJ, Lewis SE, … Smedley D. (2014). Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Research, 24(2), 340–348. 10.1101/gr.160325.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze K, Buchanan J, Taylor JC, & Wordsworth S (2018). Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genetics in Medicine, 20(10), 1122–1130. 10.1038/gim.2017.247 [DOI] [PubMed] [Google Scholar]
- Singleton MV, Guthery SL, Voelkerding KV, Chen K, Kennedy B, Margraf RL,… Yandell M. (2014). Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. American Journal of Human Genetics, 94(4), 599–610. 10.1016/j.ajhg.2014.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm AC, Knowles JW, Gidding SS, Ahmad ZS, Ahmed CD, Ballantyne CM, … Rader D. (2018). Clinical genetic testing for familial hypercholesterolemia: JACC Scientific Expert Panel. Journal of the American College of Cardiology, 72(6), 662–680. 10.1016/j.jacc.2018.05.044 [DOI] [PubMed] [Google Scholar]
- Sun Y, Ruivenkamp CA, Hoffer MJ, Vrijenhoek T, Kriek M, van Asperen CJ, … Santen GW. (2015). Next-generation diagnostics: Gene panel, exome, or whole genome? Human Mutation, 36(6), 648–655. 10.1002/humu.22783 [DOI] [PubMed] [Google Scholar]
- Trump N, McTague A, Brittain H, Papandreou A, Meyer E, Ngoh A, . Scott RH. (2016). Improving diagnosis and broadening the phenotypes in early-onset seizure and severe developmental delay disorders through gene panel analysis. Journal of Medical Genetics, 53(5), 310–317. 10.1136/jmedgenet-2015-103263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull C, Scott RH, Thomas E, Jones L, Murugaesu N, Pretty FB, … Caulfield MJ. (2018). The 100 000 Genomes Project: Bringing whole genome sequencing to the NHS. BMJ, 361, k1687 10.1136/bmj.k1687 [DOI] [PubMed] [Google Scholar]
- Vissers LE, Gilissen C, & Veltman JA (2016). Genetic studies in intellectual disability and related disorders. Nature Reviews Genetics, 17(1), 9–18. 10.1038/nrg3999 [DOI] [PubMed] [Google Scholar]
- Wieczorek D (2018). Autosomal dominant intellectual disability. Medical Genetics, 30(3), 318–322. 10.1007/s11825-018-0206-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ankala A, Wilcox WR, & Hegde MR (2015). Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: Single-gene, gene panel, or exome/genome sequencing. Genetics in Medicine, 17(6), 444–451. 10.1038/gim.2014.122 [DOI] [PubMed] [Google Scholar]
- Yang H, Robinson PN, & Wang K (2015). Phenolyzer: Phenotype-based prioritization of candidate genes for human diseases. Nature Methods, 12(9), 841–843. 10.1038/nmeth.3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.