

Abstract
Testing for variation in BRCA1 and BRCA2 (commonly referred to as BRCA1/2), has emerged as a standard clinical practice and is helping countless women better understand and manage their heritable risk of breast and ovarian cancer. Yet the increased rate of BRCA1/2 testing has led to an increasing number of Variants of Uncertain Significance (VUS), and the rate of VUS discovery currently outpaces the rate of clinical variant interpretation. Computational prediction is a key component of the variant interpretation pipeline. In the CAGI5 ENIGMA Challenge, six prediction teams submitted predictions on 326 newly-interpreted variants from the ENIGMA Consortium. By evaluating these predictions against the new interpretations, we have gained a number of insights on the state of the art of variant prediction and specific steps to further advance this state of the art.
Keywords: BRCA, BRCA1, BRCA2, Variant Interpretation, CAGI, CAGI5
Introduction
While women have a 12% lifetime risk of breast cancer on average (Howlader et al., 2017), that risk rises to roughly 70% in women with pathogenic variants in BRCA1/2 (Kuchenbaecker et al., 2017). For ovarian cancer, the average lifetime risk is approximately 1.3% for women in the general population, while the risk is 44% for BRCA1 carriers and 17% for BRCA2 carriers. These facts, and the decreasing cost of sequencing, have led to an upsurge of BRCA1/2 testing in recent years (Kolor et al., 2017). This increased rate of BRCA1/2 testing has led to an increasing discovery of new variants, and this rate of variant discovery has outpaced the rate of variant interpretation. Out of 21,695 variants currently listed at BRCA Exchange, the largest public source of BRCA1/2 variation data (Cline et al., 2018), almost half (9,225) have no clinical interpretation in either ClinVar (Landrum & Kattman, 2018) or the Leiden Open Variation Database (LOVD) (Fokkema et al., 2011). Further, only 7,225 so far have expert interpretations by the ENIGMA Consortium (Spurdle et al., 2012), the ClinGen expert panel for curation of variants in BRCA1/2. These numbers underscore the need for both developing robust, high-throughput methods for BRCA1/2 variant interpretation and gaining a clear understanding of the capabilities of the existing methods.
The CAGI5 ENIGMA Challenge provided an opportunity to evaluate the current state of the art in predicting the clinical significance of BRCA1/2 variants, leveraging blind prediction. The ENIGMA Consortium provided the CAGI organizers with not-yet-published clinical interpretations for hundreds of BRCA1/2 variants. Six research teams predicted the clinical significance of these variants, using fourteen methods altogether. In this paper, we compare results from these 14 prediction methods, as well as three widely-used reference methods from the literature, against the expert clinical interpretations, with the goals of evaluating what types of approaches were most effective and identifying areas for further improvement.
Methods
This challenge featured 326 variants that were recently interpreted by the ENIGMA Consortium, as detailed in another paper in this issue (Parsons et al, 2019). This paper also details the variant classification process used by ENIGMA researchers. Briefly, unclassified variants were prioritized by the ENIGMA Consortium for classification based on the amount of available evidence and/or prior likelihood of pathogenicity based on variant location and predicted effect (Tavtigian, Byrnes, Goldgar, & Thomas, 2008; Vallée et al., 2016). These variants were classified using multifactorial analysis (Goldgar et al., 2008, 2004). While the standard ACMG guidelines evaluate multiple lines of evidence by qualitative rules (Richards et al., 2015), multifactorial analysis combines evidence types quantitatively in a Bayesian network to estimate the overall likelihood of pathogenicity. Of the 326 variants that were shared with the prediction teams, all were exonic, and were either missense variant or in-frame deletions (ENIGMA had provided additional intronic variants that were not shared with the predictors). All of these variants were assessed as Benign, Likely Benign, Likely Pathogenic or Pathogenic at the time of submission to CAGI5. None of the variants had a population frequency of 1% or greater in any reference population studied, and none were predicted truncating variants (Parsons et al, 2019).
Table 1 summarizes the 326 ENIGMA Challenge variants included in the CAGI5 challenge, comprising 318 single-nucleotide variants and eight in-frame deletions. All BRCA1 variants reflect the NM_007294.3 transcript, and all BRCA2 variants reflect NM_000059.3. Although the ENIGMA Consortium had prioritized some variants according to prior likelihood of pathogenicity, most of the variants were either Benign or Likely Benign. This skew is consistent with the actual proportions of the different clinical significance annotations for BRCA1/2 variants (Cline et al., 2018). During the course of the CAGI experiment, the ENIGMA Consortium reclassified several of these variants with new evidence. Five variants were reclassified from Likely Benign to VUS after ENIGMA received new evidence that conflicted with previous evidence, and these variants were omitted from the assessment. Seventeen variants were reclassified from Likely Benign to Benign, and one was reclassified from Likely Pathogenic to Pathogenic. That is, the majority of the reclassifications increased certainty in the assignment. Since the CAGI5 challenge examined benign and likely benign as one group, and pathogenic and likely pathogenic as another, these reclassifications did not affect the assessment except for removing five variants from the assessment pool.
Table 1:
The summarizes the variants of the BRCA Challenge according to gene, domain region and clinical significance as interpreted by the ENIGMA Consortium. The Domain column indicates which variants were part of a clinically-significant protein domain, by the criteria of the ENIGMA Consortium (ENIGMA Consortium, 2017). The rows marked “None” indicate variants that are not part of a clinically-significant domain. 318 of the variants were single-nucleotide substitutions, while the remaining eight were in-frame deletions.
| Gene | Domain | Benign | Likely Benign | Uncertain Significance | Likely Pathogenic | Pathogenic | Total |
|---|---|---|---|---|---|---|---|
| BRCA1 | BRCT | 2 | 7 | 1 | 4 | 1 | 15 |
| None | 29 | 90 | 2 | 2 | 123 | ||
| RING | 1 | 4 | 4 | 9 | |||
| BRCA2 | DNB | 11 | 27 | 3 | 3 | 44 | |
| None | 20 | 110 | 2 | 132 | |||
| TR2/RAD5 | 1 | 2 | 3 | ||||
| Total | 64 | 240 | 5 | 7 | 10 | 326 | |
The fact that twenty-three variants were reclassified illustrates two things: all interpretations have some uncertainty, the level of which is inherent in the probability of pathogenicity and the class assigned; additional data is helpful to move variants from “likely” categories to outer categories with higher probabilities in favour of a benign or pathogenic classification
Prediction Methods
Six teams submitted blind predictions, using a total of fourteen methods. The methods are summarized in Table 2 and summarized below. Most teams have submitted methods papers to the CAGI5 publication set, and we have referenced those methods for further detail. We have also indicated which methods were executed as published; for the others, further details are available in the Supporting Methods section.
Table 2:
Summary of the BRCA Challenge blind prediction teams and methods.
| Predictor | Method | Brief Description |
|---|---|---|
| Lichtarge Lab | Evolutionary Action (EA) | Estimates pathogenicity from evolutionary and phylogenic information, and substitution likelihood. |
| Normalized EA | Normalizes EA predictions with the estimated fraction of BRCA isoforms affected by the mutation. | |
| Mooney Radivojac 2018 | MutPred2 | Machine learning predictor with features including estimated changes in structural and functional properties for single-nucleotide variants, augmented with an unpublished method for in-frame deletions. |
| TransBio-Inf | TBI_1 | Neural network trained to predict clinical significance from estimated splice site impact and sequence-based features. |
| TBI_2 | Similar to TBI_1, but with multiple linear regression prediction of functional assay scores. | |
| TBI_3 | Similar to TBI_1, but with no estimated splicing impact | |
| TBI_4 | Similar to TBI_2, but with no estimated splicing impact | |
| Bologna Bio-computing | SNPs&GO | Machine learning predictor that integrates features extracted from sequence, sequence profile and GO functional annotation of the input protein. |
| Disease Index Matrix | Statistical scale estimating the probability of a variation type to be associated with disease. | |
| AIBI | AIBI | Weakly supervised linear regression using categorized inexact labels from ClinVar, 15 selected features from MutPred2, and designed loss functions. |
| Color Genomics | LEAP 1 | Two-class logistic regression using function predictions, splicing predictions, frequency of cancer in individuals and their families, co-occurrence with pathogenic variants, andliterature and cancer associations from HGMD. |
| LEAP 2 | As LEAP 1, but with publicly-available information only | |
| LEAP 3 | As LEAP 1, but with random forest classifier rather than logistic regression | |
| LEAP 4 | As LEAP 1, but with three-class logistic regression rather than two-class. |
The Lictarge Lab submitted predictions with Evolutionary Action (EA) (as published) (Katsonis & Lichtarge, 2014) and a normalized version of EA, as detailed in the CAGI5 special issue (Katsonis & Lictarge, 2019). EA estimates variant pathogenicity through evolutionary information by using an analytic equation. The components of the equation are the likelihood that the reference and alternative amino acids substitute to each other in numerous multiple sequence alignments (MSA), and the sensitivity of the protein function to residue changes calculated by the Evolutionary Trace method (Lichtarge, Bourne, & Cohen, 1996; Mihalek, Res, & Lichtarge, 2004) using MSA and phylogenic information. The Normalized EA predictions had the EA scores adjusted to the fraction of the isoforms affected by the mutation. See Supporting Methods for further details.
The Mooney-Radivojac 2018 team submitted predictions for single-nucleotide variants with MutPred2(Pejaver et al., 2017), a machine learning predictor that incorporates contextual features from protein sequence, conservation, and homology, along with features that encode mutation-induced changes in protein structure and function, as predicted by over 50 built-in machine learning models. The in-frame deletions were scored using MutPred-indel, a neural network-based pathogenicity prediction method that incorporates similar features, representative of protein structure, function, and conservation (unpublished).
The TransBioInf (Vall d’Hebron University Hospital) team submitted four sets of predictions, as detailed in the CAGI5 special issue (Padilla et al, 2019). TBI_1 and TBI_3 predict clinical significance with neural networks, given features including sequence alignment conservation and biophysical measures of the differences between the reference and alternative amino acids. TBI_2 and TBI_4 predict functional assay scores with multiple linear regression and a similar set of input features. Additionally, TBI_1 and TBI_2 incorporate estimates of the impact of the mutation on existing splice sites, while TBI_3 and TBI_4 do not.
Bologna Biocomputing submitted predictions with SNPs&GO (Calabrese, Capriotti, Fariselli, Martelli, & Casadio, 2009) and the Disease Index Matrix (Casadio, Vassura, Tiwari, Fariselli, & Luigi Martelli, 2011), both executed as published. SNPs&GO is a machine learning predictor that estimates pathogenicity from information on the Gene Ontology terms associated with the protein, as well as features describing amino acid conservation, the local sequence environment, and the evolutionary likelihood of the reference and alternative amino acids. The Disease Index Matrix (Pd) is a scale that associates each variant type (i.e., pair of wild type and variant residues) with the probability of being related to disease. The scale has been estimated with a statistical analysis of a large dataset of disease-related and neutral variations retrieved from UniProtKB and dbSNP databases.
AIBI directly predicted the probability of pathogenicity with weakly supervised linear regression, as detailed in the CAGI5 special issue (Cao et al., 2019) as the exact probabilities are not available for supervised machine learning. They used variants annotated with the class of pathogenicity in ClinVar, selected from MutPred2 15 features about molecular impacts upon variation, and designed parabola-shaped loss functions that penalize the predicted probability of pathogenicity according to its supposed class.
- Color Genomics submitted four sets of predictions with LEAP (Lai et al., 2018), a machine learning framework that predicts variant pathogenicity according to features including:
- population frequencies from gnomAD;
- function prediction from SnpEFF (Cingolani et al., 2012), SIFT (Ng & Henikoff, 2003), PolyPhen-2 (Adzhubei, Jordan, & Sunyaev, 2013) and MutationTaster2 (Schwarz, Cooper, Schuelke, & Seelow, 2014);
- splice impact estimation from Alamut (Interactive Biosoftware, Rouen, France) and Skippy (Woolfe, Mullikin, & Elnitski, 2010);
- indications of publications mentioning the variant and cancer associations from the subscription version of HGMD, indicating whether or not the variant is included in HGMD, whether or not it is associated with one or more articles curated by HGMD, and whether HGMD associates the variant with cancer (Stenson et al., 2017);
- aggregate information from individuals who have undergone genetic testing. This information consists of frequencies of cancer in the individuals tested, and within the individuals’ families (covering many cancer types, not simply breast and ovarian cancer), and co-occurrence of pathogenic variants in the same individual.
For reference, we analyzed the variants with the popular methods SIFT Version 5.2.2 (Ng & Henikoff, 2003), PolyPhen-2 Version 2.2 (Adzhubei et al., 2013) and REVEL (Version December 2018) (Ioannidis et al., 2016). SIFT applies substitution matrices to estimate the likelihood that a variant is pathogenic. PolyPhen-2 scores variants based on substitution matrices, evolutionary information, differences in the biophysical properties of the reference and alternative amino acids, functional residue and domain annotations, and predicted secondary structure. REVEL is a meta-predictor that estimates variant pathogenicity on the basis of several individual predictors, including SIFT, PolyPhen-2, MutPred and MutationTaster. We generated scores for these methods via the Ensembl Variant Effect Predictor (McLaren et al., 2016) in December 2018.
The ENIGMA Consortium incorporates “priors” of variant pathogenicity prediction as part of its variant interpretation process (Parsons et al, 2019). This pathogenicity prediction incorporates splice site impact, protein conservation, and expert knowledge, as detailed in a recent publication (Vallée et al, 2016; Tavtigian et al, 2008). Briefly, the impact of a variant on known splice sites and the likelihood of a variant introducing an ectopic splice site, are assessed by MaxEntScan (Yeo and Burge, 2004). To estimate the impact of missense variants, the variants are binned according to two factors: whether or not the variant is inside a clinically-important protein domain (Tavtigian et al, 2008); and ranges of substitution scores from Align-GVGD (http://agvgd.iarc.fr/), which estimates substitution likelihoods from alignments of orthologous protein sequences. The variants within each bin are assigned a probability of pathogenicity which was estimated from previous analyses of disease-causing variation (Easton et al, 2007). This approach shares features with some of the predictors in this experiment; the TransBioInf and LEAP methods use similar splicing information, and many methods use genomic conservation (which is related to the protein conservation implicit in Align-GVGD). However, these similarities are minor. Moreover, ENIGMA variant interpretation is based on many lines of evidence beyond pathogenicity prediction, namely several forms of clinical observation. None of the predictors use such lines information except for LEAP, and LEAP uses similar observations that were collected independently. The LEAP predictors reflect individuals who have been tested by Color Genomics (which was founded in 2013), while ENIGMA priors reflect individuals who had been tested prior to the mid-2000s. These sets of individuals could overlap, but this overlap is likely to be minor given the gap in time. In summary, the ENIGMA priors include similar information to some of the prediction methods, but the risk of bias from this similarity is minimal.
Assessment Methods
The 326 variants that were submitted for prediction analysis in the CAGI5 ENIGMA Challenge had all been interpreted by the ENIGMA Consortium, the ClinGen-approved expert panel for BRCA1/2 variant interpretation. Supporting Table S1 lists these variants along with the ENIGMA interpretation, and the predicted probability of pathogenicity and prediction standard deviation by all fourteen prediction methods and the three reference methods (See Supp Table S1).
Of the 326 BRCA1/2 variants that were shared with the CAGI5 prediction teams, ENIGMA interpreted 64 as Benign (Class 1), 240 as Likely Benign (Class 2), 5 as VUS (Class 3), 7 as Likely Pathogenic (Class 4), and 10 as Pathogenic (Class 5). As described earlier, these were the final interpretations by ENIGMA; the consortium had interpreted these variants when they were submitted to CAGI, and subsequently revised the interpretation of several variants according to new evidence that became available during the CAGI experiment. By IARC classification criteria (Plon et al, 2008), Benign variants include those with posterior estimates of pathogenicity of less than 0.001 in multifactorial estimation, while the threshold posteriors for Likely Benign variants is 0.049; the threshold for Likely Pathogenic variants is 0.95 while that for Pathogenic variants is 0.99. Two aspects of this statistical modeling are the evidence that the variant is damaging or increases disease risk, and the strength of the evidence. For example, suppose two variants have a similar impact on the protein function, but one is observed in a very few individuals while the second is observed much more frequently. The first variant might be classified, as Likely Pathogenic while the second is Pathogenic, because the smaller amount of evidence might not reach the threshold for Pathogenic classification. The amount of evidence on a variant is not relevant to pathogenicity prediction, while the predicted impact on function is. In our assessment, we grouped the Benign and Likely Benign variants together (assigning them a target probability of 0.025), grouped the Pathogenic and Likely Pathogenic variants together (assigning them a target probability of 0.975), omitted the VUS (each of which had been classified previously as Likely Benign but were reclassified based on additional evidence), and evaluated the prediction methods on their accuracy at predicting these target probabilities.
Most predictors submitted numerical predictions of pathogenicity ranging from 0.0 (predicted benign) to 1.0 (predicted pathogenic). One team submitted class labels (Class 1 - 5), which we translated to random probabilities selected from within ENIGMA’s posterior probability ranges (ENIGMA Consortium, 2017). Most predictors submitted standard deviations to accompany their estimated probabilities, and some submitted comments on their predictions.
We approached the assessment by computing several different summary statistics, as each can offer distinct insights. These included both threshold-dependent and threshold-independent metrics. The threshold-dependent metrics included:
Precision: the ratio of true positives to true and false positives, or variants accurately predicted as pathogenic as related to all pathogenic predictions;
Recall: the ratio of true positives to true positives and false negatives, or variants accurately predicted as pathogenic as related to all pathogenic variants (also known as sensitivity);
Accuracy: the ratio of true positive and true negative predictions to all true and false predictions, or the fraction of variants accurately classified as benign or pathogenic relative to the number of variants;
F1: the harmonic mean of precision and recall.
A contrast between Accuracy and F1 is that Accuracy reflects in part the number of True Negatives, benign variants predicted as such, while F1 does not. In cases such as this, with a large skew between the positive and negative sets, F1 is generally considered more meaningful. Accordingly, we leveraged F1 for threshold selection, and empirically selected one threshold for each predictor by sampling candidate thresholds across the prediction range and selecting the threshold that yielded the largest F1. We applied these thresholds in measuring Precision, Recall, and Accuracy. Supplementary Table 2 lists these thresholds along with these performance metrics.
We applied the following threshold-independent methods:
ROC AUC: Area under the ROC curve, which relates sensitivity (recall) to specificity (which in this context represents the fraction of benign variants correctly classified as benign). ROC AUC is a widely-used classification metric, which lends itself easily to probabilistic interpretation.
P/R AUC: Area under the Precision-Recall curve. This metric is similar to ROC AUC but is more effective for datasets such as this one with a large skew between positives and negatives.
RMSD: Root-mean-squared deviation describes the numerical distance between the prediction and its target value.
Pearson Correlation: This is a standard parametric correlation metric. Like RMSD, it tends to rewards predictions that are numerically close to the target value.
We also evaluated Kendall correlation, but found that for these data, it was redundant with ROC AUC (data not shown).
To evaluate significance in predictor performance, with confidence intervals, we performed 10,000 iterations of bootstrapping. For predictors that supplied standard deviations (as most did), in each bootstrapping iteration, we added a small amount of noise, sampled at random from a normal distribution with a mean of zero and the standard deviation supplied by the predictor, and measured all summary statistics on these data. We computed the standard deviation of these bootstrapped summary statistics. We considered the difference between two prediction methods to be significant if their summary statistics differed by more than one standard deviation. When a prediction was accompanied by a large standard deviation (which communicates a high degree of uncertainty), the bootstrapping communicated wide confidence intervals around the prediction metrics; small or no standard deviations translated to greater certainty around the summary statistics. Note that the bootstrapping was used only to estimate the error bars around the summary statistics, and the summary statistics themselves were computed on the actual prediction values.
One last component of the assessment was to identify a subset of variants that had proved to be challenging in general and analyze the commonalities of these variants. To identify these difficult variants, we computed the median predicted probability from all prediction methods and selected the pathogenic variants with lower median predictions and the benign variants with higher median predictions.
All of the software used in this assessment is publicly available at https://github.com/melissacline/CAGI5-BRCA-Assessment. Supporting Table S2 provides all of the assessment statistics for each method assessed (see Supp Table S2).
Results
We evaluated results from fourteen blind prediction methods and three reference methods. With few exceptions, the blind prediction methods reported values for the same variants, so their results can be compared directly. The three reference methods did not report values for many of these variants, and due to the number of missing values, their results should be viewed as only rough approximations of their performance. Figure 1 shows a dendrogram of the predictions and indicates the missing values. As shown, there were very few missing values. Almost all predictors submitted predictions on the same variants; the results were not confounded by missing values. The dendrogram shows that, unsurprisingly, different methods by the same teams tend to cluster together.
Figure 1:
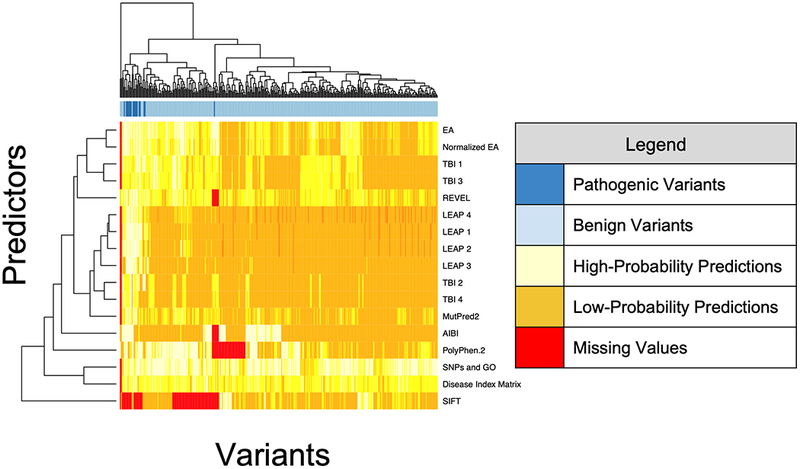
Dendrogram illustrating the predictions on all variants by all prediction methods
Supporting Figure S1 shows the distributions of probabilities estimated by each method and contrasts the probabilities for Benign and Likely Benign variants to those for Pathogenic and Likely Pathogenic variants (See Supp Figure S1). Supporting Figure S2 breaks this down further by separately showing predictions for the Benign, Likely Benign, Likely Pathogenic, and Pathogenic classes (Classes 1, 2, 4, and 5 respectively) (See Supp Figure S2). This figure illustrates that the predictions were not necessarily stronger for Pathogenic versus Likely Pathogenic variants, nor for Benign versus Likely Benign variants. This supports the assertion that the difference between Benign and Likely Benign, and between Pathogenic and Likely Pathogenic, reflects the strength of the clinical evidence rather than the expected functional impact of the variant, and is not relevant to this assessment.
Figure 2 summarizes the performance of the methods in terms of four metrics chosen as most illustrative: ROC AUC, P/R AUC, Precision and Recall. Supporting Table S2 lists the complete set of performance metrics (See Supp Table S2). While each metric has nuances, the rank order was largely consistent between the metrics. As a reflection of the overall performance accuracy, the strongest F1 accuracy was achieved by LEAP 4 at 0.83. In other words, on this particular dataset, the state of the art methods were correct in roughly four out of five cases, which illustrates that variant prediction remains a hard problem.
Figure 2:
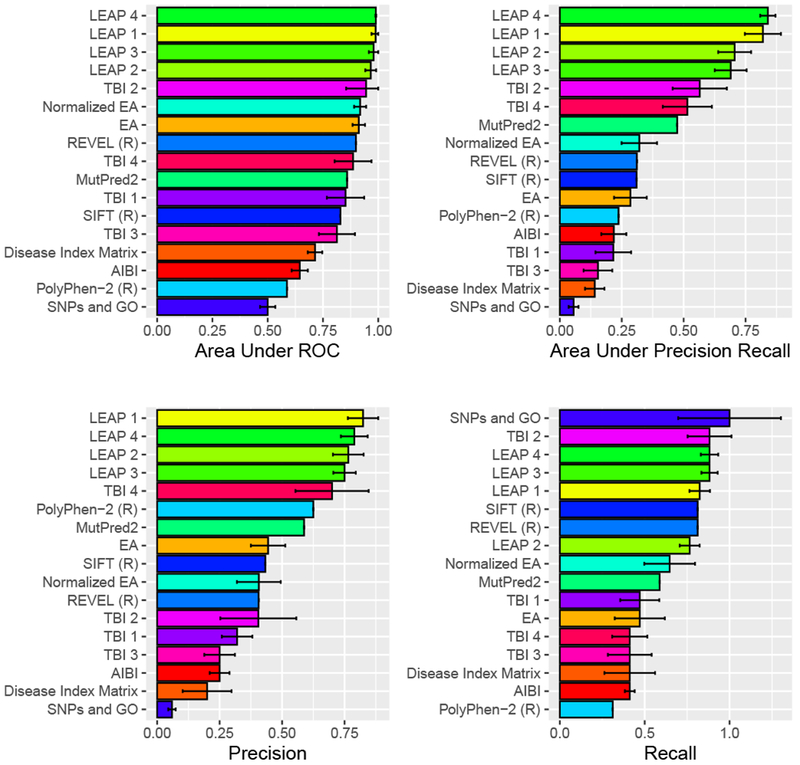
Shown is the performance of the fourteen blind prediction methods and three reference methods (denoted with R), for four selected performance metrics. The bar lengths and the error bars reflect the mean performance and standard deviation observed in random benchmarks, where each estimated probability was permuted according to standard deviation supplied by the predictor. No benchmarking was performed on methods for which the predictor supplied no standard deviation, or on the reference methods.
Overall, most methods fared better at predicting pathogenic variants as pathogenic than predicting benign variants as benign, as seen by comparing the Precision and Recall graphs in Figure 2. The LEAP methods were an exception, with strong precision as well as recall at the best empirically-selected threshold.
Interpretability was a design objective for the LEAP methods. LEAP 1 and LEAP 2, which are both regularized logistic regression methods, listed the input features which were most significant for each prediction. These include: scores from LRT, MutationTaster, SIFT, PolyPhen 2, and phastCons 100way vertebrate conservation. These features are also inputs to the reference method REVEL, which did not score quite as well on these variants. One possible explanation for LEAP’s performance advantage concerns differences in what the methods were trained to do. REVEL was trained to predict variants that are pathogenic in disease in general, and there may have been some variation in the methods that were used to interpret the variants in its training set. LEAP was trained to predict pathogenicity in cancer specifically and was trained on variants that were interpreted consistently, according to the ACMG Guidelines, by board-certified medical geneticists. In general, the methods that had been trained to identify disease variants in general did not fare as well on this challenge. This includes PolyPhen-2 and Disease Index Matrix. Arguably, LEAP addressed an easier problem by limiting its scope to cancer.
A second explanation is that features shared by LEAP and REVEL were necessary but not sufficient, and LEAP’s performance can be attributed to additional features. Important features that were distinct to LEAP included patient-derived information. Co-occurrence with known pathogenic variants was valuable in the accurate prediction of roughly one-third of the benign variants. Information on individuals who carry the variant and the frequency of cancer in these individuals and their families, was a strong predictor for a few difficult pathogenic variants (Lai et al, 2018). Since patient-level information informs clinical variant interpretation, including the ENIGMA variant interpretations, it comes as no surprise that it also valuable for improving variant pathogenicity prediction above that based on bioinformatic information alone.
Another form of information that benefitted LEAP were population frequencies from gnomAD. Higher minor allele frequencies within a distinct out-bred population is a characteristic of benign variants. While ENIGMA omitted variants with sufficiently high population frequencies to meet the ACMG Guidelines as benign, higher population frequencies still suggested benign variants. Since population frequency repositories are publicly-available, and growing in size and quality, their demonstrated value to LEAP’s performance suggests that they may be valuable to other methods as well.
While the variants interpreted by ENIGMA had no publicly-available interpretations at the time of the challenge, many of them proved to be in HGMD, where additional information is available to paid subscribers. The information in HGMD includes assigned categories (particularly, the “Disease-causing mutation” or DM category) and the presence of the variant in the literature. This proved to be a strong source of information for the methods that included data from HGMD in their input set (LEAP 1, LEAP 3, LEAP 4 and REVEL). In fact, for pathogenic variants, the data from HGMD was among the more important inputs to LEAP 1: the presence of the variant in the literature was instrumental in accurate prediction 15 of the 17 pathogenic variants, and the HGMD-assigned category of DM was instrumental in accurate prediction of 12 of these variants. There were two pathogenic variants for which LEAP 1 did not indicate HGMD features as key inputs (BRCA2 c.7819A>C and BRCA2 c.8975_9100del), and LEAP 1 mispredicted on these two variants. Since HGMD features papers on pathogenic or damaging variants, it makes sense that the mere fact that the mere mention a variant in HGMD is a strong predictor of pathogenicity. The rules of the CAGI experiment stipulate that each prediction team can use whatever information they have available, including private information. While the merits of subscription databases can be argued elsewhere, the scientific lesson is that the added information in these databases appears to be valuable. The lesson for the larger scientific community is that there exists additional data that could in theory be shared publicly (should its owner so decide), and sharing these data would advance the science of variant interpretation.
While most pathogenic variants were accurately predicted as pathogenic, there were a few that received lower predictions on average, and review of these variants was instructive. A number of mispredicted pathogenic variants were proximal to splice sites. Examples include BRCA1:c.4675G>A and BRCA1:c.4484G>C (which are adjacent to splice sites) and BRCA1:c.5144G>A and BRCA2:c.7819A>C (which are in close proximity to splice sites). The TransBioInf team further illustrated the impact of splicing information in the construction of their four methods, which themselves were a controlled experiment. Two included the predicted splicing impact, while two did not. In addition, the methods used two different learning frameworks: neural network prediction of clinical significance and multiple linear regression estimation of functional assay scores. As shown in Figure 3, including the predicted splicing impact improved prediction accuracy in both learning frameworks. Splicing-related information was also valuable to LEAP, such as the distance to the nearest splice site and exon length. Exon length is an interesting quantity that other researchers have also found to be valuable in such prediction; there appears to be valuable information encoded in exon length, beyond whether or not this length is of modulo three (Jagadeesh et al, 2019), to indicate information such as if the loss of the exon would introduce a frameshift. This further suggests that future method development may benefit from a greater emphasis on splicing information and might look beyond the splice sites themselves to additional regulatory features.
Figure 3:
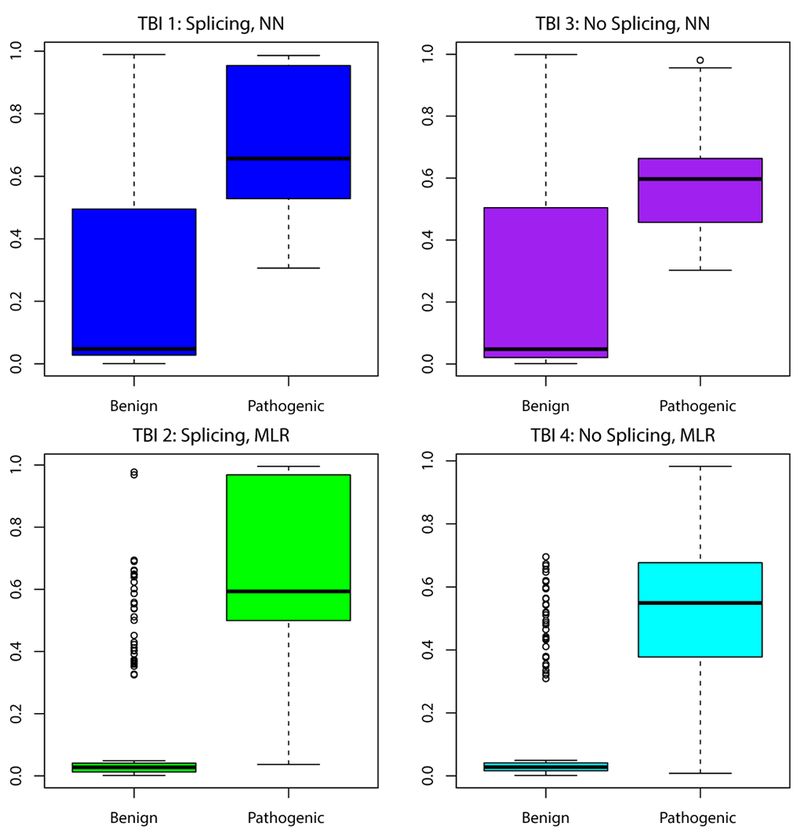
Of the four methods by the TransBioInf team, two (left) used predicted splicing information while two (right) did not. Further, the methods used two different learning frameworks and objective functions: neural network prediction of clinical significance (top), and multiple linear regression of functional assay scores (bottom). These boxplots show that in both architectures, including the splicing information improved prediction accuracy.
We reviewed a number of variants that were annotated as Benign or Likely Benign by ENIGMA, yet predicted as pathogenic by most predictors. Many of these variants were in conserved, clinically-important domains, yet in solvent-exposed, loop regions within these domains. Examples include BRCA1:c.5312C>G and BRCA2:c.8764A>G. Many predictors in this experiment use protein structure information, either directly or indirectly through predictors they incorporate, yet perhaps the protein structure information is being masked by the strong conservation signal.
Conclusion
In the CAGI5 ENIGMA Challenge, six teams submitted blind predictions with fourteen methods on a set of 326 BRCA1/2 exonic variants (mostly missense variants plus a few in-frame indels). These variants all had a minor allele frequency of less than 1%, and had recently been assessed for clinical significance by the ENIGMA Consortium using multifactorial likelihood analysis methodology. While this set of variants was skewed to the Benign and Likely Benign category, this skew is representative of the BRCA1/2 variants encountered in clinical practice. Yet it should be noted that given the small size of the data, and particularly the small number of pathogenic variants, this assessment should not be over-interpreted. For example, a predictor who predicts 100% sensitivity on a set of 17 pathogenic variants can still have a Wilson confidence interval as low as 82% (Wilson, 1927). Predicting the clinical impact of variants remains a hard problem. This experiment showed that the best methods achieved an F1 accuracy of just over 0.8, implying that variant prediction might be wrong one out of every five variants, at bestx. Variant prediction is not yet ready for clinical application in the absence of other data. With that said, this assessment may provide useful qualitative information.
A confounding factor in this experiment was that most of the pathogenic variants were in the subscription version HGMD and were predicted as pathogenic (“Disease-causing Mutations”) by HGMD. This information was only available to HGMD subscribers. In theory, paid HGMD subscriptions are available to anyone; in practice, the subscription fees are beyond the means of many academic labs and smaller institutions. This information was available to the LEAP methods (minus LEAP 2, which used publicly-available information only), and appears to have been instrumental in many correct pathogenic predictions by LEAP 1. Recognizing this potential bias, the results of this experiment should best be used as a motivation for methods development rather than a guide for direct clinical interpretation. Yet these results present a powerful lesson for the scientific community that there is private data that shows value in variant prediction. By extension, efforts to make such data more broadly-available are likely to advance the science of variant prediction.
Nonetheless, we learned several valuable lessons in this experiment, including the value of population frequency data. The LEAP methods leveraged population frequencies from gnomAD, which were instrumental in many accurate predictions. This is an information source was not used by most of the variant prediction methods, yet is available now and stands to improve as more population-scale sequencing studies become available (Lek et al, 2016).
While the pathogenic variants were few in number, they presented a clear story on the importance of splicing information. In the LEAP methods, splicing information as instrumental at predicting both pathogenic variants as pathogenic, and benign variants as benign. The results of the TransBioInf team demonstrated that splicing information improved prediction in two distinct architectures. When we assessed the pathogenic variants that were not predicted as pathogenic by many methods, many of them were proximal to splice sites. Our observations suggest that predictive methods should routinely include prediction of splicing impact. As our knowledge of splicing regulation improves, this improved knowledge may translate to further improvements in variant prediction methods.
The LEAP team from Color Genomics was able to draw upon their large database of patient-level clinical results, as well as a subscription to HGMD. They observed that the cancer frequencies of individuals and their families was a valuable input for some variants that would otherwise be difficult to classify. We observed that variant co-occurrence information was an important factor in their correctly predicting many of the benign variants as benign. It should come as no surprise that the types of information that are valuable for variant interpretation are also informative for predicting variant pathogenicity. This offers an optimistic note on how data sharing might improve the practice of variant prediction. While individual-level (or case-level) data is difficult to share for privacy reasons, case-derived information such as family history summary statistics and variant co-occurrences can be shared in ways that do not compromise patient privacy. As progress is made to share such information, those who benefit will include the developers and users of variant prediction methods.
Supplementary Material
Acknowledgements
We deeply thank the organizers of the CAGI5 experiment for their hard work and dedication. Blind prediction has had a profound benefit on the field of bioinformatics, yet it relies on the persistence and determination of the organizers, as well as many individuals who keep the infrastructure complete and intact.
The CAGI experiment coordination is supported by NIH U41 HG007346 and the CAGI conference by NIH R13 HG006650. MSC is supported by grant U54HG007990 from the National Human Genome Research Institute (genome.gov). OL and PK were supported by the NIH-GM079656 and the NIH-GM066099 grants. MTP is supported by a grant from Newcastle University, UK. ABS is supported by NMHRC Senior Research Fellowship ID 1061778. YC, YS and YS are supported by NIH R35GM124952. KP and OL are submitted by NIA R01-AG061105
Funding Information: Grant Numbers
NIH U41 HG007346
NIH R13 HG006650
NIH U54 HG007990
NIH GM 079656
NIH-GM 066099
NIH R35 GM124952.
NMHRC Senior Research Fellowship ID 1061778
NIA R01 AG061105
References
- Adzhubei I, Jordan DM, & Sunyaev SR (2013). Predicting functional effect of human missense mutations using PolyPhen-2 Current Protocols in Human Genetics / Editorial Board, Haines Jonathan L. … [et al. ], Chapter 7, Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese R, Capriotti E, Fariselli P, Martelli PL, & Casadio R (2009). Functional annotations improve the predictive score of human disease-related mutations in proteins. Human Mutation, 30(8), 1237–1244. [DOI] [PubMed] [Google Scholar]
- Cao Y, Sun Y, Karimi M, Chen H, Moronfoye O, & Shen Y (2019). Predicting Pathogenicity of Missense Variants with Weakly Supervised Regression. Human Mutation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadio R, Vassura M, Tiwari S, Fariselli P, & Luigi Martelli P (2011). Correlating disease-related mutations to their effect on protein stability: a large-scale analysis of the human proteome. Human Mutation, 32(10), 1161–1170. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, … Ruden DM (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly, 6(2), 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline MS, Liao RG, Parsons MT, Paten B, Alquaddoomi F, Antoniou A, … Spurdle AB (2018). BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genetics, 14(12), e1007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, Tavtigian SV, Monteiro AN, Iversen ES, Couch FJ, Goldgar DE (2007) A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. American Journal of Human Genetics 81(5):873–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENIGMA Consortium. (2017, June 29). ENIGMA BRCA1/2 Gene Variant Classification Criteria, Version 2.5.1. Retrieved from https://enigmaconsortium.org/wp-content/uploads/2018/10/ENIGMA_Rules_2017-06-29-v2.5.1.pdf
- Fokkema IFAC, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, & den Dunnen JT (2011). LOVD v.2.0: the next generation in gene variant databases. Human Mutation, 32(5), 557–563. [DOI] [PubMed] [Google Scholar]
- Goldgar DE, Easton DF, Byrnes GB, Spurdle AB, Iversen ES, Greenblatt MS, & IARC Unclassified Genetic Variants Working Group. (2008). Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Human Mutation, 29(11), 1265–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro ANA, Tavtigian SV, & Couch FJ (2004). Integrated Evaluation of DNA Sequence Variants of Unknown Clinical Significance: Application to BRCA1 and BRCA2. American Journal of Human Genetics, 75(4), 535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Kosary CL, … Cronin KA (Eds.). (2017, April). SEER Cancer Statistics Review, 1975-2014 (Version based on November 2016 SEER data submission, posted to the SEER web site). SEER. Bethesda, MD: National Cancer Institute; Retrieved from https://seer.cancer.gov/csr/1975_2014/ [Google Scholar]
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, … Sieh W (2016). REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. American Journal of Human Genetics, 99(4), 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagadeesh KA, Paggi JM, Ye JS, Stenson PD, Cooper DN, Bernstein JA, Bejerano G. (2019). S-CAP extends pathogenicity prediction to genetic variants that affect RNA splicing. Nature Genetics. doi: 10.1038/s41588-019-0348-4. [DOI] [PubMed] [Google Scholar]
- Katsonis P, & Lichtarge O (2019). CAGI5: Objective Performance Assessments of Predictions Based on the Evolutionary Action Equation. Human Mutation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsonis P, & Lichtarge O (2014). A formal perturbation equation between genotype and phenotype determines the Evolutionary Action of protein-coding variations on fitness. Genome Research, 24(12), 2050–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolor K, Chen Z, Grosse SD, Rodriguez JL, Green RF, Dotson WD, … Khoury MJ (2017). BRCA Genetic Testing and Receipt of Preventive Interventions Among Women Aged 18–64 Years with Employer-Sponsored Health Insurance in Nonmetropolitan and Metropolitan Areas - United States, 2009–2014. Morbidity and Mortality Weekly Report. Surveillance Summaries , 66(15), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips K-A, Mooij TM, Roos-Blom M-J, … Olsson H (2017). Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA: The Journal of the American Medical Association, 317(23), 2402–2416. [DOI] [PubMed] [Google Scholar]
- Lai C, O’Connor R, Topper S, Ji J, Stedden W, Homburger J, … Mishne G (2018, February). Using Machine Learning to Support Variant Interpretation in a Clinical Setting. Presented at the Advances in Genome Biology and Technology (AGBT) Retrieved from https://static.getcolor.com/pdfs/research/Color_AGBT_PH_Poster_2018.pdf [Google Scholar]
- Landrum MJ, & Kattman BL (2018). ClinVar at five years: Delivering on the promise. Human Mutation, 39(11), 1623–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, … Exome Aggregation Consortium. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtarge O, Bourne HR, & Cohen FE (1996). Evolutionarily conserved Galphabetagamma binding surfaces support a model of the G protein-receptor complex. Proceedings of the National Academy of Sciences of the United States of America, 93(15), 7507–7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, … Cunningham F (2016). The Ensembl Variant Effect Predictor. Genome Biology, 17(1), 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalek I, Res I, & Lichtarge O (2004). A family of evolution-entropy hybrid methods for ranking protein residues by importance. Journal of Molecular Biology, 336(5), 1265–1282. [DOI] [PubMed] [Google Scholar]
- Ng PC, & Henikoff S (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Research, 31(13), 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla N, Moles-Fernández A, Riera C, Montalban G, Özkan S, Ootes L, Bonache S, Díez O, Gutiérrez-Enríquez S, & de la Cruz X (2019). BRCA1- and BRCA2-specific in silico tools for variant interpretation in the CAGI 5 ENIGMA challenge. Human Mutation [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons MT, Tudini E, Li H, Goldgar DE, & Spurdle AB (2019). Largescale multifactorial likelihood analysis of BRCA1 and BRCA2 variants within ENIGMA: A resource to inform qualitative classification criteria. Human Mutation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejaver V, Urresti J, Lugo-Martinez J, Pagel KA, Lin GN, Nam H-J, … Radivojac P (2017). MutPred2: inferring the molecular and phenotypic impact of amino acid variants. 10.1101/134981 [DOI] [PMC free article] [PubMed]
- Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, … IARC Unclassified Genetic Variants Working Group. (2008). Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 29(11):1282–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … ACMG Laboratory Quality Assurance Committee. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 17(5), 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, & Seelow D (2014). MutationTaster2: mutation prediction for the deep-sequencing age. Nature Methods, 11(4), 361–362. [DOI] [PubMed] [Google Scholar]
- Spurdle AB, Healey S, Devereau A, Hogervorst FBL, Monteiro ANA, Nathanson KL, … ENIGMA. (2012). ENIGMA--evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Human Mutation, 33(1), 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, … Cooper DN (2017). The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Human Genetics, 136(6), 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Byrnes GB, Goldgar DE, & Thomas A (2008). Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Human Mutation, 29(11), 1342–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallée MP, Di Sera TL, Nix DA, Paquette AM, Parsons MT, Bell R, … Tavtigian SV (2016). Adding In Silico Assessment of Potential Splice Aberration to the Integrated Evaluation of BRCA Gene Unclassified Variants. Human Mutation, 37(7), 627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EB (1927). “Probable inference, the law of succession, and statistical inference”. J Am Stat Association. 22:209–212. [Google Scholar]
- Woolfe A, Mullikin JC, & Elnitski L (2010). Genomic features defining exonic variants that modulate splicing. Genome Biology, 11(2), R20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Burge CB. (2004) Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 11(2–3):377–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.