

Abstract
Thermodynamic stability is a fundamental property shared by all proteins. Changes in stability due to mutation are a widespread molecular mechanism in genetic diseases. Methods for the prediction of mutation-induced stability change have typically been developed and evaluated on incomplete and/or biased data sets. As part of the Critical Assessment of Genome Interpretation (CAGI), we explored the utility of high-throughput variant stability profiling (VSP) assay data as an alternative for the assessment of computational methods and evaluated state-of-the-art predictors against over 7,000 non-synonymous variants from two proteins. We found that predictions were modestly correlated with actual experimental values. Predictors fared better when evaluated as classifiers of extreme stability effects. While different methods emerged as top-performers depending on the metric, it is non-trivial to draw conclusions on their adoption or improvement. Our analyses revealed that only 16% of all variants in VSP assays could be confidently defined as stability-affecting. Furthermore, it is unclear to what extent VSP abundance scores were reasonable proxies for the stability-related quantities that participating methods were designed to predict. Overall, our observations underscore the need for clearly defined objectives when developing and using both computational and experimental methods in the context of measuring variant impact.
Keywords: CAGI, variant stability profiling, VAMP-seq, Phosphatase and tensin homolog, PTEN, Thiopurine S-methyl transferase, TPMT
Introduction
The thermodynamic stability of a protein is the net balance of forces determining whether a protein is in its native folded conformation or is denatured/unfolded (Gromiha, 2010). Mutations that lead to changes in protein stability are important mechanisms of disease and have thus been subject to intensive research (Pejaver, et al., 2017b; Wang and Moult, 2001; Yue, et al., 2005). Experimental techniques for the identification of destabilizing and/or stabilizing mutations include circular dichroism (Greenfield, 2006), differential scanning calorimetry (Bruylants, et al., 2005), and fluorescence and UV absorbance spectroscopy (Pace and Scholtz, 1997). Most commonly, these techniques are used to establish stability of a wild-type protein and its in vitro-mutated version. The difference between these is computed as the signed change in Gibbs free energy (unfolding energy). That is, (1) if this difference is negative, i.e. mutant unfolding energy is higher than wild-type, the mutation is stabilizing, (2) if it is zero, the mutation has no effect, and (3) otherwise it is destabilizing. While these techniques provide valuable insights, they do not scale up to a large number of variants per protein across many different proteins. Thus, two complementary approaches have been explored: (1) high-throughput mutagenesis, including denaturation experiments or the profiling of other indicators of protein stability, and (2) computational methods for prediction of stability changes due to mutation.
Computational methods are broadly based on the features of protein structure (e.g. FoldX (Schymkowitz, et al., 2005) or PoPMuSIC (Dehouck, et al., 2011)) or sequence (e.g. INPS (Fariselli, et al., 2015) or EASE-MM (Folkman, et al., 2016)). Some methods, for example I-Mutant (Capriotti, et al., 2005) and INPS-MD (Savojardo, et al., 2016), among others, can run in both sequence and structure modes. Integrative methods combining features from sequence, structure, experimental details, and/or conservation/evolutionary history have also been developed. Methods also vary in provided output; e.g. FoldX and PoPMuSIC predict actual values of the change in Gibbs free energy – a regression task. On the other hand, e.g. I-Mutant and MUpro (Cheng, et al., 2006), can frame the prediction problem as a classification task (destabilizing or stabilizing).
Due to their ease of use, computational methods play an important role in advancing our understanding of protein folding, stability, and function. However, the ability of these methods to perform their assigned tasks has yet to be thoroughly evaluated. In fact, most methods are developed and evaluated using data from the same handful of sources, e.g. ProTherm (Kumar, et al., 2006) or the Protein Mutant Database (Kawabata, et al., 1999). As an ideal gold-standard, large-scale experimental analysis of protein variants’ stability is still lacking. Even independent method assessments have relied on newer versions of the same training databases for their evaluations (Khan and Vihinen, 2010; Potapov, et al., 2009). Therefore, it remains unclear how accurate the predictions made for larger data sets spanning thousands of mutations and proteins are.
High-throughput methods to measure mutation-induced stability changes have proven insightful. Protein alanine-scans, for example, included substituting amino acids at a range of protein positions by alanine one at a time, followed by denaturation experiments with free energy change measurements (Blaber, et al., 1995; Milla, et al., 1994). These experiments, although painstaking and limited in variability, have been particularly useful in identifying critical residues (stability, and otherwise) (Morrison and Weiss, 2001). Other approaches have opted to measure more accessible proxies of stability changes; e.g. lowered steady-state abundance of a protein may reflect increased degradation due to instability. For instance, Yen et al. demonstrated that the global protein stability (GPS) profiling technique was able to detect the increase in stability due to the p.Thr380Ala mutation in cyclin E (Yen, et al., 2008). Combined with deep mutational scanning approaches, GPS profiling could facilitate stability measurements of all possible protein variants (Fowler and Fields, 2014).
The recently developed multiplexed variant stability profiling (VSP) assay (Matreyek, et al., 2018) uses the fluorescent reporter system (EGFP; to measure the protein variant abundance) and mCherry (expressed co-transcriptionally or co-translationally from the same construct) on cells carrying mutated proteins. The per-cell EGFP/mCherry ratios are calibrated with respect to cells carrying the wild-type protein or known destabilized proteins to define a (high to low, stable to unstable) range. Cells are sorted into bins according to their EGFP/mCherry ratios and sequenced to quantify each variant’s frequency in each bin. A variant stability score is calculated on the basis of these bin frequencies, with 0=unstable, 1=wild-type, and >1=more stable than wild-type protein. A key advantage of VSP assays is that they are applicable to a wide range of proteins and can measure effects of all possible mutations at all positions. They particularly help overcome issues of overrepresentation of to-alanine (Magliery, 2015) and destabilizing mutations (Pucci, et al., 2018), the underrepresentation of “inverse” mutations (Thiltgen and Goldstein, 2012), and general errors in curation (Yang, et al., 2018). Thus, VSP assays are an attractive alternative to existing data sources for the development and validation of computational methods.
One of the main objectives of the Critical Assessment of Genome Interpretation (CAGI) is to objectively assess computational methods for the prediction of the impact of genetic variation through task-specific challenges in community-wide experiments. The recent CAGI5 edition included a prediction challenge, in which variants from multiplexed VSP assays of two biomedically important proteins were made available to the CAGI participants: phosphatase and tensin homolog (PTEN) and thiopurine S-methyl transferase (TPMT). PTEN is a ubiquitously expressed protein that dephosphorylates phosphatidylinositol (3,4,5)-triphosphate (PIP3), a secondary messenger molecule promoting cell growth and survival (Lee, et al., 2018). PTEN missense mutations have been observed in cancers, including glioma, endometrial cancer, and melanoma. Germline variation in PTEN results in PTEN Hamartoma Tumor Syndromes (PHTS), a group of developmental abnormalities (Hobert and Eng, 2009) and is associated with autism spectrum disorders (Butler, et al., 2005). TPMT is a key enzyme in the metabolism of thiopurine drugs, which suppress the immune system and have been used to treat acute-lymphoblastic leukemia, autoimmune diseases, and to prevent organ rejection after transplants (Burchenal, et al., 1953; Weinshilboum, 2001). Thiopurine overdose may lead to treatment interruptions that cause poor health outcomes and, in some cases, a life-threatening myelosuppression and hepatotoxicity (Weinshilboum, 2001).
The PTEN and TPMT variants were provided to CAGI participants, who were allowed to use any data and resources to make predictions of the associated VSP scores. In this study, we performed the assessment of these predictions using these experimental scores as the ground truth. Furthermore, we used these variant sets to evaluate existing methods (not submitted to CAGI), which have emerged as de facto standards. Importantly, we also provide some discussion on the topic of the value of gold-standard experiments in evaluation of computational methods.
Materials and Methods
Data sets.
Multiplexed variant stability profiling (VSP) assays for PTEN and TPMT yielded variant stability scores for 7,954 protein variants (4,002 PTEN variants and 3,952 TPMT variants; Table 1). For most variants (99%), reference values were obtained by calculating the mean of these scores, along with the standard deviation, and lower and upper confidence interval (CI) limits. For 73 variants (1%), no replicate data was available so the scores from the lone experiments were included “as is”.
Table 1:
Summary of data set pre-processing and the resulting variant numbers
| Data set | #variants (PTEN) | #variants (TPMT) | #variants (Total) | Range of exp. values [min, max] |
|---|---|---|---|---|
| Original data set | 4,000 | 3,952 | 7,952 | [−0.975, 2.198] |
| Nonsense | 120 | 196 | 316 | [−0.975, 2.198] |
| Negative score | 20 | 143 | 163 | [−0.975, 0.000] |
| Final data set | 3,860 | 3,613 | 7,473 | [0.001, 2.198] |
| Effect | 2,448 | 1,853 | 4,301 | [0.001, 2.198] |
| Stabilizing | 852 | 787 | 1,639 | [0.841, 2.198] |
| Destabilizing | 1,596 | 1,066 | 2,662 | [0.001, 1.338] |
| Wild-Type | 539 | 698 | 1,237 | [0.900, 1.099] |
| Synonymous | 144 | 129 | 273 | [0.130, 2.010] |
| Classification data set | 2,987 | 2,551 | 5,538 | [0.001, 2.198] |
The stability scores from VSP assays are typically calibrated according to the scores for synonymous variants (no effect) on one extreme and nonsense variants on the other extreme. Here, they ranged between −0.975 and 2.198, with the negative values arising from the exclusion of nonsense variants near the termini in score calibration. Initially, variants with negative scores were deemed to be outside the interpretable range and were, therefore, excluded. Note that an alternative approach, in which scores for these variants were set to zero, yielded similar trends (Supp. Table S1). We also excluded nonsense variants from further consideration because their destabilizing effects are expected to arise largely from protein truncation. Note that synonymous variants in our set were described as wild-type to wild-type substitutions of amino acids as opposed to actual DNA-level mutations. As such they were retained for predictor evaluations. The Final reference data set used for evaluation consisted of 3,860 and 3,613 missense variants in PTEN and TPMT, respectively (Table 1).
Fitting the distribution of variant scores.
Given the bimodal nature of the distribution of non-synonymous variants (see below), we assumed a Gaussian mixture model (GMM) and estimated the parameters of two possible non-synonymous variant score distributions (effect vs. no effect) using a positive-unlabeled learning approach. In this approach, the experimental values for the synonymous variants contributed to learning the parameters for the synonymous-like (no effect) variants in the non-synonymous distribution. More specifically, an extension of the expectation maximization algorithm that estimates parameters of the mixing components from two mixtures, referred to as the multi-sample Gaussian mixture model (MSGMM), was used. We note that here, the synonymous distribution was also assumed to be a mixture of correctly and incorrectly labeled variants (Jain, et al., 2016).
Predictors and baseline methods.
The rules of the challenge were as follows: predictors assigned continuous scores to each variant such that a score=0 indicates an unstable protein (destabilizing), a score=1 indicates wild-type stability (no effect), and a score>1 indicates stability greater than that of wild-type (stabilizing). Participating groups were allowed up to six submissions, and were asked to provide brief descriptions of the methods underlying each of their submissions (Supplementary Methods). We note that although the true variant stability scores are now published (Matreyek, et al., 2018), they were not made available to the participating groups until after the challenge was closed.
For the purpose of comparison, three general-purpose variant effect prediction methods were also included in this evaluation: SIFT (Ng and Henikoff, 2001), SNAP (Bromberg and Rost, 2007) and PolyPhen-2 (Adzhubei, et al., 2010). While these were not developed for the specific task of predicting effects of variants on protein stability, the VSP assay itself was originally developed as a general-purpose technique for the high-throughput identification of deleterious and benign variants and may thus be somewhat comparable with the effects these methods were developed to predict. For the baseline methods, score transformation was carried out (heuristically) to match the expected score range as follows (rounded to three decimal points):
Additionally, Envision, a machine learning model trained on large-scale mutagenesis data sets (similar to this study) to predict the impact of missense variants on protein function, was also included as a newer baseline method (Gray, et al., 2018). No score transformation was necessary for Envision. Finally, a naïve random predictor was implemented by randomly reassigning all stability scores from the assay for a given protein to all variants in that protein. This was motivated by the need for a baseline when comparing classification accuracies on imbalanced data sets (see below).
Evaluation design.
The continuous nature of the experimental and prediction scores motivated the need for three different evaluation approaches. First, methods were evaluated on their ability to predict the actual experimental values of the Final set variants. Specifically, we calculated the Pearson and Spearman correlation coefficients (ρ) between the predicted and experimental values (Rosner, 2015). Second, the challenge was treated as a three-class classification task, in which methods were evaluated on their ability to assign variants to one of the three classes – most destabilizing, wild-type, and most stabilizing. These classes were defined by thresholding the experimental value distribution corresponding to the bottom 25% (most destabilizing) and top 25% (most stabilizing) of all values. In other words, a variant was treated as most destabilizing if the value of its lower CI limit was less than 0.468 and most stabilizing if the value of its upper CI limit was greater than 1.200. We note that these criteria were generous and allowed for variants with borderline scores but high experimental variability to be assigned to the extreme classes. Variants with scores in the range [0.9, 1.1] were assigned to the wild-type group. These three classes formed the Classification data set (Table 1). Finally, the challenge was also treated as a binary classification task, in which methods were assessed in terms of their ability to correctly classify variants into two classes, effect or no effect, by combining the destabilizing and stabilizing variants into a single effect class. Classification accuracy (the fraction of correct class predictions) was chosen as the evaluation metric in both tasks.
Results
Eight groups participated in the challenge and 16 submissions were received; while four of the groups contributed only one submission, one group submitted five predictions. Methods relied on combinations of features derived from the proteins’ sequence, structure, local physicochemical environment, and evolutionary history, integrated via a variety of algorithms and/or machine learning models (Table 2; Supplementary Methods). The practical predictor-distinguishing aspects included training protocols, output calibration, the choice of underlying methods (e.g. to build alignments), etc. Some submissions did not make predictions for all reference set variants (Supp. Figure S1).
Table 2:
Summary of prediction methods submitted by participating groups.
| Prediction method | Design objective | Approach | Features | Customization | Reference |
|---|---|---|---|---|---|
| Group 1 | |||||
| Submission 1 | Predict free energy change | Support vector machine | Sequence + Evolutionary + Structure | Calibrated to known variants in TPMT Sub. 1 + calibrated to known variants in PTEN Sub. 2 + applied penalty for specific positions | Fariselli, et al. (2015); Savojardo, et al. (2016) |
| Submission 2 | |||||
| Submission 3 | |||||
| Group 2 | |||||
| Submission 1 | Classify into three classes of stability change | Random forest | Sequence + Evolutionary | Transformed output to match expected score range | In review |
| Group 3 | |||||
| Submission 1 | Predict free energy change | Support vector machine | Structure | Transformed output to match expected score range | Yang, et al. (2013) |
| Group 4 | |||||
| Submission 1 | Predict change in protein flexibility | Coarse grain molecular mechanics | Structure | Transformed output to match expected score range | NA |
| Group 5 | |||||
| Submission 1 | Predict effect on fitness | Evolutionary action equation | Evolutionary | Down-weighed predictions according to solvent accessibility No down-weighing; transformed output to match expected score range | Katsonis and Lichtarge (2014) |
| Submission 2 | |||||
| Group 6 | |||||
| Submission 1 | Predict protein activity levels | Support vector machine ensemble | Predictions from pathogenicity predictors | ||
| Submission 2 | Predict free energy change | Conformational sampling (Rosetta) | Structure | ||
| Submission 3 | Classify into two classes of stability change + Sub. 1 | Knowledge-base (SNPs3D) for certain variants + Sub. 1 for rest | Structure + Features from Sub. 1 | Calibrated to known variants; transformed output to match expected score range | Yin, et al. (2017); Yue, et al. (2005) |
| Submission 4 | Sub. 1 + Sub. 2 | Sub. 1 for core and Sub. 2 for surface residues | Features from Sub. 1 + features from Sub. 2 | ||
| Submission 5 | Sub. 1 + Sub. 3 | Sub. 1 for core and Sub. 3 for surface residues | Features from Sub. 1 + features from Sub 3 | ||
| Group 7 | |||||
| Submission 1 | Predict free energy change | Empirical forcefield (FoldX) Conformational sampling (Rosetta) | Structure | Transformed output to match expected score range |
Schymkowitz, et al. (2005) Kellogg, et al. (2011) |
| Submission 2 | |||||
| Group 8 | |||||
| Submission 1 | Predict free energy change | Support vector machine | Sequence + Evolutionary | Transformed output to match expected score range | Folkman, et al. (2016) |
Distribution of experimental scores highlights difficulty in differentiating variant effect.
The destabilizing variants in our set were fairly evenly distributed throughout the protein structures, with modest preference towards buried regions (Supp. Figure S2). For any given position, stabilizing effects were infrequent.
We explored the distribution of the experimentally-derived VSP scores for non-synonymous variants in the Final set (Figure 1A, blue). This distribution was arguably bimodal (peaks at ~0.3 and ~0.9) and asymmetric, i.e. there were more destabilizing than stabilizing variants – an observation in line with previous deep mutational scanning studies (Araya, et al., 2012). Notably, a large number of the apparently neutral variants scored between 0.7 and 1 (just slightly lower than wild-type stability), suggesting either a preponderance of mild destabilizing effects in the reference data set or, more likely, a limit to experimental resolution. The latter was also in line with the observations made for the nonsense and synonymous variant distributions; these fit expectations from VSP assay calibration (peaks at ~0 and ~1, respectively), but had exceedingly high variances (Figure 1A, red and yellow, respectively). The distribution overlaps highlight a considerable experimental uncertainty in calling variants “effect” and “no effect”.
Figure 1. Experimental VSP score distributions.
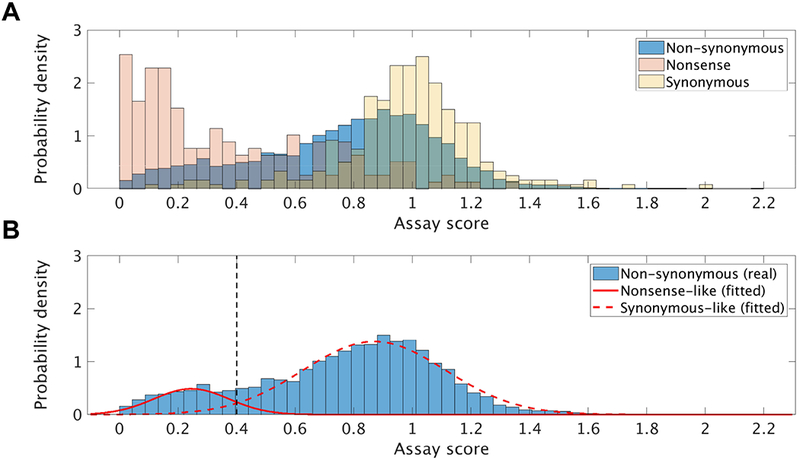
For consistency with the MSGMM algorithm, probability density functions were plotted instead of raw counts or frequencies. (A) Distribution of all variant types (bar heights correspond to the frequency of variants from each bin divided by the bin-width, 0.044) and (B) Gaussians fitted to the non-synonymous distribution using the MSGMM algorithm (with parameters learned from the synonymous distribution). The black dashed line corresponds to the estimated threshold that separates likely stability-impacting variants from synonymous-like ones.
To estimate the proportion of variants that are most likely to affect stability, we modeled the non-synonymous distribution as a mixture of two Gaussian distributions (Figure 1B) – a “synonymous-like” (closer to score=1) and “nonsense-like” (closer to score=0). We observed that the scores of 84.3% of all non-synonymous variants fell within the estimated score range for synonymous variants (synonymous-like, mean=0.87 and standard deviation=0.24). Note that over 99% of the synonymous variants are also in this range. It remains unclear whether these results emerge from experimental noise or real (mild) effects of synonymous variants; i.e. in depth experimental follow-up is necessary to clarify these observations. The remaining 15.7% variants represented the most likely “effect” variants (score≤0.40, mean=0.25, standard deviation=0.13; Figure 1B).
Prediction score distributions differ from experimental ones.
We compared the scores from three popular (baseline) prediction methods chosen for this assessment (SIFT, SNAP, and PolyPhen-2) to the experimental measurements. The distributions of the three methods’ scores were clearly bimodal, with peaks closer to the extremes and a relatively flat middle (Figure 2). This is unsurprising as these methods were trained on binary class labels of variant impact, not specifically continuous stability changes in this task. Thus, for SNAP, for example, the score is a measure of prediction accuracy, which correlates with effect severity, but is not meant to replace it (Bromberg, et al., 2009; Mahlich, et al., 2017). We also considered an additional baseline model trained on deep mutational scanning data sets called Envision. While the shape of Envision’s score distribution was similar to that of the experimental data, no scores below 0.4 were observed.
Figure 2. Distribution of the baseline method predictions.
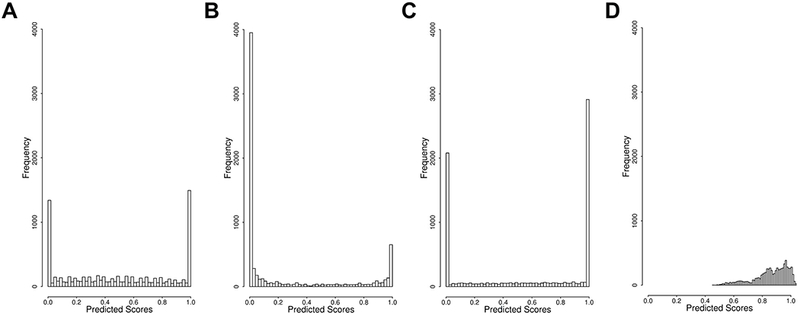
SIFT, SNAP and PolyPhen-2 predictions, standardized to fit the [0,1] score range, and Envision predictions (without standardization).
Prediction score distributions of the challenge submissions varied. Broadly, the distributions could be grouped into four types: (1) similar to the experimental distribution with an additional peak close to 0, i.e. excess destabilizing mutations (Group 1, Group 6-Submission 2 and Group 7), (2) normal or skew-normal with a peak close to 1, i.e. mostly no-effect (Groups 4 and 8), (3) distributions with peaks set around specific thresholds, e.g. 0, 0.5, and/or 1 (Group 2 and Group 6-Submission 3), and (4) others (Groups 3 and 5 and the remaining Group 6 submissions).
Predictions correlate modestly with experimental values.
We first assessed the ability of computational methods to predict the VSP experimental scores. This was essentially a regression task, in which both the predictors and the assays assigned real numbers greater than or equal to zero to each variant. We used (value-based) Pearson and (rank-based) Spearman correlation coefficients as measures of agreement between predictions and the ground truth. We also calculated correlations between predictors because we observed that predictors generally rely on a similar pool of features, varying more explicitly in the technical aspects of their development, e.g. prediction algorithms, training sets, and use of feature selection techniques. We were, thus, particularly interested in comparing and contrasting method construction and predictive performance.
Positive non-zero correlations between predicted and experimental values were observed for all but one method, clearly exceeding that of the random predictor (Figures 3A and 3B). However, there was no method with a correlation coefficient that exceeded 0.5. Among the baseline methods, SIFT performed the best. However, a number of submissions performed better than SIFT, suggesting recent improvements in the state-of-the-art. Interestingly, while these submissions generally repurposed previously developed methods, they varied substantially in their approaches, e.g. support vector regression models using sequence- and structure-based features, evolutionary analytical models, and protein-folding models, among others. Specifically, the top-performing methods (from Groups 5 and 6) either explicitly or implicitly accounted for the solvent accessibility of residues in the 3D structure, and down-weighed the effects of surface variants more than of those that were buried.
Figure 3. Performance of predictors in predicting experimental variant scores.
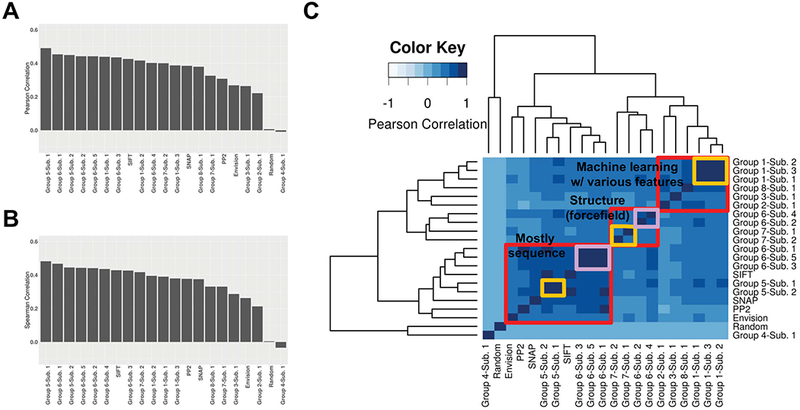
Method performance was measured using (A) Pearson correlation and (B) Spearman correlation. In (C) Correlation (Pearson) between methods was also computed. Red boxes indicate clusters as defined by the dendrogram in the figure. Pink and yellow boxes indicate sub-groupings of submissions from the same participating group, within each cluster.
We next calculated all-pairwise correlations between predictors. Correlations between methods, even across groups, were comparable to or higher than predictor correlation to experimental scores, e.g. Pearson ρ=0.740 between Group 8–Submission 1 and Group 1-Submission 1 and ρ=0.807 between Group 6-Submission 1 and PolyPhen-2. These results reflect similarities in training data and method development. However, the mean correlation coefficient among all pairs of predictors (ρ=0.416) was not significantly different from the mean correlation coefficient between predicted and experimental values (ρ=0.371, two-sample t-test P = 0.1619). Correlation of scores grouped predictors into three clusters that could be broadly classified as: (1) methods that use direct or derived sequence information, (2) structure-based methods, using stability prediction (FoldX) and protein folding (Rosetta) methods, and (3) integrative methods using machine learning (Figure 3C). Curiously, the first cluster contained the top-performing methods (as per Figure 3A,B) as well as the baseline methods, although SNAP, PolyPhen-2 and Envision were arguably outliers.
Method performance changes when variant effect prediction is a classification task.
An alternative view of our prediction task considers the effect of each variant as belonging to one of three possible classes. Variants with experimental scores at ~0 can be thought of as destabilizing, those with scores at ~1 as wild-type (no effect), and those with scores >1 as stabilizing. Variants that did not fit the class definition criteria were excluded from consideration in this evaluation (Methods; Table 1, Classification set). Predictors were assessed on their ability to accurately predict the class of the variant effect, regardless of prediction proximity to the experimental score. We further simplified the prediction task by combining the destabilizing and stabilizing variants into one class so that the methods were assessed in a familiar binary, effect or no-effect, setting.
We observed that in the three-class setting, no prediction method exceeded 50% overall accuracy (Figure 4A). The binary class setting was easier with accuracy reaching 70% -- a number much more in line with the expected performance of predictors designed to make extreme class predictions (Figure 4B). Here, a different set of method leaders emerged in both three- and two-class settings (Groups 2 and 3); these were machine learning methods integrating multiple sequence and structure features. Furthermore, Group 5-Submission 1 (the weighted evolutionary action method) was the best submitted method in terms of accuracy-to-correlation tradeoff (the mean of the Pearson correlation co-efficient and accuracy).
Figure 4. Performance of predictors in assigning a class to each variant.
Performance in the (A) three-class and (B) two-class setting. Numbers next to each point indicate the submission number.
Prediction performance varies between PTEN and TPMT.
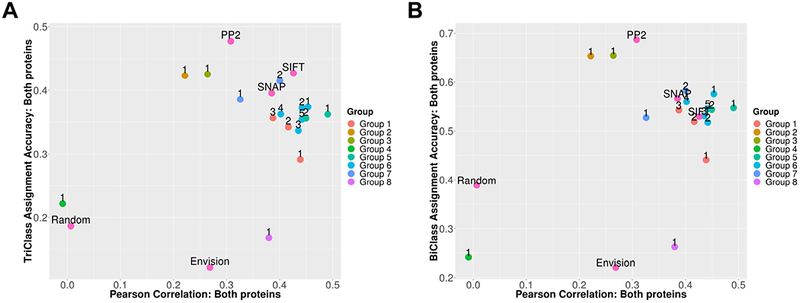
Finally, we investigated whether there were any differences in predictive performance between the two proteins in this study. For simplicity, we restricted our analyses to calculating Pearson correlation coefficients for the regression task and binary class accuracy for the classification task (Figure 5). However, results for Spearman correlation coefficients were similar (Supp. Table S2). We observed that for some methods there indeed were differences between PTEN and TPMT. For instance, Group 7-Submission 1 and Group 5-Submission 1 exceeded a Pearson ρ of 0.5 for PTEN, but no methods achieved this in TPMT. Furthermore, only two of the top-five performing methods for PTEN also featured in the top-five for TPMT (Group 5-Submission 1 and Group 6-Submission 1). In some cases, the differences in correlation were drastic, e.g. Group 7-Submission 1, PTEN ρ=0.531 and TPMT ρ=0.310 (Figures 5A and 5B). Differences persisted for binary classification (Figures 5C and 5D). Notably, submissions from Group 7 were the best in terms of three-class accuracy for both proteins. Group 7-Submission 1 (FoldX) was the most accurate, among all methods, for PTEN and the same group’s Submission 2 (Rosetta) did the best for TPMT. However, in line with the per-protein performance differences these submissions were only exceptional for one of the proteins, moving overall (two protein) performance of each submission drastically lower. Interestingly, although Envision performed relatively poorly on both the regression and classification tasks, its performance was highly protein-specific. In terms of correlation, it was among the bottom three methods for PTEN but among the top three methods for TPMT (with the random predictor excluded).
Figure 5. Per protein predictor performance.
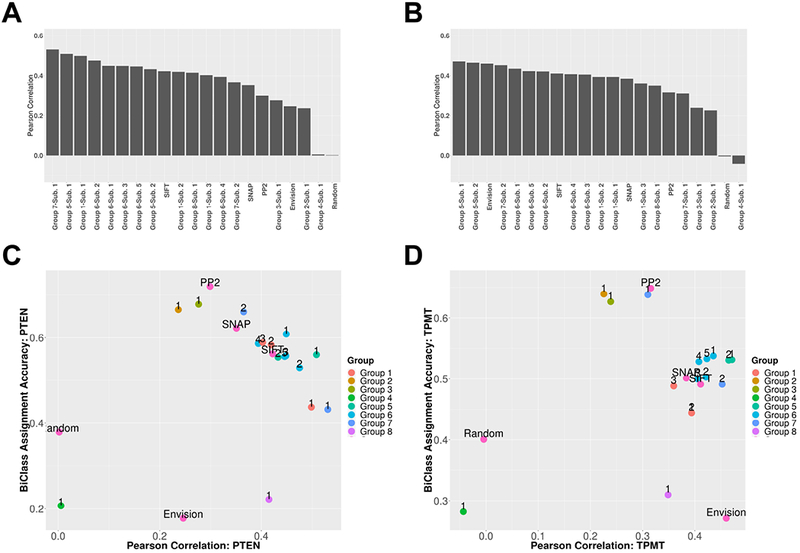
PTEN performance as (A) the Pearson correlation of predictions with experimental values for variants in the Final set and (C) the Pearson correlation of predictions with experimental values for variants in the Classification set mapped vs. binary classification accuracy. TPMT performance as (B) the Pearson correlation of predictions with experimental values for variants in the Final set and (D) the Pearson correlation of predictions with experimental values for variants in the Classification set mapped vs. binary classification accuracy.
Discussion
We assessed the ability of computational methods to predict the effects of individual non-synonymous variants on the VSP assay-derived stability of two proteins. Our assessment differs from previous assessments (Khan and Vihinen, 2010; Potapov, et al., 2009; Pucci, et al., 2018; Thiltgen and Goldstein, 2012) in two ways. First, the use of high-throughput VSP assay data as the gold standard enabled evaluations free from ascertainment bias; i.e. mutations were not explicitly targeted to protein positions of interest. Second, the scope of methods evaluated was not restricted to stability predictors, highlighting the overlap between functional, structural, and disease-driving variant effects.
Uncertainty in the experimental data.
We used VSP assay scores as the gold standard to evaluate computational predictions. When simply evaluating the experimentally-derived variant impact scores, one of two observations is most salient (Figure 1). Either (1) the assay is essentially noise free, and a large number of synonymous variants have a significant effect on protein abundance and stability or (2) the experimental system carries high levels of variability. If the latter is true, the correlation between predictor scores and experimental values becomes exceedingly difficult to interpret. Assessment in a (two or three class) classification setting, may be better suited to address this concern, but more sophisticated methods for selecting experimental thresholds to define impact class are necessary to account for these uncertainties.
Overall, the limitations of using the VSP assay data as a gold standard for evaluating generalized computational methods are as follows: (1) as noted above, many synonymous variants have high VSP scores in PTEN and TPMT, making straightforward mapping of effect/no-effect difficult, (2) VSP scores reflect changes in both stability and protein abundance, but do not directly account for changes in protein function, which may unevenly impact performance of computational methods, (3) the use of a heuristic cutoff to discretely classify continuous VSP scores may affect evaluation, as can the data providers’ post-experiment score transformation/normalization to create the set of final values. Despite these flaws, VSP assays constitute a novel, independent, and important way to assess performance of computational predictors.
Different predictors emerge as top-performing in different evaluation settings.
We evaluated all methods in both the value prediction (regression) and classification settings. When considering the correlation between experimental and prediction scores, the best submission came from Group 5. However, in the three and two class classification settings, methods from Groups 2 and 3 (both from the same research team) emerged as the most accurate. Apart from classification accuracy, the optimal tradeoff between correlation in the regression setting and accuracy in the classification setting is a desirable metric. By this measure, methods from Groups 5 and 6 emerged as the best-performing for the full data set. Group 7 was the best in terms of three-class accuracy on each protein individually. Note that no submission exceeded a correlation co-efficient (Pearson and Spearman) or a three-class accuracy of 0.5 or binary classification accuracy of 0.7 for both proteins together, highlighting a still existing need for improved methods.
The dependence of performance on the evaluation metric suggests that different methods are optimized for different settings. However, the regression and classification settings represent distinct use-cases, and methods should be chosen accordingly.
With the exception of outliers, most predictors perform similarly.
Previous efforts to compare and contrast predictors of general variant impact have found that methods are correlated in their predictions, largely due to similar design objectives, overlapping training sets and/or feature sets (Ioannidis, et al., 2016; Mahlich, et al., 2017). Interestingly, for predictors of mutation-induced stability change, correlations were found to be small in previous evaluations (Khan and Vihinen, 2010; Potapov, et al., 2009). Due to the broader scope of methods considered, our study revealed trends similar to the former situation rather than the latter, with methods generally grouping according to their design principles. Despite these commonalities, top-performing methods were generally distinguished by the customization made for this particular prediction task. For instance, almost all the top-performing methods adjusted their scores to the variant solvent accessibility. This feature was indeed found to be the most correlated with assay scores in the recently published study on the PTEN-TPMT data set (Matreyek, et al., 2018) and perhaps provided these methods with a predictive advantage. We note that it is also possible that methods were similar to each other in their deviation from the requirements of the prediction challenge, i.e., there was a mismatch between method objectives and the nature of the experimental data (see next section for more details).
Defining the scope of enquiry when studying and predicting variant effects.
Many publications do not explicitly define their variant impact prediction goals except by the data sets they choose for method development and evaluation. However, it is safe to say that most have been designed to prioritize variants that affect protein structure or function (both global and local) or are implicated in disease (pathogenicity prediction). Note here: (1) the misuse of the word “deleterious,” which can imply stabilizing or gain of function variants but is most often used to identify de-stabilizing or loss of function effects and (2) the misuse of the word “pathogenic,” which is usually established for variants that cause monogenic disease but is most often used to define those that may have some (not necessarily causative) impact in pathogenicity pathways.
While the various variant impacts (as defined above) are correlated it is not clear how transferable method predictions are across these different tasks; although some methods have shown promise (Pejaver, et al., 2017a; Schaefer, et al., 2012). Some issues are important to consider here: First, note that VSP assays measure the steady-state abundance of mutated proteins as a reflection of stability. In fact, protein abundance may be affected by other means, e.g. altered post-transcriptional regulation, translational speeds, and disrupted trafficking (Matreyek, et al., 2018). Similarly, note that the experiment-specific (here, VSP assay-specific) resolution limits and variation between replicates complicate the use of individual experimental data sets as a fixed gold standard to evaluate computational method performance. Second, since baseline methods did not perform as well as expected from previous studies, the VSP stability score likely does not reflect functional effects. As function-changing mutations may have no effects on protein stability and not all stability changes lead to functional disruptions (Bromberg and Rost, 2009; Lugo-Martinez, et al., 2016; Sahni, et al., 2015), function prediction methods may be at a disadvantage. Third, state-of-the-art stability prediction tools may not generalize across all proteins and variants within a protein. The best example here are the two submissions from Group 7, each of which performed best on one protein and failed on another (three class accuracy; Supplementary Materials). Finally, the stabilizing effects of variants in this study must be interpreted cautiously. We note that most baseline methods were not designed to predict stabilizing effects. This is also true for most submissions, which generally repurposed previously developed methods for this challenge. Moreover, the link between protein abundance and thermodynamic stability is weaker in the stabilizing range and it is, thus, unclear to what extent these experimental scores are meaningful.
As more VSP assay data accumulates, insights into the associated uncertainties will emerge, enabling a more rigorous evaluation of the generalizability of predictors for this particular task. However, as it must be clear by now, it is not trivial to make recommendations of the best prediction methods for general use. The issues presented here highlight the need for the users and developers of both computational and experimental methods to clearly define design goals, state caveats when such methods are repurposed for related tasks, and provide evidence for the validity of this repurposing.
Supplementary Material
Acknowledgments
We thank Dr. Steven E. Brenner and other organizers of the CAGI challenges for their guidance and support. We also thank Zishuo Zeng and Yanran Wang at Rutgers University for their help with the assessment and Dr. Kenneth Matreyek at the University of Washington for clarifications on the experimental work. VP was supported by the Washington Research Foundation Fund for Innovation in Data-Intensive Discovery and the Moore/Sloan Data Science Environments Project at the University of Washington and NIH K99 LM012992. JM was supported in part by NIH R01 GM104436 and R01GM120364. OL and PK were supported by the NIH R01 GM079656 and the NIH R01 GM066099 grants. YB and MM were supported by the NIH U01 GM115486 and NIH U24 MH06845 grants. The CAGI experiment coordination is supported by NIH U41 HG007346 and the CAGI conference by NIH R13 HG006650.
Funding information: NIH U41 HG007346, NIH R13 HG006650, NIH K99 LM012992, NIH R01 GM104436, NIH R01 GM120364, NIH R01 GM079656, NIH R01 GM066099, NIH U01 GM115486 and NIH U24 MH06845.
Footnotes
The authors declare no conflict of interest.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nature Methods 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya CL, Fowler DM, Chen W, Muniez I, Kelly JW, Fields S. 2012. A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proceedings of the National Academy of Sciences of the United States of America 109:16858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaber M, Baase WA, Gassner N, Matthews BW. 1995. Alanine scanning mutagenesis of the alpha-helix 115–123 of phage T4 lysozyme: effects on structure, stability and the binding of solvent. Journal of Molecular Biology 246:317–30. [DOI] [PubMed] [Google Scholar]
- Bromberg Y, Overton J, Vaisse C, Leibel RL, Rost B. 2009. In silico mutagenesis: a case study of the melanocortin 4 receptor. Federation of American Societies for Experimental Biology Journal 23:3059–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg Y, Rost B. 2007. SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Research 35:3823–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg Y, Rost B. 2009. Correlating protein function and stability through the analysis of single amino acid substitutions. BMC Bioinformatics 10 Suppl 8:S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruylants G, Wouters J, Michaux C. 2005. Differential scanning calorimetry in life science: thermodynamics, stability, molecular recognition and application in drug design. Current Medicinal Chemistry 12:2011–20. [DOI] [PubMed] [Google Scholar]
- Burchenal JH, Murphy ML, Ellison RR, Sykes MP, Tan TC, Leone LA, Karnofsky DA, Craver LF, Dargeon HW, Rhoads CP. 1953. Clinical evaluation of a new antimetabolite, 6-mercaptopurine, in the treatment of leukemia and allied diseases. Blood 8:965–99. [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R and others 2005. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. Journal of Medical Genetics 42:318–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capriotti E, Fariselli P, Casadio R. 2005. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Research 33:W306–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Randall A, Baldi P. 2006. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 62:1125–32. [DOI] [PubMed] [Google Scholar]
- Dehouck Y, Kwasigroch JM, Gilis D, Rooman M. 2011. PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics 12:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fariselli P, Martelli PL, Savojardo C, Casadio R. 2015. INPS: predicting the impact of non-synonymous variations on protein stability from sequence. Bioinformatics 31:2816–21. [DOI] [PubMed] [Google Scholar]
- Folkman L, Stantic B, Sattar A, Zhou Y. 2016. EASE-MM: sequence-based prediction of mutation-induced stability changes with feature-based multiple models. Journal of Molecular Biology 428:1394–1405. [DOI] [PubMed] [Google Scholar]
- Fowler DM, Fields S. 2014. Deep mutational scanning: a new style of protein science. Nature Methods 11:801–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray VE, Hause RJ, Luebeck J, Shendure J, Fowler DM. 2018. Quantitative missense variant effect prediction using large-scale mutagenesis data. Cell Systems 6:116–124.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield NJ. 2006. Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nature Protocols 1:2527–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromiha MM. 2010. Protein bioinformatics: from sequence to function: Academic Press. [Google Scholar]
- Hobert JA, Eng C. 2009. PTEN hamartoma tumor syndrome: an overview. Genetics in Medicine 11:687–94. [DOI] [PubMed] [Google Scholar]
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D and others 2016. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. American Journal of Human Genetics 99:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, White M, Radivojac P. Estimating the class prior and posterior from noisy positives and unlabeled data Advances in Neural Information Processing Systems; 2016; Barcelona, Spain: p 2693–2701. [Google Scholar]
- Katsonis P, & Lichtarge O (2014). A formal perturbation equation between genotype and phenotype determines the Evolutionary Action of protein-coding variations on fitness. Genome research, 24(12), 2050–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata T, Ota M, Nishikawa K. 1999. The Protein Mutant Database. Nucleic Acids Research 27:355–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg EH; Leaver-Fay A; Baker D, Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 2011, 79 (3), 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Vihinen M. 2010. Performance of protein stability predictors. Human Mutation 31:675–84. [DOI] [PubMed] [Google Scholar]
- Kumar MD, Bava KA, Gromiha MM, Prabakaran P, Kitajima K, Uedaira H, Sarai A. 2006. ProTherm and ProNIT: thermodynamic databases for proteins and protein-nucleic acid interactions. Nucleic Acids Research 34:D204–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YR, Chen M, Pandolfi PP. 2018. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nature Reviews Molecular Cell Biology 19:547–562. [DOI] [PubMed] [Google Scholar]
- Lugo-Martinez J, Pejaver V, Pagel KA, Jain S, Mort M, Cooper DN, Mooney SD, Radivojac P. 2016. The loss and gain of functional amino acid residues Is a common mechanism causing human inherited disease. PLOS Computational Biology 12:e1005091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magliery TJ. 2015. Protein stability: computation, sequence statistics, and new experimental methods. Current Opinion in Structural Biology 33:161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlich Y, Reeb J, Hecht M, Schelling M, De Beer TAP, Bromberg Y, Rost B. 2017. Common sequence variants affect molecular function more than rare variants? Scientific Reports 7:1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matreyek KA, Starita LM, Stephany JJ, Martin B, Chiasson MA, Gray VE, Kircher M, Khechaduri A, Dines JN, Hause RJ and others 2018. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nature Genetics 50:874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milla ME, Brown BM, Sauer RT. 1994. Protein stability effects of a complete set of alanine substitutions in Arc repressor. Nature Structural & Molecular Biology 1:518–23. [DOI] [PubMed] [Google Scholar]
- Morrison KL, Weiss GA. 2001. Combinatorial alanine-scanning. Current Opinion in Chemical Biology 5:302–7. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. 2001. Predicting deleterious amino acid substitutions. Genome Research 11:863–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace CN, Scholtz JM. 1997. Measuring the conformational stabiity of a protein. In: Creighton TE, editor. Protein structure: a practical approach; p 299–321. [Google Scholar]
- Pejaver V, Mooney SD, Radivojac P. 2017a. Missense variant pathogenicity predictors generalize well across a range of function-specific prediction challenges. Human Mutation 38:1092–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejaver V, Urresti J, Lugo-Martinez J, Pagel KA, Lin GN, Nam H-J, Mort M, Cooper DN, Sebat J, Iakoucheva LM and others 2017b. MutPred2: inferring the molecular and phenotypic impact of amino acid variants. bioRxiv 134981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapov V, Cohen M, Schreiber G. 2009. Assessing computational methods for predicting protein stability upon mutation: good on average but not in the details. Protein Engineering, Design & Selection 22:553–60. [DOI] [PubMed] [Google Scholar]
- Pucci F, Bernaerts KV, Kwasigroch JM, Rooman M. 2018. Quantification of biases in predictions of protein stability changes upon mutations. Bioinformatics 34:3659–3665. [DOI] [PubMed] [Google Scholar]
- Rosner B 2015. Fundamentals of biostatistics: Nelson Education.
- Sahni N, Yi S, Taipale M, Fuxman Bass JI, Coulombe-Huntington J, Yang F, Peng J, Weile J, Karras GI, Wang Y and others 2015. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 161:647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savojardo C, Fariselli P, Martelli PL, Casadio R. 2016. INPS-MD: a web server to predict stability of protein variants from sequence and structure. Bioinformatics 32:2542–4. [DOI] [PubMed] [Google Scholar]
- Schaefer C, Bromberg Y, Achten D, Rost B. 2012. Disease-related mutations predicted to impact protein function. BMC Genomics 13 Suppl 4:S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. 2005. The FoldX web server: an online force field. Nucleic Acids Research 33:W382–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiltgen G, Goldstein RA. 2012. Assessing predictors of changes in protein stability upon mutation using self-consistency. PLOS One 7:e46084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Moult J. 2001. SNPs, protein structure, and disease. Human Mutation 17:263–70. [DOI] [PubMed] [Google Scholar]
- Weinshilboum R 2001. Thiopurine pharmacogenetics: clinical and molecular studies of thiopurine methyltransferase. Drug Metabolism and Disposition 29:601–5. [PubMed] [Google Scholar]
- Yang Y, Chen B, Tan G, et al. Structure-based prediction of the effects of a missense variant on protein stability.[J]. Amino Acids, 2013, 44(3):847–855. [DOI] [PubMed] [Google Scholar]
- Yang Y, Urolagin S, Niroula A, Ding X, Shen B, Vihinen M. 2018. PON-tstab: protein variant stability predictor. importance of training data quality. International Journal of Molecular Sciences 19:E1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen HC, Xu Q, Chou DM, Zhao Z, Elledge SJ. 2008. Global protein stability profiling in mammalian cells. Science 322:918–23. [DOI] [PubMed] [Google Scholar]
- Yin Y, Kundu K, Pal LR and Moult J Ensemble variant interpretation methods to predict enzyme activity and assign pathogenicity in the CAGI4 NAGLU (Human N-Acetyle Glucosaminidase) and UBE2I (Human SUMO-ligase) challenges. Human Mutation. 2017; 38:1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue P, Li Z, Moult J. 2005. Loss of protein structure stability as a major causative factor in monogenic disease. Journal of Molecular Biology 353:459–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.