

Abstract
GM1 gangliosidosis is an autosomal recessive neurodegenerative disorder caused by the deficiency of lysosomal β-galactosidase (β-gal) and resulting in accumulation of GM1 ganglioside. The disease spectrum ranges from infantile to late onset and is uniformly fatal, with no effective therapy currently available. Although animal models have been useful for understanding disease pathogenesis and exploring therapeutic targets, no relevant human central nervous system (CNS) model system has been available to study its early pathogenic events or test therapies. To develop a model of human GM1 gangliosidosis in the CNS, we employed CRISPR/Cas9 genome editing to target GLB1 exons 2 and 6, common sites for mutations in patients, to create isogenic induced pluripotent stem (iPS) cell lines with lysosomal β-gal deficiency. We screened for clones with <5% of parental cell line β-gal enzyme activity and confirmed GLB1 knockout clones using DNA sequencing. We then generated GLB1 knockout cerebral organoids from one of these GLB1 knockout iPS cell clones. Analysis of GLB1 knockout organoids in culture revealed progressive accumulation of GM1 ganglioside. GLB1 knockout organoids microinjected with AAV9-GLB1 vector showed a significant increase in β-gal activity and a significant reduction in GM1 ganglioside content compared with AAV9-GFP–injected organoids, demonstrating the efficacy of an AAV9 gene therapy-based approach in GM1 gangliosidosis. This proof-of-concept in a human cerebral organoid model completes the pre-clinical studies to advance to clinical trials using the AAV9-GLB1 vector.
Abbreviations: 4MU, 4-methylumbelliferyl; AAV, adeno-associated virus; AAV9, AAV serotype 9; β-gal, β-galactosidase; BSA, bovine serum albumin; CNS, central nervous system; CPB, citrate phosphate buffer; EB, embryoid body; GFP, green fluorescent protein; hiPSC, human induced pluripotent stem cells; HPTLC, high-performance thin-layer chromatography; iPS, induced pluripotent stem; PBS, phosphate-buffered saline; RT-qPCR, real-time quantitative polymerase chain reaction; SD, standard deviation; X-gal, 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside
1. Introduction
GM1 gangliosidosis is an autosomal recessive neurodegenerative disorder caused by deficiency of the lysosomal hydrolase β-galactosidase (β-gal) [1]. Mutations in the gene GLB1 result in reduced or absent activity of β-gal, leading to lysosomal accumulation of GM1 ganglioside, a membrane glycosphingolipid important for neuronal function [2]. The estimated incidence of GM1 gangliosidosis is 1 in 100,000–200,000 live births [3,4],. Currently no effective treatments are available. The disease is a continuum, with decreasing residual β-gal activity leading to increasing clinical severity and where the amount of residual enzyme is determined by the specific GLB1 mutation(s) [5]. The continuum has been historically classified into three types. Type I (OMIM 230500), Type II (OMIM 230600), and Type III (OMIM 230650) and Type II has been recently subdivided into late-infantile (IIa) and juvenile (2b) subtypes [1].
GM1 gangliosidosis is predominantly a central nervous system (CNS) disorder and has been difficult to study in vivo. Several naturally occurring animal models recapitulate the phenotype to varying degrees, including cat [[6], [7], [8], [9]], sheep [[10], [11], [12]], dog [[13], [14], [15], [16]], calves [17], and bears [18], as well as engineered mouse models [[19], [20], [21], [22]]. Phenotypes presented by these animal models include GM1 ganglioside storage in neurons, progressive neurologic dysfunction, and a shortened lifespan. Although these models have been beneficial in understanding disease pathogenesis and exploring potential therapies, they lack human-specific features of the disease. Thus far, human studies have been generally limited to cranial imaging and post-mortem end-stage histologic analyses.
Human induced pluripotent stem cells (hiPSCs) are a powerful tool to model human disorders. They can be readily differentiated into relevant cell types and thus provide an in vitro model of disease [23]. By combining the versatile nature of hiPSCs with the CRISPR/Cas9 genome editing technique [24], it is possible to create isogenic hiPSC lines to analyze the effects of a monogenic disorder while minimizing differences in genetic backgrounds. Since the first reports in 2013 [25], steady advances have been made in differentiating hiPSCs into three-dimensional cerebral organoids to study neurodevelopment as well as neurologic and neuropsychiatric disorders [26]. Initial studies to develop a model system for microcephaly have expanded to include other disorders of early brain development, including lissencephaly and cortical folding. Genetic syndromes and monogenic disorders, autism spectrum disorder, and environmental insults to the developing brain (such as Zika virus) have also been modeled (reviewed in [26]).
Despite the lack of effective treatments for GM1 gangliosidosis, several studies have investigated the use of adeno-associated virus (AAV)-mediated gene therapy for the disease. Intracranial injections of AAV encoding lysosomal β-gal in mouse and cat models reduced GM1 ganglioside content throughout the CNS, extended stable behavioral functions, and increased lifespan [[27], [28], [29], [30]]. GLB1 delivered intravenously using AAV serotype 9 (AAV9) in GM1 gangliosidosis mice has been shown to traverse the blood-brain barrier, producing widespread transduction of multiple brain regions, reduced GM1 ganglioside storage in the CNS, and improved survival [31]. Here we describe a GLB1 knockout hiPSC line generated using CRISPR/Cas9 genome editing, demonstrate the advantage of a fluorescence-based enzyme assay to screen large numbers of clones for targeted genome editing, establish an in vitro human GLB1 knockout cerebral organoid model of GM1 gangliosidosis, and demonstrate the efficacy of AAV9-GLB1 gene therapy in reducing GM1 ganglioside content in GLB1 knockout cerebral organoids.
2. Results
2.1. Generation of a GLB1 knockout hiPSC line
We used hiPSCs from an unaffected individual, referred to as the parental cell line, to develop isogenic GLB1 knockout cell lines using CRISPR/Cas9 genome editing. Because exons 2 and 6, along with exon 15, of GLB1 contain the majority of reported nonsense and missense mutations in human GM1 gangliosidosis patients [3], we designed sgRNAs each to exon 2 (Exon 2-1) and exon 6 (Exon 6-1 and 6-2). The hiPSCs were transfected with a plasmid vector carrying Cas9 and one of the sgRNAs, and successfully transfected cells were selected with puromycin. Colonies that maintained undifferentiated morphology were assayed for enzyme activity using a 4-methylumbelliferyl (4MU) substrate specific for β-gal (expressed as a percent of activity of the parental cell line). Less than 5% activity was set as the criteria for successful genome editing. In total, 107 colonies were screened: 44 colonies transfected with sgRNA Exon 2-1, 44 colonies transfected with sgRNA Exon 6-1, and 19 colonies transfected with sgRNA Exon 6-2 (Fig. 1A). Most of the colonies (78%) had ≤10% of the enzyme activity found in the parental cell line. Two colonies produced by each guide with <5% activity and undifferentiated morphology, were selected for sequencing confirmation of GLB1 mutations. One clone (asterisk, Fig. 1A) contained bi-allelic GLB1 mutations that were targeted by sgRNA Exon 6-2 (i.e., a one base-pair insertion and a one base-pair deletion [Fig. 1B]); this clone was chosen for further studies.
Fig. 1.

GLB1 knockout clone screening and mutation confirmation.
(A) Screening of CRISPR/Cas9-edited clones using a β-gal enzyme activity assay. Three different sgRNAs, one targeting exon 2 (Exon 2-2) and two targeting exon 6 (Exon 6-1 and Exon 6-2), were designed and used for editing the GBL1 gene in hiPSCs. The resulting 107 colonies were screened for β-gal enzyme activity and plotted: 44 for sgRNA Exon 2-1 (blue bars), 44 for sgRNA Exon 6-1 (orange bars) and 19 for sgRNA Exon 6-2 (green bars). Of those, two clones produced by each sgRNA, with undifferentiated morphology and <5% enzyme activity (compared with the parental cell line [value set at 100%]) were sequenced. An asterisk marks the clone selected for further experiments. (B) Characterization of the mutations in the selected knockout hiPSC clone. The reference sequence of the GLB1 gene in the edited region is shown. Allele 1 contains a 1 base-pair insertion, an A, and allele 2 contains a 1 base-pair deletion, both highlighted in yellow. Boxes outline the location of the protospacer adjacent motif (PAM) sequence. (C) Predicted proteins resulting from the mutations in alleles 1 and 2 compared with β-gal protein domains(top;, adapted from [48]). Both mutations are predicted to produce truncated proteins prior to E368, an important catalytic nucleophile. (D) Relative mRNA expression of GLB1 normalized to GAPDH. KO, GBL1 knockout organoids. Data represent the mean ± standard deviation (SD), n = 3 for each line) (⁎⁎p < .01, t-test analysis between control and KO values). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Both GLB1 mutations in the selected clone result in premature stop codons and are predicted to encode truncated proteins (Fig. 1C). To evaluate the validity of the GLB1 knockout hiPSC line as an isogenic model to the parental cell line, we measured GLB1 mRNA expression by real-time quantitative polymerase chain reaction (RT-qPCR) and sequenced potential Cas9 off-target sites. Using a 3′-specific TaqMan probe to GLB1, we determined that GLB1 mRNA expression was negligible in the GLB1 knockout line compared with the parental cell line (Fig. 1D). The top five predicted off-target sites (CRISPRtool, historically: http://crispr.mit.edu) were sequenced for evidence of off-target editing and none were found.
2.2. Generation and characterization of GLB1 knockout cerebral organoids with β-galactosidase deficiency and GM1 ganglioside storage
Cerebral organoids were generated from the parental cell line, henceforth referred to as the isogenic control, and the selected CRISPR edited hiPSCs by embedding cell-aggregated embryoid bodies (EBs) in the artificial extracellular matrix, Matrigel, to promote radial growth, then maintained in bioreactors [32] (Fig. S1). Histologic evaluation of control organoids showed ventricle-like structures lined with cilia, (Fig. S2A and B), and immunohistochemistry revealed regions of neural progenitors (SOX2) and astrocytes (GFAP) within the organoids (Fig. S2C and D).
Fig. S1.

Progressive growth from hiPSCs to cerebral organoids.
(A) Timeline of cerebral organoid differentiation. (B) Day 1, 24 h of EB formation in Aggrewell™400 plate. (C) Day 1, ultra-low adherence (ULA) plate. (D) Day 2. (E) Day 4. (F) Day 5, neural induction. (G) Day 6, 24 h in neural induction media. Visible lighter edge indicating formation of ectodermal layer. (H) Day 7. (I) Day 8, visible translucent edges of ectodermal buds. (J) Day 9. (K) Day 10, prior to insertion of EB into Matrigel droplet and neural differentiation media (B-27–). (L) Day 11, 24 h after insertion into Matrigel. (M) Day 12, evidence of neuroepithelial budding and non-neuroepithelial growth is visible. (N) Day 14, organoid with neuroepithelial budding and non-neuroepithelial cells migrating into the Matrigel. Ready to place into the bioreactor with neural differentiation media (B-27+). (O) Glass bioreactor in incubator. (P) 4-week organoid harvest. (Q) 20-week organoid harvest. Scale bars, 1000 μm.
Fig. S2.
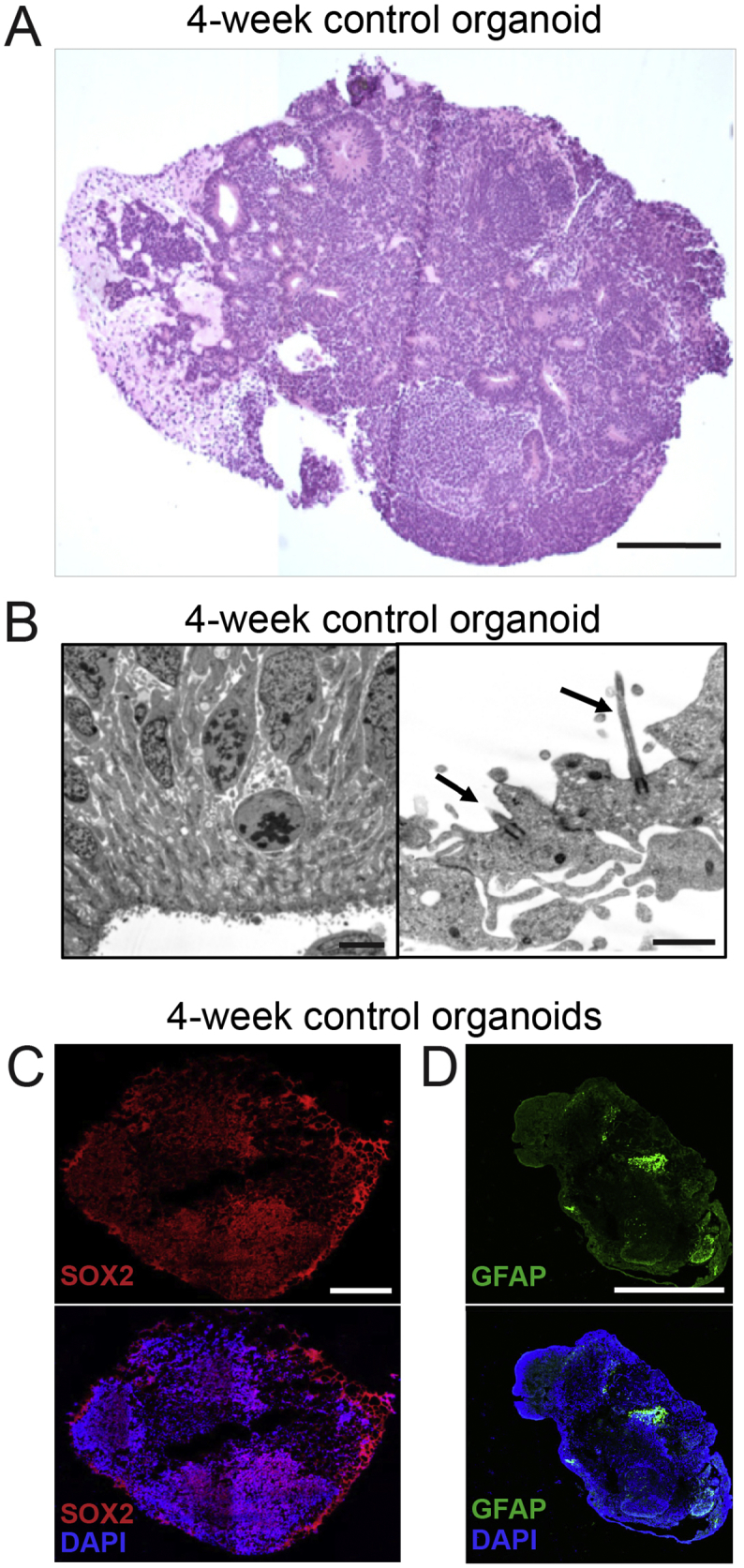
Histologic characterization of cerebral organoids.
(A) Paraffin-embedded 4-week-old control cerebral organoid sectioned and stained with H&E. Scale bar, 250 μm. (B) Electron microscopy of 4-week-old control organoid shows columnar cells lining the ventricle-like structures, with cilia protruding into the ventricle space (arrows). Scale bars, 5 μm (left image); 1 μm (right image). (C) Representative immunofluorescence images of 4-week old control organoids stained for SOX2, a marker for neural progenitors (top panel) and SOX2/DAPI-merged staining (bottom panel). Scale bars, 500 μm. (D) Representative immunofluorescence images of 4-week old control organoids stained for GFAP, a marker for astrocytes (top panel) and GFAP/DAPI-merged staining (bottom panel). Scale bars, 500 μm.
In GM1 gangliosidosis, mutations in the GLB1 gene result in reduction or loss of lysosomal β-gal enzyme activity, leading to the accumulation of the GM1 ganglioside in lysosomes. To analyze GM1 ganglioside accumulation in organoids over time, immunohistochemistry was performed using an anti-GM1 monoclonal antibody (validated in Fig. S3) on GLB1 knockout and isogenic control organoids at 5-week intervals. At 20 weeks in culture, a significant increase of GM1 ganglioside storage in GLB1 knockout organoids compared to the isogenic control organoids was detectable by immunofluorescence (Fig. 2A and B). The fluorescent signals from anti-GM1 ganglioside and anti-LAMP1 (lysosomal marker) immunostaining of GLB1 knockout organoids were co-localized, indicating that the GM1 ganglioside storage occurred in lysosomes (Fig. S4). X-gal staining of whole cerebral organoids at 15 weeks showed decreased β-gal activity throughout the GLB1 knockout organoids compared with the isogenic control organoids from the parental line (Fig. 2C).
Fig. S3.

Validation of GM1 ganglioside monoclonal antibody.
Immunostaining with anti-GM1 ganglioside antibody (green) of normal and end-stage GM1 gangliosidosis mouse (A) and cat brain tissue (B) and normal and GM1 gangliosidosis patient human fibroblasts (C). Nuclei were visualized with DAPI staining (blue). Arrows indicate areas of GM1 ganglioside storage. Scale bars, 50 μm (A) and 100 μm (B, C).
Fig. 2.

GM1 Ganglioside Storage and β-Gal Enzyme Activity in GLB1 knockout Cerebral Organoids.
(A) Representative images of isogenic control and GLB1 knockout cerebral organoids harvested at 5, 10, 15, and 20 weeks and stained with anti-GM1 ganglioside (green) and anti-β3 tubulin (red) antibodies. Nuclei were visualized with DAPI staining (blue). Arrows indicate areas of GM1 storage. Scale bars: white, 500 μm; yellow, 10 μm. (B) Quantification of GM1 fluorescent signal normalized to DAPI of GLB1 knockout organoids compared with isogenic control organoids. Data represent the mean relative GM1 expression ± SD, n ≥ 3 for both control and knockout organoids at each time point (⁎⁎⁎p < .001, two-way Anova analysis between control and knockout values). (C) X-gal staining of isogenic control and GLB1 knockout cerebral organoids harvested at 15 weeks. Blue staining on the organoids indicates β-gal activity. Scale bars, 500 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. S4.
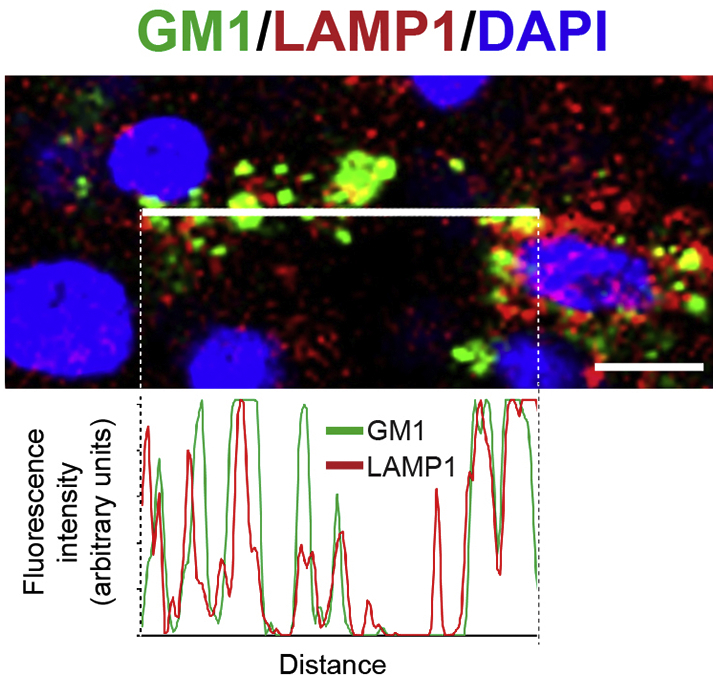
Localization of GM1 ganglioside in lysosomes of GLB1 knockout cerebral organoids.
(Top panel) Cryo-sectioned 20-week-old GLB1 knockout cerebral organoids co-immunostained with anti-GM1 ganglioside (green) and anti-LAMP1 (red), used as a marker for lysosomes. Nuclei were visualized with DAPI staining (blue). Scale bar (lower right corner), 33 μm. (Bottom panel) Co-localization of GM1 ganglioside and LAMP1 was analyzed by quantification of the fluorescence intensities of the GM1 ganglioside signal in green with and LAMP1 signal in red along a 1 pixel-wide line (shown in the center of the top panel).
Ganglioside content in the cerebral organoids was determined by high-performance thin-layer chromatography (HPTLC) (Fig. 3A); GM1 ganglioside content was significantly elevated in the GLB1 knockout organoids at 10 and 20 weeks, but not earlier, suggesting a progressive accumulation of GM1 when compared with isogenic control organoids (Fig. 3B).
Fig. 3.

GM1 Ganglioside Storage in GLB1 Knockout Cerebral Organoids.
(A) Representative HPTLC analysis of gangliosides extracted from isogenic control and GLB1 knockout cerebral organoids harvested at 20 weeks. Gangliosides extracted from 3 pooled organoids (control and knockout) were applied per lane. Std, Monosialogangliosides used as standards. (B) Quantification of GM1 ganglioside content of control and GLB1 knockout organoids harvested at 10, 15, and 20 weeks. GM1 content was determined by densitometry of the HPTLC plate and normalized with the corresponding phospholipid concentrations. Data represent mean GM1 content at each time point relative to control content at week 10 ± SD, n = 3 lanes (⁎p < .05, t-test analysis between control and knockout values).
2.3. Reduction in GM1 ganglioside content in GLB1 knockout cerebral organoids following AAV9-GLB1 injection
AAV9-GLB1 was injected into the center of 10-week-old GLB1 knockout cerebral organoids and β-gal activity and GM1 ganglioside content were analyzed 5 weeks after injection. Controls included uninjected GLB1 knockout organoids, GLB1 knockout organoids injected with an equivalent titer of AAV9-green fluorescent protein (AAV9-GFP), and uninjected isogenic control organoids. All GLB1 knockout organoids used were from the same culture to minimize variability. Relative β-gal activity (expressed as a percent of activity of isogenic controls) was significantly increased in AAV9-GLB1–injected organoids compared with the uninjected and AAV9-GFP–treated organoids (Fig. 4A and B). GM1 ganglioside content was significantly decreased in AAV9-GLB1–treated organoids compared with AAV9-GFP–treated organoids when examined by both immunohistochemistry (Fig. 4C and D) and HPTLC analysis (Fig. 4E and F).
Fig. 4.

Treatment of GLB1 Knockout Cerebral Organoids with AAV9-GLB1 Gene Therapy.
GLB1 knockout cerebral organoids were injected with AAV9-GFP or AAV9-GLB1 at 10 weeks and harvested for analysis 5 weeks later. (A) Representative X-gal staining of AAV9-GFP–injected and AAV9-GLB1–injected organoids. Scale bars, 500 μm. (B) β-gal enzyme activity of whole cerebral organoid lysates (compared to uninjected isogenic controls whose value was set at 100%). Data represent the mean relative β-gal activity ± SD (n = 5 organoids for each treatment); ⁎p < .05, t-test analysis). (C) Representative images of AAV9-GFP– and AAV9-GLB1–injected organoids stained with anti-GM1 (green) and anti-β3 tubulin (red) antibodies. Nuclei were visualized with DAPI staining (blue). Arrows indicate areas of GM1 storage. Scale bars: white, 500 μm; yellow, 65 μm. (D) Quantification of GM1 fluorescent signal normalized to DAPI of AAV9-GLB1–injected organoids compared with AAV9-GFP–injected organoids. Data represent mean GM1 content normalized with DAPI ± SD, n = 3 injected organoids per treatment and 16 immunostained fields per analysis (⁎⁎⁎p < .001, t-test analysis between AAV9-GFP-injected and AAV9-GLB1-injected organoids). (E) Representative HPTLC plate of gangliosides extracted from AAV9-GFP–injected and AAV9-GLB1–injected organoids. Gangliosides extracted from 3 organoids were applied per lane for AAV9-GFP- and AAV9-GLB1-injected organoids. Std, monosialogangliosides used as standards. (F) Quantification of GM1 content of AAV9-GFP–injected and AAV9-GLB1–injected organoids by HPTLC. GM1 content was determined by densitometry and normalized with the corresponding phospholipid concentrations. Data represent mean GM1 content of AAV9-GLB1–injected organoids relative to AAV9-GFP–injected organoids ± SD, n = 3 lanes (⁎⁎p < .01, t-test analysis between AAV9-GFP-injected and AAV9-GBL1-injected values). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3. Discussion
Since their initial description in 2013, the study of stem cell-derived cerebral organoids has expanded rapidly [25] and has filled an important niche in the study of genetic and acquired CNS disorders [26]. Using organoids produced by CRISPR/Cas9 genome editing and comparing the edited organoids to isogenic controls, it is now possible to study disease pathogenesis flowing from mutations in a single gene.
GM1 gangliosidosis is a monogenic, progressive, uniformly fatal neurodegenerative disorder with no effective therapy, thereby limiting treatment to supportive care. Acquiring relevant human primary tissue to verify findings in animal model systems is extremely difficult. Here, we have leveraged the advances in CRISPR/Cas9 editing and hiPSC-based three-dimensional cultures to provide a human CNS model system for GM1 gangliosidosis to investigate the molecular pathogenesis of and therapy for this devastating disease. Through isolation of a GLB1 knockout hiPSC line and establishment of GLB1 knockout cerebral organoids, we established an in vitro human GM1 gangliosidosis model system that demonstrated the hallmarks of the disease (i.e., loss of β-gal enzyme activity and progressive storage of GM1 ganglioside).
Quantitative enzyme analysis is one of the most widely used methods for diagnosing lysosomal storage disorders in clinical laboratories. Activity is usually determined using a fluorescent, 4MU substrate specific to the enzyme of interest. We applied this simple enzymatic detection method for screening and identification of hiPSC clones that were successfully targeted by our CRISPR/Cas9 sgRNAs to introduce GLB1 mutations and therefore negate β-gal activity. Generally, clones are first selected after Sanger sequencing of PCR-amplified genomic DNA [24], but this method is time-consuming, especially if the frequency of successfully targeted editing is low. We were able to rapidly screen a large number of clones using the enzymatic assay and then sequence only those found to be essentially devoid of enzyme activity. This procedure is both time and cost efficient. Although sequencing is still required to confirm resulting mutations, the screening greatly reduces the pool of candidate clones before that step. Because 4MU substrates are available for most lysosomal enzymes, this method would be widely applicable to other lysosomal diseases [33].
In the current study, GLB1 knockout cerebral organoids demonstrated GM1 ganglioside accumulation detectable by HPTLC beginning as early as 10 weeks in culture (Fig. 4) and visible histologically at 20 weeks (Fig. 3). Using a similar system, our lab has demonstrated that cerebral organoids derived from an infantile Sandhoff disease (GM2 gangliosidosis) patient show GM2 ganglioside accumulation beginning at 4 weeks [38]. Whereas GM1 ganglioside is an abundant constituent of neuronal plasma membranes [34], GM2 ganglioside is an intermediate in the ganglioside metabolic pathway and is present in low quantities in normal human brain [35]. This low background may allow earlier detection of abnormal accumulation in the Sandhoff organoids [36]. The discrepancy in the timing of ganglioside accumulation in the Sandhoff and GLB1 knockout organoids may be also be related to intrinsic differences in the recycling and metabolism of the two different gangliosides.
Lysosomal storage of GM1 and GM2 ganglioside in GM1 and GM2 gangliosidosis, respectively, has been demonstrated in human fetuses as early as the second trimester [35,37]. Cerebral organoids have been shown to model the human fetal neocortex [34,36,38]. In Sandhoff disease organoids, the enzyme deficiency and GM2 ganglioside accumulation impaired neuronal differentiation in developmental parallel to the early fetal period [36]. Given that GM1 ganglioside storage also begins in fetal life in infantile GM1 gangliosidosis [39], neuronal disturbances might also be expected during fetal development, suggesting that effective therapy for infantile onset disease may require in utero treatment (ref).
Intravenous gene therapy with AAV9-GLB1 has been effective in treating murine and feline models of GM1 gangliosidosis [31]. Here, treating GLB1 knockout cerebral organoids with comparable doses of AAV9-GLB1 vector significantly increased β-gal enzyme activity and significantly decreased GM1 ganglioside content, indicating its efficacy in a relevant human model system. This method of testing therapeutics could be applied to other monogenic CNS disorders and provides a bridge between animal models and clinical trials.
The pathogenesis of many lysosomal storage disorders has also been shown to involve the activation of bone marrow-derived microglial cells [40]. Recently described “assembloids” composed of both cerebral and microglial organoids may be an important next step in elucidating pathogenesis in these disorders [41]. So too, vascularization of cerebral organoids as recently demonstrated [42,43] may facilitate prolonged growth and differentiation of organoids and may provide further clues to the mechanism of disease.
In conclusion, we have generated a human CNS model of GM1 gangliosidosis using GLB1 knockout hiPSCs differentiated to cerebral organoids grown in parallel with isogenic control cerebral organoids. We have demonstrated that the GLB1 knockout organoids recapitulate the hallmark phenotype of GM1 ganglioside storage that can be reversed with AAV9-GLB1 gene therapy. This demonstration was the final step in the pre-clinical development of the AAV9-GLB1 vector for a phase 1/2 clinical trial for children with type II GM1 gangliosidosis (NCT03952637).
4. Materials and methods
4.1. hiPSC culture
The unaffected parental hiPSC line was obtained from Alstem (Episomal, HFF: #iPS11), and was verified pluripotent and contamination free. It is a footprint-free iPS cell line derived from human foreskin fibroblasts by ectopic expression of OCT4, SOX2, KLF4, and cMYC genes using episomal plasmids. hiPSCs were maintained feeder-free in mTeSR1 (STEMCELL Technologies, 05850) on Matrigel (Corning, 354277)-coated plates. To prevent cell death, 10 μM Y-27632 (STEMCELL Technologies, 72304) was added to culture media after each passage or detachment.
4.2. Genome editing using CRISPR/Cas9
sgRNAs were designed with the CRISPRtool (historically: http://crispr.mit.edu) to maximize on-target binding and minimize potential off-target editing. Two sgRNAs were designed for exons 2 and 6 of GLB1: sgRNA Exon 2-1: CGGTCCTTCCAGTAGAAGCG; sgRNA Exon 2-2: CTTCCTGAGATGTAGCGAAA; sgRNA Exon 6-1: CAAAGTCCACCGTGGTGTAG; and sgRNA Exon 6-2: GAAGCGCTTTCGCCACCATC. sgRNAs were cloned into the CRISPR-Cas9 plasmid, pSpCas9(BB)-2A-Puro (PX459), a gift from Feng Zhang (Addgene, Plasmid #48139) [24]. The hiPSCs from two wells of a 6-well plate at 70% confluency were detached with 1:1 Accutase:phosphate-buffered saline (PBS) and after that, combined, filtered using a 40 μm cell strainer to obtain a uniform cell suspension and pelleted. Cells were electroporated with 20 μg of CRISPR-Cas9 plasmid with inserted sgRNA, using the Human Stem Cell Nucleofector Kit 1 and the Nucleofector II Device (Lonza). Selection for edited cells started 24 h post-transfection by treating with 0.5 μg/ml puromycin for 48 h. The hiPSCs were later maintained in media containing a 1:1 ratio of fresh mTeSR1 and filtered, conditioned media collected from established hiPSCs cultures, until stable colonies were formed.
4.3. Clone screening
Individual hiPSC colonies were picked, cultured in individual 48-well plates, and screened for β-gal activity as previously described [44] to determine whether editing occurred. Briefly, hiPSC were lysed in 0.1 M citric buffer (pH 4.2) containing 0.1% Triton X-100 on ice for 30 min. β-gal activity was measured with 4 mM 4-methylumbelliferyl (4MU) β-D-galactopyranoside (Sigma-Aldrich) and was normalized with β-hexosaminidase activity using 20 mM 4MU N-acetyl-β-D-glucosaminide (Sigma-Aldrich). Parental, untransfected hiPSCs were subjected to the same analysis to serve as a reference value for normal (100%) β-gal activity. Both reactions were performed at 37 °C for 30 min and stopped with 200 μl 0.2 M glycine (pH 9.6). Fluorescence was measured on a Perkin Elmer Victor 1420 Multilabel Counter (excitation: 360 nm, emission: 446 nm).
To validate the targeted mutation sites, genomic DNA was isolated and PCR-amplified using primers to encompass each exon (Table S1) and cloned into a TOPO-TA sequencing vector (Invitrogen, 45-0030). DNA was isolated from 10 bacterial colonies and analyzed by Sanger sequencing (outsourced to Eurofins Genomics).
4.4. Off-target screening
The top five off-target sequences were extracted from the CRISPRtool (historically: http://crispr.mit.edu) when designing the guides. Genomic DNA was isolated and PCR-amplified using primers to encompass each off-target site (Table S2). PCR products were purified using the Qiagen QIAquick PCR Purification Kit (Qiagen, 28106) and sequenced.
4.5. Multiplex RT-qPCR
Total RNA was extracted from hiPSCs using the RNeasy Plus Micro kit (Qiagen, 74034). cDNA was synthesized using the SuperScript® IV First-Strand Synthesis System (Invitrogen, 18091050) following the manufacturer's instructions. Taqman probes (GLB1 [Hs01035168]-FAM and GAPDH [Hs02758991_g1]-VIC) were purchased from Thermo Fisher Scientific. The PCR reaction was initialized with 2 min at 50 °C and 10 min at 95 °C followed by 40 cycles of 15 s at 95 °C and 60 s at 60 °C.
4.6. Generation of cerebral organoids
Cerebral organoids were generated using the parental (isogenic control) and GBL1-edited hiPSC lines as described previously [25,32], with the following minor modifications. Single-cell suspensions were produced by detaching cells with Accutase for 10 min at 37 °C. Aggrewell™400 plates (STEMCELL Technologies) and Aggrewell™ EB Formation Medium (STEMCELL Technologies) were used following the manufacturer's protocols during days 0–3 of EB formation. On days 2–10, EBs were transferred to 24-well ultra-low adherence plates and cultured in neural induction media, beginning on day 5, with media changes every other day. The resulting organoids were embedded in Matrigel matrix droplets on day 10 and grown in neural differentiation media supplemented with B-27 without vitamin A (B-27-) for 4 days in 10 mm dish. After this, organoids were grown in neural differentiation media supplemented with B-27 plus vitamin A (B-27+)in suspension in 125 ml spinner flasks, on a low-speed microstirrer (Wheaton) at 25 rpm in a 5% CO2 tissue-culture incubator. Organoids were harvested at weeks 5, 10, 15, and 20 for analysis.
4.7. X-gal staining
Whole cerebral organoids were stored at −80 °C and, without thawing, fixed in 2% glutaraldehyde/2% formaldehyde in PBS for 10 min at room temperature. Organoids were incubated in ice-cold citrate phosphate buffer (CPB, pH 4.2) for 20 min, then incubated in X-gal working solution containing 40 mg/ml 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal, Invitrogen, 15520-018), 20 mM K3Fe(CN)6 (Sigma-Aldrich), 20 mM K4Fe(CN)6 (Sigma-Aldrich), 0.02% IGEPAL (Sigma-Aldrich), 0.01% deoxycholic acid (Sigma-Aldrich), and 2 mM MgCl2 (Mallinckrodt) in CPB at 37 °C for 30 min to 2 h. The tissue was rinsed first with CPB and then with PBS, and imaged using a dissecting microscope.
4.8. Organoid β-galactosidase activity
Whole organoids were stored at −80 °C and, without thawing, homogenized in 0.1 M citric buffer (pH 4.2) containing 0.1% Triton X-100 and placed on ice for 30 min. β-gal activity was measured with 4 mM 4MU β-D-galactopyranoside (Sigma-Aldrich) and was normalized with β-hexosaminidase activity using 20 mM 4MU N-acetyl-β-D-glucosaminide (Sigma-Aldrich). Isogenic control organoid lysate was subjected to the same analysis to serve as a reference value for normal (100%) β-gal activity. Both reactions were performed at 37 °C for 30 min and stopped with 200 μl 0.2 M glycine (pH 9.6). Fluorescence was measured on a Perkin Elmer Victor 1420 Multilabel Counter (excitation: 360 nm, emission: 446 nm).
4.9. Histological analysis
Human fibroblasts were cultured in chamber slides and fixed in 4% paraformaldehyde in PBS at 4 °C for 15 min and then washed with PBS. Cerebral organoids were fixed in 4% paraformaldehyde in PBS at 4 °C for 15 min, washed with PBS, and then equilibrated overnight in 30% sucrose prior to OCT compound embedding and sectioning. Frozen OCT-embedded blocks of normal and GM1 gangliosidosis cat brain tissue were obtained from Dr. Douglas R. Martin, College of Veterinary Medicine, Auburn University. Frozen OCT-embedded blocks of normal and GM1 gangliosidosis mouse brain were obtained from Dr. Alessandra d'Azzo, St. Jude Children's Research Hospital. Fixed cells or brain sections were incubated with PBS for 15 min and permeabilized with 0.2% Triton in PBS for 10 min, washed with PBS, and then blocked with 1% bovine serum albumin (BSA) and 5% horse serum in PBS at room temperature for 1 h. Sections were then incubated in 1% BSA and 5% horse serum in PBS overnight with the following primary antibodies: anti-GM1 ganglioside (mouse monoclonal antibody, Amsbio clone GMB16, 1:50); anti-β3 tubulin (mouse, previously Convance, clone TUJ1, 1:750); anti-SOX2 (rabbit, Chemicon, AB5603, 1:300); anti-GFAP (rabbit, Abcam, ab7260, 1:500); or anti-LAMP1 (mouse, Santa Cruz Biotechnology, H5G11, 1:200). Secondary antibodies were goat Alexa Fluor 488- and 594-conjugates (Invitrogen, 1:500 or 1:1000). Antibody-stained cryosections were mounted with Fluoroshield Mounting Medium with DAPI (Abcam) and observed using a spectral confocal microscope (LSM 780; Carl Zeiss, Inc.). Fluorescent micrographs were captured using Zen 2012 software (Carl Zeiss, Inc.). Immunofluorescence was quantified by extracting mean fluorescence for each colour channel using Fiji [45,46] (ImageJ) and GM1 ganglioside fluorescence intensity was normalized to DAPI fluorescence intensity. Co-localization analysis of GM1 ganglioside and LAPM1 expression was performed using the plot profile analysis tool in Fiji on images of organoid sections stained with antibodies against GM1 ganglioside and LAMP1. A single pixel-wide line was drawn across areas of GM1 ganglioside signal and the intensity for GM1 ganglioside fluorescence (green channel) and LAMP1 (red channel) along the line were determined and plotted.
Organoids were sectioned and stained with hematoxylin eosin (H&E) or processed for transmission electron microscopy (EM) as described in [37].
4.10. Ganglioside extraction and purification
Total gangliosides were extracted from pooled organoids (n = 3) by mechanically disrupting the organoids in 19 volumes of chloroform:methanol (2:1, v:v) and shaking for 1 h at room temperature. A second extraction was done with half the original extraction volume and shaking for another hour at room temperature. The two extraction solvents were combined and dried under nitrogen. The dried lipids were re-dissolved in chloroform:methanol (2:1, v:v) and partitioned with 0.1 M KCl (1:0.2, v:v). The organic phase was reserved for phospholipid quantitation as described previously [47] to normalize GM1 content. The aqueous phase containing the gangliosides was de-salted using reverse phase C18 Sep-Pak columns (Waters, WAT023590). The column eluate containing the gangliosides was dried under nitrogen and re-dissolved in chloroform:methanol (1:1, v:v).
4.11. HPTLC and ganglioside analysis
HPTLC was performed with pre-coated 10 × 20 cm, 250 μm thick silica gel HPTLC plates (Merck). Each plate was loaded with mixed gangliosides, including GM1, GM2, and GM3, as a reference marker (catalogue #: 1508, Matreya). Total gangliosides extracted from pooled cerebral organoids (n = 3) were applied in 0.7 cm lanes using a 10 μl syringe. The HPTLC plates were developed in a glass running chamber containing chloroform:methanol:0.25% CaCl2 (60:35:8, v:v:v) previously equilibrated for 1 h. Plates were allowed to dry completely and then sprayed with 2% resorcinol, covered with glass, and baked at 125 °C for 10 min.
Densitometry was performed to determine GM1 content using the gel analysis tool in Fiji [45,46]. Relative GM1 content was determined by normalizing the area under the GM1 peak to the corresponding total phospholipid concentrations in the organic phase.
4.12. AAVrh9 injections
AAVrh9 (AAV9) vectors carrying the human β-gal gene (AAV9-GLB1) or GFP (AAV9-GFP) were prepared as previously described [31]. Using a 32-gauge Hamilton 5 μl syringe (Hamilton), 1 μl of AAV9-GLB1 (3 × 1013 vg/ml), or AAV9-GFP vector (3 × 1013 vg/ml) as control, was injected into the center of each cerebral organoid at 10 weeks. Organoids were cultured and analyzed 5 weeks later, at 15 weeks. To control for possible GFP expression during immunohistochemistry, DAPI-only stained sections of AAV9-GFP-injected organoids were used to set the threshold for background fluorescence to exclude GFP.
4.13. Statistics
Statistical analyses were performed using the Student's t-test or one-way ANOVA to compare results between groups. P ≤ .05 was considered statistically significant.
The following are the supplementary data related to this article.
Supplementary material
Funding
This work was supported by the Intramural Research Programs of the National Human Genome Research Institute; and the National Institute of Diabetes and Digestive and Kidney Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Acknowledgments
Acknowledgements
The authors thank Dr. Douglas Martin for GM1 feline brain tissue and Dr. Alessandra D'Azzo for GM1 mouse brain tissue.
Declaration of competing interests
The authors declare no conflicts of interest.
References
- 1.Regier D.S., Tifft C.J., Adam M.P. In: Seattle (WA): University of Washington, Seattle; 1993–2017. Pagon R.A., Ardinger H.H., editors. 2013. https://www.ncbi.nlm.nih.gov/books/NBK164500/ Available from: GeneReviews® [Internet], in press. [Google Scholar]
- 2.Yu R.K., Tsai Y.T., Ariga T. Functional roles of gangliosides in neurodevelopment: an overview of recent advances. Neurochem. Res. 2012;37:1230–1244. doi: 10.1007/s11064-012-0744-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunetti-Pierri N., Scaglia F. GM1 gangliosidosis: review of clinical, molecular, and therapeutic aspects. Mol. Genet. Metab. 2008;94:391–396. doi: 10.1016/j.ymgme.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 4.Caciotti A., Garman S.C., Rivera-Colón Y., Procopio E., Catarzi S., Ferri L., Guido C., Martelli P., Parini R., Antuzzi D. GM1 gangliosidosis and Morquio B disease: an update on genetic alterations and clinical findings. Biochim. Biophys. Acta. 2011;1812:782–790. doi: 10.1016/j.bbadis.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki Y., Nanba E., Matsuda J., Higaki K., Oshima A. The McGraw-Hill Companies, Inc.; New York, NY: 2014. The Online Metabolic and Molecular Bases of Inherited Disease. Beaudet, A.L., Vogelstein, B., Kinzler, K.W., Antonarakis, S.E., Ballabio, A., Gibson, K.M. and Mitchell, G. (eds.) (in press) [Google Scholar]
- 6.Blakemore W. GM1 gangliosidosis in a cat. J. Comp. Pathol. 1972;82:179–185. doi: 10.1016/0021-9975(72)90061-8. [DOI] [PubMed] [Google Scholar]
- 7.Handa S., Yamakawa T. Biochemical studies in cat and human Gangliosidosis1, 2. J. Neurochem. 1971;18:1275–1280. doi: 10.1111/j.1471-4159.1971.tb00226.x. [DOI] [PubMed] [Google Scholar]
- 8.Farrell D.F., Baker H.J., Herndon R.M., Lindsey J.R., McKHANN G.M. Feline GM1 gangliosidosis: biochemical and ultrastructural comparisons with the disease in man. J. Neuropathol. Exp. Neurol. 1973;32:1–18. [PubMed] [Google Scholar]
- 9.Baker H.J., Lindsey J.R., McKhann G.M., Farrell D.F. Neuronal GM1 gangliosidosis in a Siamese cat with β-galactosidase deficiency. Science. 1971;174:838–839. doi: 10.1126/science.174.4011.838. [DOI] [PubMed] [Google Scholar]
- 10.Skelly B.J., Jeffrey M., Franklin R.J., Winchester B.G. A new form of ovine GM1-gangliosidosis. Acta Neuropathol. 1995;89:374–379. doi: 10.1007/BF00309632. [DOI] [PubMed] [Google Scholar]
- 11.Ahern-Rindell A.J., Prieur D.J., Murnane R.D., Raghavan S.S., Daniel P.F., McCluer R.H., Walkley S.U., Parish S.M., Opitz J.M., Reynolds J.F. Inherited lysosomal storage disease associated with deficiencies of β-galactosidase and α-neuraminidase in sheep. Am. J. Med. Genet. A. 1988;31:39–56. doi: 10.1002/ajmg.1320310108. [DOI] [PubMed] [Google Scholar]
- 12.Murnane R., Ahern-Rindell A., Prieur D. Ovine GM1 gangliosidosis. Small Rumin. Res. 1991;6:109–118. [PubMed] [Google Scholar]
- 13.Shell L.G., Potthoff A., Katherman A., Saunders G.K., Wood P.A., Giger U., Carithers R. Neuronal-visceral GM1 gangliosidosis in Portuguese water dogs. J. Vet. Intern. Med. 1989;3:1–7. doi: 10.1111/j.1939-1676.1989.tb00320.x. [DOI] [PubMed] [Google Scholar]
- 14.Saunders G., Wood P., Myers R., Shell L., Carithers R. GM1 gangliosidosis in Portuguese water dogs: pathologic and biochemical findings. Vet. Pathol. 1988;25:265–269. doi: 10.1177/030098588802500403. [DOI] [PubMed] [Google Scholar]
- 15.Read D.H., Harrington D.D., Keenana T., Hinsman E.J. Neuronal-visceral GM1 gangliosidosis in a dog with beta-galactosidase deficiency. Science. 1976;194:442–445. doi: 10.1126/science.824730. [DOI] [PubMed] [Google Scholar]
- 16.Alroy J., Orgad U., Ucci A.A., Schelling S.H., Schunk K.L., Warren C.D., Raghavan S.A., Kolodny E.H. Neurovisceral and skeletal GM1-gangliosidosis in dogs with beta-galactosidase deficiency. Science. 1985;229:470–473. doi: 10.1126/science.3925555. [DOI] [PubMed] [Google Scholar]
- 17.Donnelly W., Sheahan B., Rogers T. GM1 gangliosidosis in Friesian calves. J. Pathol. 1973;111:173–179. doi: 10.1002/path.1711110305. [DOI] [PubMed] [Google Scholar]
- 18.Muthupalani S., Torres P.A., Wang B.C., Zeng B.J., Eaton S., Erdelyi I., Ducore R., Maganti R., Keating J., Perry B.J. G M1-gangliosidosis in American black bears: clinical, pathological, biochemical and molecular genetic characterization. Mol. Genet. Metab. 2014;111:513–521. doi: 10.1016/j.ymgme.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Hahn C.N., del Pilar Martin M., Schröder M., Vanier M.T., Suzuki K., Hara Y., Suzuki K., d'Azzo A. Generalized CNS disease and massive GM1-ganglioside accumulation in mice defective in lysosomal acid β-galactosidase. Hum. Mol. Genet. 1997;6:205–211. doi: 10.1093/hmg/6.2.205. [DOI] [PubMed] [Google Scholar]
- 20.Jeyakumar M., Thomas R., Elliot-Smith E., Smith D.A., van der Spoel A.C., d'Azzo A., Hugh Perry V., Butters T.D., Dwek R.A., Platt F.M. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain. 2003;126:974–987. doi: 10.1093/brain/awg089. [DOI] [PubMed] [Google Scholar]
- 21.Matsuda J., Suzuki O., Oshima A., Ogura A., Noguchi Y., Yamamoto Y., Asano T., Takimoto K., Sukegawa K., Suzuki Y. β-Galactosidase-deficient mouse as an animal model for GM1-gangliosidosis. Glycoconj. J. 1997;14:729–736. doi: 10.1023/a:1018573518127. [DOI] [PubMed] [Google Scholar]
- 22.Matsuda J., Suzuki O., Oshima A., Ogura A., Naiki M., Suzuki Y. Neurological manifestations of knockout mice with β-galactosidase deficiency. Brain Dev. 1997;19:19–20. doi: 10.1016/s0387-7604(96)00077-0. [DOI] [PubMed] [Google Scholar]
- 23.Avior Y., Sagi I., Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016;17:170–182. doi: 10.1038/nrm.2015.27. [DOI] [PubMed] [Google Scholar]
- 24.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lancaster M.A., Renner M., Martin C.-A., Wenzel D., Bicknell L.S., Hurles M.E., Homfray T., Penninger J.M., Jackson A.P., Knoblich J.A. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amin N.D., Pasca S.P. Building models of brain disorders with three-dimensional organoids. Neuron. 2018;100:389–405. doi: 10.1016/j.neuron.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 27.McCurdy V.J., Johnson A.K., Gray-Edwards H., Randle A.N., Brunson B.L., Morrison N.E., Salibi N., Johnson J.A., Hwang M., Beyers R.J. Sustained normalization of neurological disease after intracranial gene therapy in a feline model**. Sci. Transl. Med. 2014;6:231ra248. doi: 10.1126/scitranslmed.3007733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broekman M.L.D., Baek R.C., Comer L.A., Fernandez J.L., Seyfried T.N., Sena-Esteves M. Complete correction of enzymatic deficiency and neurochemistry in the GM1-gangliosidosis mouse brain by neonatal adeno-associated virus–mediated gene delivery. Mol. Ther. 2007;15:30–37. doi: 10.1038/sj.mt.6300004. [DOI] [PubMed] [Google Scholar]
- 29.Baek R.C., Broekman M.L.D., Leroy S.G., Tierney L.A., Sandberg M.A., d'Azzo A., Seyfried T.N., Sena-Esteves M. AAV-mediated gene delivery in adult GM1-gangliosidosis mice corrects lysosomal storage in CNS and improves survival. PLoS One. 2010;5 doi: 10.1371/journal.pone.0013468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Broekman M.L.D., Tierney L.A., Benn C., Chawla P., Cha J.H., Sena-Esteves M. Mechanisms of distribution of mouse [beta]-galactosidase in the adult GM1-gangliosidosis brain. Gene Ther. 2008;16:303–308. doi: 10.1038/gt.2008.149. [DOI] [PubMed] [Google Scholar]
- 31.Weismann C.M., Ferreira J., Keeler A.M., Su Q., Qui L., Shaffer S.A., Xu Z., Gao G., Sena-Esteves M. Systemic AAV9 gene transfer in adult GM1 gangliosidosis mice reduces lysosomal storage in CNS and extends lifespan. Hum. Mol. Genet. 2015;24:4353–4364. doi: 10.1093/hmg/ddv168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lancaster M.A., Knoblich J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014;9:2329–2340. doi: 10.1038/nprot.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Civallero G., Michelin K., de Mari J., Viapiana M., Burin M., Coelho J.C., Giugliani R. Twelve different enzyme assays on dried-blood filter paper samples for detection of patients with selected inherited lysosomal storage diseases. Clin. Chim. Acta. 2006;372:98–102. doi: 10.1016/j.cca.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 34.Ledeen R.W., Wu G. The multi-tasked life of GM1 ganglioside, a true factotum of nature. Trends Biochem. Sci. 2015;40:407–418. doi: 10.1016/j.tibs.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 35.Rosengren B., Mansson J.E., Svennerholm L. Composition of gangliosides and neutral glycosphingolipids of brain in classical Tay-Sachs and Sandhoff disease: more lyso-GM2 in Sandhoff disease? J. Neurochem. 1987;49:834–840. doi: 10.1111/j.1471-4159.1987.tb00969.x. [DOI] [PubMed] [Google Scholar]
- 36.Allende M.L., Cook E.K., Larman B.C., Nugent A., Brady J.M., Golebiowski D., Sena-Esteves M., Tifft C.J., Proia R.L. Cerebral organoids derived from Sandhoff disease-induced pluripotent stem cells exhibit impaired neurodifferentiation. J. Lipid Res. 2018;59:550–563. doi: 10.1194/jlr.M081323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kobayashi T., Goto I., Okada S., Orii T., Ohno K., Nakano T. Accumulation of lysosphingolipids in tissues from patients with GM1 and GM2 gangliosidoses. J. Neurochem. 1992;59:1452–1458. doi: 10.1111/j.1471-4159.1992.tb08460.x. [DOI] [PubMed] [Google Scholar]
- 38.Camp J.G., Badsha F., Florio M., Kanton S., Gerber T., Wilsch-Bräuninger M., Lewitus E., Sykes A., Hevers W., Lancaster M. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. 2015;112:15672–15677. doi: 10.1073/pnas.1520760112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lowden J.A., Cutz E., Conen P.E., Rudd N., Doran T.A. Prenatal diagnosis of GM1-gangliosidosis. N. Engl. J. Med. 1973;288:225–228. doi: 10.1056/NEJM197302012880502. [DOI] [PubMed] [Google Scholar]
- 40.Bosch M.E., Kielian T. Neuroinflammatory paradigms in lysosomal storage diseases. Front. Neurosci. 2015;9:417. doi: 10.3389/fnins.2015.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pasca S.P. The rise of three-dimensional human brain cultures. Nature. 2018;553:437–445. doi: 10.1038/nature25032. [DOI] [PubMed] [Google Scholar]
- 42.Mansour A.A., Goncalves J.T., Bloyd C.W., Li H., Fernandes S., Quang D., Johnston S., Parylak S.L., Jin X., Gage F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018;36:432–441. doi: 10.1038/nbt.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pham M.T., Pollock K.M., Rose M.D., Cary W.A., Stewart H.R., Zhou P., Nolta J.A., Waldau B. Generation of human vascularized brain organoids. Neuroreport. 2018;29:588–593. doi: 10.1097/WNR.0000000000001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wendeler M., Sandhoff K. Hexosaminidase assays. Glycoconj. J. 2009;26:945–952. doi: 10.1007/s10719-008-9137-5. [DOI] [PubMed] [Google Scholar]
- 45.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stewart J.C.M. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 1980;104:10–14. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]
- 48.Ohto U., Usui K., Ochi T., Yuki K., Satow Y., Shimizu T. Crystal structure of human β-galactosidase: structural basis of GM1 gangliosidosis and Morquio B diseases. J. Biol. Chem. 2012;287:1801–1812. doi: 10.1074/jbc.M111.293795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material