

Abstract
The synergistic nature of bicomponent catalysts remains a challenging issue, due to the difficulty in constructing well-defined catalytic systems. Here we study the origin of synergistic effects in CoOx-Pt catalysts for selective hydrogenation by designing a series of closely contacted CoOxPt/TiO2 and spatially separated CoOx/TiO2/Pt catalysts by atomic layer deposition (ALD). For CoOx/TiO2/Pt, CoOx and platinum are separated by the walls of titania nanotubes, and the CoOx-Pt intimacy can be precisely tuned. Like CoOxPt/TiO2, the CoOx/TiO2/Pt shows higher selectivity to cinnamyl alcohol than monometallic TiO2/Pt, indicating that the CoOx-Pt nanoscale intimacy almost has no influence on the selectivity. The enhanced selectivity is ascribed to the increased oxygen vacancy resulting from the promoted hydrogen spillover. Moreover, platinum-oxygen vacancy interfacial sites are identified as the active sites by selectively covering CoOx or platinum by ALD. Our study provides a guide for the understanding of synergistic nature in bicomponent and bifunctional catalysts.
Subject terms: Catalysis, Catalyst synthesis, Catalytic mechanisms, Heterogeneous catalysis
The development of catalysts with high activity and selectivity for hydrogenation remains a challenge. Here the authors report cobalt oxide-platinum catalysts with increased oxygen vacancy resulting from the promoted hydrogen spillover with platinum-oxygen vacancies as active sites.
Introduction
Bicomponent catalysts have received considerably increasing interests in the recent decades due to the enhanced catalytic properties compared with their single-component counterparts, arising from direct contact between the two components, which can be called as synergistic effects1,2. However, due to the limitations in controlling the catalyst microstructures and precisely tuning bicomponent intimacy by the traditional methods, it is still a challenging issue to understand the origin of synergistic effects (i.e., short- or long-range interactions) and identify the active sites.
The general belief is that the bicomponents should be as close as possible (i.e., short-range interactions of bicomponents) to promote electron interaction and/or construct interfaces to achieve effective catalysis. In this case, the interaction of a metal oxide promoter with a metal particle is puzzling due to the presence of multiple potential catalytic active sites under the reaction conditions. In contrast, some recent reports revealed the effects of intimacy on the catalyst performance when the bicomponents are separated and tuned at nanoscale and even millimeter-scale distance (i.e., long-range interactions of bicomponents)3–10. For example, Coville and coworkers investigated the effect of Ru and Co intimacy on the activity and selectivity of Co catalysts using conventional incipient wetness impregnation in the typical Fischer–Tropsch reaction5. Bokhoven and coworkers prepared a series of model catalyst samples with precisely controlled Pt–FeOx intimacy at nanoscale distances using the more precise nanolithography technique, and investigated the hydrogen spillover effects on TiO2 and Al2O3 supports7. But no reaction (not possible for such structure) is conducted to verify the proposed hydrogen spillover mechanism under the real reaction conditions. Therefore, well-defined bicomponent catalytic systems are highly desirable to unravel the origin of synergistic effects under the real reaction conditions.
Atomic layer deposition (ALD) is a powerful thin-film technique for synthesis of conformal thin films and highly dispersed nanoparticles11–16. Many advanced nanocatalysts have been designed and synthesized by ALD in the past decade17–26. Herein, we investigate the effects of CoOx–Pt intimacy on the selective hydrogenation of cinnamaldehyde (CALD) and the hydrogen spillover effects based on CoOxPt/TiO2 and well-designed spatially separated structures of CoOx/TiO2/Pt catalysts by a highly controllable and reliable ALD approach. Compared with the CoOxPt/TiO2, the CoOx/TiO2/Pt catalysts exhibit similar catalytic activity and hydrogenation selectivity for the CALD hydrogenation, though the CoOx–Pt intimacy of the two catalysts is dramatically different. This long-range promoter effect is not broken even when we selectively deposit additional coating layers on the CoOx surfaces by ALD. The enhanced selectivity is ascribed to the promoted hydrogen spillover and thus increased oxygen vacancies (Ov). The well-designed structures can be synthesized and used to study the synergetic natures of other bicomponent and bifunctional catalysts with enhanced performance for other reactions.
Results
Synthesis and characterization of the catalysts
The closely contacted CoOxPt/TiO2 and spatially separated CoOx/TiO2/Pt catalysts were synthesized by a template-assisted ALD strategy (Fig. 1 and Supplementary Scheme 1)26. For the synthesis of CoOxPt/TiO2, an amorphous TiO2 film was first deposited on carbon nanocoils (CNCs) used as templates by ALD, obtaining TiO2/CNCs, and then Pt nanoparticles were deposited on the TiO2/CNCs by Pt ALD, obtaining Pt/TiO2/CNCs. Subsequently, the CNCs were removed by calcination under an air atmosphere, producing Pt/TiO2 catalysts with anatase nanotubes. Lastly, CoOx nanoparticles were deposited on the outer surface of the Pt/TiO2 through CoOx ALD, producing CoOxPt/TiO2 catalysts with the closest CoOx–Pt intimacy (Supplementary Scheme 1a). The CoOx/TiO2/Pt catalysts were prepared by exchanging the deposition sequence of TiO2 and Pt (Supplementary Scheme 1b). The CoOx–Pt intimacy can be precisely controlled by adjusting the thickness of TiO2 layer. In addition, the CoOx/TiO2/Pt/TiO2 and Al2O3/CoOx/TiO2/Pt catalysts (Fig. 1) were also prepared by selectively covering Pt with TiO2 and CoOx nanoparticles with Al2O3, respectively, to identify the real active sites in the bicomponent catalysts.
Fig. 1.

Schematic illustration of the catalysts. Semi-sectional and cross-sectional views of the different catalysts prepared by ALD. The yellow and black balls represent Pt and CoOx, respectively
Figure 2 shows the transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) images of the CoOxPt/TiO2 and CoOx/TiO2/Pt catalysts, prepared with 150, 20 and 300 cycles for CoOx, Pt and TiO2 deposition, respectively. The Pt and CoOx nanoparticles, with average sizes of 2.9 nm and 4.3 nm, respectively (Supplementary Figs. 1 and 2), are highly dispersed on the outer surface of the anatase TiO2 nanotubes with a wall thickness of ~13.7 nm, in which CoOx is deposited on/next to Pt forming CoOx–Pt interfaces (Fig. 2a, b). The measured lattice distances of the nanoparticles are ~0.223 nm and ~0.246 nm (Fig. 2b), which correspond to the Pt(111) and CoO (111) planes, respectively27,28. The larger lattice distance of 0.350 nm matches well with (101) planes of anatase TiO226. For the CoOx/TiO2/Pt catalyst (Fig. 2c, d), Pt nanoparticles (indicated by yellow circles) with an average particle size of 3.1 nm (Supplementary Fig. 3) are uniformly deposited on the inner surface of the TiO2 nanotubes, while CoOx nanoparticles are uniformly deposited on the outer surface of the TiO2 nanotubes (Supplementary Fig. 4). In CoOxPt/TiO2 catalysts, the Pt nanoparticles are closely contacted with CoOx nanoparticles, while in CoOx/TiO2/Pt catalysts, the average CoOx–Pt distance is corresponded to the TiO2 thickness. Thus the average CoOx–Pt distance in CoOxPt/TiO2 is much smaller than that of CoOx/TiO2/Pt. The sample was further characterized by using high-angle annular dark-field scanning TEM (HAADF-STEM), and energy-dispersive X-ray spectroscopy (EDS) mapping (Fig. 2e and Supplementary Figs. 5 and 6), further confirming that the Pt and CoOx nanoparticles are separately decorated on the inner and outer surfaces of the TiO2 nanotubes, respectively.
Fig. 2.
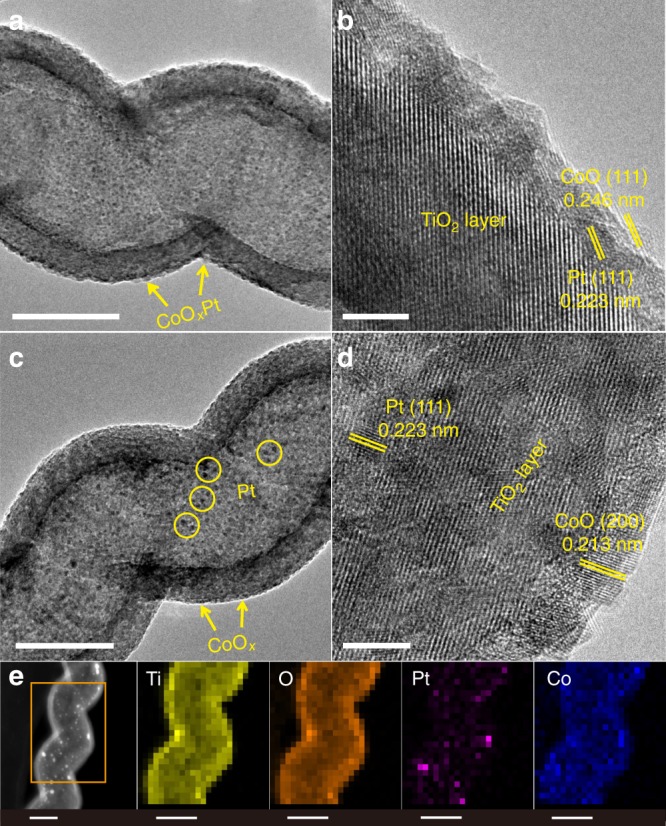
Structural characterizations of the two catalysts. TEM and HRTEM images of the CoOxPt/TiO2 (a, b) and CoOx/TiO2/Pt (c, d) catalysts. e STEM image of the CoOx/TiO2/Pt catalysts and the corresponding EDS mapping profiles in rectangular area. Scale bars: a, c and e 50 nm; b and d 5 nm
Catalytic performance
The selective hydrogenation of C=O in α, β-unsaturated aldehydes (e.g., CALD) to value-added unsaturated alcohols has been of increasing interest for the production of fine chemicals and pharmaceutical precursors29–38. However, it remains challenging to achieve high-yield unsaturated alcohols since the hydrogenation of C=C is thermodynamically more favorable than that of C=O. To address the issue, various catalysts have been designed and synthesized, and Pt-based catalysts were found to be more desirable in the selective reduction of C=O than other metals29–38. Bitter and coworkers found that Pt particle size and oxygen groups on carbon nanofiber supports have a vital influence on the selectivity to cinnamyl alcohol (CALC)29. Chen and coworkers reported that Co–Pt/SiO2 bimetallic catalysts exhibit better catalytic activity and selectivity than Pt/SiO2 and Cu–Pt/SiO2 catalysts due to the electronic property modification of Pt30,31. Similar results were also reported by Tsang et al., i.e., the selective C=O hydrogenation can be achieved with high activity using Co-decorated Pt nanocrystals as nanocatalysts38.
To unravel the synergetic mechanism, the selective hydrogenation of CALD is selected as an example to evaluate the catalytic properties of the as-prepared catalysts, and the results are summarized in Table 1. Note that the Pt nanoparticles are located at different positions for the Pt/TiO2 and TiO2/Pt catalysts. For the monometallic TiO2/Pt catalyst with Pt loading of 3.7 wt.% (Supplementary Table 1), the main product is hydrocinnamaldehyde (HALD) obtained from hydrogenation of the C=C and the selectivity to HALD amounts to 62.5%, while the selectivity to the desired product CALC, obtained from hydrogenation of the C=O, is only 16.2% (Entry 1). For the CoOx/TiO2 reference catalyst, only trace conversion (0.9%) is detected under the same reaction conditions (Entry 2). For the CoOxPt/TiO2 catalysts with closely contacted Pt and CoOx, the selectivity to CALC is remarkably improved after addition of CoOx (Entry 3). Moreover, the product of excessive hydrogenation to hydrocinnamyl alcohol (HALC) is also obviously suppressed. Similar phenomenon is also found for the CoOx-supported Pt-based catalysts, and the Pt40/CoOx catalysts exhibit the best catalytic performance with 90.2% conversion and 80.3% selectivity to CALC (Supplementary Fig. 7). The improved selectivity to CALC is usually ascribed to the promoter effects, originating from electronic or structural modifications35–41. Unexpectedly, similar hydrogenation results are also achieved over the CoOx/TiO2/Pt (13.7 nm, 300-cycle TiO2) catalysts with nanoscale CoOx–Pt intimacy (Entry 4 and Supplementary Fig. 8). Moreover, only slight decrease in hydrogenation activity and selectivity is observed over the CoOx/TiO2(900)/Pt catalysts (Entry 5) when the TiO2 layer becomes much thicker (40.5 nm, 900-cycle TiO2, Supplementary Figs. 9 and 10).
Table 1.
Selective hydrogenation results of CALD over the different catalystsa
| Entry | Catalysts | Conversion (%) (CALD) | Selectivity (%) | ||
|---|---|---|---|---|---|
| (CALC) | (HALD) | (HALC) | |||
| 1 | TiO2/Pt | 93.1 | 16.2 | 62.5 | 21.7 |
| 2 | CoOx/TiO2 | 0.9 | – | – | – |
| 3 | CoOxPt/TiO2 | 92.4 | 82.1 | 14.5 | 3.3 |
| 4 | CoOx/TiO2/Pt | 91.3 | 81.5 | 13.9 | 4.3 |
| 5 | CoOx/TiO2(900)/Ptb | 89.8 | 80.3 | 14.3 | 4.7 |
| 6 | CoOx/TiO2/Pt/TiO2 | 8.8 | 52.1 | 47.4 | – |
| 7 | Al2O3/CoOx/TiO2/Ptc | 90.2 | 77.8 | 15.9 | 5.8 |
| 8 | Al2O3/Ptd | 31.8 | – | 100 | – |
| 9 | CoOx/Al2O3/Pt | 18.5 | 39.1 | 60.7 | – |
| 10 | CoOxPt/Al2O3 | 27.4 | 69.4 | 30.2 | – |
| 11 | CoOx/Al2O3 | 0.2 | – | – | – |
aReaction conditions: 30 mL of 0.8 mmol cinnamaldehyde in ethanol and 2 MPa H2 were placed in 50 mL stainless-steel autoclave (65 °C for 1.5 h)
bThe cycle number of TiO2 is 900 for the catalysts. For all other catalysts, the cycle number of TiO2 is 300
cDense Al2O3 layer is used to guarantee the complete covering of CoOx species
dThe cycle number of Al2O3 is 100 for the Al2O3-based catalysts (Entries 8–11)
Furthermore, control experiments were also carried out by selectively exposing Pt or CoOx to confirm the roles played by CoOx and Pt during the reaction. The hydrogenation conversion and selectivity to the desired CALC product decrease remarkably when Pt is selectively covered by two-layered TiO2 (CoOx/TiO2/Pt/TiO2, Entry 6). When CoOx is selectively covered by an outer dense Al2O3 layer of ~11.5 nm (Al2O3/CoOx/TiO2/Pt), no visible change is observed in the catalytic performance (Entry 7). These results indicate that Pt–TiO2 interface regions should be the main active sites for the CALD hydrogenation reaction, instead of CoOx. It should be noted that the selectivity of CALC hydrogenation is guaranteed to improve with CoOx addition due to the long-range promoter effect and that this effect is not broken even when the surface of CoOx promoter is entirely covered by a dense Al2O3 layer (Al2O3/CoOx/TiO2/Pt, Entry 7). This effect is altered when the TiO2 layer is changed to Al2O3 (~11.5 nm), i.e., the catalytic performance is considerably influenced by the CoOx–Pt intimacy (Entries 8–11). Closely contacted CoOxPt/Al2O3 catalysts exhibit higher activity and selectivity to the desired product than those of the separated CoOx/Al2O3/Pt catalysts with nanoscale intimacy. This should be related to the poor capability of spillover hydrogen transfer on the nonreducible Al2O37. In addition, the reusability of CoOx/TiO2/Pt for selective hydrogenation of CALD was also tested (Supplementary Fig. 11). There is no obvious decrease in the conversion after five catalytic cycles, indicating a good stability of the catalysts.
X-ray absorption fine structure measurements
The electronic states of the TiO2/Pt, CoOx/TiO2/Pt and CoOxPt/TiO2 catalysts before and during the hydrogenation reaction processes were investigated by ex situ and in situ X-ray absorption fine structure measurements (Fig. 3). Figure 3a shows the normalized Pt L3-edge ex situ X-ray absorption near-edge structure (XANES) spectra of the as-prepared catalysts, and the reference spectra of Pt foil and PtO2. The white line intensity of the as-prepared catalysts falls in the range between the Pt foil and PtO2, suggesting that the as-prepared Pt nanoparticles consist of a certain amount of PtOx in addition to metallic Pt0. Expanded view of the white lines (Supplementary Fig. 12) indicates the different electronic states of Pt for TiO2/Pt, CoOx/TiO2/Pt and CoOxPt/TiO2 catalysts. Figure 3b presents the normalized Co K-edge XANES spectra of the as-prepared catalysts before the reaction, and the reference spectra of Co foil, CoO and Co3O4. Obviously, the CoOx/TiO2/Pt catalysts also exhibit different white line shapes in comparison with the CoOxPt/TiO2 catalysts, indicating the different electronic states of the CoOx species between the two catalysts. These results demonstrate that before the reaction, the electronic interaction between CoOx promoter and Pt should exist (Supplementary Fig. 13). During the reaction process, the PtOx species of TiO2/Pt, CoOx/TiO2/Pt and CoOxPt/TiO2 are fully reduced to metallic Pt0 due to the presence of H2 (Fig. 3d). The Fourier transform spectra of the Pt L3-edge extended X-ray absorption fine structure (EXAFS) oscillations for these three catalysts also confirm the full reduction of PtOx during the reaction process (Supplementary Fig. 14a, b). For the CoOx/TiO2 reference catalysts, the white line shifts to a low energy during the reaction process (Fig. 3b and e), indicating that the cobalt oxide species at high valence could be reduced to relatively low valence states (e.g., from Co3O4 to CoO) under a H2 atmosphere without Pt assistance (Supplementary Fig. 14c, d). Lower energy shifts of the white lines can be observed for the CoOxPt/TiO2 and CoOx/TiO2/Pt catalysts during the reaction process, demonstrating that more cobalt oxide species with lower valence states are formed in the two catalysts compared with the CoOx/TiO2 catalysts, which can be ascribed to the presence of Pt contributing to the reduction of CoOx due to the hydrogen spillover effect. In addition, the CoOxPt/TiO2 and CoOx/TiO2/Pt catalysts exhibit almost the same white line shapes during the reaction process (Fig. 3e), i.e., the valence states of the formed cobalt oxide species of the two catalysts are nearly identical; this is due to the strong reduction ability of the spillover hydrogen (H·), generated on Pt, toward CoOx, which is nearly independent of the CoOx–Pt intimacy on reducible TiO2 supports in the nanoscale range7.
Fig. 3.

XANES spectra of the catalysts. a Ex situ and d in situ Pt L3-edge XANES spectra of TiO2/Pt, CoOx/TiO2/Pt, CoOxPt/TiO2, and reference Pt foil and PtO2; b Ex situ and e in situ Co K-edge XANES spectra of TiO2/Pt, CoOx/TiO2/Pt, CoOxPt/TiO2, and reference Co foil, CoO and Co3O4; c Ex situ and f in situ Ti K-edge XANES spectra of TiO2/Pt, CoOxPt/TiO2, CoOx/TiO2/Pt and reference TiO2
Figure 3c and f shows the normalized Ti K-edge ex situ and in situ XANES spectra of TiO2/Pt, CoOx/TiO2/Pt, CoOxPt/TiO2 and the reference spectra of TiO2. Before the reaction, the Ti K-edge white line curves of all the three catalysts coincide, indicating the same electronic states of their TiO2 supports. During the hydrogenation reaction conditions, in situ XANES spectra show reduction of Ti4+ to Ti3+ for all three catalysts, and CoOx/TiO2/Pt and CoOxPt/TiO2 possess more reduced Ti3+ than that of the TiO2/Pt catalyst. The k3-weighted EXAFS spectra of the three catalysts and the reference TiO2, and their fitted curves are presented in Supplementary Fig. 15. The fitted results (Supplementary Table 2) are consistent with the XANES results.
Chemisorption
Figure 4a shows the H2 temperature programmed reduction (H2-TPR) profiles of the catalysts. From the H2-TPR profiles of the CoOxPt/TiO2, CoOx/TiO2/Pt and CoOx/TiO2 catalysts (also see Supplementary Fig. 10), both a low temperature (110–220 °C, Co3O4 to CoO) and a high temperature (245–345 °C, CoO to Co0) reduction peak can be observed. No peaks related to platinum oxides are observed, probably because the amounts of H2 used for the reduction of PtOx species in the catalysts are beyond the lower detection limits of the detector. In addition, the reduction peak at higher temperature (~450 °C) could be assigned to the partial reduction of TiO2, indicating that TiO2 is more difficult to reduce42. Compared with the CoOx/TiO2 catalysts, the temperatures needed for CoOx reduction are much lower for the CoOxPt/TiO2 and CoOx/TiO2/Pt catalysts due to the presence of Pt, further revealing the effect of hydrogen spillover from Pt to CoOx31. Note that the reduction temperature needed for the CoOxPt/TiO2 catalyst is slightly lower than that of the CoOx/TiO2/Pt catalyst due to the closer CoOx–Pt intimacy (Fig. 4a). Figure 4b shows the H2 temperature programmed desorption (H2-TPD) profiles of the catalysts. The TiO2/Pt and CoOx/TiO2/Pt catalysts exhibit a much higher H2 adsorption ability than that of the CoOx/TiO2 and CoOxPt/TiO2 catalysts, indicating that Pt atoms are the adsorption and dissociation sites of H2, which is further verified by the following density functional theory (DFT) simulation.
Fig. 4.

Chemisorption characterizations. a H2-TPR and b H2-TPD profiles of the catalysts
Discussion
In this work, we discovered a hydrogen spillover effect for the CALD hydrogenation reactions. Hydrogen spillover is a reversible and dynamic equilibrium process. Modulation of the spillover conditions, such as temperature and gas partial pressure, hinders or facilitates the spillover process and can be used for controlling the oxide reduction7,43. Generally, the activation and dissociation of H2 molecules on Pt is quite easy and generally considered to be barrierless7,43–45, i.e., the energy needed to activate H2 molecules on Pt is much lower than that on CoOx and TiO2 (Supplementary Fig. 16). For the TiO2/Pt catalyst under the reaction conditions, hydrogen spillover occurs (Fig. 5a). Active hydrogen species migrate from Pt onto TiO2 supports, leading to the partial reduction of TiO2 supports (Ti4+ is reduced to Ti3+) with the formation of Ov around the interface regions7,46–50. When CoOx is added, in situ XAFS results show that CoOx can be reduced to lower valent states through hydrogen spillover under the reaction conditions. More hydrogen species are consumed by CoOx, thus the original equilibrium of hydrogen spillover is disturbed (Fig. 5a). The generation of active hydrogen species and their transfer process (from Pt to CoOx directly, or via TiO2 supports) are promoted, until new equilibrium is reached. The consumption of hydrogen species can be seen as the pumps for the migration of atomic hydrogen from Pt toward TiO2. The enhanced hydrogen spillover facilitates the reduction of TiO2 supports, which is proved by in situ Ti K-edge XANES spectra (Fig. 3c and f), accompanying the increased formation of Ov sites.
Fig. 5.

DFT calculation and proposed reaction mechanisms. a The effect mechanism of CoOx on the hydrogen spillover over TiO2-supported Pt catalysts. b The optimized structure for adsorption of CALD molecule using C=O (top) or C=C (bottom) double bond on the Ov of TiO2. Atom coloring: Ti, gray; O, red; H, white; C, black. c The possible enhancement mechanism for selective CALD hydrogenation reaction. (1), (2) and (3) represent TiO2/Pt, CoOxPt/TiO2 and CoOx/TiO2/Pt catalysts, respectively. For TiO2/Pt, the number of Ov is limited, leading to its low selectivity. For CoOxPt/TiO2 and CoOx/TiO2/Pt, the addition of CoOx promotes the formation of Ov sites through hydrogen spillover and thus improves the selectivity, regardless of whether CoOx and Pt are separated by a TiO2 layer or not
The adsorption energies of different adsorption configurations of a CALD molecule on the Ov formed on TiO2 were calculated (Fig. 5b). The calculated adsorption energy of the C=O bond adsorbed on the oxygen vacancies is −2.38 eV, which is larger than that of the C=C bond on the oxygen vacancies (−1.68 eV), indicating that the adsorption configurations of the CALD molecule through the C=O bond on the Ov are more stable.
Based on the above discussions, the possible enhancement mechanism is proposed. For the TiO2/Pt catalyst, the number of Ov at the Pt–TiO2 interface is limited under the reaction conditions, leading to its low selectivity (Fig. 5c). When CoOx promoters are added, the selectivity to CALC is improved. The promotion can be ascribed to the CoOx promoter-induced increase of Ov sites through hydrogen spillover effect. For CoOx/TiO2/Pt catalysts, although CoOx is separated from the Pt catalyst by a TiO2 layer, this long-range CoOx promoter can still enhance the catalytic selectivity to CALC. XAFS results demonstrate that the electronic states of CoOx of CoOxPt/TiO2 and CoOx/TiO2/Pt catalysts are almost the same during the reaction process, meaning that the active hydrogen species generated on Pt nanoparticles are still consumed by CoOx, though CoOx is far from Pt. The energy barrier for the transfer of an electron and a proton among the different TiO2 sites can be easily overcome under the reaction conditions7, and thus the distance of hydrogen spillover across TiO2 layer almost has no influence on the catalytic performance. The addition of CoOx results in the increased generation of Ov sites on the Pt–TiO2 interface regions and the remarkably improved selectivity to CALC.
To further clarify the long-range promoter effect (i.e., the role of CoOx played), we selectively deposit additional oxide layers on the surfaces of CoOx or Pt by ALD. From the XAFS results, it can be concluded that CoOx species in the catalysts can be reduced in two ways. Firstly, the CoOx species can be reduced under H2 atmosphere; secondly, the CoOx species can be further reduced to a lower valence state by the active H species spilled from Pt nanoparticles through TiO2 support. When Pt is selectively covered by TiO2 (CoOx/TiO2/Pt/TiO2), the access of CALD molecules to Pt is nearly blocked due to the diffusion limitation (Supplementary Figs. 17 and 18, Supplementary Table 3), leading to the decreased catalytic performance (Entry 6, Table 1). On the contrary, when CoOx is selectively covered by a dense Al2O3 layer (Al2O3/CoOx/TiO2/Pt), the first way is blocked (Supplementary Fig. 18), while the second way is still preserved and CALD molecules can still have access to Pt via open the ends of the TiO2 nanotubes (with a diameter of ~70–90 nm), leading to a nearly unchanged catalytic performance (Entry 7, Table 1). These results further demonstrate that H spillover way is the main path to improve selectivity, and that Pt–Ov interface instead of CoOx should be the active site for the CALD hydrogenation reaction.
In summary, we have successfully developed a general method based on template-assisted ALD to synthesize closely contacted CoOxPt/TiO2 and spatially separated CoOx/TiO2/Pt catalysts, achieving the precise tuning of CoOx–Pt intimacy by varying the deposition sequence and the wall thickness of the TiO2 nanotubes. We discover that hydrogen spillover effect generated by the addition of CoOx can cause the increase in Ov, providing the adsorption sites for CALD via C=O bond and correspondingly resulting in the enhancement of selectivity to CALC in the CALD hydrogenation reactions. This hydrogen spillover effect is not broken even when we selectively deposit additional oxide layers on the CoOx promoter by ALD to cover its surface entirely. Our work has demonstrated an efficient strategy based on ALD for the design of various bicomponent catalysts with distinct promoter–metal intimacy and the assembly of metals and oxide supports, which is helpful to reveal the origin of bicomponent synergy, the enhancement mechanism and the real active sites.
Methods
Synthesis of CNCs and the ALD process
The detailed CNC synthesis process and the ALD process can be found in our previous reports24–26.
Synthesis of TiO2/Pt catalysts
CNCs were firstly decorated with Pt nanoparticles by Pt ALD (20 cycles) and then coated with a TiO2 layer (300 cycles) by TiO2 ALD, producing TiO2/Pt/CNCs. After the ALD processes, TiO2/Pt/CNCs were calcinated at 500 °C for 2 h in air to remove the CNC templates, obtaining TiO2/Pt catalysts, in which Pt nanoparticles were confined in TiO2 nanotubes.
Synthesis of CoOx/TiO2/Pt catalysts
The above-obtained TiO2/Pt catalysts were then deposited with cobalt oxides (denoted as CoOx) by CoOx ALD (150 cycles), obtaining shell-isolated CoOx/TiO2/Pt catalysts.
Synthesis of CoOxPt/TiO2 catalysts
CNCs were firstly coated with a TiO2 amorphous film by TiO2 ALD, and then Pt nanoparticles were deposited on the TiO2/CNCs by Pt ALD obtaining Pt/TiO2/CNCs composites. Subsequently, CNCs were removed by calcination under an air atmosphere producing Pt/TiO2 catalysts with porous anatase nanotubes. Lastly, CoOx nanoparticles were deposited on the outer surface of Pt/TiO2 through CoOx ALD, producing CoOxPt/TiO2 catalysts with the closest CoOx–Pt intimacy.
Synthesis of CoOx/TiO2/Pt/TiO2 catalysts
CNCs were first coated with a TiO2 layer (150 cycles) by TiO2 ALD and then decorated with Pt nanoparticles by Pt ALD and another TiO2 layer (300 cycles) producing TiO2/Pt/TiO2/CNCs. After the ALD processes, TiO2/Pt/TiO2/CNCs were calcinated at 500 °C for 2 h in air to remove the CNC templates, obtaining sandwich-like TiO2/Pt/TiO2 catalysts, in which Pt nanoparticles are coated by the two-layer TiO2. Lastly, the obtained TiO2/Pt/TiO2 catalysts were decorated with CoOx nanoparticles by CoOx ALD producing CoOx/TiO2/Pt/TiO2.
Synthesis of Al2O3/CoOx/TiO2/Pt catalysts
CNCs were sequentially decorated with Pt nanoparticles by CoOx ALD, coated with a TiO2 layer (300 cycles) by TiO2 ALD, decorated with Pt nanoparticles by Pt ALD and then coated with an Al2O3 layer (100 cycles) by Al2O3 ALD producing Al2O3/CoOx/TiO2/Pt/CNCs, which were calcinated at 550 °C for 2 h in air to remove the CNC templates obtaining Al2O3/CoOx/TiO2/Pt catalysts, in which CoOx nanoparticles were covered by the outer Al2O3 layer.
Catalyst characterizations
TEM and HRTEM images were taken with a JEOL-2100F field-emission transmission electron microscope operated at 200 kV. HAADF-STEM images and EDS mapping profiles were collected on a JEOL ARM-200F field-emission transmission electron microscope operated at 200 kV. The X-ray diffraction (XRD) patterns were recorded by using a Philips X’Pert Pro Super X-ray diffractometer with Cu Kα radiation (λ = 1.540 Å) in the 2θ range from 10° to 90°. The X-ray photoelectron spectra (XPS) were collected on an ESCALab-250 X-ray photoelectron spectrometer with an Al Kα source (1486.6 eV). The XANES and EXAFS spectra of the Pt L3-edge and Co K-edge were measured on the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF), Shanghai Institute of Applied Physics (SINAP), China, operated at 3.5 GeV. A Si (111) double-crystal monochromator was used to reduce the harmonic component of the monochrome beam. Pt foil, PtO2, Co foil, CoO and Co3O4 were used as reference samples and measured in the transmission mode, and all the catalysts were also measured in the transmission mode. The diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS, CO chemisorption) measurements were performed on a Bruker Vector 22 spectrometer. After the sample was loaded, it was firstly reduced by 10%H2/Ar at 150 °C for 1.5 h. Then the sample was cooled to 30 °C under 10%H2/Ar atmosphere, on which a background spectrum was collected. Subsequently, CO was introduced onto the sample until saturation, followed by Ar purge to remove the physically adsorbed gaseous CO. Finally, CO-DRIFTS spectra were collected with 250 scans. H2-TPR experiments were performed in a tubular quartz reactor (TP-5080, Tianjin Xianquan, China), into which 50 mg sample was loaded. The reduction was conducted in a 10% H2/N2 atmosphere at a heating rate of 10 °C/min. H2-TPD experiments were performed in the same apparatus. A 50-mg sample was firstly reduced in situ at 250 °C for 1 h in a 10% H2/N2 flow and then cooled to 30 °C in the same atmosphere. Subsequently, the sample was swept with nitrogen at a flow rate of 30 sccm for 30 min to remove physisorbed or weakly bound species. TPD was performed by heating the sample from room temperature to 800 °C at a ramp rate of 10 °C/min in nitrogen. N2 adsorption–desorption experiments were performed on a BELSORP-Mini system at 77 K. The specific surface area was determined using the Brunauer–Emmett–Teller (BET) method, and the pore size distributions were calculated by the Barrett–Joyner–Halenda (BJH) method according to the desorption branches. The Pt and Co metal content of the samples was determined by inductively coupled plasma mass spectrometry (ICP-MS) analysis (Thermo ICAP 6300).
Catalytic activity measurements
CALD hydrogenation reactions were performed on the above catalysts in a 50-mL stainless-steel autoclave reactor. The reaction was carried out at 65 °C and 2.0 MPa H2 with a certain amount of catalysts in 30 mL of ethanol, and 100 μL of CALD. After the reaction, the reactor was cooled and then slowly depressurized. Finally, the reaction mixture was separated by centrifugation in order to remove the solid catalysts. The reaction products were analyzed and quantified by gas chromatographic mass spectrometry (GC-MS, Agilent, 7890A). The reaction conversion and selectivity were determined by the product analysis.
Computational method
Periodic DFT calculations within the generalized gradient approximation (GGA) were conducted with the Vienna ab initio Simulation Package (VASP 5.3.5) with considering the spin-polarization, in which a projector-augmented potential (PAW) method is implemented. The Perdew–Burke–Ernzerhof (PBE) functional at the GGA level was used, and the plane wave basis set was cut off at the energy of 400 eV. For our ALD-prepared cobalt-based catalysts, the obtained cobalt oxides consist of Co3O4 and CoO and the main species is CoO. Therefore, CoO was selected for DFT calculations. The CoO surface was modeled with a six GGA extension atomic layer 3 × 3 CoO(111) slab. DFT + U corrections (an effective onsite Coulomb interaction parameter) were employed to mitigate the self-interaction errors of Co 3d orbital with U-J of 7.1 eV. The main exposed crystal plane for ALD-prepared TiO2 is the thermodynamically most stable (101) surface, and thus (101) surface was chosen as a model for the anatase TiO2 interface. The TiO2 surface was modeled with a six atomic layer 3 × 3 slab. The slab and its image were separated by a vacuum region of 15 Å. The reactant molecular and the top two layers of these model systems were relaxed, whereas other atoms were fixed in their initial lattice sizes.
Supplementary information
Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (21773282, U1832208 and 21673269), National Science Fund for Distinguished Young Scholars (21825204), the National Key R&D Program of China (2017YFA0700101), Natural Science Foundation of Shanxi Province (201801D211011), Excellent Youth Scholars of State Key Laboratory of Coal Conversion (2016BWZ004), Youth Innovation Promotion Association of the Chinese Academy of Sciences (2018208) and National Postdoctoral Program for Innovative Talents (BX20180323). We are grateful to Shanghai Institute of Applied Physics for the X-ray absorption spectroscopy measurement.
Author contributions
J.Z. synthesized the catalysts and performed the activity tests. S.W. performed the theoretical calculation. G.W. helped to perform the XAFS measurement. X.G., B.Z., S.X. and S.Z. assisted in the synthesis and characterizations of the catalysts. Z.G. and Y.Q. conceived the idea and supervised the work. J.Z., Z.G. and Y.Q. wrote the manuscript. All authors contributed to the manuscript.
Data availability
All the relevant data are available from the authors upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks Neil Coville and other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Zhe Gao, Email: gaozhe@sxicc.ac.cn.
Yong Qin, Email: qinyong@sxicc.ac.cn.
Supplementary information
Supplementary Information accompanies this paper at 10.1038/s41467-019-11970-8.
References
- 1.Tao FF. Synthesis, catalysis, surface chemistry and structure of bimetallic nanocatalysts. Chem. Soc. Rev. 2012;41:7977–7979. doi: 10.1039/c2cs90093a. [DOI] [PubMed] [Google Scholar]
- 2.Sankar M, et al. Designing bimetallic catalysts for a green and sustainable future. Chem. Soc. Rev. 2012;41:8099–8139. doi: 10.1039/c2cs35296f. [DOI] [PubMed] [Google Scholar]
- 3.Behrens Malte, Zander Stefan, Kurr Patrick, Jacobsen Nikolas, Senker Jürgen, Koch Gregor, Ressler Thorsten, Fischer Richard W., Schlögl Robert. Performance Improvement of Nanocatalysts by Promoter-Induced Defects in the Support Material: Methanol Synthesis over Cu/ZnO:Al. Journal of the American Chemical Society. 2013;135(16):6061–6068. doi: 10.1021/ja310456f. [DOI] [PubMed] [Google Scholar]
- 4.Briggs NM, et al. Identification of active sites on supported metal catalysts with carbon nanotube hydrogen highways. Nat. Commun. 2018;9:3827. doi: 10.1038/s41467-018-06100-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phaahlamohlaka TN, et al. Effects of Co and Ru intimacy in Fischer-Tropsch catalysts using hollow carbon sphere supports: assessment of the hydrogen spillover processes. ACS Catal. 2017;7:1568–1578. doi: 10.1021/acscatal.6b03102. [DOI] [Google Scholar]
- 6.Zhang H, et al. Revealing the role of interfacial properties on catalytic behaviors by in situ surface-enhanced raman spectroscopy. J. Am. Chem. Soc. 2017;139:10339–10346. doi: 10.1021/jacs.7b04011. [DOI] [PubMed] [Google Scholar]
- 7.Karim W, et al. Catalyst support effects on hydrogen spillover. Nature. 2017;541:68–71. doi: 10.1038/nature20782. [DOI] [PubMed] [Google Scholar]
- 8.Samad JE, Blanchard J, Sayag C, Louis C, Regalbuto JR. The controlled synthesis of metal-acid bifunctional catalysts: the effect of metal:acid ratio and metal-acid proximity in Pt silica-alumina catalysts for n-heptane isomerization. J. Catal. 2016;342:203–212. doi: 10.1016/j.jcat.2016.08.004. [DOI] [Google Scholar]
- 9.Cheng K, et al. Direct and highly selective conversion of synthesis gas into lower olefins: design of a bifunctional catalyst combining methanol synthesis and carbon-carbon coupling. Angew. Chem. Int. Ed. Engl. 2016;128:4803–4806. doi: 10.1002/ange.201601208. [DOI] [PubMed] [Google Scholar]
- 10.Gläser R. Catalysis: the complexity of intimacy. Nature. 2015;528:197–198. doi: 10.1038/528197a. [DOI] [PubMed] [Google Scholar]
- 11.Lu J, Elam JW, Stair PC. Synthesis and stabilization of supported metal catalysts by atomic layer deposition. Acc. Chem. Res. 2013;46:1806–1815. doi: 10.1021/ar300229c. [DOI] [PubMed] [Google Scholar]
- 12.Lee H-B-R, Bent SF. Formation of continuous Pt films on the graphite surface by atomic layer deposition with reactive O3. Chem. Mater. 2015;27:6802–6809. doi: 10.1021/acs.chemmater.5b03076. [DOI] [Google Scholar]
- 13.O’Neill BJ, et al. Catalyst design with atomic layer deposition. ACS Catal. 2015;5:1804–1825. doi: 10.1021/cs501862h. [DOI] [Google Scholar]
- 14.Liu X, et al. Oxide‐nanotrap‐anchored platinum nanoparticles with high activity and sintering resistance by area‐selective atomic layer deposition. Angew. Chem. Int. Ed. Engl. 2017;129:1670–1674. doi: 10.1002/ange.201611559. [DOI] [PubMed] [Google Scholar]
- 15.Meng X, et al. Atomic layer deposition for nanomaterial synthesis and functionalization in energy technology. Mater. Horiz. 2017;4:133–154. doi: 10.1039/C6MH00521G. [DOI] [Google Scholar]
- 16.Gao Z, Qin Y. Design and properties of confined nanocatalysts by atomic layer deposition. Acc. Chem. Res. 2017;50:2309–2316. doi: 10.1021/acs.accounts.7b00266. [DOI] [PubMed] [Google Scholar]
- 17.Dendooven J, et al. Independent tuning of size and coverage of supported Pt nanoparticles using atomic layer deposition. Nat. Commun. 2017;8:1074. doi: 10.1038/s41467-017-01140-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu J, et al. Coking- and sintering-resistant palladium catalysts achieved through atomic layer deposition. Science. 2012;335:1205–1208. doi: 10.1126/science.1212906. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J, et al. Highly dispersed Pt nanoparticles supported on carbon nanotubes produced by atomic layer deposition for hydrogen generation from hydrolysis of ammonia borane. Catal. Sci. Technol. 2017;7:322–329. doi: 10.1039/C6CY01960A. [DOI] [Google Scholar]
- 20.Gould TD, et al. Controlling nanoscale properties of supported platinum catalysts through atomic layer deposition. ACS Catal. 2015;5:1344–1352. doi: 10.1021/cs501265b. [DOI] [Google Scholar]
- 21.Cheng N, et al. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction. Nat. Commun. 2016;7:13638. doi: 10.1038/ncomms13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Bui H, Grillo F, Helmer R, Goulas A, van Ommen JR. Controlled growth of palladium nanoparticles on graphene nanoplatelets via scalable atmospheric pressure atomic layer deposition. J. Phys. Chem. C. 2016;120:8832–8840. doi: 10.1021/acs.jpcc.6b10026. [DOI] [Google Scholar]
- 23.Wang X, et al. Atomic layer deposited Pt-Co bimetallic catalysts for selective hydrogenation ofa, α,β-unsaturated aldehydes to unsaturated alcohols. J. Catal. 2018;366:61–69. doi: 10.1016/j.jcat.2018.07.031. [DOI] [Google Scholar]
- 24.Gao Z, et al. Multiply confined nickel nanocatalysts produced by atomic layer deposition for hydrogenation reactions. Angew. Chem. Int. Ed. Engl. 2015;54:9006–9010. doi: 10.1002/anie.201503749. [DOI] [PubMed] [Google Scholar]
- 25.Ge H, et al. A tandem catalyst with multiple metal oxide interfaces produced by atomic layer deposition. Angew. Chem. Int. Ed. Engl. 2016;128:7197–7201. doi: 10.1002/ange.201600799. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, et al. Porous TiO2 nanotubes with spatially separated platinum and CoOx cocatalysts produced by atomic layer deposition for photocatalytic hydrogen production. Angew. Chem. Int. Ed. Engl. 2017;56:816–820. doi: 10.1002/anie.201611137. [DOI] [PubMed] [Google Scholar]
- 27.Ramachandran RK, et al. Atomic layer deposition route to tailor nanoalloys of noble and non-noble metals. ACS Nano. 2016;10:8770–8777. doi: 10.1021/acsnano.6b04464. [DOI] [PubMed] [Google Scholar]
- 28.Mao S, et al. High-performance bi-functional electrocatalysts of 3D crumpled graphene-cobalt oxide nanohybrids for oxygen reduction and evolution reactions. Energy Environ. Sci. 2014;7:609–616. doi: 10.1039/C3EE42696C. [DOI] [Google Scholar]
- 29.Plomp AJ, et al. Particle size effects for carbon nanofiber supported platinum and ruthenium catalysts for the selective hydrogenation of cinnamaldehyde. Appl. Catal. A Gen. 2008;351:9–15. doi: 10.1016/j.apcata.2008.08.018. [DOI] [Google Scholar]
- 30.Yu W, Porosoff M, Chen JG. Review of Pt-based bimetallic catalysis: from model surfaces to supported catalysts. Chem. Rev. 2012;112:5780–5817. doi: 10.1021/cr300096b. [DOI] [PubMed] [Google Scholar]
- 31.Zheng R, et al. Controlling hydrogenation of C=O and C=C bonds in cinnamaldehyde using silica supported Co-Pt and Cu-Pt bimetallic catalysts. Appl. Catal. A Gen. 2012;419-420:126–132. doi: 10.1016/j.apcata.2012.01.019. [DOI] [Google Scholar]
- 32.Nongwe I, et al. Pt supported nitrogen doped hollow carbon spheres for the catalysed reduction of cinnamaldehyde. Appl. Catal. A Gen. 2016;517:30–38. doi: 10.1016/j.apcata.2016.02.025. [DOI] [Google Scholar]
- 33.Wu B, et al. Selective hydrogenation of α,β-unsaturated aldehydes catalyzed by amine-capped platinum-cobalt nanocrystals. Angew. Chem. Int. Ed. Engl. 2012;51:3440–3443. doi: 10.1002/anie.201108593. [DOI] [PubMed] [Google Scholar]
- 34.Ji X, et al. Selective hydrogenation of cinnamaldehyde to cinnamal alcohol over platinum/graphene catalysts. ChemCatChem. 2014;6:3246–3253. doi: 10.1002/cctc.201402573. [DOI] [Google Scholar]
- 35.Wang H, et al. A strongly coupled ultrasmall Pt3Co nanoparticle-ultrathin Co(OH)2 nanosheet architecture enhances selective hydrogenation of α,β-unsaturated aldehydes. ACS Catal. 2019;9:154–159. doi: 10.1021/acscatal.8b03471. [DOI] [Google Scholar]
- 36.Li C, et al. The remarkable promotion of in situ formed Pt-cobalt oxide interfacial sites on the carbonyl reduction to allylic alcohols. Mol. Catal. 2018;455:78–87. doi: 10.1016/j.mcat.2018.05.028. [DOI] [Google Scholar]
- 37.Guo Z, et al. Carbon nanotube-supported Pt-based bimetallic catalysts prepared by a microwave-assisted polyol reduction method and their catalytic applications in the selective hydrogenation. J. Catal. 2010;276:314–326. doi: 10.1016/j.jcat.2010.09.021. [DOI] [Google Scholar]
- 38.Tsang SC, et al. Engineering preformed cobalt-doped platinum nanocatalysts for ultraselective hydrogenation. ACS Nano. 2008;2:2547–2553. doi: 10.1021/nn800400u. [DOI] [PubMed] [Google Scholar]
- 39.Kennedy G, Melaet G, Han H-L, Ralston WT, Somorjai GA. In situ spectroscopic investigation into the active sites for crotonaldehyde hydrogenation at the Pt nanoparticle-Co3O4 interface. ACS Catal. 2016;6:7140–7147. doi: 10.1021/acscatal.6b01640. [DOI] [Google Scholar]
- 40.Zhao M, et al. Metal-organic frameworks as selectivity regulators for hydrogenation reactions. Nature. 2016;539:76–80. doi: 10.1038/nature19763. [DOI] [PubMed] [Google Scholar]
- 41.Zheng R, Zhu Y, Chen JG, et al. Promoting low-temperature hydrogenation of C=O bonds of acetone and acetaldehyde by using Co–Pt bimetallic catalysts. ChemCatChem. 2011;3:578–581. doi: 10.1002/cctc.201000268. [DOI] [Google Scholar]
- 42.Zhu Y, Liu D, Meng M. H2 spillover enhanced hydrogenation capability of TiO2 used for photocatalytic splitting of water: a traditional phenomenon for new applications. Chem. Commun. 2014;50:6049–6051. doi: 10.1039/C4CC01667J. [DOI] [PubMed] [Google Scholar]
- 43.Spreafico C, et al. Hydrogen adsorption on nanosized platinum and dynamics of spillover onto alumina and titania. J. Phys. Chem. C. 2017;121:17862–17872. doi: 10.1021/acs.jpcc.7b03733. [DOI] [Google Scholar]
- 44.Kyriakou G, et al. Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations. Science. 2012;335:1209–1212. doi: 10.1126/science.1215864. [DOI] [PubMed] [Google Scholar]
- 45.Zhan G, Zeng H. Hydrogen spillover through Matryoshka-type (ZIFs@)n-1ZIFs nanocubes. Nat. Commun. 2018;9:3778. doi: 10.1038/s41467-018-06269-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prins R. Hydrogen spillover. Facts and fiction. Chem. Rev. 2012;112:2714–2738. doi: 10.1021/cr200346z. [DOI] [PubMed] [Google Scholar]
- 47.Im J, et al. Maximizing the catalytic function of hydrogen spillover in platinum-encapsulated aluminosilicates with controlled nanostructures. Nat. Commun. 2014;5:3370. doi: 10.1038/ncomms4370. [DOI] [PubMed] [Google Scholar]
- 48.Ruiz Puigdollers A, Schlexer P, Tosoni S, Pacchioni G. Increasing oxide reducibility: the role of metal/oxide interfaces in the formation of oxygen vacancies. ACS Catal. 2017;7:6493–6513. doi: 10.1021/acscatal.7b01913. [DOI] [Google Scholar]
- 49.Conner WC, Jr, Falconer JL. Spillover in heterogeneous catalysis. Chem. Rev. 1995;95:759–788. doi: 10.1021/cr00035a014. [DOI] [Google Scholar]
- 50.An K, et al. Enhanced CO oxidation rates at the interface of mesoporous oxides and Pt nanoparticles. J. Am. Chem. Soc. 2013;135:16689–16696. doi: 10.1021/ja4088743. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the relevant data are available from the authors upon request.