

SUMMARY
Although double-strand break (DSB) repair is essential for a cell’s survival, little is known about how DSB repair mechanisms are affected by age. Here we characterize the impact of cellular aging on the efficiency of single-strand annealing (SSA), a DSB repair mechanism. We measure SSA repair efficiency in young and old yeast cells and report a 23.4% decline in repair efficiency. This decline is not due to increased use of non-homologous end joining. Instead, we identify increased G1 phase duration in old cells as a factor responsible for the decreased SSA repair efficiency. Expression of 3xCLN2 leads to higher SSA repair efficiency in old cells compared with expression of 1xCLN2, confirming the involvement of cell-cycle regulation in age-associated repair inefficiency. Examining how SSA repair efficiency is affected by sequence heterology, we find that heteroduplex rejection remains high in old cells. Our work provides insights into the links between single-cell aging and DSB repair efficiency.
In Brief
Young et al. demonstrate that the efficiency of double-strand break repair by single-strand annealing (SSA) declines with replicative age in yeast cells. This decline is associated with changes in cell-cycle progression and not due to increased use of non-homologous end joining. Heteroduplex rejection due to a 3% mismatch in SSA repair substrates does not decline with replicative age.
Graphical Abstract
INTRODUCTION
DNA damage has long been hypothesized to be both a driver and a consequence of aging (Burhans and Weinberger, 2007; Freitas and de Magalhães, 2011). Old tissues and cells accumulate DNA damage and mutations (McMurray and Gottschling, 2003; Adams et al., 2015; Lu et al., 2004). Such mutations can negatively affect tissue homeostasis and organismal function and may even increase the risk for cancer (McMurray and Gottschling, 2004; Aparicio et al., 2014; White et al., 2015). The DNA double-strand break (DSB) represents a major category of DNA damage. Inability to repair a DSB can lead to cell death or genomic instability (Morgan et al., 1998). Even when a DSB is repaired, there are mutagenic repair mechanisms that can both increase genetic variability and affect cellular fitness (Symington et al., 2014). Because of the potential for DSB repair to affect so many cellular functions that intersect with the aging process, the question of how cellular aging affects DSB repair efficiency in single cells is an important area of research with several open questions.
A useful model system to understand how cellular aging affects DSB repair efficiency is that of replicative aging of the budding yeast Saccharomyces cerevisiae (Steinkraus et al., 2008). Replicative lifespan is the number of times a yeast mother cell produces daughters (Steinkraus et al., 2008). This number varies across genetic backgrounds and growth conditions and is connected to several metabolic pathways (McCormick et al., 2015; Kaeberlein et al., 2005). The connection between DSB repair and replicative lifespan is important for several reasons. Mutations in DSB repair genes result in shortened replicative life-span, so proper repair of DSBs is necessary for a normal replicative lifespan (Delaney et al., 2013). In the other direction, continued cell division after mutagenic DSB repair results in propagation of these mutations to future generations with potentially negative consequences on cell fitness.
Multiple mechanisms exist to repair DSBs, and their efficiency and ability to function properly could change with replicative age. Among these mechanisms, the non-homologous end joining (NHEJ) pathway involves ligation of the free ends flanking the DSB (Ceccaldi et al., 2016). The other main class of DSB repair mechanisms is the set of homology-directed repair (HDR) mechanisms, which make use of sequence homology between the break site and a repair template. Regulation of HDR can have significant consequences for the genome. This is due to the ability of different HDR mechanisms to change allele copy numbers and lead to recombination between chromosomes (Symington et al., 2014). The relative use and efficiency of different DSB repair mechanisms depend on factors including cell-cycle stage, ploidy, and cell type (Ceccaldi et al., 2016; Kadyk and Hartwell, 1992; Trovesi et al., 2011; Karanam et al., 2012). Previous studies have detected changes in repair pathway use between chronologically old and young tissues, which could indicate age-related changes in repair efficiency (Preston et al., 2006; Delabaere et al., 2017; Sukup-Jackson et al., 2014). Other studies have also reported declines in the efficiency and fidelity of NHEJ and HDR in senescent mammalian cells (Seluanov et al., 2004; Mao et al., 2012). However, how the repair efficiency of specific DSB repair pathways longitudinally changes in mitotically aging single cells remains unexplored.
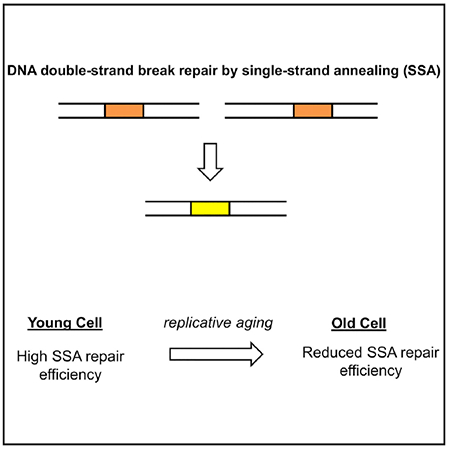
Here we assess whether the efficiency of DSB repair via the single-strand annealing (SSA) pathway changes with the age of the host cell. The SSA pathway repairs DSBs occurring between direct repeats of an identical sequence, resulting in deletion of the intermediate sequence (Figure 1A) (Ceccaldi et al., 2016). Repetitive sequences play important roles in cellular function, with the rDNA locus being one prominent example (Sinclair and Guarente, 1997; Paredes and Maggert, 2009). An age-related change in the efficiency of SSA, which would lead to differences in the copy numbers of the repeated sequence, would be expected to have important consequences for the cell. Because SSA and other HDR pathways share regulatory aspects such as end resection and Rad52 recruitment to the repair sites, understanding how SSA efficiency changes with age can provide key insight into whether the efficiency of other HDR mechanisms would also change with age. Using a single-cell longitudinal approach (Song et al., 2018) in a haploid genetic background, we measure the efficiency of SSA repair in young and older cells. We further explore age-related changes in SSA repair efficiency as they relate to age-related changes in cell cycle and NHEJ pathway activity and in terms of the amount of heterology between the SSA repeats.
Figure 1. Schematics of the Experimental System and SSA Repair Measurements.
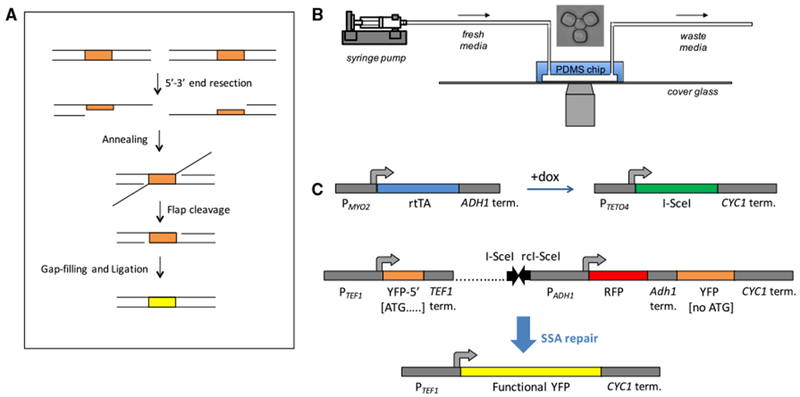
(A) Diagram showing the steps of single-strand annealing based repair. The direct repeats are highlighted in orange. The upper row shows the situation immediately after the double-strand break.
(B) Schematic showing the setup of the experiment. The yeast replicator device allows replicative aging of yeast to be observed using a microscope. Fresh media is supplied, and waste is removed over the course of the movie. A microscope image of a trapped yeast cell is shown above the chip.
(C) Genetic constructs used to measure SSA. rtTA is expressed from the constitutive PMYO2 promoter. In the presence of doxycycline, rtTA activates expression of I-SceI from a non-leaky PTETO4 promoter. The I-SceI cut site consists of a pair of inverted 18 bp I-SceI sites (black arrows) placed within the SSA reporter (rc, reverse complemented). The non-fluorescent 5′ YFP repeat contains only the 5′ 192 bp of YFP. The 3′ YFP repeat is not expressed because it consists of the entire YFP ORF except for the start codon. Between the TEF1 terminator and 3′ YFP repeat is a 2.1 kb stretch of DNA (dotted line), with inverted I-SceI cut sites at a distance of 0.5 kb from the TEF1 terminator. Between the I-SceI cut site and the 3′ non-functional YFP is an ADH1 promoter driving mCherry (abbreviated as “RFP”). Degron-tagged RFP expression below a threshold was used as a reporter for DNA cutting. After SSA repair occurs between the repeats, the resulting product is a full-length YFP with a start codon.
RESULTS
Developing a Single-Cell SSA Repair Reporter and Experimental Design
Cells were aged in a microfluidic chip (Figure 1B) to obtain either young or old cells in which to measure SSA efficiency. To control the age at which SSA repair is assessed, we used a haploid strain in which the endonuclease I-SceI is expressed under the doxy-cycline-inducible PTETO4 promoter. To measure SSA, we developed an SSA reporter containing an I-SceI cut site between two non-functional YFP repeats integrated at the chromosomal ura3 locus. If SSA repair occurs using the two YFP repeats of the reporter, then a functional YFP is formed and detected in single cells aging in real time (Figure 1C). For direct detection of whether cutting occurred or not, an RFP reporter is constitutively expressed from the genomic region between the two nonfunctional YFP repeats; cutting is detected by the dilution of RFP concentration that results from a halt in RFP expression during continued cell growth. The I-SceI cut site consists of two adjacent 18 bp I-SceI recognition sequences in inverted orientation so that after two I-SceI cleavages the DNA ends flanking the break are incompatible (Mao et al., 2008). This situation is more representative of naturally occurring DSBs (Mao et al., 2008). We also used a control strain identical to the strain carrying the SSA repair cassette, except for the absence of the I-SceI cut site.
Under the scenario of highly efficient I-SceI cutting between the non-functional YFP repeats, SSA is expected to be the main repair mechanism for our reporter (Sugawara et al., 2004). Nevertheless, we considered other repair mechanisms for the potential production of functional YFP and deletion of RFP. These alternative repair mechanisms come into play when a cell with two sister chromatids containing the YFP reporter is inefficiently cut by I-SceI in only one of the sister chromatids. Although SSA can still occur between the YFP repeats of the cut sister chromatid, the uncut sister chromatid could instead be used as a repair template for the gene conversion repair or a crossover-based repair mechanism. If the repair template were aligned perfectly with the cut cassette, these repair mechanisms would repair the cut cassette perfectly to its original state. If, instead, only one of the non-functional YFP repeats is used as the repair template, then the above mechanisms would result in a product identical to that produced by SSA. However, with highly efficient cutting and end resection of both sister chromatids, an exposed YFP repeat can only anneal to the opposite, single-stranded repeat on the other side of the DSB. In this case, the opposite YFP repeat can be on the same sister chromatid or a different one. The repair mechanism is SSA regardless of which of these scenarios hold. As will be shown in later parts of this study, we were able to verify highly efficient I-SceI cutting in our experimental system. Therefore, we refer to our reporter as a reporter for SSA.
For inducing I-SceI expression and cutting, we chose a 4 h time window during which cells were exposed to doxycycline (dox). SSA repair efficiency was measured in separate experiments targeting young or older cells. Repair efficiency was quantified using the fraction of initially YFP− cells that became YFP+ within the 9 h period (4 h of dox treatment plus 5 h of post-dox inspection) after addition of dox. A cutoff-based approach was used to determine whether a cell was YFP− or YFP+ at any given time (Figure S1A). Only cells that were observed to be alive within the 5 h post-dox time window were considered.
To confirm that our reporter is specific to SSA, we performed control experiments by measuring reporter efficiency in the absence of Rad51 or Rad52. Rad51 is not necessary for SSA repair, while Rad52 is required (Fishman-Lobell et al., 1992; Ivanov et al., 1996; Sugawara and Haber, 1992). Consistent with our expectations, we quantified a 96.4% repair efficiency in young rad51Δ cells, while 0% efficiency was observed in young rad52Δ cells (Figure S2). Performing another experiment by taking a mixed sample (without age specificity and enrichment) from overnight-grown cultures not treated with dox, we again saw that the fraction of YFP+ cells was 0% in the rad52Δ case, while we observed a non-zero and wild-type (WT)-like YFP+ fraction in the rad51Δ case (Figure S2).
To assess whether the genotypic consequences of the SSA reporter activity match our expectations, we performed sequencing experiments on our reporter by using isogenic colonies grown on plates after dox treatment (I-SceI cutting) of mixed cell cultures (STAR Methods; Figure S3A; Table S1). Colonies were picked to initiate overnight growth of cultures. These cultures were analyzed by flow cytometry to check for the YFP and RFP outcomes, as well as genome-prepped to PCR-amplify and sequence the SSA reporter locus. We saw that, for 9 (of 12) cultures in which no YFP was produced (Table S1), SSA had occurred but the mutations responsible for the non-functionality of the YFP repeats were preserved in the SSA repair product. These SSA-positive but YFP− cases were all identifiable by a lack of RFP expression. Because all SSA-positive cultures (on the basis of sequencing) were identifiable by a lack of RFP expression, we further measured the YFP+ percentage within the RFP− fraction of un-induced cultures (no dox treatment). The resulting percentages had a mean of 99.2% ± 0.2% (SEM; n = 4), indicating that the cases in which SSA occurs but no YFP fluorescence is detected are quite rare (Figures S3B and S3C). Additionally, induction of I-SceI expression by dox treatment for 4 h in cultures yielded nearly 100% YFP+ cells at 9 h after dox addition (Figure S3D). Therefore, it is highly unlikely that the YFP-inactivating mutations are preserved in the SSA repair product.
For the SSA repair experiments in young cells, the average age of those cells was 2.6 generations at the beginning of the dox treatment. In the experiments with old cells, the average age was 18.1 generations at the beginning of the dox treatment (Figure 2A; Figure S4). The average replicative lifespan of haploid yeast of the same strain background used in this study is 29.3 generations (Liu et al., 2015). The two expected phenotypes of YFP appearance, or its absence, were observed and quantified across individual cells longitudinally (Figure 2B; Data S1; Figure S5).
Figure 2. Measuring SSA Repair Efficiency under Perfect Sequence Homology.
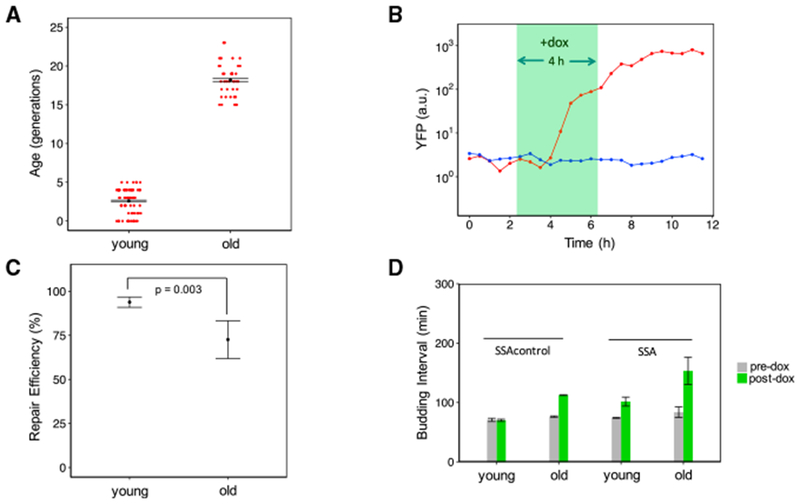
(A) Error bars correspond to mean ± SEM of the average age within replicates. Red points represent the age of each cell at the beginning of the doxycycline treatment. The mean age of old cells (n = 2 replicates) was 18.1 generations, while the mean age of the young cells (n = 2 replicates) was 2.6 generations.
(B) YFP trajectories are shown for two young sample cells, with the cell with the red trajectory representing successful SSA repair.
(C) The fraction of cells with successful SSA repair was calculated for each experimental replicate. Shown are mean ± SEM (n = 2) by age group for the fraction of repaired cells. The old cell replicates had a mean repair efficiency of 71.8% compared with a mean repair efficiency of 93.8% for the young cell replicates. The p value of 0.003 is for a two-sided Fisher’s exact test for association between repair and age on pooled data.
(D) Shown are the mean ± SEM (n = 2) of the replicate-based averages of single-cell average budding intervals for time windows before and after doxycycline addition. Pre-dox corresponds to the 4 h time window before doxycycline addition. Post-dox corresponds to the 9 h time window after doxycycline addition. At both young and old ages, the strain carrying the SSA repair reporter exhibits increased budding interval relative to the control strain, indicative of DNA damage-induced cell-cycle arrest (from 74 ± 1 min to 102 ± 8 min in young cells with the cut site; from 84 ± 9 min to 153 ± 23 min in old cells with the cut site).
Measuring SSA Repair Efficiency in Young and Old Cells
The SSA repair efficiency for the old cells was significantly lower than that of the young cells (71.8% versus 93.8%), indicating an age-associated decline in SSA repair (Figure 2C; p = 0.003, Fisher’s exact test). The fraction of unrepaired old cells that were repaired in the subsequent 5 h time window after dox removal was also found to be low (1 of 11) (Figure S6). Therefore, the absence of repair at the 9 h time point was not due simply to a small reduction in the speed of repair. Additionally, the average time after dox addition at which YFP first appeared was similar between the old and young repaired cells, indicating that aging does not significantly affect the speed of SSA repair in cells that are able to carry out SSA repair (Figure S5). To assess whether cells were arresting their cell cycle because of cutting by I-SceI, we calculated the average budding interval experienced by cells in the 4 h time interval before dox addition and the 9 h time interval after dox addition. We compared this with the corresponding average budding intervals calculated for the control strain lacking an I-SceI cut site. In the time window prior to dox treatment, the two strains show similar average budding intervals (Figure 2D) for the same age groups. Consistent with I-SceI cleavage resulting in cell-cycle arrest, average budding intervals after dox addition are higher in the strain containing the cut site compared with the control strain without the cut site. After dox addition, the strain containing the cut site had an average budding interval of 153 ± 23 min in old cells compared with 112 ± 1 min for the old cells of the control strain (Figure 2D). Therefore, induction of the DSB leads to longer cell-cycle durations on average.
Although TEF1 is a very strong promoter with a maximal expression level approximately 100-fold above the expression level corresponding to the YFP+ threshold (Data S1), we nevertheless performed a control experiment to show that the lower YFP− fraction in old cells was not due to a major reduction in PTEF1-YFP expression in old cells. For this, we measured YFP fluorescence during replicative aging of YFP+ single cells. We observed that the YFP expression stayed high across all generations experienced by the aging cells (Figure S7A).
To determine whether the unrepaired YFP− cells were arrested in their cell cycle consistent with cutting and failure to repair, we considered the time that had passed since these cells last started forming a daughter bud. Because the first appearance of a daughter bud corresponds to early S phase, the amount of time since the previous bud initiation can include the G1 phase of the subsequent cell cycle. If the cells are arrested in G2/M phase, on the other hand, this time period includes only the S/G2/M phase of the same cell cycle. Compared with the YFP+ repaired cells, cells that were YFP− at the 9 h time point after dox addition had not budded for a longer period of time (Figures S8A and S8B). These unrepaired cells are therefore either arrested for a long time in G2/M or went through mitosis before experiencing a long G1 in the subsequent cell cycle.
To determine whether the cells that failed to produce YFP by SSA repair exhibited any cell-cycle delays consistent with a DSB-induced cell-cycle arrest, we measured the average budding interval of each cell in three non-overlapping time windows: the 4 h window prior to dox treatment, the 4 h window coinciding with dox treatment, and the 5 h window after dox treatment. The old YFP− cells exhibited slightly longer average budding intervals prior to dox addition compared with the old cells that were eventually repaired (Figure S8C). During the 4 h of dox exposure and the 5 h period after dox exposure, both YFP+ and YFP− cells showed an increased average budding interval compared with the control strain (Figures S8D and S8E). Average budding intervals in the 5 h post-dox time window for young YFP− cells were far longer than those for young cells of the SSA control strain lacking a cut site (Figure S8E). These results suggest that cutting efficiency is 100% efficient in young cells. For old cells, on the other hand, the considerable overlap in average budding interval for YFP− cells and control cells lacking a cut site makes budding interval a poor measure of cutting efficiency (Figure S8E), necessitating the use of more direct approaches, such as the RFP reporter used in this study, for determining cutting efficiency.
Direct Measurement of I-SceI Cutting Efficiency in Old Cells
The age-associated decline in SSA repair efficiency could be due to several factors. First, the cells could be cut and failing to complete repair using SSA. Second, weaker induction of I-SceI in old cells could result in a lower rate of cutting. A third possibility is that the cells in which YFP fails to appear are actually cut by I-SceI but repaired by a different repair pathway like NHEJ or MMEJ (microhomology-mediated end joining) that does not generate functional YFP. To address the possibility of decreased cutting efficiency by I-SceI in old cells, we constructed a strain in which the RFP originally introduced between the two non-functional YFP repeats was further tagged with a degron. The degron tag we used is a truncation of the C-terminal destabilization motif from the cyclin protein Cln2, and it leads to the fast degradation of Cln2 and RFP. The stability of the original non-tagged RFP results in poor detection of any cutting due to the longer time and continued growth requirement for the detection of RFP loss after cutting. In the strain carrying the degron-tagged RFP, a halt in RFP transcription due to cutting by I-SceI is expected to result in a rapid and prolonged decline in RFP transcription. At the 9 h time point after dox addition, we measured the length of time that each cell had been RFP− using a fixed cutoff for RFP (Figure 3A; Figure S1B). Soon after addition of dox, young and old cells of this strain showed a rapid and lasting decline in RFP expression that did not occur in old cells of a control strain containing the RFP-degron fusion but without a cut site (Figure 3B; Data S1). Because old cells of the cuttable and control strains, both containing the SSA cassette and degron-tagged RFP, have similar RFP expression levels prior to the decline in RFP expression, we conclude that the difference in the metric of “RFP− duration” is not due to differences in promoter activity (Figure 3C). Only 2 of 58 old cells (3.4%) of the cut site-free strain containing the RFP-degron fusion lost RFP expression for more than 4.5 h. Among all old, initially YFP− cells from the SSA strain carrying degron-tagged RFP, 96.2% showed either a decline in RFP signal to background levels for more than 4.5 h or production of functional YFP. On the basis of these results, we concluded that I-SceI cutting in old cells was very efficient. Therefore, the observed decline in SSA repair efficiency in old cells compared with young cells is due to either decreased SSA repair efficiency or increased efficiency of competing repair pathways.
Figure 3. Directly Measuring I-SceI Cutting Efficiency Using a Degron-Tagged mCherry (“RFP”) in the SSA Repair Cassette.
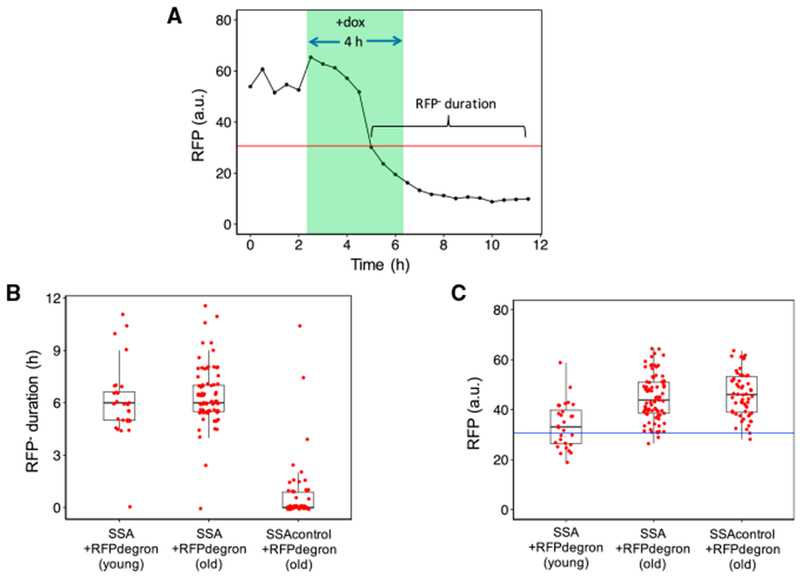
(A) An RFP cutoff was applied to cells from two strains containing a degron-tagged mCherry (abbreviated “RFP”) within an SSA repair reporter: one strain contained an I-SceI cut site while the other lacked the cut site. RPF measurements from one such cut-site-positive cell are shown along with the RFP cutoff (red line). For each cell analyzed, the duration of RFP absence was quantified between the first occurrence of below-cutoff RFP level and the end of the 9 h post-doxycycline time window.
(B) The duration of RFP absence is shown for old and young cells of the strain containing a degron-tagged RFP within an SSA repair reporter and old cells of the same strain missing the I-SceI cut site. Forty-one of the 58 old cells (71%) analyzed from the second strain missing the cut site had a duration of RFP absence quantified as 0 min, compared with 1 of the 81 old cells from the first strain carrying the cut site. Using pooled data from two independent experiments, the boxplots show the distribution of the single-cell RFP− durations for each cell.
(C) For each cell that was missing RFP (expression below the cutoff) at the 9 h time point after doxycycline addition, all RFP values prior to the drop below the cutoff were averaged for that cell. Red points correspond to these single-cell averages. Using pooled data from two independent experiments, the boxplots show the distribution of the single cell-averaged RFP values corresponding to each cell’s RFP+ period. The RFP values corresponding to the RFP+ period are similar between old cells of the two degron-tagged SSA reporter strains, with and without the cut site.
Measuring Age-Associated SSA Repair Efficiency in the Absence of NHEJ
We hypothesized that the age-related repair deficiency in the SSA repair strain was due to increased competition from the NHEJ repair pathway in old cells. Previous work performed in young cells has shown that DSB repair is more biased toward NHEJ during G1 phase, when there is no identical sister chromatid for homology-based repair (Aylon et al., 2004). Deletion of the key NHEJ proteins Ku and Dnl4 has also been shown to increase DSB end resection, an important step of homologous recombination-based repair pathways (Clerici et al., 2008). These examples illustrate ways in which NHEJ can compete with and inhibit homology-based repair pathways like SSA.
If the decline we observed in the fraction of YFP+ cells was due to NHEJ outcompeting SSA in old cells, and SSA itself is still fast and efficient, then inhibition of NHEJ would increase the SSA repair efficiency in old cells back to that of young cells. To test for this possibility, we generated a perfect-homology SSA repair strain identical to the previous strain except with the NHEJ gene DNL4 deleted. The Dnl4 protein is a DNA ligase that is an essential component of the NHEJ mechanism. Deleting DNL4 does not affect replicative lifespan (Kaeberlein et al., 1999). Using this strain, we performed the same type of dox induction experiments by targeting young (average age 2.4 generations) or older cells (average age 19.3 generations) (Figure S4) and measured the fraction of cells with YFP fluorescence as a sign of SSA repair (Data S1). SSA repair efficiency in young dnl4Δ cells was 90.4% (Figure 4A), essentially unaffected by the deletion compared with the non-deleted strain’s 93.8% repair efficiency. In old dnl4Δ cells, we measured a 65.0% SSA repair efficiency compared with the 71.8% SSA repair efficiency in the old cells of the non-deleted strain (Figure 4A). As with the DNL4 intact strain, only a small fraction of the dnl4Δ cells that were unrepaired at the 9 h post-dox time point were repaired in the subsequent 5 h (Figure S6). In the 9 h after dox addition, the average budding interval was 156 ± 16 min for old cells and 103 ± 16 min for young cells, similar to the corresponding values for the strain with intact DNL4 (Figure 4B). In this strain, the ratio of the repair efficiency in old cells to the one in young cells was 0.72, very similar to the ratio of 0.77 measured in the SSA strain with intact DNL4 (Figure S9A). Consistent with this, the ratio of average budding interval in the 9 h post-dox time window between old and young cells of this strain was also similar to the ratio for the strain with intact DNL4 (Figures S9B and S9C). The lack of increase in the SSA repair efficiency when the NHEJ repair mechanism is inactive suggests that increased use of NHEJ is not the cause of the age-specific inefficiency in SSA-based repair.
Figure 4. The Age-Associated Decline in SSA Repair Efficiency Is Due to Changes in Cell-Cycle Progression in Old Cells.
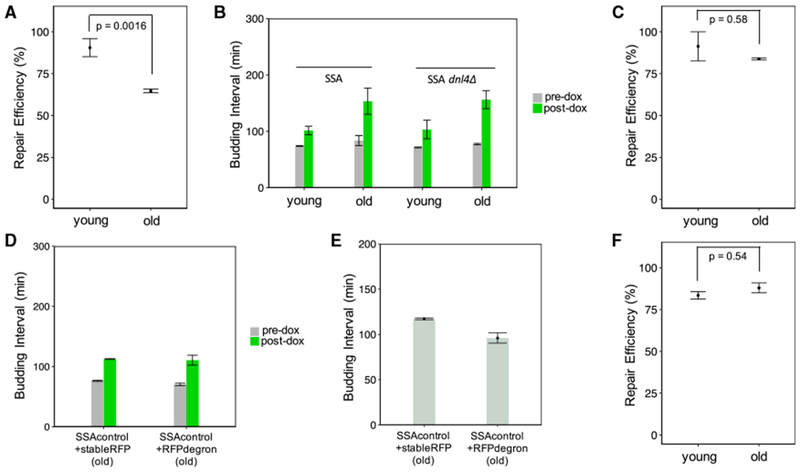
(A) dnl4Δ cells fail to show an increase in SSA repair efficiency when old. The mean repair efficiency for old cells (n = 2 replicates) was 65.0%, while for young replicates (n = 2), it was 90.4%. The error bars show the mean ± SEM (n = 2) of these replicate-based statistics. The p value of 0.0016 is for a two-sided Fisher’s exact test to test for association between repair and age on pooled data.
(B) The dnl4Δ strain shows a large increase in budding interval after doxycycline treatment regardless of age (from 71 ± 1 minto 103 ± 16 min in young cells; from 77 ± 1 min to 156 ± 16 min in old cells). Shown are the mean ± SEM (n = 2) of replicate-based averages of single-cell average budding intervals during the 4 h pre-doxycycline addition and 9 h post-doxycycline addition time window.
(C) The strain containing a degron-tagged mCherry (“RFP”) within the SSA repair reporter has an SSA repair efficiency of 84.0% for old cells (n = 3 replicates) and 89.2% for young cells (N = 2 replicates). Shown are mean ± SEM of replicate-based repair efficiencies. The p value of 0.58 is for a two-sided Fisher’s exact test to test for association between repair and age on the pooled data.
(D) Average budding intervals during the 9 h post-doxycycline time window are similar in old cells of the SSAcontrol strain (no cut site) carrying degron-tagged mCherry (“SSAcontrol+RFPdegron”) and old cells of the SSAcontrol strain carrying stable mCherry (“SSAcontrol+stableRFP”). Shown are mean ± SEM (n = 2) of replicate-based averages of single-cell average budding intervals, pre-dox (during the 4 h time window before doxycycline addition) and post-dox (during the 9 h time window after doxycycline addition). In old cells, the average post-dox budding interval was 115 ± 6 min for the RFPdegron-carrying control strain and 111 ± 8 min for the stable RFP-carrying control strain.
(E) Average budding intervals occurring after generation 15, measured for old cells of the SSA control strains (no cut site), with stable RFP or degron-tagged RFP. Only cells of age 15 generations or older at the time of doxycycline addition were used. Mean ± SEM (n = 2) of replicate-based averages of single cell-averaged budding intervals are shown. The mean values are 117 ± 1 min for the strain with the stable RFP and 96 ± 6 min for the strain with the degron-tagged RFP.
(F) SSA repair efficiency is increased in old cells of the strain carrying three copies of the CLN2 gene. Shown are the mean ± SEM (n = 2) by age group for the fraction of repaired cells. The old cell replicates had a mean repair efficiency of 88.7% (n = 2). The young cell replicates had a mean repair efficiency of 83.8% (n = 2). The p value of 0.54 is for a two-sided Fisher’s exact test to test for association between repair and age on pooled data.
The Role of Changes in Cell-Cycle Progression on SSA Repair Efficiency in Young versus Old Cells
Because eliminating Dnl4 did not restore SSA repair efficiency in old cells to the levels observed in young cells, we next considered whether some aspect of SSA repair itself was deficient in old cells. SSA falls under the class of HDR mechanisms that are normally inactive in the G1 phase and activated during other phases of the cell cycle. It has been shown that several generations prior to the end of their replicative lifespan, cells undergo a lengthening of the cell cycle (Liu and Acar, 2018; Neurohr et al., 2018; Sarnoski et al., 2018). This increase in cell-cycle duration correlates at the single-cell level with an increase in the duration of G1 phase and lower expression of key G1/S transition proteins, including the cyclin Cln2 (Neurohr et al., 2018). Outside of G1 phase, the cyclin dependent kinase subunit Cdc28 is activated by S- and G2-phase cyclins (Clbs) to promote HDR. Clb-activated Cdc28 promotes HDR by inhibiting the binding of the NHEJ-promoting Ku proteins to the DSB break site and by promoting end resection and recruitment of HR proteins to the site (Symington et al., 2014; Clerici et al., 2008; Aylon et al., 2004). An age-related increase in G1 phase duration could result in a decrease in SSA repair efficiency due to reduced levels of Clbs that activate Cdc28’s HDR-related activities (Symington et al., 2014). Because the Ku complex prevents end resection in the absence of Cdc28 activity, deletion of DNL4 under this scenario would be expected to fail to activate HDR by SSA, which was observed (Figure 4A). The old cells experiencing longer G1 durations are expected to have longer budding intervals and to be worse at SSA repair. Consistent with this hypothesis, we saw that old cells containing the SSA cassette that failed to repair YFP had longer average budding intervals in the 4 h time window prior to dox treatment compared with old cells that were able to repair YFP (Figures S8C and S9D).
Interestingly, we observed that SSA repair efficiency in old cells of the SSA strain containing the degron-tagged RFP was 84.0%, a number greater than the efficiency observed in the old cells of the SSA strain containing the stable RFP without the degron (Figure 4C). Additionally, the speed of SSA repair in old cells of this degron-carrying strain was fast: YFP was detected slightly sooner in old cells of this strain than old cells of the SSA strain with stable RFP (Figure S5). The fractional increase in average post-dox budding interval measured for this strain was also reduced compared with the increase in budding intervals measured for the strains containing stable RFP, which is consistent with reduced cell-cycle arrests induced by the DSB (Figures S9B and S9C). We attribute these observations to the presence of the degron we tagged to the RFP. The degron we used is a truncated Cln2 C-terminal destabilization sequence found in cyclin Cln2. Cln2 is one of three G1 cyclins that together with Cdc28 activate the transcription factor SBF (Doncic et al., 2011), leading to expression of genes involved in the G1/S transition. Once the cell is out of G1 phase, the expressed S and G2 cyclins activate Cdc28 to promote HDR (Aylon et al., 2004). The Cln2 C-terminal destabilization sequence encodes protein domains that allow Cln2 to be recognized and targeted for proteasomal degradation by the SCF E3 ubiquitin-ligase complex containing F box protein Grr1 (Berset et al., 2002). Given the low abundance of Grr1 (Berset et al., 2002), constitutive expression of the Cln2 degron tagged to RFP could lead to competition with endogenous Cln2 for recognition by Grr1 and subsequent degradation, which would be expected to result in higher levels of endogenous Cln2. Indeed, deletion of Grr1 has been shown to result in increased Cln2 levels (Landry et al., 2012). A higher level of Cln2 in the degron-carrying strain would be expected to speed up the G1/S transition, resulting in a greater percentage of cells with HDR-promoting Clb-Cdc28 activity. This would explain the higher SSA repair efficiency observed in the strain containing the degron-tagged RFP.
Given the above mechanism, and the previously observed single-cell level correlation between G1 phase duration and total budding interval in old cells (Neurohr et al., 2018), old cells expressing the degron-tagged RFP would be expected to have shorter budding intervals than old cells expressing stable RFP. To assess whether this was the case, we compared average budding intervals in two strains carrying the SSA reporter cassette and lacking the I-SceI cut site. One strain contained the RFP without the degron tag, while the other strain contained the degron-tagged RFP. During the 9 h post-dox time window, old cells from the two strains displayed similar average budding intervals (Figure 4D). However, at the time of dox exposure, the average age of the degron-tagged RFP containing strain was two generations greater than that of the stable RFP containing strain (Figure S4). To take into consideration this age difference, we next compared average budding intervals between the two strains by using old cells going through more similar ages. For this, we measured the average of all budding intervals occurring after the 15th generation for each cell and saw that cells of the degron-containing strain had shorter budding intervals (Figure 4E). At the single-cell level, the right tail of the average budding interval distributions we measured after the 15th generation was longer for the stable RFP containing strain compared with the strain carrying the degron-tagged RFP (Figure S9E). Therefore, the difference in SSA repair efficiency between the two strains can be attributed, at least in part, to the changes in Cln2 levels.
Despite the differences in SSA repair efficiency and budding intervals between the strains containing the degron-tagged RFP and stable RFP, the replicative lifespans of the strains are very similar (Figures S7B and S7C). This indicates that the differences in SSA repair efficiency and cell-cycle progression that result from expression of the degron-tagged RFP do not increase cell longevity.
In an attempt to directly measure the effect of degron-tagged RFP expression on G1 duration in old cells, we constructed a strain in which Whi5 is tagged to YFP. Whi5 enters the nucleus at the end of mitosis, remains there during G1 phase and exits just prior to S phase and budding (Figure S10A) (Costanzo et al., 2004; de Bruin et al., 2004). Therefore, the duration of nuclear Whi5 localization can be used as a measure of G1 duration (Neurohr et al., 2018). Using the Whi5-YFP reporter, we performed automated localization analysis using a quantitative metric and measured G1 duration in old cells carrying the control SSA (no I-SceI cut site) cassette with stable RFP or degron-tagged RFP. As previously observed, the budding intervals of the strain carrying the stable RFP were on average longer compared with those of the strain carrying the degron-tagged RFP (Figure S10D). However, because of limited sensitivity of the localization metric used, the results from the G1 duration measurements between the two strains were not conclusive: while mean G1 duration was 6 min longer in the stable RFP strain, its median value was ~1 min shorter, and the 6 min difference between mean G1 durations would be eliminated if the two largest outliers were removed (Figure S10E).
To independently assess how increased CLN2 expression affects SSA repair efficiency, we constructed a strain with three copies of the CLN2 gene integrated at its endogenous locus. In addition to the full open reading frame, each copy of the CLN2 gene contained its full upstream intergenic region and the downstream intergenic region, so that CLN2 regulatory sequences would be preserved. If delayed G1/S transitions in old cells are preventing repair, then increasing CLN2 copy number should speed up the G1/S transition to result in higher SSA repair efficiency. Indeed, in old cells, the three-copy CLN2 strain showed a higher SSA repair efficiency than the one-copy CLN2 strain (88.7% compared with 71.8%) (Figure 4F). Additionally, the post-dox-addition budding interval was shortened in this strain relative to the strain carrying one copy of CLN2, consistent with faster repair (Figure S9C).
Assessing Age-Associated SSA Repair Efficiency Using a Reporter with Imperfect Sequence Homology
Homologous recombination is dependent on there being a high degree of homology between the DSB site and repair template. In the context of SSA, strong homology between the direct repeats flanking the DSB is important for repair. Previous work showed that in the presence of as little as 3% heterology, there can be a 6-fold reduction in the ability of cells to complete SSA repair, a phenomenon called heteroduplex rejection (Sugawara et al., 2004). Several mismatch repair proteins and the Sgs1 helicase play an important role in heteroduplex rejection (Sugawara et al., 2004). An age-related change in the rate of heteroduplex rejection would have consequences for the mutation rate in older cells, because heteroduplex rejection prevents recombination between homologous sequences with high levels of sequence heterology.
To determine whether the efficiency of heteroduplex rejection changes with age, we modified the SSA reporter to have 3% heterology between the non-functional YFP repeats. Using the strain carrying this modified reporter, we measured the rate of YFP repair in young and old cells as before (Data S1; Figure 5A). As with the strain containing perfect homology (Figure 2C), the repair efficiency in old cells was lower than it was in young cells (27.5% versus 34.9%; p = 0.2787, Fisher’s exact test) (Figure 5A). Among the young and old cells that were unrepaired at the 9 h post-dox addition time point, only a small fraction repaired within the next 5 h (Figure S6), consistent with robust heteroduplex rejection in cells from both age groups. In fact, despite the expected reduction in the absolute fraction of repaired cells in this strain relative to the strain with perfect homology, the ratio of the repaired-cell fraction in old age to the one in young age (0.79) was very similar to the same ratio measured in the strain with perfect homology (0.77) (Figure S9A). Consistent with the decline in repair efficiency when there is sequence heterology, the average budding interval of the 3% heterology strain in the 9 h interval after dox addition was increased to 192 ± 4 min for old cells and 190 ± 19 min for young cells (Figure 5B). The ratio of post-dox budding intervals between old and young cells of this strain was also reduced relative to that of the strain carrying perfect sequence homology in the SSA repair reporter cassette (Figures S9B and S9C). The increase in cell-cycle delays experienced by the young cells due to 3% heterology between the non-functional YFP repeats is not matched by a pro-portional increase in the delays experienced by the old cells. In summary, our results show that there is no replicative age-related decline in heteroduplex rejection in the context of SSA. The processes normally responsible for heteroduplex rejection work just as well in old cells as they do in young cells.
Figure 5. Measuring SSA Repair Efficiency under 3% Sequence Heterology.
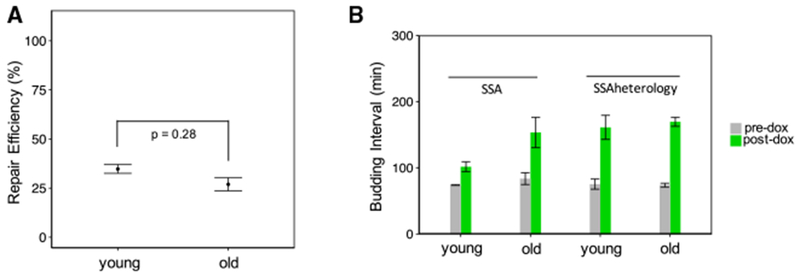
(A) Old cells show a decline in SSA repair efficiency relative to young cells. Repaired cell fraction was measured for each replicate to quantify SSA repair efficiency. The mean and SEM (n = 2 or 5) of these replicate-based statistics are shown. The replicates (n = 5) of the old cells had a mean repair efficiency of 27.5%, while the replicates (n = 2) of the young cells had a mean repair efficiency of 34.9%. The p value of 0.28 is for a two-sided Fisher’s exact test to test for association between repair and age on pooled data.
(B) Shown are the mean ± SEM (n = 2 or 5) of replicate-based averages of single-cell average budding intervals for the 4 h time window before and the 9 h time window after doxycycline addition. Cells exhibit an especially large increase in cell-cycle duration after doxycycline treatment (from 75 ± 8 min to 161 ± 18 min for young cells; from 74 ± 3 min to 169 ± 7 min for old cells).
DISCUSSION
Repairing DNA DSBs is essential for cells to maintain genome stability and homeostasis. Repair pathway dysfunction can have drastic consequences on a variety of phenotypes in both young and old cells. Despite this, we have very limited understanding about how much the efficiency of specific DNA repair pathways changes in an age-dependent manner in single cells. The lack of extensive knowledge on this topic is due in part to the historical challenges associated with longitudinally tracking aging cells and simultaneously measuring repair events in them. In this study, using a reporter for detecting SSA-based repair, we compared the efficiency of SSA repair for young and replicatively aged old cells and discovered an age-dependent decline in SSA repair efficiency in yeast. Measurement of SSA repair efficiency in strains lacking a key NHEJ protein, expressing RFP fused to a CLN2-PEST degron, or expressing three copies of CLN2 provided additional insights into repair pathway use and efficiency in the context of aging. Finally, we further characterized the change in SSA repair efficiency between young and old cells in the presence of 3% sequence heterology.
An aging-related decline in the efficiency of SSA-based repair has several important implications. Because the SSA mechanism shares molecular steps, such as end resection, with other homology-based repair mechanisms, the molecular reasons behind the reduced SSA repair efficiency in old cells are expected to lead to reductions in the efficiency of other homology-based repair mechanisms as well. In line with this expectation, previous work has shown that homologous recombination-based DSB repair is impaired in replicatively aged yeast cells and this repair defect is corrected by overexpression of homologous recombination proteins Rad51 and Mre11 (Pal et al., 2018).
The fact that the deletion of the NHEJ protein Dnl4 fails to increase the efficiency of SSA repair suggests that the age-dependent decline in SSA efficiency is not due to increased competition between NHEJ and SSA mechanisms. Given that the repair templates are located close to one another in SSA repair (2.6 kb for the YFP reporter), deficient homolog search cannot easily explain the observed age-related decline in SSA efficiency either. The observation that old cells of a strain containing an SSA repair reporter with RFP fused to a Cln2 degron have higher SSA repair efficiency than old cells of a strain containing an SSA reporter with stable RFP lends support to the mechanism that an increase in G1 duration is partly responsible for the decline in SSA repair efficiency. Although Whi5-YFP localization measurements showed that a strain expressing stable RFP has a mean G1 duration 6 min longer than a strain expressing RFP fused to the Cln2 degron, the results were not conclusive, for the reasons previously specified. However, the single-cell level correlation previously shown (Neurohr et al., 2018) between the overall cell-cycle duration and G1 phase duration, and our observation of shorter budding intervals for the old cells of the degron-carrying strain, further support the mechanism in which shorter G1 durations are behind the higher repair efficiency measured in the old cells of the degron-carrying strain. Consistent with the proposed mechanism, old cells carrying three copies of the CLN2 gene had increased SSA repair efficiency compared with the old cells carrying one copy of the CLN2 gene. Taken together, the age-dependent decline in SSA repair efficiency (Figure 2C) is due to the old cells’ experiencing longer G1 durations resulting in their inability to activate the steps of the SSA repair mechanism that normally occur outside of G1 (Ceccaldi et al., 2016; Symington et al., 2014), such as end resection, annealing, flap cleavage, gap filling, or ligation. Although our results and the model we propose elucidate DSB repair in the context of replicative aging of single cells, the broad idea of cell cycle-dependent regulation of DSB repair and the potential role of cyclins had been previously proposed in non-aging contexts (Aylon et al., 2004).
Our experiments with an SSA strain containing 3% heterology between the direct YFP repeats showed a repair efficiency decline (between young and old cells) similar to the decline observed in the cells of the SSA strain containing perfect homology (Figure S9A). Because heteroduplex rejection in SSA is dependent on both recognition of mismatches by mismatch repair proteins Msh2 and Msh6, and unwinding of the heteroduplex by the Sgs1 helicase, this result implies that these three proteins do not show a significant decline in activity in old cells (Sugawara et al., 2004). Given that an increased mutation rate from recombination between non-allelic sequences would be detrimental to cellular fitness, it is not surprising that heteroduplex rejection does not decline with replicative age. For the cells of the SSA strain containing 3% heterology, the similar drop of repair efficiency in the old cells relative to the young cells is also consistent with a model in which the factor that is responsible for the general decline in SSA efficiency acts independently of heteroduplex rejection.
The relationship between replicative aging and genomic stability has great relevance to our understanding of human health and disease. An increase in genomic instability with age has been hypothesized as a potentially reason for the increased rate of cancer in older individuals (Adams et al., 2015). In this study, we used a simple eukaryotic model system to learn whether replicative aging actually coincides with defective DNA repair. We show that replicative aging results in a cell-cycle progression-related decrease in the efficiency of SSA repair, but not heteroduplex rejection. These results raise the possibility that although old cells show a decline in the efficiency of homology-based repair, they do not show a decline in the efficiency of the mechanisms that prevent aberrant recombination. The age-associated decline we observed in SSA repair efficiency also raises questions about the repair outcome in old cells that are unable to perform SSA repair, because alternative outcomes such as MMEJ repair or checkpoint adaptation in the absence of repair (Toczyski et al., 1997) could adversely affect the genome and limit replicative lifespan. Determining what repair outcomes increase in frequency at the expense of SSA and other HDR repair pathways is therefore an exciting research area for future exploration.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for strains, reagents, and recombinant DNA should be directed to and will be fulfilled by the Lead Contact, Murat Acar (murat.acar@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All Saccharomyces cerevisiae strains in this study were constructed in a BY4742 haploid strain background with MET15 deleted and LYS2 intact. Table S2 shows the complete strain descriptions and genotypes.
The growth conditions used for the DSB induction experiments in aging cells were as follows. To study the effect of replicative age on SSA repair, cells were aged in a microfluidic chip (Figure 1B) designed to trap virgin mother cells and wash away budded daughter cells. The day before each experiment, a single colony of the strain to be tested was picked to 2 mL CSM 2% glucose and vortexed. 30 μl of the suspended cells was added to 5 mL CSM 2% glucose. After 14 hours of growth in a shaker-incubator (225 rpm) set to 30 C, the culture was diluted and grown further so that at the time of loading the chip, the OD600 was between 0.25 and 0.3. For strains carrying an I-SceI cut site, the cultures were also FACS’d to determine which culture had a large fraction of unrepaired, YFP− cells. Immediately prior to loading cells to the microfluidic chip, the cultures were diluted to OD600 0.1. Per lane of the chip, 50-70 μl of the OD600 0.1 culture was loaded at 20 μl/min media flow speed. The base media used throughout the shaker-growth phases and the microfluidic-chip experiments were always CSM minimal media containing 2% glucose as the carbon source. After starting the movie, media flow rate was set to alternate between at 30 μl/min for 2 min and 2.0 μl/min for 8.05 min.
METHOD DETAILS
Construction of plasmids and yeast strains
For all SSA repair reporter constructs, the 5′ non-functional mCitrine repeat consists of the 5′ 192 bp of mCitrine followed by a TAG stop codon. The 3′ non-functional mCitrine repeat consists of the full 717 bp mCitrine sequence except for the start codon. A 420 bp TEF1 promoter drives expression of the 5′ non-functional mCitrine, with a 507 bp TEF1 terminator. A 455 bp segment of DNA from the pRS401 plasmid (lacking homology to the rest of the repair cassette) lies between the TEF1 terminator and I-SceI cutsite. In total, 1 kb separates the 5′ mCitrine and the I-SceI cutsite. Directly downstream of the I-SceI cut site, a 720 bp ADH1 promoter drives mCherry with a 207 bp ADH1 terminator. Immediately downstream of the ADH1 terminator is the 3′ ATG-less mCitrine followed by a CYC1 terminator. Altogether, 1.6 kb separates the I-SceI cutsite from the 3′ mCitrine. The TEF1 and ADH1 promoters, and TEF1 and ADH1 terminators are all from the S288C strain background. While mCitrine and YFP may be used interchangeably throughout the paper, they both refer to mCitrine. Also, while mCherry and RFP may be used interchangeably throughout the paper, they both refer to mCherry.
The mCitrine repeats described in the above paragraph were derived from the yeast codon optimized mCitrine on plasmid pKT0211(Sheff and Thorn, 2004). pKT0211 was a gift from Kurt Thorn (Addgene plasmid # 8734). The mCherry was also yeast codon optimized and amplified from plasmid pFA6-link-yomCherry-CaURA3(Lee et al., 2013). pFA6-link-yomCherry-CaURA3 was a gift from Wendell Lim & Kurt Thorn (Addgene plasmid # 44876).
The SSA control construct is identical to the SSA repair reporter construct described above, except that each 18bp I-SceI sequence has been replaced with the randomly-generated sequence TTGACAATTATTAGTACA. The SSA repair reporter construct carrying 3% heterology is identical to the SSA repair reporter except for 6 wobble position changes in the 5′ non-functional mCitrine. These changes occur at positions 27, 54, 81, 111, 132, and 174.
The mCherry degron (abbreviated ‘RFPdegron’) variants of the SSA reporter constructs contain a 426 bp CLN2-C-terminal destabilization sequence tagged to the C terminus of mCherry. This sequence is a truncation of the CLN2-C-terminal destabilization sequence found in a plasmid containing dsGFP(Salama et al., 1994). It encodes amino acids 368-487 of Cln2 immediately followed by amino acids 525-546 of Cln2. The truncated sequence still contains the PEST and D domains of Cln2 that are involved in Cln2 degradation. The plasmid containing dsGFP, PTEF1-yEGFPCLN2PEST-pRS406, was a gift from Claudia Vickers (Addgene plasmid #64406).
All SSA constructs were constructed in pRS306 plasmids carrying the URA3 marker. The SSA construct was located at the multi-cloning site with a reverse orientation relative to the URA3 marker. For integrating the construct, long homologies to the ura3 locus were cloned upstream of the URA3 and upstream of the SSA reporter (distal to the URA3 marker). A1280 bp ura3 upstream homology with a natural BseRI cutsite at the end was cloned upstream of the plasmid’s URA3 marker. A 669 bp ura3 downstream homology with a natural AatII cutsite at the end was cloned after the PTEF1 driving the 5′ mCitrine repeat. Digestion at the ends of these long homologies using BseRI and AatII generated a linear fragment containing [URA3 marker + SSA reporter] that was gel-purified and transformed to the ura3 locus. Integration of the SSA construct was confirmed by colony PCR and sequencing.
Regarding the system of doxycycline-inducible I-SceI expression, a variant of rtTA (rtTA-SEG72P) was used with a less leaky PTETO4 promoter(Roney et al., 2016). The yeast strain used to amplify the rtTA variant and PTETO4 were provided by Mads Kaern. I-SceI driven by PTETO4 contains an N-terminal NLS and HA tag. The I-SceI DNA sequence was codon optimized for yeast and designed to produce the protein sequence described in previous work(Böttcher et al., 2014).
CLN2 gene copy number was tripled using a pRS400 backbone plasmid containing the CLN2 ORF with full upstream and downstream intergenic regions in between the KpnI and BamHI sites at the multicloning site. This plasmid was linearized within the unique PshAI site in the CLN2 sequence prior to transformation into yeast. qPCR was performed on transformant colonies to quantify the number of integrations.
Whi5 was tagged in a markerless manner with mCitrine (‘Whi5-YFP’) using a pop-in, pop-out strategy with URA3. The C-terminal linker GGATCCGTTTCT connecting Whi5 to mCitrine was identical to that used in the strain KSE108-1(Schmoller et al., 2015). The description of this linker sequence was kindly provided by Kurt Schmoller.
Sequencing-based genotypic characterization of the SSA reporter activity
3 cultures of the SSA strain (yTY125a) were grown overnight in CSM 2% glucose media to the cell-densities (OD600) of 0.27, 0.31, and 0.13. In the morning, 2 mL of the overnight cultures was added to 3ml of doxycycline-containing media (to a final doxycycline concentration of 10 ug/ml). The doxycycline-containing cultures were then returned to the shaker and grown for another 4 hours. Then, 250 ul of doxycycline-treated culture was diluted 20-fold in 5 mL media to a final doxycycline concentration of 0.5 ug/ml and grown for another 5 hours. Cells were sorted based on high or low YFP fluorescence in a FACSAria (Becton Dickinson) cell sorter. The sorted cells were then plated and allowed to grow for another 2 days to form colonies. To characterize SSA repair outcomes in the colonies, colonies were picked to CSM 2% glucose liquid media, and allowed to grow overnight. The overnight-grown cultures were genome prepped and analyzed by flow cytometry.
PCRs were performed on the genome-prepped cultures to amplify the SSA reporter locus for sequencing (Figure S3A). Primers for these PCRs and subsequent sequencing reactions are listed in the Key Resources Table. Presence of PADH1 at the ura3 locus was checked using primers oTY204 and pAdh1r (Figure S3A). Presence of “TEFf terminator PADH1” was checked using primers oTY176 and pAdh1r (Figure S3A). If a PCR product was produced, it was then sequenced with the primer pAdh1r to check the I-SceI cutsite.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae: Strain background: BY4742 haploid with MET15 deleted and LYS2 intact. | Mark Hochstrasser | N/A |
| S. cerevisiae: Strains constructed for this paper, see Table S2 | This Paper | N/A |
| Oligonucleotides | ||
| oTY204 (5′- > 3′): GTGAGCGTCAAGGTCTGTTGAG | This paper | N/A |
| oTY176 (5′ - > 3′): TTATCCCAGTCCGATTCATCAGG | This paper | N/A |
| pAdh1r (5′ - > 3′): TCATTGCGGATATGGTGAGACAAC | This paper | N/A |
| oTY126 (5′ - > 3′): CATGGGTAATACCAGCAGCAG | This paper | N/A |
| oTY18rc (5′ - > 3′): CACCTTCAAACTTGACTTCAGC | This paper | N/A |
| Recombinant DNA | ||
| pKT0211 (source of mCitrine) | Sheff and Thorn, 2004 | Addgene #8374 |
| pFA6-link-yomCherry-CaURA3 (source of mCherry) | Lee et al., 2013. Wendell Lim & Kurt Thorn | Addgene #44876 |
| PTEF1-yEGFPCLN2PEST-pRS406 (Cln2 destabilization sequence used to tag RFP in SSA reporter) | Claudia Vickers | Addgene #64406 |
| Yeast codon-optimized I-SceI with N-terminal NLS and HA tag | Coding sequence identical to the one in Böttcher et al., 2014. | N/A |
| rtTA-SEG72P amplified from yeast strain IR223 | Roney et al., 2016 | N/A |
| PTETO4 promoter amplified from yeast strain IR223 | Roney et al., 2016 | N/A |
| C-terminal linker between Whi5 and mCitrine | Schmoller et al., 2015 | N/A |
| pRS400 (with KanMX4 marker backbone) used for cloning multiple copies of the Cln2 gene | Jef Boeke | N/A |
| Software and Algorithms | ||
| NIS Elements Advanced Research (software) | Nikon Instruments Inc | N/A |
| BD FACSDiva (software) | BD Biosciences | N/A |
| Other | ||
| BD FACSAria Flow Cytometer | BD Biosciences | N/A |
| Andor DU-897 camera | Oxford Instruments | N/A |
| Plan Apo λ 60x Oil Ph3 DM objective | Nikon Instruments Inc | N/A |
| YFP filter cube | Chroma | Chroma 49003 - ET-EYFP |
| mCherry filter cube | Chroma | Chroma 49008 - ET-mCherry, Texas Red |
Presence of PTEF1-mCitrine at the ura3 locus was checked using a PCR reaction with primers oTY204 and oTY126. oTY126 only binds at the 3′ end of mCitrine with reverse orientation. It does not bind in the truncated YFP initially adjacent to PTEF1. oTY204 binds at the SSA reporter locus, outside of the integration site. For this Taq-based PCR reaction, a shorter amplification time of 2 minutes was used to avoid amplifying from the ATG-less YFP in the unrepaired SSA template. If SSA had occurred, the region between the two YFP repeats would be deleted so that the oTY126 primer binding site is 2.9 kb closer to the oTY204 primer binding site. Due to this increased proximity, the PCR reaction is able to amplify a product despite the short extension time.
To make sure that the oTY204 + oTY126 PCR reaction was not able to amplify from the ATG-less YFP in unrepaired SSA templates, the PCR reaction was performed on templates that were positive for “TEF1terminator….PADH1” based on the oTY204+oTY176 PCR reaction. As expected, these templates did not produce product.
The oTY204+oTY126 PCR products were sequenced with oTY18rc and oTY126 to check the interior of mCitrine and PTEF1.
Assuming loss of the I-SceI cutsite prior to colony formation, the overnight cultures were expected to be genetically homogeneous at the SSA locus. Columns 1 and 2 of Table S1 show the FACS-based characterization of different cultures (Percent YFP+, Percent RFP−). The remaining columns show the results of PCR and sequencing-based characterization performed on the genomic DNA extracted from each overnight culture.
Column 3 of Table S1, titled ‘TEF1t…PADH1-RFP present?’, summarizes the results from the PCR reaction with primers oTY176 and pAdh1r. oTY176 binds the TEF1 terminator with forward orientation, while pAdh1r binds the ADH1 promoter with reverse orientation. Amplification of PCR product implies the continued presence of the TEF1t…PADH1-RFP region of the SSA reporter (‘yes’ in column 3). The presence of TEF1t…PADH1-RFP at the SSA reporter locus was confirmed using a PCR reaction with primers oTY204 and pAdh1r. Because the oTY204 primer binds at the SSA reporter locus (ura3), outside of the reporter integration site, formation of a PCR product indicates presence of an ADH1 promoter at the ura3 locus. If SSA occurred between the YFP repeats, no PCR product would be produced. The results of the PCR reaction using primers oTY204+pAdh1r agreed with the results of the PCR reaction using primers oTY176+pAdh1r (column 3).
Column 4 of Table S1, titled ‘I-SceI cutsite’, summarizes the results from the sequencing of the PCR products obtained using the oTY176 + pAdh1r primers, as described in the previous paragraph. The PCR products were sequenced with the primer pAdh1r to check the presence of the I-SceI cutsite. The two outcomes that were observed were: 1. A perfectly intact I-SceI cutsite (‘intact’), and 2. A deleted I-SceI cutsite (‘deleted’).
Column 5 of Table S1, titled ‘URA3locus-PTEF1-YFP present?’, summarizes the results of the PCR reaction with primers oTY204 and oTY126. oTY126only binds the 3′ end of mCitrine. Prior to SSA repair, this binding site is only present in the nonfunctional YFP at the 3′ end of the SSA reporter sequence. This is too distant from the oTY204 binding site for any product to be amplified when a short PCR extension time is used (outcome ‘no’ in the table). SSA repair results in loss of the sequence between the two non-functional YFP repeats. As a result, the oTY126 primer’s binding/annealing site is close enough to the oTY204 primer’s binding site for a PCR product to be amplified (outcome ‘yes’ in the table). To verify that joining of the direct YFP repeats was not occurring in the PCR reaction itself, the reaction was performed on genomic DNA templates that had previously been confirmed to be SSA negative. These were the templates that had produced a PCR product from the use of the oTY176 + pAdh1r primers (outcome ‘yes’ in column 3). As expected, these templates did not produce a PCR product when oTY204 + oTY126 primers were used (all ‘no’ in the table).
Column 6 of Table S1, titled ‘Mutations in PTEF1-YFP?’, refers to the results from the sequencing of the PCR products obtained using the oTY204 + oTY126 primers (outcome ‘yes’ in column 5); the sequencing primers were oTY18rc and oTY126. These sequencing primers bind to the mCitrine sequence with reverse orientation, so that the PTEF1-YFP region could be sequenced. Each of the sequencing results obtained from the use of the sequencing primer oTY126 confirmed that PTEF1 was directly upstream of a full length YFP. Two types of mutations were identified in the YFP− cultures (YFP− nature was characterized based on FACS). The first was the absence of a start codon for mCitrine (‘no start codon’ outcome in the table). The second was a premature stop codon within the mCitrine at the position 193 bp from the 5′ end of the mCitrine (outcome ‘TAG stop codon (193th bp of YFP)’ in the table).
Column 7 of Table S1, titled ‘Number of Cultures’, lists the number of cultures falling into categories defined by various combinations of outcomes for the previous columns. Column 8, titled “SSA”?, summarizes the previous columns’ results by stating whether SSA repair occurred between the YFP repeats. ‘Yes’ in column 8 means the data summarized in columns 3,5, and 6 is consistent with SSA repair having occurred in the SSA reporter. For example, all 6 cultures described in row 1 of Table S1 were confirmed to have undergone SSA repair in the reporter, while the 4 cultures described in the bottom row of Table S1 did not undergo SSA repair.
Due to dilution of RFP over many generations, cultures derived from a cell that had undergone SSA repair between the non-functional YFP repeats should be RFP−. Sequencing and FACS measurement showed the expected SSA-repaired sequence in the 6 YFP+, RFP− cultures. Nine YFP−, RFP− cultures were also confirmed to have undergone SSA repair between the YFP repeats based on the results of columns 3, 5, and 6 of Table S1. Absence of YFP in these cultures was due to preservation of one of the YFP inactivating mutations found in the original non-functional YFP repeats. In these cultures, the SSA locus was found to have a single PTEF1-YFP in which the YFP was either lacking a start codon (like the 3′ non-functional YFP repeat), or interrupted by a stop codon at the same location as that of the original 5′ non-functional YFP repeat (position 193 from the start of the YFP sequence).
There were four cultures that were 1%–10% YFP+ and 1%–10% RFP−, with an intact I-SceI cutsite. These results can be explained by the scenario in which the cell that produced the colony was not cut in the I-SceI site. Under this scenario, leaky I-SceI expression and cutting at the I-SceI cutsite, followed by repair by SSA, occurs in only a subset of cells during colony formation and growth of the overnight culture. This leads to an intermediate percentage of YFP+, RFP− cells.
A single culture had an intact I-SceI cutsite but0% YFP+ and RFP− cells. Given the previous discussion of cutting due to leaky I-SceI expression, a possible explanation for this culture is an I-SceI inactivating mutation.
Finally, there were two 0% YFP+, 0% RFP− cultures that were found to have lost the I-SceI cutsite via small deletions of 26 and 28 bp. Such a deletion may have occurred via NHEJ. The lack of YFP+ or RFP− cells in these cultures is easily explainable by the absence of the I-SceI cutsite. Suppose that the cell that gave rise to the picked colony had lost the I-SceI cutsite via a deletion. Then, every cell in the colony and every cell in the overnight culture would have inherited the I-SceI-deleted genotype. This would have shut down any opportunity for generation of functional YFP and loss of RFP via SSA repair.
Microfluidic chip construction, its operation, and RLS measurements
Detailed information about the PDMS chip construction, its operation, and RLS measurements are provided in a previous paper from our laboratory(Liu et al., 2015). As the growth condition in the chip, CSM minimal media (containing 2% glucose) was used.
Measurement and comparison of age-related changes in SSA repair efficiency
Only cells that were initially YFP− (below threshold) before doxycycline treatment were kept for determination of SSA repair efficiency. Cells obtained from the experiments targeting the old age were required to be > = 15 generations old at the beginning of the doxycycline exposure. Cells obtained from the experiments targeting the young age were required to be ≤ 5 generations old at the beginning of the doxycycline exposure. Only cells that were alive (un-bursted) and observable at or beyond the 5-hour time window after doxycycline removal were included in analysis. The relevant cell-counts per experimental replicate are shown in Table S3. For each experimental replicate, the SSA repair efficiency was calculated as: [(the number of cells repaired within 5 hours after dox removal) / (total number of cells in analysis)].
For each strain, statistical significance of the observed age-related SSA repair efficiency differences was assessed using Fisher’s Exact Test with the R function fisher.test. Single cells from the young cell replicates were pooled together; the same was separately done for the single cells from the old cell replicates. Fisher’s Exact Test was applied to the 2×2 contingency table, with the rows specifying whether the cell was in an old or young SSA repair experiment, and the columns specifying whether the cell was repaired or not repaired based on YFP. The default arguments of the fisher.test function were used, including a two-sided alternative hypothesis.
An estimate for the ratio of old cells’ SSA repair efficiency to young cells’ SSA repair efficiency was obtained by taking the ratio of the old repaired cell fraction to the young repaired cellfraction(Katz et al., 1978). Let ro be the number of repaired old cells out of no old cells, and ry be the number of repaired young cells out of ny young cells. The estimate is then given by the Metric: [(ro/no)/(ry/ny)](Katz et al., 1978). Assuming that the log of this metric is approximately normally distributed, we obtain a 95% confidence interval (CI) for it(Katz et al., 1978). To calculate the standard error (SE) of the log of the Metric, we assume that the old cell outcomes arise from a binomial distribution with no trials and the probability of repair pold(repaired) (Katz et al., 1978). Similarly, we assume that the young cell outcomes arise from an independent binomial distribution with ny trials and the probability of repair pyoung(repaired) (Katz et al., 1978). Then our log(Metric) is an estimator for log(pold(repaired) / pyoung(repaired)). Applying the delta method to estimate the variance of our estimator for log(pold(repaired) / pyoung(repaired)), we obtain a approximate variance of 1/ro + 1/ry − 1/no − 1/ny(Katz et al., 1978). The square root of this approximate variance is the standard error(SE). The 95% CI for the log(Metric) is then given by [(ro / no) / (ry / ny)] ± 1.96*SE. The error bars on Figure S9 show the antilog of this CI.
DSB induction experiments in aging cells
To test SSA repair in young cells, the cells were aged for 7 hours before I-SceI induction. For old cells, the cells were aged for 28 hours before I-SceI induction. Due to differences in cell-division rate, a range of replicative ages are present in the chip before I-SceI induction. Nonetheless, most cells at 7 hours are less than or equal to 5 generations old, and most cells at 28 hours are greater than or equal to 15 generations old (the average age of cells are given in the main text). Only cells in these age ranges are selected for age-based comparison of SSA repair.
After the initial aging period without doxycycline, cells are treated with 10 ug/ml doxycycline media for 4 hours. Doxycyline-free media is restored for the remainder of the movie. SSA repair in each cell is assessed by comparing YFP levels prior to doxycycline treatment and after doxycycline treatment; the assessment is made based on whether or not the YFP level of each cell increases over the threshold level during the 9-hour time period after the addition of doxycycline (4 hours of doxycycline treatment and 5 hours afterward).
To avoid differences in photo-toxicity prior to I-SceI induction, old and young cells are subjected to the same period of fluorescence snapshots before doxycycline treatment. The fluorescence part of the movie is started 140 minutes before the addition of doxycycline-containing media. YFP/mCitrine (5% Sola intensity, 50 ms exposure time) and mCherry (5% Sola intensity, 200 ms exposure time) snapshots were taken every 30 minutes for the remainder of the movie. PhaseG images were taken every 10 minutes throughout the entire movie. For the degron-tagged mCherry containing strains (referred to as ‘RFPdegron’), the same imaging protocol was used except that for mCherry the Sola intensity was 4% with a 200 ms exposure time.
Average budding interval measurements
To have a measure of I-SceI DSB-induced cell-cycle arrests, the average budding interval for 3 different time windows was measured: the 4-hour window prior to doxycycline addition (‘pre-dox’), the 4-hour window during doxycycline treatment, and the 5-hour window after doxycycline removal. The average budding interval for the 9-hour time window after doxycycline addition (including the 5-hour time window after doxycycline removal) was also measured. Here, budding interval refers to the difference in time between the appearances of consecutive daughter buds from the same mother cell. For each cell, the budding intervals fully contained within the time window of interest were measured. The full budding interval beginning before, but ending within, the time window of interest was also measured. For each cell and time window of interest, the final partial budding interval was defined as the time between the appearance of the last daughter bud in the time window of interest and the end of that time window. The purpose of measuring this partial budding interval was to be able to detect cell-cycle arrested cells. The average of the final partial budding interval and the full budding intervals measured in the time window of interest was then calculated.
G1 duration measurement experiments in aging cells
Strains yTY159b and yTY160a, containing Whi5-YFP, were grown and loaded into the aging chip in a manner identical to the one described for the SSA repair measurement experiments performed for old cells. Prior to fluorescence measurements, the cells were aged in the chip while bright-field images were taken for 25 h 40 min using the same settings as the SSA repair measurement experiments. Fluorescence measurements consisted of 3 YFP snapshots taken per frame (5% Sola, 50 ms exposure, Normal 1 Mhz readout); these snapshots were taken at the main imaging plane, and 1 um above and below the main imaging plane. These snapshots were taken every 10 minutes for 800 minutes (> 9 hours after doxycycline addition). Because of the increased time perxy-location due to acquiring z stacks, changes to the microscopy protocol were introduced to avoid overlap between the fast media-flow period of the chip(Liu et al., 2015) and image acquisition, improving image quality and localization detection. More specifically, this was needed because fast media-flow through the chip could lead to changes in z-height of the cells that could prevent observation of nuclear Whi5-YFP. To help reduce the overlap, total image acquisition time was reduced by choosing only a subset of all available x-y locations for fluorescent measurements. To make up for the reduction in total number of cells analyzed, frame locations with sufficiently high numbers of trapped cells were selected for z stack imaging. Media flow settings(Liu et al., 2015) were also changed during the z stacked fluorescence imaging part of the movie to avoid interference with image acquisition. The slow phase of the media flow was increased from 8 min (16.1 ul at 2.0ul/min) to 18 min (36.1 ul at 2.0ul/min). The fast phase of the media flow (60 ul at 30ul/min) was unchanged. For consistency with SSA repair measurement experiments, cells were treated with doxycycline-containing media at the same time point and duration after initiating the movie, and by using the same concentration of doxycycline (10 ug/ml), but with the modified flow rate settings as described above.
PTEF1-YFP measurements in aging cells
For the aging experiment performed using the strain carrying the PTEF1-YFP reporter (Figure S7A), we saw a 100% YFP+ and 100% RFP− cell population. We attributed this outcome to leaky cutting and SSA repair in the cell that gave rise to the colony we picked for the cell culture used to initialize the aging experiment. The strain corresponding to the picked colony was identical to the main strain (yTY125a) used for SSA repair measurements in this study except that the 5′ non-functional mCitrine sequence was replaced with a full mCitrine (‘YFP’) sequence containing a stop codon and I-SceI cutsite inserted into its interior. The interior stop codon is located at the same position where it is found in the 5′ mCitrine repeat of yTY125a. Two I-SceI cutsites (in opposite orientation) immediately follow the interior stop codon, and the strain lacks the I-SceI cutsites immediately upstream of the ADH1 promoter, as in the main strain yTY125a. Due to the interior stop codon, the 5′ mCitrine repeat of the strain does not produce full length, fluorescent protein. The strain still contains an mCherry (‘RFP’) downstream of the ADH1 promoter. Following cutting by I-SceI in the strain, SSA repair between the mCitrine repeats results in generation of functional mCitrine (‘YFP’) and loss of mCherry (‘RFP’). The resulting sequence is identical to what the main SSA strain (yTY125a) would produce at the SSA reporter locus following SSA repair. Despite the initial sequence of the reporter differing from that of the SSA repair reporter contained in yTY125a, the fact that the culture used in the experiment was 100% YFP+ and 100% RFP− means that all cells had been repaired by SSA.
The settings used for the PTEF1-YFP fluorescence measurement experiments were slightly different from those used in the SSA repair experiments. Doxycycline was added at 21.8 h into the movie rather than at 28 h. Media containing 4 ug/ml doxycycline was used for one hour instead of treating the cells for 4 hours with media containing 10 ug/ml doxycycline. Additionally, fluorescence measurements were taken every 2 hours as opposed to every 0.5 hours. A fluorescence measurement was assigned to a generation only if that measurement event occurred during the generation interval, and if the fluorescence measurement was the first to occur after the budding event marking the beginning of that generation. Therefore, for a specific cell, it was possible not to have fluorescence measurements for some generations.
QUANTIFICATION AND STATISTICAL ANALYSIS
Cell-by-cell assessment of YFP repair
After YFP background correction, SSA repair was assessed. A YFP cutoff was first defined, so that cells crossing and remaining above this threshold could be classified as repaired. To determine the cutoff, cells from the SSA control strain were aged to young or old ages (Figure S1A), and imaged by microscopy using the protocol described in the relevant sections above. The mean and standard deviation of YFP levels in the cells of the control strain (not having a cut site) was measured for the 5 time-points prior to dox treatment, to determine the YFP signal due to autofluorescence. This procedure was carried out separately for 2 replicates of young or old cells. The mean + 5*SD was calculated and it less than 6 AU (arbitrary units) for each of the 4 replicates. Based on this, we chose a YFP threshold of 7 AU, values above which indicate the presence of functional YFP.
An R-script was used to identify cells that crossed this 7 AU threshold and remained above it for the remainder of the movie, and the times at which this occurred. The script also recorded cells that crossed and remained above a 300 AU threshold, and the times that this occurred. Cells that crossed and remained above this 300 AU threshold were automatically classified as repaired. Since bright neighboring cells can push the YFP signal of a cell above the 7 AU threshold, cells in which only the 7 AU threshold was crossed were manually inspected movie-by-movie. If no bright neighboring cells were nearby the cell to analyze, the cell was confirmed as repaired. For cells for which the crossing of the threshold was assessed to be likely due to a bright neighbor, the later time points were inspected. If at some later time point, YFP was above the threshold and no bright cells were nearby, then the cell was classified as repaired and this time was recorded. If not, the cell was recorded as unrepaired.
Additionally, the distribution of lag times between crossing of the 7 AU and 300 AU threshold was measured. Based on this distribution, lags > 4 fluorescent measurement durations (120 min) were considered abnormal. For such cells, the crossing of the lower threshold was considered a possible false-positive, and manually inspected to make sure no bright neighbors were nearby. If the crossing of the threshold was due to bright neighbor cells, the later time points were manually scanned to find the first above-threshold YFP measurement that was clearly due to the cell of interest’s internal fluorescence.
Cell-by-cell assessment of RFP absence in the degron-carrying strain
To determine a cutoff for detecting the absence of mCherry (abbreviated ‘RFP’) expression in cells containing the the degron-tagged RFP within the SSA repair reporter cassette, we measured single-cell RFP levels by quantifying average RFP pixel intensities in YFP+ cells. Formation of functional YFP is contingent on loss of the mCherry+degron cassette after cutting. RFP measurements from these cells provide a measure of the intracellular RFP signal as well as cellular autofluorescence and background fluorescence. The RFP cutoff was chosen to be the 95% quantile of the single-cell RFP measurements pooled from the cells that were always observed to be YFP+ (Figure S1B). Any RFP expression level below this cutoff was treated as RFP negative while any level above the cutoff was treated as RFP positive.
Image processing
Fluorescence frames were aligned based on non-fluorescent PhaseG images using NIKON NIS Elements software’s image-alignment method, and exported as tifs. A custom MATLAB script was used to automatically draw a standard polygon within each trap of the microfluidic chip and save the corresponding binary masks. Masks were manually inspected to make sure the polygons were placed correctly within each trap, so that they would fall within the tracked cells. The masks were applied to the fluorescent images to obtain average mCitrine (‘YFP’) and mCherry (‘RFP’) pixel intensities within each polygon for all time frames in the movie. For situations in which the polygonal region of interest was not fully contained within the cell, a polygon was manually drawn within the cell of interest.
Background fluorescence was subtracted from the cell-specific fluoresence measurements to obtain a more accurate measure of RFP and YFP levels in the cell. Due to spatial and temporal homogeneity in background fluorescence, background fluorescence was obtained for a local, rectangular region around each cell of interest. A custom MATLAB GUI was written to identify any non-background region (cell or PDMS) in each rectangle for all time-points of observation. These pixels were excluded in calculating the local background. The average pixel intensity of all background pixels in the local rectangle was defined as the background signal.
A source of error would be present when very bright cells/objects are directly adjacent to the cell of interest. This is not accounted for by the local background pixel average method described above, which includes further away pixels not influenced by the fluorescence of the bright object. To identify these situations, adjacent, non-daughter objects were circled manually using a custom MATLAB GUI. If the average pixel intensity of these adjacent objects was greater than 200 AU and if the objects were less than 15 pixels away from the center of the polygonal region of interest, then the local background quantification method described above was not used. Instead, if possible, a test ROI to measure the effect of the bright neighbor object on background was drawn at the same relative distance as the trapped cell ROI. Then, for a given mother cell, the average pixel intensities of these test ROIs were averaged and used as the background to be subtracted.
For the measurement of PTEF1-YFP expression over generations (Figure S7A), a modified background-subtraction method was used. For each image frame, a single rectangular region, 3800-10000 pixels in area, was selected at a location of the image frame lacking cells. The mean fluorescence within this cell-free rectangular region was defined as the background fluorescence for the entire image frame. For cells in that image frame, this background fluorescence was subtracted from each circled cell’s mean fluorescence measured across the circled area/pixels.
For G1 duration measurements in the Whi5-YFP strain (Figure S10), trapped cells were first circled across relevant time frames. A maximum intensity projection (z direction) of the three YFP snapshots was then taken. For each circled cell, the total YFP value (from the maxIP image) in each 5x5 pixel-square fitting entirely within the circled region was calculated. The maximum and the mean of these total YFP values across all allowed 5x5 pixel locations were recorded. If Whi5-YFP is nuclear-localized, we expect the maximum value to be high, and to occur for a 5x5 pixel-square with a high level of nuclear overlap. If Whi5-YFP is not nuclear-localized, we expect the maximum value to be lower and its location to fail to overlap with the location of the nucleus. The mean 5x5 pixel value serves as a measure of the background fluorescence in the cell. The difference between the maximum and mean of the 5x5 pixel YFP totals was used as the Whi5-YFP localization score for that cell. A thresholding procedure, described in Figure S10, was applied to this YFP localization score to measure G1 duration.
Flow cytometry data analysis
Raw flow cytometry data were gated using a gate selected based on the forward scattering (FSC) and side scattering (SSC) measurements. The gate chosen was a rectangle with edges parallel to the FSC-SSC axes and with coordinates FSCmin: 28000a.u., FSCmax: 48000a.u., SSCmin: 8000a.u., SSCmax: 28000a.u.. Each sample had about ~5800 cells on average after gating. Cells with YFP > 1000a.u. were defined to be YFP+. Cells with RFP < 1000a.u. were defined to be RFP−.
DATA AND CODE AVAILABILITY
The published article includes all data and results generated or analyzed during this study. Data analysis details are included in the published article (no special computational algorithms or code was used).
Supplementary Material
Highlights.
Efficiency of DNA repair by single-strand annealing is lower in old yeast cells
This decline is not due to increased use of non-homologous end joining
Cell-cycle regulation is involved in the age-associated DNA repair inefficiency
Heteroduplex rejection remains high in old cells
ACKNOWLEDGMENTS
We thank Valerie Horsley, Mark Hochstrasser, Weimin Zhong, Ruijie Song, and Acar laboratory members for useful discussions and feedback on this study. T.Z.Y. acknowledges support through a Gruber Science Fellowship. T.Z.Y. was in part supported by a predoctoral training grant from the NIH (T32 GM007499). M.A. acknowledges funding from the Ellison Medical Foundation (AG-NS-1015-13) and the NIH (1DP2AG050461-01, 1R01GM127870-01, and 1U54CA209992-01).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.082.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PD, Jasper H, and Rudolph KL (2015). Aging-induced stem cell mutations as drivers for disease and cancer. Cell Stem Cell 16, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio T, Baer R, and Gautier J (2014). DNA double-strand break repair pathway choice and cancer. DNA Repair (Amst.) 19, 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Liefshitz B, and Kupiec M (2004). The CDK regulates repairof double-strand breaks by homologous recombination during the cell cycle. EMBO J. 23, 4868–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berset C, Griac P, Tempel R, La Rue J, Wittenberg C, and Lanker S (2002). Transferable domain in the G(1) cyclin Cln2 sufficient to switch degradation of Sic1 from the E3 ubiquitin ligase SCF(Cdc4) to SCF(Grr1). Mol. Cell. Biol 22, 4463–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher R, Hollmann M, Merk K, Nitschko V, Obermaier C, Philippou-Massier J, Wieland I, Gaul U, and Förstemann K (2014). Efficient chromo-somal gene modification with CRISPR/cas9 and PCR-based homologous recombination donors in cultured Drosophila cells. Nucleic Acids Res. 42, e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burhans WC, and Weinberger M (2007). DNA replication stress, genome instability and aging. Nucleic Acids Res. 35, 7545–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Rondinelli B, and D’Andrea AD (2016). Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 26, 52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Mantiero D, Guerini I, Lucchini G, and Longhese MP (2008). The Yku70-Yku80 complex contributes to regulate double-strand break processing and checkpoint activation during the cell cycle. EMBO Rep. 9, 810–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Nishikawa JL, Tang X, Millman JS, Schub O, Breitkreuz K, Dewar D, Rupes I, Andrews B, and Tyers M (2004). CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 117, 899–913. [DOI] [PubMed] [Google Scholar]
- de Bruin RA, McDonald WH, Kalashnikova TI, Yates J 3rd, and Wittenberg C (2004). Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 117, 887–898. [DOI] [PubMed] [Google Scholar]
- Delabaere L, Ertl HA, Massey DJ, Hofley CM, Sohail F, Bienenstock EJ, Sebastian H, Chiolo I, and LaRocque JR (2017). Aging impairs double-strand break repair by homologous recombination in Drosophila germ cells. Aging Cell 16, 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney JR, Chou A, Olsen B, Carr D, Murakami C, Ahmed U, Sim S, An EH, Castanza AS, Fletcher M, et al. (2013). End-of-life cell cycle arrest contributes to stochasticity of yeast replicative aging. FEMS Yeast Res. 13, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncic A, Falleur-Fettig M, and Skotheim JM (2011). Distinct interactions select and maintain a specific cell fate. Mol. Cell 43, 528–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman-Lobell J, Rudin N, and Haber JE (1992). Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol. Cell. Biol 12, 1292–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas AA, and de Magalhães JP (2011). A review and appraisal of the DNA damage theory of ageing. Mutat. Res 728, 12–22. [DOI] [PubMed] [Google Scholar]
- Ivanov EL, Sugawara N, Fishman-Lobell J, and Haber JE (1996). Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics 142, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadyk LC, and Hartwell LH (1992). Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 132, 387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, and Guarente L (1999). The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, Powers RW 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, and Kennedy BK (2005). Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310, 1193–1196. [DOI] [PubMed] [Google Scholar]
- Karanam K, Kafri R, Loewer A, and Lahav G (2012). Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell 47, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz D, Baptista J, Azen SP, and Pike MC (1978). Obtaining confidence intervals for the risk ratio in cohort studies. Biometrics 34, 469–474. [Google Scholar]
- Landry BD, Doyle JP, Toczyski DP, and Benanti JA (2012). F-box protein specificity for g1 cyclins is dictated by subcellular localization. PLoS Genet. 8, e1002851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lim WA, and Thorn KS (2013). Improved blue, green, and red fluorescent protein tagging vectors for S. cerevisiae. PLoS ONE 8, e67902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, and Acar M (2018). The generational scalability of single-cell replicative aging. Sci. Adv 4, o4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Young TZ, and Acar M (2015). Yeast Replicator: a high-throughput multiplexed microfluidics platform for automated measurements of single-cell aging. Cell Rep. 13, 634–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, and Yankner BA (2004). Gene regulation and DNA damage in the ageing human brain. Nature 429, 883–891. [DOI] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, and Gorbunova V (2008). Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst.) 7, 1765–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Tian X, Van Meter M, Ke Z, Gorbunova V, and Seluanov A (2012). Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc. Natl. Acad. Sci. U S A 109, 11800–11805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick MA, Delaney JR, Tsuchiya M, Tsuchiyama S, Shemorry A, Sim S, Chou AC, Ahmed U, Carr D, Murakami CJ, et al. (2015). A comprehensive analysis of replicative lifespan in 4,698 single-gene deletion strains uncovers conserved mechanisms of aging. Cell Metab. 22, 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, and Gottschling DE (2003). An age-induced switch to a hyper-recombinational state. Science 301, 1908–1911. [DOI] [PubMed] [Google Scholar]
- McMurray MA, and Gottschling DE (2004). Aging and genetic instability in yeast. Curr. Opin. Microbiol 7, 673–679. [DOI] [PubMed] [Google Scholar]
- Morgan WF, Corcoran J, Hartmann A, Kaplan MI, Limoli CL, and Ponnaiya B (1998). DNA double-strand breaks, chromosomal rearrangements, and genomic instability. Mutat. Res 404, 125–128. [DOI] [PubMed] [Google Scholar]
- Neurohr GE, Terry RL, Sandikci A, Zou K, Li H, and Amon A (2018). Deregulation of the G1/S-phase transition is the proximal cause of mortality in old yeast mother cells. Genes Dev. 32, 1075–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Postnikoff SD, Chavez M, and Tyler J (2018). Impaired cohesion and homologous recombination during replicative aging in budding yeast. Sci. Adv 4, eaaq0236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes S, and Maggert KA (2009). Ribosomal DNA contributes to global chromatin regulation. Proc. Natl. Acad. Sci. U S A 106, 17829–17834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston CR, Flores C, and Engels WR (2006). Age-dependent usage of double-strand-break repair pathways. Curr. Biol 16, 2009–2015. [DOI] [PubMed] [Google Scholar]
- Roney IJ, Rudner AD, Couture JF, and Kærn M (2016). Improvement of the reverse tetracycline transactivator by single amino acid substitutions that reduce leaky target gene expression to undetectable levels. Sci. Rep 6, 27697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama SR, Hendricks KB, and Thorner J (1994). G1 cyclin degradation: the PEST motif of yeast Cln2 is necessary, but not sufficient, for rapid protein turnover. Mol. Cell. Biol 14, 7953–7966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnoski EA, Song R, Ertekin E, Koonce N, and Acar M (2018). Fundamental characteristics of single-cell aging in diploid yeast. iScience 7,96–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmoller KM, Turner JJ, Kõivomägi M, and Skotheim JM (2015). Dilution of the cell cycle inhibitor Whi5 controls budding-yeast cell size. Nature 526, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seluanov A, Mittelman D, Pereira-Smith OM, Wilson JH, and Gorbunova V (2004). DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc. Natl. Acad. Sci. USA 101, 7624–7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff MA, and Thorn KS (2004). Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast 21, 661–670. [DOI] [PubMed] [Google Scholar]
- Sinclair DA, and Guarente L (1997). Extrachromosomal rDNA circles-a cause of aging in yeast. Cell 91, 1033–1042. [DOI] [PubMed] [Google Scholar]
- Song R, Sarnoski EA, and Acar M (2018). The systems biology of single-cell aging. iScience 7, 154–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinkraus KA, Kaeberlein M, and Kennedy BK (2008). Replicative aging in yeast: the means to the end. Annu. Rev. Cell Dev. Biol 24, 29–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, and Haber JE (1992). Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol. Cell. Biol 12, 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N, Goldfarb T, Studamire B, Alani E, and Haber JE (2004). Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc. Natl. Acad. Sci. USA 101,9315–9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukup-Jackson MR, Kiraly O, Kay JE, Na L, Rowland EA, Winther KE, Chow DN, Kimoto T, Matsuguchi T, Jonnalagadda VS, et al. (2014). Rosa26-GFP direct repeat (RaDR-GFP) mice reveal tissue- and age-dependence of homologous recombination in mammals in vivo. PLoS Genet. 10, e1004299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Rothstein R, and Lisby M (2014). Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics 198, 795–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toczyski DP, Galgoczy DJ, and Hartwell LH (1997). CDC5 and CKII control adaptation to the yeast DNA damage checkpoint. Cell 90, 1097–1106. [DOI] [PubMed] [Google Scholar]
- Trovesi C, Falcettoni M, Lucchini G, Clerici M, and Longhese MP (2011). Distinct Cdk1 requirements during single-strand annealing, noncrossover, and crossover recombination. PLoS Genet. 7, e1002263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RR, Milholland B, de Bruin A, Curran S, Laberge RM, van Steeg H, Campisi J, Maslov AY, and Vijg J (2015). Controlled induction of DNA double-strand breaks in the mouse liver induces features of tissue ageing. Nat. Commun 6, 6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all data and results generated or analyzed during this study. Data analysis details are included in the published article (no special computational algorithms or code was used).