

Abstract
Although genome-wide association studies (GWAS) have identified hundreds of risk loci for breast and prostate cancer, only a few studies have characterized the GWAS association signals across functional genomic annotations with a particular focus on single nucleotide polymorphisms (SNPs) located in DNA regulatory elements. In this study, we investigated the enrichment pattern of GWAS signals for breast and prostate cancer in genomic functional regions located in normal tissue and cancer cell lines. We quantified the overall enrichment of SNPs with breast and prostate cancer association p values < 1 × 10−8 across regulatory categories. We then obtained annotations for DNaseI hypersensitive sites (DHS), typical enhancers, and super enhancers across multiple tissue types, to assess if significant GWAS signals were selectively enriched in annotations found in disease-related tissue. Finally, we quantified the enrichment of breast and prostate cancer SNP heritability in regulatory regions, and compared the enrichment pattern of SNP heritability with GWAS signals. DHS, typical enhancers, and super enhancers identified in the breast cancer cell line MCF-7 were observed with the highest enrichment of genome-wide significant variants for breast cancer. For prostate cancer, GWAS signals were mostly enriched in DHS and typical enhancers identified in the prostate cancer cell line LNCaP. With progressively stringent GWAS p value thresholds, an increasing trend of enrichment was observed for both diseases in DHS, typical enhancers, and super enhancers located in disease-related tissue. Results from heritability enrichment analysis supported the selective enrichment pattern of functional genomic regions in disease-related cell lines for both breast and prostate cancer. Our results suggest the importance of studying functional annotations identified in disease-related tissues when characterizing GWAS results, and further demonstrate the role of germline DNA regulatory elements from disease-related tissue in breast and prostate carcinogenesis.
Introduction
Breast and prostate cancer are the most commonly diagnosed cancer types among women and men in US, respectively, with an estimated 250,000 and 160,000 newly diagnosed cases in 2017 (Siegel et al. 2017). Twin studies have demonstrated that excess familial risk play an important role in development of both cancers, with heritability estimates of 31% (breast cancer) and 57% (prostate cancer) (Hjelmborg et al. 2014; Lichtenstein et al. 2000; Mucci et al. 2016; Page et al. 1997). Multiple genome-wide association studies (GWASs) have identified hundreds of risk loci (Al Olama et al. 2014; Eeles et al. 2013; Michailidou et al. 2013; Michailidou et al. 2017; Schumacher et al. 2018; Thomas et al. 2008; Turnbull et al. 2010). Previous studies have characterized GWAS signals of breast and prostate cancer across tissue-specific functional genomic regions (Al Olama et al. 2015; Dadaev et al. 2018; Han et al. 2015; Hazelett et al. 2014; Li et al. 2013; Michailidou et al. 2017; Quiroz-Zarate et al. 2017; Rhie et al. 2013). A recent study explored the contribution of multiple genomic annotations to cancer single nucleotide polymorphism (SNP) heritability and found a significant enrichment of conserved and regulatory regions (Jiang et al. 2019). However, there is a lack of studies that comprehensively and systematically quantify the contribution of SNPs located in DNA regulatory elements across multiple tissues to breast and prostate cancer risk.
The ENCODE and Roadmap Epigenomics projects have revealed that more than 80% of the human genome participates in at least one type of biochemical event in at least one cell type (Consortium EP 2012). However, regulatory elements of an individual cell type usually cover only 1–2% of the human genome, which suggests a high cell type selectivity of regulatory DNA (Heinz et al. 2015; Song et al. 2011). It is problematic to ignore the cell type when interpreting GWAS findings using information about functional data, and DNA regulatory elements identified in disease-related tissue or pathogenic cell types have been reported to harbor enriched GWAS signals for various traits, from cardiovascular traits to immune diseases (Gusev et al. 2014; Maurano et al. 2012; Trynka et al. 2013). Previous studies have shown that the majority of prostate cancer SNP heritability lies in H3K27ac-marked regions as measured in the prostate adenocarcinoma cell line LNCaP or in DNaseI hypersensitive sites (DHS) measured in cancer cell lines (Gusev et al. 2016; Hazelett et al. 2014). It is reasonable to hypothesize that SNPs significantly associated with breast and prostate cancer risk may be disproportionally clustered in DNA regulatory elements specifically identified in corresponding tissue types.
Here, we used GWAS summary statistics published by the BCAC (Michailidou et al. 2017; Milne et al. 2017) and PRACTICAL (Schumacher et al. 2018) consortia to investigate the enrichment of GWAS signals among 800 types of regulatory genomic regions identified from tissue or cell lines, including DHS, histone modifications H3K27ac and H3K4me1, typical enhancers and super enhancers. By utilizing a newly published statistical approach, GARFIELD 2.0 (Chen et al. 2016; Iotchkova et al. 2019; Trynka et al. 2013), we first quantified the overall enrichment of genome-wide significant signals (p value < 1 × 10−8) across regulatory categories, regardless of tissue type. We then obtained data on DHS, typical enhancers, and super enhancers from individual tissues to assess if significant GWAS signals were selectively enriched in annotations found in disease-related cell lines. To detect any subtype-specific enrichments in breast cancer associations, we further assessed the enrichment in regulatory regions by breast cancer subtype, as defined by estrogen receptor (ER) status: ER+/ER−. Finally, we used stratified LD score regression (Bulik-Sullivan et al. 2015; Finucane et al. 2015) to quantify the enrichment of breast and prostate cancer SNP heritability, respectively, in regulatory regions, to compare patterns of enrichment based on SNP heritability (which assesses all common SNPs) and independent GWAS signals (which only assesses genome-wide significant SNPs).
We found that genome-wide significant breast cancer associations were mostly enriched in DHS, typical enhancers, and super enhancers specific to the breast carcinoma cell line MCF-7. For prostate cancer, GWAS signals were observed to be mostly enriched in DHS and typical enhancers identified in the prostate carcinoma cell line LNCaP as well as super enhancers specific to colorectal carcinoma cell line VACO-9M. These enrichments became stronger with more stringent association p value thresholds. Results from heritability enrichment analysis supported the selective enrichment pattern of functional genomic regions in disease-related cell lines for both breast and prostate cancer.
Materials and methods
Collection of DNA regulatory elements
We collected four categories of DNA regulatory elements: DNasel hypersensitive sites (DHS), histone modification H3K27ac and H3K4me1, typical enhancers, and super enhancers (Supplementary Table 1). For each category of regulatory elements, multiple annotations were generated based on different cell, tissue or tissue donors. In total, 403 types of DHS, 127 types of H3K27ac-marked enhancers, 98 types of H3K4me1-marked enhancers, and 86 types of typical enhancers as well as super enhancers were included.
DHSs were collected from two resources as part of ENCODE (Consortium EP 2012) and the Roadmap Epigenomics Program (Roadmap Epigenomics C et al. 2015) (Supplementary Table 1). The first source included genomewide DHS mapping performed by Maurano et al. (2012) from 349 different cells or tissues, including cultured primary cells (n = 56); immortalized, malignancy derived or pluripotent cell lines (n = 26); primary or differentiated hematopoietic cells (n = 15); multipotent progenitor and pluripotent cells (n = 19); and fetal tissue samples (n = 233). In addition, we further collected 54 types of cell or tissue-specific DHS reported by Thurman et al. (2012), which included immortalized primary cells (n = 16), malignancy-derived cell lines (n = 30), and multipotent and pluripotent progenitor cells (n = 8). In total, we collected DHS from 403 different cells or tissues. Active enhancers marked by histone modification H3K4me1 were collected from Roadmap Epigenomics Program (n = 111) and the ENCODE Project (n = 16) as one of the five core histone marks. The Roadmap Epigenomics Program only tested a proportion of tissue types for non-core histone marks, resulting in data on 98 tissue types (82 from Roadmap; 16 from ENCODE) for the H3K27ac marker. Data on typical enhancers and super enhancers were collected from a previous publication reported by Hnisz et al. (Hnisz et al. 2013), which included primary cells (n = 10), malignancy-derived cell lines (n = 18), blood cells (n = 22), adult tissue samples (n = 31), and fetal tissue samples (n = 5). Annotation data were downloaded as.bed files. DHS data (published by Maurano et al. and Thurman et al.) were downloaded from https://github.com/joepickrell/1000-genomes; typical enhancer and super enhancer data were downloaded as supporting material for Hnisz et al. (2013) cell; histone mark data were downloaded from https://data.broadinstitute.org/alkesgroup/ANNOTATIONS/PRCA/.
GWAS data for breast and prostate cancer
GWAS summary statistics of overall breast cancer, ER+ breast cancer and ER−breast cancer were obtained from the meta-analysis of GWAS published by the Breast Cancer Association Consortium (BCAC), a collaboration involving more than 50 case–control studies (Michailidou et al. 2017; Milne et al. 2017). In total, there were 122,977 breast cancer cases and 105,974 controls of European ancestry. For ER+ breast cancer, data were based on 69,501 cases and 95,042 controls and for ER− breast cancer, data were based on 21,468 cases and 100,594 controls combined with 18,908 BRCA1 mutation carriers (9414 with breast cancer) of European ancestry. GWAS summary statistics for prostate cancer were based on the meta-analysis of GWAS published by the Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome (PRACTICAL) consortium (Schumacher et al. 2018). The meta-analysis was based on 79,194 European ancestry prostate cancer cases and 61,112 controls. Both the breast and prostate cancer GWAS were imputed using the Phase 3 version 5 October 2014 release of 1000 Genome Project as reference panel. We only included SNPs with minor allele frequency (MAF) ≥ 0.1% and imputation quality score r2 ≥ 0.3. In total, the number of SNPs included in our study was 12.4 M for breast cancer and 17.7 M for prostate cancer.
Functional enrichment
To evaluate the contribution of the included functional annotations to breast and prostate cancer GWAS associations, we utilized GARFIELD 2.0 (GWAS analysis of regulatory and functional information enrichment with LD correction), which estimates enrichments of GWAS signals (Iotchkova et al. 2019). Based on GWAS summary statistics, GARFIELD first performs greedy pruning of SNPs eliminating all variants with linkage disequilibrium (LD) r2 > 0.1. Those independent SNPs that pass the pruning process are then annotated according to the functional annotation of interest. GARFIELD then employs a generalized linear model (GLM) to quantify the enrichment of GWAS signals in each functional annotation at various p value cutoffs. To address major sources of confounding, GARFIELD matches for minor allele frequency (MAF), distance to the nearest transaction start site (TSS), and number of LD proxies (LD r2 > 0.8) in the GLM framework. Essential reference files, such as LD matrix, MAF, and distance to the nearest TSS were based on sequence data from a European ancestry population in the UK10K study (Consortium UK et al. 2015) as implemented in the GARFIELD software package. GARFIELD is also able to calculate the effective number of annotations (Neff) for adjustment of multiple testing. In this analysis, we used p = 0.05/Neff as the criteria for statistical significance.
Enrichment of breast and prostate cancer heritability was estimated using stratified LD score regression, which uses GWAS summary statistics to partition the heritability of traits by functional categories (Bulik-Sullivan et al. 2015; Finucane et al. 2015; Gazal et al. 2017). It relies on the fact that for a polygenic trait, SNPs with a high LD score will have larger effect than SNPs with a low LD score, since the χ2 association statistic for a given SNP includes the effects of all SNPs that it tags. If SNPs are grouped into functional categories that contribute differentially to the heritability of a trait, then LD to a category that is enriched for heritability will increase the χ2 statistic of a SNP more than LD to a category that does not contribute to heritability. Contribution of heritability from a certain functional category can be estimated from the regression model , where N is sample size, C the indexes categories, ℓ(j, C) the LD score of SNP j with respect to category C, α the term that measures the contribution of confounding biases, and τC is the per-SNP heritability in category C. Enrichment of heritability within a functional category is defined to be the proportion of SNP heritability in the category divided by the proportion of SNPs. Standard errors and corresponding p values are then estimated with a block jackknife approach. To provide an unbiased estimation of heritability enrichment for each cell type-specific annotation, we utilized a ‘full baseline model’ that controlled for 53 annotations non-specific to any cell type (Gazal et al. 2017).
Results
Enrichment of GWAS signals in four categories of functional annotations
We first estimated the enrichment of GWAS association signals at various p value thresholds (p < 10−5, p < 10−6, p < 10−7, p < 10−8) for breast and prostate cancer in five categories of DNA regulatory elements, including DHS, histone marks H3K27ac and H3K4me1 (marking active enhancers), typical enhancers, and super enhancer across multiple cell and tissue types (Fig. 1). In total, we had data for 403 cell types, tissue types or donors for DHS, 127 for H3K27acmarked enhancers, 98 for H3K4me1-marked enhancers, and 86 for both typical enhancers and super enhancers.
Fig. 1.

Overall enrichment of GWAS signals of breast and prostate cancer across five categories of DNA regulatory elements. Each box represents the first through third quartile of the enrichment fold of GWAS signals at specific thresholds for each functional category type, with whiskers covering 1.5-fold of interquartile range from the box. Tissue-specific annotations with extremely high or low enrichment in the category are marked as outliers. All tissue- or cell-specific functional regions were considered (n = 800)
In general, we observed slightly higher median enrichments across cell types in breast cancer compared to prostate cancer for all four GWAS p value thresholds. The distribution of enrichments across cell types did not differ depending on GWAS p value threshold. Of interest, we observed a wider spread of cell type-specific enrichments for DHS, typical enhancer, and super enhancer as compared to histone marks, with some tissue types showing enrichment estimates higher than sixfold at an association p value threshold of 10−8.
Breast and prostate cancer GWAS signals are mostly enriched in regulatory elements of disease-related cell lines
We then investigated the enrichment of GWAS associations at the p < 10−8 threshold for breast and prostate cancer in tissue or cell type-specific DHS (N = 403), typical enhancer (N = 86), and super enhancer (N = 86), as carcinoma cell line annotations were only available for those three categories (Fig. 2, Supplementary Tables 2–7). For breast cancer GWAS signals, we observed the highest enrichment for DHS, typical enhancer, and super enhancer in the breast carcinoma cell line MCF-7 (DHS: enrichment fold = 4.63, p value = 9.6 × 10−23; typical enhancers: enrichment fold = 6.19, p value = 2.3 × 10−10; super enhancer: enrichment fold = 13.39, p value = 2.8 × 10−11). To investigate the role of DHS in breast cancer GWAS signals, we further identified active DHS by including data on histone marks H3K4me1 and H3K27ac (both mark active enhancers) in MCF-7 from ENCODE. We found that breast cancer GWAS association signals at p value < 10−8 were enriched in DHS overlapping with enhancer histone marks H3K27ac and H3K4me1 (enrichment fold = 3.57, p value = 4.4 × 10−4), although this enrichment was slightly attenuated compared to DHS in MCF-7 overall (enrichment fold = 4.63, p value = 9.6 × 10−23). We were not able to do corresponding analysis in prostate cancer since ENCODE has not released histone marks data in LNCaP. For prostate cancer, the highest enrichments for both DHS and typical enhancers were observed in the prostate carcinoma cell line LNCaP (DHS: enrichment fold = 3.49, p value = 1.4 × 10−19; typical enhancers: enrichment fold = 4.13, p value = 7.8 × 10−13). For super enhancers, the highest enrichment was observed in the colorectal cancer cell line VACO 9M (enrichment fold = 7.67, p value = 3.48 × 10−23). The enrichment in prostate cancer GWAS signals for super enhancers in LNCaP was 3.01 (p value = 6.3 × 10−4), which was not significant after adjusting for number of tests.
Fig. 2.
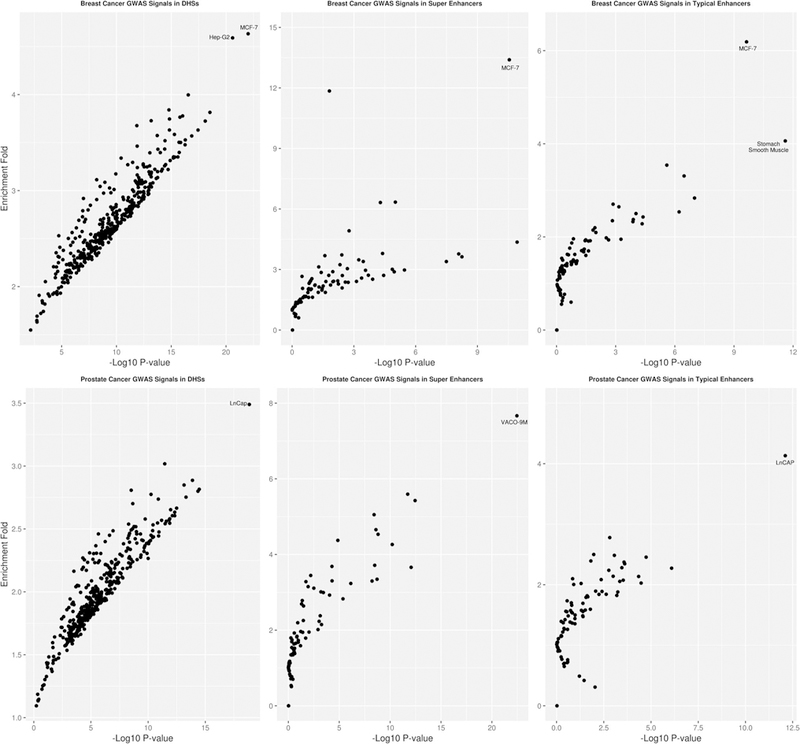
Enrichment of GWAS significant signals with p value < 10−8 of breast (upper panel) and prostate cancer (lower panel) in DHS (left panel), super enhancer (middle panel), and typical enhancer regions (right panel). Each observation in the plot corresponds to an annotation type
Sensitivity analysis increasing the MAF threshold from 0.1 to 1% did not appreciably change our results. Breast cancer GWAS signals at the p value < 10−8 threshold were enriched by 4.32-fold (p value = 1.60 × 10−21) for DHS, 5.49-fold (p value = 1.10 × 10−8) for typical enhancers, and by 11.54-fold (p value = 5.57 × 10−9) for super enhancers identified in the MCF-7 cell line. Prostate cancer GWAS signals at the p value < 10−8 threshold were enriched by 7.26-fold (p value = 1.73 × 10−10) for DHS, 4.86-fold (p value = 7.63 × 10−4) for typical enhancers, and 1.52-fold (p value = 0.68) for super enhancers identified in the LNCaP cell line.
To assess if enrichments observed in non-breast and non-prostate tissues were due to SNP overlaps with disease-related tissues, we removed any SNPs located in functional annotations found in breast and prostate cancer tissue, respectively, from the corresponding annotations in other tissues and reran the analysis (Tables 1, 2). Removing SNPs located in annotations for MCF-7 did not qualitatively change the enrichments for significant non-breast tissue DHS including the liver carcinoma cell line Hep-G2. Similarly, for both typical and super enhancers, some tissues, including stomach smooth muscle tissue, retained significant enrichments (Table 1, Supplementary Fig. 1, Supplementary Tables 8–10). In contrast, regulatory elements in prostate cancer showed much higher tissue specificity. For both DHS and typical enhancers, after removing SNPs located in LNCaP, we only observed statistically significant enrichments in fetal intestine (DHS) and some blood traits (typical enhancers) (Table 2, Supplementary Fig. 2, Supplementary Tables 11–13). This result suggests that the observed enrichments in DHS and typical enhancers across tissue types were mainly driven by the SNP overlap with LNCaP. In contrast, removing SNPs that were observed in super enhancers in LNCaP had little effect on the enrichment for non-relevant tissues.
Table 1.
Enrichment of breast cancer (BrCa) GWAS signals with p value < 10−8 for non-breast-related annotations before and after removing SNPs overlapping with corresponding annotation in MCF-7
| Non-breast DHS with top enrichment of BrCa GWAS signals | % of SNPs overlap with MCF-7 DHS | Enrichment (95% CI) before removal | p value before removal | Enrichment (95% CI) after removal | p value after removal |
|---|---|---|---|---|---|
| Hep-G2 cell line, liver cancer | 37.65 | 4.59 (3.35–6.29) | 2.51E−21 | 4.69 (2.90–7.59) | 3.19E−10 |
| Fetal large intestine tissue | 33.67 | 3.82 (2.85–5.11) | 2.95E−19 | 4.06 (2.64–6.26) | 1.94E−10 |
| GM06990 cell line, lymphoblastoid | 26.04 | 3.78 (2.76–5.17) | 9.09E−17 | 4.09 (2.57–6.52) | 3.11E−09 |
| T helper 2 cells, lymphocytes | 28.01 | 3.77 (2.75–5.16) | 1.61E−16 | 3.91 (2.43–6.29) | 1.86E−08 |
| CD3 cord blood | 32.44 | 3.75 (2.71–5.18) | 1.45E−15 | 3.56 (2.16–5.88) | 6.88E−07 |
| T helper 17 cells, lymphocytes | 36.23 | 3.73 (2.64–5.26) | 6.79E−14 | 4.71 (2.78–7.96) | 7.82E−09 |
| Fetal stomach tissue | 26.02 | 3.72 (2.78–4.98) | 7.90E−19 | 3.49 (2.26–5.39) | 1.75E−08 |
| GM19239 cell line, lymphoblastoid | 30.99 | 3.68 (2.56–5.27) | 1.38E−12 | 3.24 (1.78–5.88) | 1.16E−04 |
| Fetal small intestine tissue | 30.17 | 3.63 (2.65–4.98) | 1.47E−15 | 4.18 (2.62–6.66) | 1.93E−09 |
| Non-breast typical enhancer with top enrichment of BrCa GWAS signals | % of SNPs overlap with MCF-7 typical enhancer | Enrichment (95% CI) before removal | p value before removal | Enrichment (95% CI) after removal | p value after removal |
| Stomach smooth muscle tissue | 7.11 | 4.06 (2.74–6.01) | 2.51E−12 | 3.75 (2.49–5.65) | 2.96E−10 |
| Brain angular gyrus | 5.33 | 3.54 (2.09–6.00) | 2.55E−06 | 3.18 (1.81–5.59) | 5.49E−05 |
| Non-breast super enhancer with top enrichment of BrCa GWAS signals | % of SNPs overlap with MCF-7 super enhancer | Enrichment (95% CI) before removal | p value before removal | Enrichment (95% CI) after removal | p value after removal |
| Bladder tissue | 2.95 | 6.34 (2.80–14.37) | 9.69E−06 | 6.60 (2.91–14.97) | 6.21E−06 |
| CD34 cell, blood | 1.03 | 6.32 (2.59–15.43) | 5.17E−05 | 6.46 (2.65–15.78) | 4.20E−05 |
| Brain anterior caudate tissue | 1.50 | 4.36 (2.85–6.67) | 1.19E−11 | 4.42 (2.89–6.76) | 7.77E−12 |
| VACO 9M cell line, colorectal cancer | 2.87 | 3.59 (2.63–4.90) | 7.59E−16 | 3.14 (1.55–6.37) | 1.56E−04 |
| Brain middle hippocampus tissue | 0.93 | 3.63 (2.35–5.60) | 5.66E−09 | 3.67 (2.38–5.66) | 4.24E−09 |
Only annotations with statistical significance and 3.5+ fold of enrichment before removal are listed
Table 2.
Enrichment of prostate cancer (PrCa) GWAS signals with p value < 10−8 for non-prostate-related annotations before and after removing SNPs overlapping with corresponding annotation in LNCaP
| Non-prostate DHS with top enrichment of PrCa GWAS signals | % of SNPs overlap with LNCaP DHS | Enrichment (95% CI) before removal | p value before removal | Enrichment (95% CI) after removal | p value after removal |
|---|---|---|---|---|---|
| Fetal large intestine tissue | 41.02 | 3.02 (2.21–4.12) | 3.51E−12 | 3.00 (1.63–5.54) | 4.3E−04 |
| CACO-2 cell line, colorectal cancer | 23.10 | 2.89 (2.20–3.78) | 1.33E−14 | 1.94 (1.15–3.25) | 0.01 |
| Fetal small intestine tissue | 35.19 | 2.85 (2.17–3.75) | 7.21E−14 | 2.72 (1.63–4.54) | 1.4E−04 |
| Hep-G2 cell line, liver cancer | 41.38 | 2.78 (2.05–3.77) | 5.57E−11 | 1.51 (0.74–3.07) | 0.26 |
| MCF-7 cell line, breast cancer | 44.57 | 2.70 (1.95–3.74) | 2.19E−09 | 2.26 (1.07–4.78) | 0.03 |
| Fetal stomach tissue | 31.26 | 2.61 (2.01–3.39) | 7.58E−13 | 2.14 (1.31–3.50) | 0.002 |
| GM12878 cell line, lymphocyte | 29.79 | 2.58 (1.93–3.45) | 1.80E−10 | 1.22 (0.64–2.33) | 0.55 |
| CD3 cell, blood | 37.41 | 2.54 (1.86–3.47) | 5.07E−09 | 0.77 (0.31–1.93) | 0.58 |
| hTH2 cell, blood | 32.14 | 2.52 (1.87–3.40) | 1.69E−09 | 1.10 (0.52–2.31) | 0.80 |
| GM06990 cell line, lymphocyte | 29.89 | 2.52 (1.87–3.39) | 1.19E−09 | 1.13 (0.56–2.28) | 0.74 |
| Non-prostate super enhancer with top enrichment of PrCa GWAS signals | % of SNPs overlap with LNCaP super enhancer | Enrichment (95% CI) before removal | p value before removal | Enrichment (95% CI) after removal | p value after removal |
| VACO 9M cell line, colorectal cancer | 8.30 | 7.67 (5.13–11.46) | 3.48E−23 | 8.06 (5.27–12.34) | 7.63E−22 |
| VACO 400 cell line, colorectal cancer | 9.95 | 5.59 (3.46–9.03) | 1.87E−12 | 6.01 (3.62–9.98) | 3.77E−12 |
| Colorectal cancer crypt tissue | 13.07 | 5.43 (3.44–8.56) | 3.52E−13 | 4.03 (2.78–5.84) | 1.71E−13 |
| Thymus tissue | 5.64 | 5.05 (2.95–8.66) | 3.76E−09 | 5.57 (3.25–9.55) | 4.10E−10 |
| Pancreas tissue | 17.76 | 4.66 (2.81–7.72) | 2.47E−09 | 5.77 (3.37–9.89) | 1.86E−10 |
| CD19 cell, blood | 3.08 | 4.53 (2.78–7.40) | 1.52E−09 | 4.76 (2.92–7.78) | 4.44E−10 |
| GM12878 cell line, leukemia | 2.07 | 4.37 (2.25–8.50) | 1.37E−05 | 4.55 (2.34–8.86) | 7.90E−06 |
| HCT-116 cell line, colorectal cancer | 7.19 | 3.68 (1.96–6.93) | 5.20E−05 | 3.47 (1.72–7.02) | 5.25E−04 |
| Gastric tissue | 16.15 | 3.66 (2.57–5.22) | 8.61E−13 | 4.03 (2.78–5.84) | 1.71E−13 |
| CD20 cell, blood | 3.21 | 3.35 (2.26–4.96) | 1.92E−09 | 3.49 (2.36–5.18) | 5.01E−10 |
| Sigmoid colon tissue | 12.78 | 3.30 (2.21–4.93) | 5.97E−09 | 3.58 (2.36–5.43) | 2.04E−09 |
| HCC-1954 cell line, breast cancer | 4.27 | 3.30 (1.85–5.89) | 5.07E−05 | 2.99 (1.59–5.62) | 6.59E−04 |
| Duodenum smooth muscle tissue | 6.75 | 3.23 (2.03–5.15) | 7.31E−07 | 3.35 (2.08–5.40) | 6.54E−07 |
| CD56 cell, blood | 4.42 | 2.93 (1.71–5.01) | 8.89E−05 | 3.10 (1.81–5.30) | 3.65E−05 |
| Fetal large intestine tissue | 8.16 | 2.83 (1.82–4.40) | 4.28E−06 | 2.87 (1.81–4.57) | 8.51E−06 |
Only annotations with statistical significance and 2.5+ fold of enrichment before removal are listed
We observed that with progressively stringent GWAS p value threshold, the enrichment of GWAS signals increased among the annotations identified from the disease-related tumor cell lines, but not for corresponding normal cells, other carcinoma cell lines, or other normal cells (Fig. 3, Supplementary Table 14). For breast cancer, this trend was most strongly observed for DHS, typical enhancers, and super enhancers found in MCF-7. The enrichment of breast cancer GWAS signals at p value thresholds of 10−5, 10−6, 10−7, and 10−8 was 2.6, 3.4, 4.0, and 4.6 for DHS, 2.2, 3.2, 5.0, and 5.9 for typical enhancers, and 8.1, 11.8, 11.9, and 13.4 for super enhancers. For prostate cancer, an increasing trend was observed in DHS and typical enhancers identified in the LNCaP cell line (DHS: 2.8, 3.0, 3.2, and 3.5-fold enrichment; typical enhancers: 3.3, 3.6, 3.7, 4.1-fold enrichment for prostate cancer GWAS signals p value thresholds of 10−5, 10−6, 10−7, and 10−8) as well as some other carcinoma cell lines (e.g., super enhancers in the colorectal carcinoma cell line VACO 9M), but not in normal prostate cells or other normal cells. We further dropped the p value threshold and still found that GWAS signals at p value < 0.0001, < 0.001, and < 0.01 thresholds were significantly enriched across DHS, super enhancers, and typical enhancers in MCF-7 (breast cancer) and LNCaP (prostate cancer), respectively (Supplementary Table 14). However, we observed a mono-tone decreasing trend in enrichment with a more relaxed p value threshold. For example, for DHS, the breast cancer GWAS signal enrichment in MCF-7 ranged from 4.63 (p value = 9.62 × 10−23) for a p value threshold of 10−8 to 1.24 (p value = 9.91 × 10−14) for a p value threshold of 0.01. For prostate cancer, the GWAS signal enrichment of DHS in LNCaP ranged from 3.49 (p value = 1.42 × 10−19) for a p value threshold of 10−8 to 1.40 (p value = 1.27 × 10−38) for a p value threshold of 0.01. Interestingly, for prostate cancer GWAS signals, we observed a significant enrichment in super enhancers in LNCaP as the p value threshold decreased, although the enrichment estimate decreased with increasing p value threshold. This likely reflects the small proportion of the genome that contains super enhancers in LNCaP and suggests that super enhancers located in LNCaP are indeed enriched for prostate cancer, but we did not have enough statistical power to detect a significant signal for stringent p value thresholds.
Fig. 3.
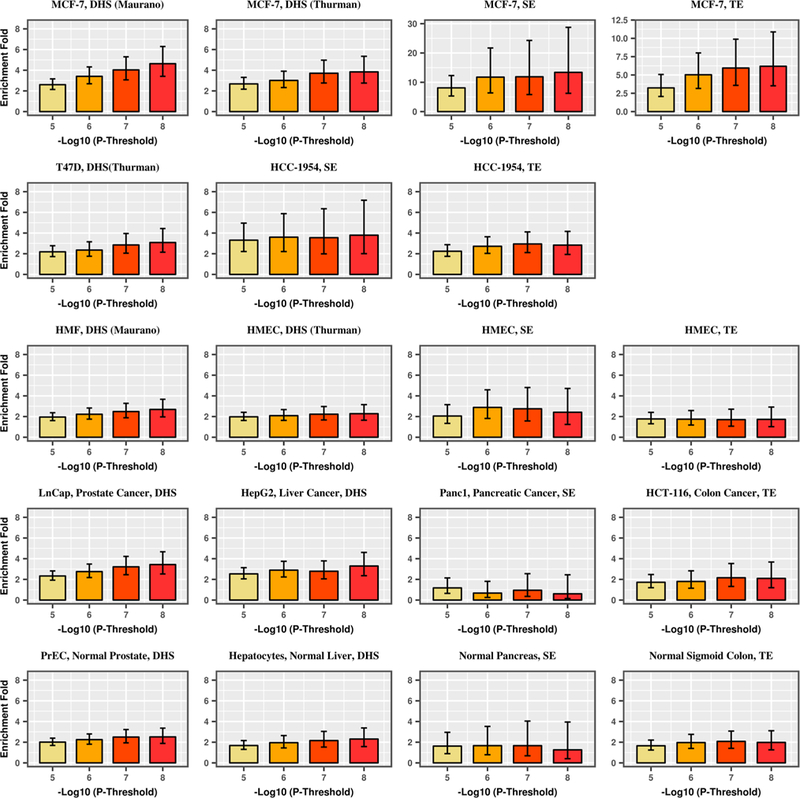
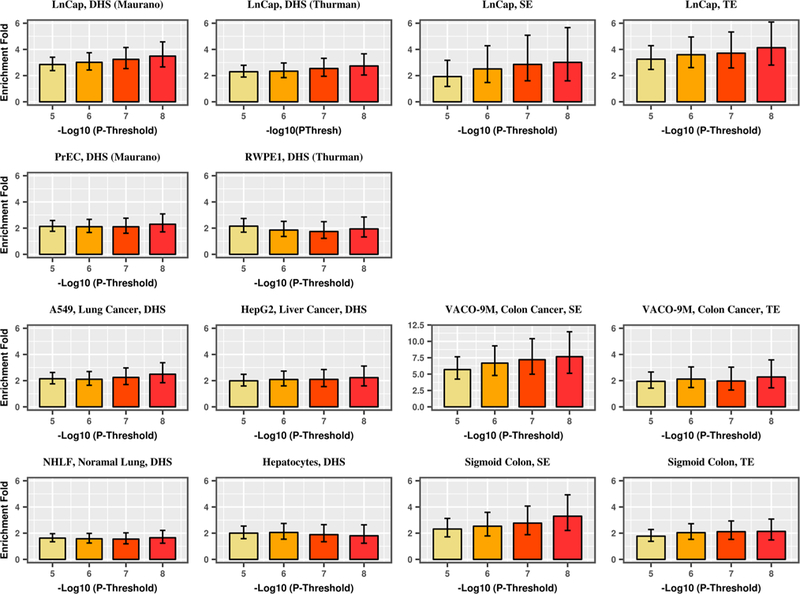
Enrichment of DHS, typical enhancers (TE), and super enhancers (SE) for breast (a) and prostate (b) cancer GWAS signals by GWAS p value threshold for disease-related carcinoma cell lines, disease-related normal tissues, unrelated carcinoma cell lines, and unrelated normal tissues. a-first row: MCF-7: ER-positive breast cancer cell line; a-second row: T47D: ER-positive breast cancer cell line; HCC-1954: ER-negative breast cancer cell line. a-third row: HMEC (2 and 4): primary mammary epithelial cell; HMF: human mammary fibroblast; vHMEC: variant human mammary epithelial cell; a-fourth row: LNCaP: prostate cancer cell line; HepG2: liver cancer cell line; Panc1: pancreatic cancer cell line; HCT-116: colorectal cancer cell line; a-fifth row: PrEC: primary prostate epithelial cell; hepatocyte: normal liver cell; normal pancreas tissue; normal sigmoid colon tissue. b-first row: LNCaP: prostate cancer cell line; b-second row: PrEC: primary prostate epithelial cell; RWPE1: prostate epithelial cell; b-third row: A549: lung cancer cell line; HepG2: liver cancer cell line; VACO-9M: colorectal cancer cell line; b-fourth row: NHLF: lung fibroblast; hepatocyte: normal liver cell; normal sigmoid colon tissue
We also studied the enrichment pattern of GWAS significant signals for ER+ and ER− breast cancer separately (Fig. 4, Supplementary Tables 15–19). ER+ breast cancer GWAS signals at p < 10−8 were mostly enriched in DHS, typical enhancers, and super enhancers identified in MCF-7 (which is an ER+ breast cancer cell line). Super enhancers located in MCF-7 were enriched for ER− breast cancer GWAS signals (enrichment fold: 80.3, p value: 2.1 × 10−22). In contrast, we observed the strongest enrichment for fetal intestine tissue for DHS enrichment of ER− breast cancer (enrichment fold: 4.99, p value: 1.1 × 10−5), and no significant enrichment of typical enhancers of ER− breast cancer for any tissue.
Fig. 4.
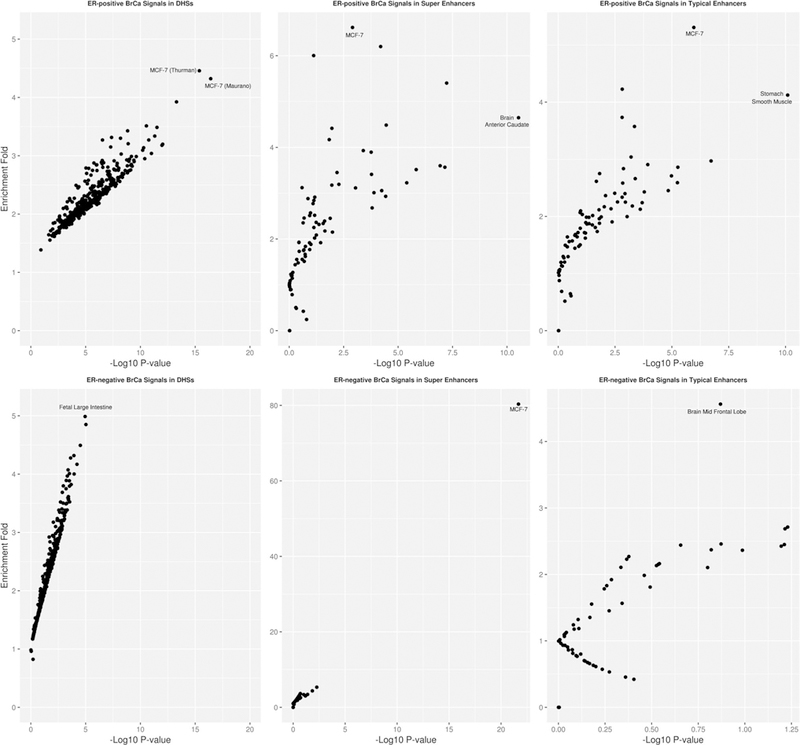
Enrichment of GWAS significant signals with p value < 10−8 of ER-positive (upper panel) and ER-negative (lower panel) breast cancer in DHS (left panel), super enhancer (middle panel), and typical enhancer regions (right panel). Each observation in the plot corresponds to an annotation type
Estimation of breast and prostate cancer heritability enrichment in functional regions specific to carcinoma cell lines and corresponding normal tissue
We utilized stratified LD score regression to study the SNP heritability of breast and prostate cancer for DHS, typical enhancers, and super enhancers identified in normal and carcinoma cell lines across breast and prostate tissues (Fig. 5, Supplementary Tables 20, 21). Regulatory elements identified in the breast cancer cell lines MCF-7 and HCC1954 showed the highest enrichment of overall breast cancer heritability. No enrichment of breast cancer heritability was observed for the regulatory elements located in the breast cancer cell line T-47D, prostate cancer cell lines, and normal breast and prostate cells. The highest enrichment for prostate cancer heritability was observed for regulatory elements identified in the LNCaP cell line, whereas no enrichment in heritability was observed in normal prostate tissue. Significant enrichment of prostate cancer heritability was also found for DHS and typical enhancers identified in MCF-7. To assess if these results are due to overlap in annotations for breast and prostate tissue, we studied the overlap in annotations between tissue types for the significant results (DHS: LNCaP vs. MCF7; Typical Enhancers: LNCaP vs. HCC1954; LNCaP vs. MCF7). For each annotation, after removing all SNPs that were present in both tissue types, we observed a decrease in the enrichment z score in our heritability analysis for prostate cancer (Supplementary Table 22). Thus, we believe that the enrichment of prostate cancer heritability observed for annotations in breast cancer cell lines is partly driven by their overlap with the LNCaP, however, it does not completely explain the enrichment signal.
Fig. 5.
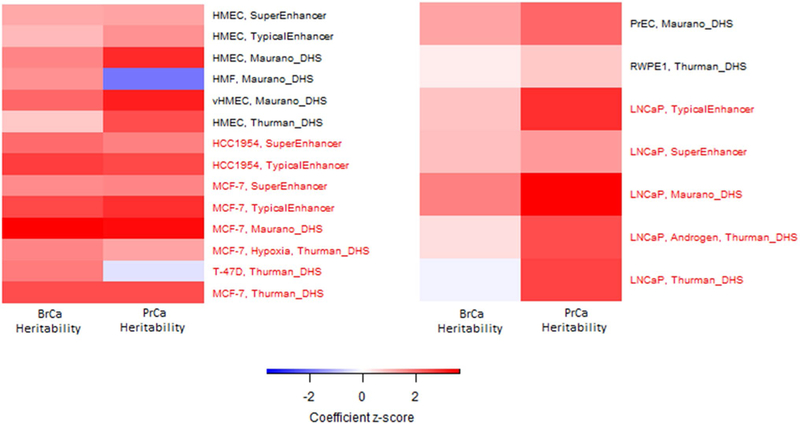
Heritability enrichment of breast cancer and prostate cancer in DHS, typical enhancer, and super enhancer regions identified from breast or prostate carcinoma cell lines (marked in red) and corresponding primary cell or normal tissue (marked in black)
Discussion
In this study, we assessed the contribution of SNPs located in regulatory regions to breast and prostate cancer GWAS associations. We found that genome-wide significant SNPs were especially enriched in regulatory regions specific to disease-related carcinoma cell lines for both diseases. Specifically, DHS, typical enhancers, and super enhancers identified in the breast cancer cell line MCF-7 were observed with the highest enrichment of genome-wide significant variants for breast cancer. For prostate cancer, genome-wide significant variants were mostly enriched in DHS and typical enhancers identified in the prostate cancer cell line LNCaP as well as super enhancers identified in colorectal cancer cell line VACO 9M. An increasing trend of enrichment was also observed in disease-related annotations as we applied a more stringent association p value threshold for inclusion, arguing that the strongest GWAS signals were particularly enriched for regulatory regions in relevant tissues. For ER+ breast cancer, DHS, typical enhancers, and super enhancers in MCF-7 had the highest enrichment of genome-wide significant SNPs across tissues. For ER− breast cancer, super enhancers located in MCF-7 had the highest enrichment, while for DHS, fetal large intestine was the most enriched tissue. We did not observe any enrichments among typical enhancers for ER−breast cancer, regardless of tissue type. Results of heritability enrichment analysis further supported our findings, as certain regulatory elements identified in MCF-7 and LNCaP had the highest enrichment for breast and prostate cancer, respectively.
A majority of GWAS-identified risk loci for common disease are located in non-coding regions and have been hypothesized to act through an enhancer function (Corradin and Scacheri 2014). Only 1.5% and 2.2% of the susceptible SNPs with p value for association < 10−8 for breast cancer and prostate cancer are located in coding regions. Further, it has been shown that GWAS-identified SNPs are located within enhancer elements specific to relevant cell types, in both cancer and other complex traits (Ernst et al. 2011). Further, a disproportional enrichment of disease-relevant cell type-specific super enhancers has been shown among disease susceptibility variants (Hnisz et al. 2013; Whyte et al. 2013). For example, SNPs associated with plasma low-density lipoprotein concentrations were significantly co-localized with active enhancers marked by H3K4me3 in liver (Trynka et al. 2013); enhancers identified in pancreatic islet clusters were enriched among type 2 diabetes risk-associated variants (Pasquali et al. 2014; Stitzel et al. 2010); and a majority of genome-wide significant SNPs for colorectal cancer are located in H3K4me1-marked enhancer regions in colon crypts (Akhtar-Zaidi et al. 2012). Hnisz et al. investigated the density of trait-associated non-coding SNPs linked to eight diseases or traits, including Alzheimer’s disease, type I diabetes, and rheumatoid arthritis, in super enhancer domains identified in twelve human cell and tissue types. Similar to our results, they found that disease-associated SNPs tended to occur in super enhancers of disease-relevant cells or tissues.
Similarly, our study provided evidence that variants associated with breast and prostate cancer were especially concentrated in DHS, typical enhancer, and super enhancer regions discovered in corresponding cancer cell lines. This is consistent with a previous finding that the genomic distribution of chromatin features identified in carcinoma cells is more strongly associated with local mutation density as compared to the distribution observed in non-cancer cell types (Huyghe et al. 2019; Michailidou et al. 2017; Polak et al. 2015). Polak et al. reported that DHS specific to melanocytes can explain a substantially larger fraction of the variance in melanoma mutation density than DHS from any other tissue or cell types. They also showed that the mutation density for melanoma and liver cancer genomes was highly specific to annotations measured in the cell of origin, even though annotations in melanocytes and hepatocytes were highly correlated. Michailidou et al. reported that candidate target genes of GWAS susceptible loci of breast cancer were strongly overlapped with the somatic driver genes in the breast tumor. In contrast, Huyghe et al. suggested that the GWAS significant signals of colorectal cancer were more strongly enriched in DHS identified in the fetal tissue and normal cells, rather in the colorectal cancer cell lines including HCT-116. Our findings, along with those previous studies, strengthened our understanding of DNA annotations and their role in carcinogenesis via transcription regulation and highlight the importance of considering both tumor and normal tissue when integrating functional annotation data in cancer GWAS.
When stratifying the results for DHS by ER status, we surprisingly found that genome-wide significant signals in the ER−breast cancer were not enriched for breast tissue. The number of genome-wide significant SNPs for ER−breast cancer is few, likely due to the relatively smaller sample size. After the greedy pruning process implemented by GARFIELD, only 52 independent SNPs with p value less than 10−8 were kept. This low number of SNPs results in large variability in enrichment estimation of specific annotations, especially when annotations are small. DHS located in fetal large intestine and other non-disease-related tissues were observed with only marginally significant enrichment for ER-negative breast cancer signals. We note that approximately a third of all SNPs located in the most enriched tissue for DHS of fetal large intestine are also found in DHS in MCF-7. Thus, the combination of relatively weak GWAS signal and overlap in annotations across tissue types might identify non-relevant tissue types by chance. Previous studies have assessed tissue-specific enrichments of regulatory regions by looking at the fraction of SNPs located in tissue-specific DHS with a GWAS p value below a specified cutoff, divided by the total number of SNPs located in that annotation (Maurano et al. 2012). In contrast, GARFIELD utilized a generalized linear model, which further adjusted for minor allele frequency, distance to nearest transcription start site, and number of LD proxies. This improvement assured that our enrichment estimates were robust and unbiased. Gusev et al. (2016) partitioned the heritability of prostate cancer by tissue-specific genomic functional categories using a restricted maximum likelihood (REML) approach. They showed that the majority of SNP heritability was located in DHS and enhancers marked by histone mark H3K27ac identified from the prostate cancer cell line LNCaP. Super enhancers identified in LNCaP were not found with disproportionally high heritability of prostate cancer. Their results were consistent with our results although we used slightly different data source and study designs. Although both studies leveraged data from the PRACTICAL consortium, we based our analysis on the more recent 79,194 cases and 61,112 controls, as compared to Gusev et al. who used data over a much smaller sample size of 59,089 (cases and controls). In addition, Gusev et al. had access to individual level GWAS data and estimated the heritability enrichment of the functional categories using the restricted maximum likelihood (REML) as implemented in the GCTA software (Yang et al. 2011). In contrast, we only had access to GWAS summary statistics and used LD score regression (Bulik-Sullivan et al. 2015) to estimate the enrichment of prostate cancer heritability among functional categories. We also observed an enrichment of prostate cancer heritability in functional annotations identified in breast cancer cell lines. After removing overlap between prostate and breast across annotations, we observed a decreased enrichment heritability for prostate cancer. Thus, we believe that the enrichment of prostate cancer heritability observed for annotations in breast cancer cell lines is partly driven by their overlap with the LNCaP, however, it does not completely explain the enrichment signal. Thus, it is possible that the previous observed pleiotropy between breast and prostate cancer (Jiang et al. 2019; Kar et al. 2016) can explain some of these enrichment patterns.
Our study has several strengths. The GWAS results used in this study came from the most recent and largest published GWAS for both breast and prostate cancer, providing increased statistical power to observe significant enrichments. We obtained publicly available data on DNA regulatory regions from large collaborative consortia with comprehensive protocols and quality control pipelines, ensuring robust data. But it is also important to acknowledge the limitations of our study. Disease-relevant enhancers marked by histone marks H3K27ac and H3K4me1 were not collected by the Roadmap projects, prohibiting us to assess if the observed enrichments of those histone marks are driven by tissue-specific (breast cancer and prostate cancer, respectively) annotations. Several non-relevant (i.e., other than breast or prostate) tissue and cell types showed statistically significant enrichment of GWAS signals for both breast and prostate cancer, implying non-specificity. To address potential non-specificity, we reran the analysis, excluding all SNPs located in breast or prostate-specific annotations, respectively. While our results for breast cancer did not materially change, no unrelated annotation type showed significant enrichment after removing prostate-specific DHS and super enhancer SNPs. It is not clear if other annotations that overlap with the non-breast tissue annotations and not assessed here harbor SNPs associated with breast cancer and thus, are confounding our results, leading to an apparent enrichment of regulatory regions not expressed in breast tissue.
In conclusion, our study revealed high enrichment of breast and prostate cancer risk variants in DHS, typical enhancer, and super enhancer regions, with particularly high enrichment for regulatory regions in disease-specific carcinoma cell lines (MCF-7 (breast) and LNCaP (prostate)). We found evidence for a significant contribution of disease-related annotations for both genome-wide significant SNPs and across all common SNPs in the genome, arguing that there is likely to be more causal SNPs located in regulatory regions that have yet not been identified in disease-specific GWAS. Our results suggest the importance of functional annotations in understanding GWAS results, and further demonstrate the role that DNA regulatory elements from disease-related tissue play in breast and prostate carcinogenesis.
Supplementary Material
Acknowledgements
This work was supported by CA194393. The breast cancer genome-wide association analyses were supported by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, the ‘Ministère de l’Économie, de la Science et de l’Innovation du Québec’ through Genome Québec and Grant PSR-SIIRI-701, The National Institutes of Health (U19 CA148065, X01HG007492), Cancer Research UK (C1287/A10118, C1287/A16563, C1287/A10710) and The European Union (HEALTH-F2-2009-223175 and H2020 633784 and 634935). All studies and funders are listed in Michailidou et al. (2017). The Prostate cancer genome-wide association analyses are supported by the Canadian Institutes of Health Research, European Commission’s Seventh Framework Programme Grant Agreement No. 223175 (HEALTH-F2-2009-223175), Cancer Research UK Grants C5047/A7357, C1287/ A10118, C1287/A16563, C5047/A3354, C5047/A10692, C16913/A6135, and The National Institute of Health (NIH) Cancer Post-Cancer GWAS initiative Grant: No. 1 U19 CA 148537–01 (the GAME-ON initiative). We would also like to thank the following for funding support: The Institute of Cancer Research and The Everyman Campaign, The Prostate Cancer Research Foundation, Prostate Research Campaign UK (now Prostate Action), The Orchid Cancer Appeal, The National Cancer Research Network UK, The National Cancer Research Institute (NCRI) UK. We are grateful for support of NIHR funding to the NIHR Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust. The Prostate Cancer Program of Cancer Council Victoria also acknowledge Grant support from The National Health and Medical Research Council, Australia (126402, 209057, 251533, 396414, 450104, 504700, 504702, 504715, 623204, 940394, 614296), VicHealth, Cancer Council Victoria, The Prostate Cancer Foundation of Australia, The Whitten Foundation, PricewaterhouseCoopers, and Tattersall’s. EAO, DMK, and EMK acknowledge the Intramural Program of the National Human Genome Research Institute for their support. Genotyping of the OncoArray was funded by the US National Institutes of Health (NIH) [U19 CA 148537 for Elucidating Loci Involved in Prostate cancer Susceptibility (ELLIPSE) project and X01HG007492 to the Center for Inherited Disease Research (CIDR) under contract number HHSN268201200008I] and by Cancer Research UK Grant A8197/A16565. Additional analytic support was provided by NIH NCI U01 CA188392 (PI: Schumacher). Funding for the iCOGS infrastructure came from: the European Community’s Seventh Framework Programme under Grant Agreement No. 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A 10710, C12292/A11174, C1281/ A12014, C5047/A8384, C5047/A15007, C5047/A10692, C8197/A16565), the National Institutes of Health (CA128978) and Post-Cancer GWAS initiative (1U19 CA148537, 1U19 CA148065 and 1U19 CA148112—the GAME-ON initiative), the Department of Defence (W81XWH-10–1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund. The BPC3 was supported by the U.S. National Institutes of Health, National Cancer Institute (cooperative agreements U01-CA98233 to D.J.H., U01-CA98710 to S.M.G., U01-CA98216 toE.R., and U01-CA98758 to B.E.H., and Intramural Research Program of NIH/National Cancer Institute, Division of Cancer Epidemiology and Genetics). CAPS GWAS study was supported by the Swedish Cancer Foundation (Grant No. 09-0677, 11–484, 12–823), the Cancer Risk Prediction Center (CRisP; http://www.crispcenter.org), a Linneus Centre (Contract ID 70867902) financed by the Swedish Research Council, Swedish Research Council (Grant No. K2010-70X-20430-04-3, 2014-2269). PEGASUS was supported by the Intramural Research Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s00439-019-02041-5) contains supplementary material, which is available to authorized users.
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Conflict of interest On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Akhtar-Zaidi B et al. (2012) Epigenomic enhancer profiling defines a signature of colon cancer. Science 336:736–739. 10.1126/science.1217277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Olama AA et al. (2014) A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet 46:1103–1109. 10.1038/ng.3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Olama AA et al. (2015) Multiple novel prostate cancer susceptibility signals identified by fine-mapping of known risk loci among Europeans. Hum Mol Genet 24:5589–5602. 10.1093/hmg/ddv203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan BK et al. (2015) LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47:291–295. 10.1038/ng.3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L et al. (2016) Genetic drivers of epigenetic and transcriptional variation in human immune cells. Cell 167(1398–1414):e1324 10.1016/j.cell.2016.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium UK et al. (2015) The UK10K project identifies rare variants in health and disease. Nature 526:82–90. 10.1038/nature14962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradin O, Scacheri PC (2014) Enhancer variants: evaluating functions in common disease. Genome Med 6:85 10.1186/s13073-014-0085-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadaev T et al. (2018) Fine-mapping of prostate cancer susceptibility loci in a large meta-analysis identifies candidate causal variants. Nat Commun 9:2256 10.1038/s41467-018-04109-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeles RA et al. (2013) Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet 45:385–391. 10.1038/ng.2560(391e381–382) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J et al. (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473:43–49. 10.1038/nature09906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK et al. (2015) Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 47:1228–1235. 10.1038/ng.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazal S et al. (2017) Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat Genet 49:1421–1427. 10.1038/ng.3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A et al. (2014) Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet 95:535–552. 10.1016/j.ajhg.2014.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A et al. (2016) Atlas of prostate cancer heritability in European and African-American men pinpoints tissue-specific regulation. Nat Commun 7:10979 10.1038/ncomms10979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y et al. (2015) Integration of multiethnic fine-mapping and genomic annotation to prioritize candidate functional SNPs at prostate cancer susceptibility regions. Hum Mol Genet 24:5603–5618. 10.1093/hmg/ddv269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazelett DJ et al. (2014) Comprehensive functional annotation of 77 prostate cancer risk loci. PLoS Genet 10:e1004102 10.1371/journal.pgen.1004102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Glass CK (2015) The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol 16:144–154. 10.1038/nrm3949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjelmborg JB et al. (2014) The heritability of prostate cancer in the Nordic Twin Study of Cancer. Cancer Epidemiol Biomark Prev 23:2303–2310. 10.1158/1055-9965.EPI-13-0568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D et al. (2013) Super-enhancers in the control of cell identity and disease. Cell 155:934–947. 10.1016/j.cell.2013.09.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyghe JR et al. (2019) Discovery of common and rare genetic risk variants for colorectal cancer. Nat Genet 51:76–87. 10.1038/s41588-018-0286-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iotchkova V et al. (2019) GARFIELD classifies disease-relevant genomic features through integration of functional annotations with association signals. Nat Genet 51:343–353. 10.1038/s41588-018-0322-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X et al. (2019) Shared heritability and functional enrichment across six solid cancers. Nat Commun 10:431 10.1038/s41467-018-08054-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar SP et al. (2016) Genome-wide meta-analyses of breast, ovarian, and prostate cancer association studies identify multiple new susceptibility loci shared by at least two cancer types. Cancer Discov 6:1052–1067. 10.1158/2159-8290.CD-15-1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q et al. (2013) Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell 152:633–641. 10.1016/j.cell.2012.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein P et al. (2000) Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 343:78–85. 10.1056/NEJM200007133430201 [DOI] [PubMed] [Google Scholar]
- Maurano MT et al. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science 337:1190–1195. 10.1126/science.1222794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidou K et al. (2013) Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat Genet 45:353–361. 10.1038/ng.2563(361e351–352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidou K et al. (2017) Association analysis identifies 65 new breast cancer risk loci. Nature 551:92–94. 10.1038/nature24284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne RL et al. (2017) Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat Genet 49:1767–1778. 10.1038/ng.3785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucci LA et al. (2016) Familial risk and heritability of cancer among twins in Nordic countries. JAMA 315:68–76. 10.1001/jama.2015.17703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page WF, Braun MM, Partin AW, Caporaso N, Walsh P (1997) Heredity and prostate cancer: a study of World War II veteran twins. Prostate 33:240–245 [DOI] [PubMed] [Google Scholar]
- Pasquali L et al. (2014) Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet 46:136–143. 10.1038/ng.2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P et al. (2015) Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 518:360–364. 10.1038/nature14221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz-Zarate A et al. (2017) Expression quantitative trait loci (QTL) in tumor adjacent normal breast tissue and breast tumor tissue. PLoS One 12:e0170181 10.1371/journal.pone.0170181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie SK, Coetzee SG, Noushmehr H, Yan C, Kim JM, Haiman CA, Coetzee GA (2013) Comprehensive functional annotation of seventy-one breast cancer risk Loci. PLoS One 8:e63925 10.1371/journal.pone.0063925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics C et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature 518:317–330. 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher FR et al. (2018) Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat Genet 50:928–936. 10.1038/s41588-018-0142-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A (2017) Cancer statistics, 2017. CA Cancer J Clin 67:7–30. 10.3322/caac.21387 [DOI] [PubMed] [Google Scholar]
- Song L et al. (2011) Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res 21:1757–1767. 10.1101/gr.121541.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitzel ML et al. (2010) Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab 12:443–455. 10.1016/j.cmet.2010.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G et al. (2008) Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet 40:310–315. 10.1038/ng.91 [DOI] [PubMed] [Google Scholar]
- Thurman RE et al. (2012) The accessible chromatin landscape of the human genome. Nature 489:75–82. 10.1038/nature11232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trynka G, Sandor C, Han B, Xu H, Stranger BE, Liu XS, Raychaudhuri S (2013) Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet 45:124–130. 10.1038/ng.2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull C et al. (2010) Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet 42:504–507. 10.1038/ng.586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA et al. (2013) Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153:307–319. 10.1016/j.cell.2013.03.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88:76–82. 10.1016/j.ajhg.2010.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.