

Abstract
Steroid hormones are key gene regulators in breast cancer cells. While estrogens stimulate cell proliferation, progestins activate a single cell cycle followed by proliferation arrest. Here, we use biochemical and genome‐wide approaches to show that progestins achieve this effect via a functional crosstalk with C/EBPα. Using ChIP‐seq, we identify around 1,000 sites where C/EBPα binding precedes and helps binding of progesterone receptor (PR) in response to hormone. These regions exhibit epigenetic marks of active enhancers, and C/EBPα maintains an open chromatin conformation that facilitates loading of ligand‐activated PR. Prior to hormone exposure, C/EBPα favors promoter–enhancer contacts that assure hormonal regulation of key genes involved in cell proliferation by facilitating binding of RAD21, YY1, and the Mediator complex. Knockdown of C/EBPα disrupts enhancer–promoter contacts and decreases the presence of these architectural proteins, highlighting its key role in 3D chromatin looping. Thus, C/EBPα fulfills a previously unknown function as a potential growth modulator in hormone‐dependent breast cancer.
Keywords: breast cancer, C/EBPα, cell proliferation, hormone‐dependent gene regulation, progesterone receptor
Subject Categories: Cancer; Chromatin, Epigenetics, Genomics & Functional Genomics; Transcription
Introduction
The steroid hormones estrogens and progesterone acting via their intracellular receptors (ER and PR, respectively) control the proliferation of breast cancer cells in a very different way. While estrogens are inducers of cell proliferation that activate cyclin D1 (Musgrove et al, 1994; Planas‐Silva & Weinberg, 1997; Planas‐Silva et al, 1999) and decrease the expression of CDK inhibitors (Prall et al, 1997), progestins promote a single cell cycle followed by proliferation arrest at G1/S, that correlates with a delayed activation of CDK inhibitor p21WAF1 (Owen et al, 1998). Studies using PR‐positive breast cancer cell lines have demonstrated a biphasic cellular response to progestins, with an immediate proliferative burst followed by a sustained growth arrest (Groshong et al, 1997; Musgrove et al, 1998; Skildum et al, 2005).
C/EBPα is a member of the C/EBP family of transcription factors, which plays a critical role in the regulation of mitotic growth arrest and differentiation in numerous cell types, including pre‐adipocytes, myeloid cells, hepatocytes, keratinocytes, and pneumocytes (Cao et al, 1991; Freytag et al, 1994; Wang et al, 1995; Flodby et al, 1996; Radomska et al, 1998). Despite the fact that C/EBPα is expressed in many tissues, its function has been best characterized only in adipocytes and in the hematopoietic system. Regulation of C/EBPα expression is fundamental for maintaining homeostasis of both embryonic and adult tissues (Schuster & Porse, 2006). Deletion of C/EBPα in mice results in pleiotropic disorders and the mice die shortly after birth due to the failure of the liver to store glycogen (Wang et al, 1995). Animals lacking C/EBPβ are viable but sterile and more susceptible to infections due to defects in the immune system (Screpanti et al, 1995). These mice exhibit also multiple defects in mammary gland development, including cystic, enlarged mammary ducts with decreased secondary branching (Seagroves et al, 1998). In addition, interactions between C/EBPα and CBP/p300 histone acetyltransferases or SWI/SNF chromatin remodeling complexes have been shown to regulate C/EBPα target genes involved in tissue specification (Erickson et al, 2001; Pedersen et al, 2001).
Though it is known that C/EBPs are involved in the regulation of cell proliferation and cell differentiation of the mammary gland (Johnson, 2005), the role of C/EBPs in breast cancer cells has not been characterized. Although functional C/EBPα mutations have not been found in solid tumors, C/EBPα levels are found down‐regulated in various types of cancer, indicating that inactivation of C/EBPα might be a requirement for tumor development (Lourenco & Coffer, 2017). In contrast, some studies performed in hepatocellular carcinomas and breast cancer cells have shown that increased expression of C/EBPα correlated with increased proliferation and disease progression (Tomizawa et al, 2007; Lu et al, 2010; Ming et al, 2015). However, the mechanism by which C/EBPα fulfills this function in hormone‐dependent processes and in particular in breast cancer cells is unknown.
Regulation of enhancer–promoter interactions is a fundamental mechanism underlying differential transcriptional control. Cell‐specific 3D chromatin folding brings specific enhancers in contact with their target promoters in cis to regulate gene expression in a cell type‐specific manner. Genome‐wide studies have shown that the architectural proteins CTCF and its frequent associated partner cohesin are important for partition of the genome into largely conserved topologically associating domains or TADs on the megabase (Dixon et al, 2012; Nora et al, 2012; Phillips‐Cremins et al, 2013). These architectural proteins along with the large mediator complex contribute directly to enhancer–promoter communication by mediating loop formation (Malik & Roeder, 2010; Ong & Corces, 2014). In embryonic stem (ES) cells and in mouse embryonic fibroblasts, RNAi studies showed that cohesin and the mediator complex are necessary for enhancer–promoter interactions of pluripotency genes (Kagey et al, 2010).
Although the role of transcription factors in mediating association between specific regulatory elements in the genome is well characterized, the nature of the protein complexes required for establishing and maintaining these interactions has remained elusive and identifying the functional interactions in the context of the large number of noisy contacts remains a real challenge. Changes in the levels of architectural proteins have been associated with disease. For instance, RAD21 expression was shown to be significantly lower in invasive breast cancers compared with their in situ counterparts (Xu et al, 2011). Moreover, transcription factors can interact with architectural proteins and contribute to the cell‐specific 3D genome topology (Faure et al, 2012; Yan et al, 2013).
In this study, we describe a functional crosstalk between PR and C/EBPα, which acts as inhibitor of breast cancer cells proliferation. Progestins induced the expression of C/EBPα, which participates in hormone regulation in different ways. In around 1,000 DNA enhancer regions C/EBPα assists PR binding by maintaining the chromatin in an open conformation. In these sites, C/EBPα is also required to establish and to maintain promoter–enhancer contacts that assure the progestin regulation of key genes involved in cell proliferation, such as DUSP1. C/EBPα fulfills this function via interactions with RAD21, YY1, CTCF, and the Mediator complex. Moreover, we identify topoisomerase IIα (Top2α) as a previously unknown factor in C/EBPα function, required for both mechanisms of cooperation with PR. Our results demonstrate that PR and C/EBPα cooperate in a gene expression program that enables controlled cell growth in the presence of progestins.
Results
C/EBPα is a hormone‐target gene that modulates hormonal gene regulation
To address the possible role of C/EBPα in breast cancer cells, we measure its expression in the ER+ and PR+ T47D cell line exposed to hormones. In cells cultured for 4 h in medium containing 10% serum deprived of hormone by treatment with dextran‐coated charcoal (CS‐FBS), we found an 8‐fold increase in C/EBPα expression after 6‐h exposure to the progestin R5020 (Fig 1A, top left panel). Time curve experiments showed that the levels of C/EBPα mRNA increased already after 3 h, reached a peak at 6 h, and decreased gradually thereafter (Fig 1A, top right panel). ChIP‐seq of PR after exposure to hormone for 60 min showed two strong peaks around the C/EBPα gene region located at −8.2 kb and −5.4 kb from the TSS, as well as two weak peaks inside the gene (Fig 1A, lower panel), indicating that the C/EBPα gene could be a direct target of PR. The C/EBPα protein levels increased after 6 h of hormone exposure and reached a peak after 19 h (Fig EV1A, left panel). The levels of the related protein C/EBPβ also increase in the presence of hormone, but this increment is moderate and delayed compared to that of C/EBPα (Fig EV1A, right panel and Fig EV1B).
Figure 1. In breast cancer cells, C/EBPα is a hormone‐target gene and modulates hormonal gene regulation and cell proliferation.
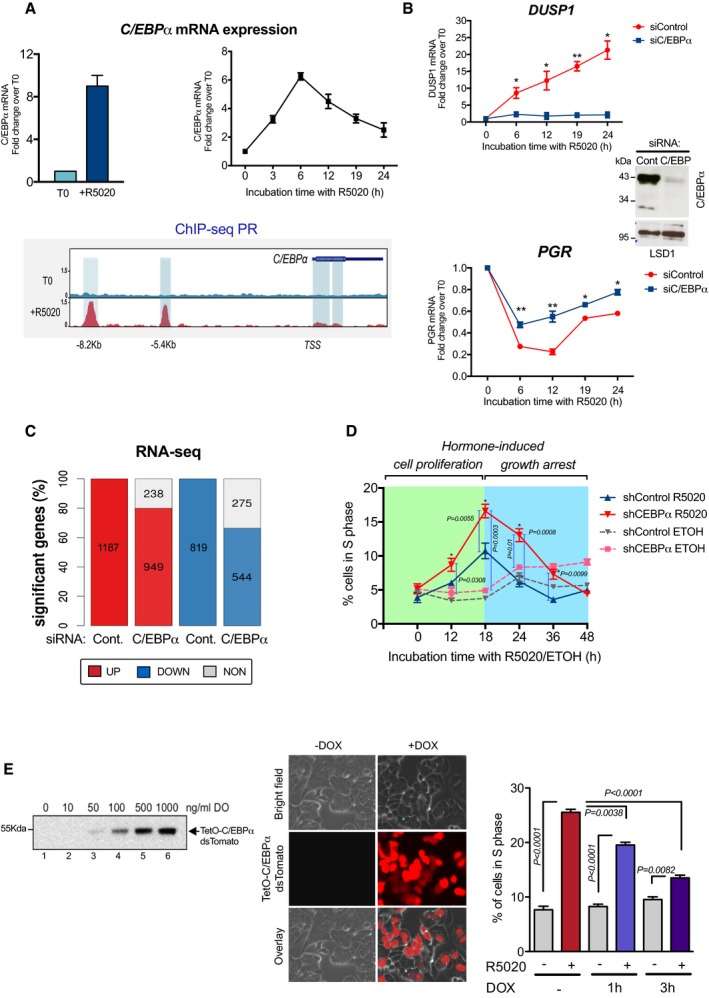
- Top left panel: Hormone‐induced fold change in the expression of mRNA for C/EBPα in T47D cells exposed to vehicle (T0) or to progestin (10 nM R5020) for 6 h (+R5020). The fold change relative to T0 is expressed as mean ± SD from three experiments performed in duplicate. Top right panel: Time kinetics of C/EBPα mRNA expression after hormone induction in T47D cells. The fold change relative to T0 is expressed as mean ± SD from three experiments performed in duplicate. Lower panel: Snapshot of the genome browser showing the profile of PR ChIP‐seq at T0 and after 60 min of hormone exposure (+R5020) around the C/EBPα gene shown in the upper right corner. The direction of transcription indicated.
- T47D cells transfected with control or C/EBPα siRNAs were treated with 10 nM R5020 for the indicated time periods; cDNA was generated and used as template for real‐time PCR with DUSP1 (upper panel)‐specific and PGR (lower panel)‐specific primers. The values are given as mean ± SD from three experiments performed in duplicate. The inset shows the level of C/EBPα depletion by Western blot, using LSD1 as loading control. P‐values were obtained by Student's t‐test and are relative to time zero (*P ≤ 0.05 and **P ≤ 0.01)
- Effect of C/EBPα knockdown on global hormonal gene regulation. T47D cells transfected with control or C/EBPα siRNAs were incubated with 10 nM R5020 for 6 h, and RNA‐seq experiments were performed as described in the Materials and Methods. The number of significant (q < 0.01) differentially expressed genes upon exposure to hormone identified in each of the siControl and siC/EBPα systems is shown. Differential expression analysis between treated and control conditions was done with R (https://www.R-project.org/) package DESeq2 (Love et al, 2014), selecting as significant those genes with an adj. P < 0.01 and a fold change > 2 between conditions.
- Effect of C/EBPα depletion in T47D cells on hormone‐induced entry in S phase. Cells were treated with Ethanol or R5020 for different time periods and subjected to flow cytometric analysis. Data are represented as mean ± SD from three experiments performed in duplicate. The P‐values were obtained using ANOVA followed by Tukey test.
- Left panel: Levels of C/EBPα expression in cells transfected with a Dox‐inducible TetO‐C/EBPα vector (T47DindC/EBPα) incubated with different concentrations of doxycycline, as detected by Western blot. Middle panel: T47DindC/EBPα were induced with doxycycline (1 μg/ml Dox) for 16 h and the expression of C/EBPα was detected by immune fluorescence microscopy. Right panel: Quantitation of the percentage of T47DindC/EBPα cells entering in S phase upon exposure to Dox (1 μg/ml) for 1 or 3 h followed by hormone induction for 18 h. Data are represented as mean ± SD from three experiments performed in duplicate. The P‐values were obtained using ANOVA followed by Tukey test.
Source data are available online for this figure.
Figure EV1. Hormone induces the expression of C/EBPα in breast cancer cells.
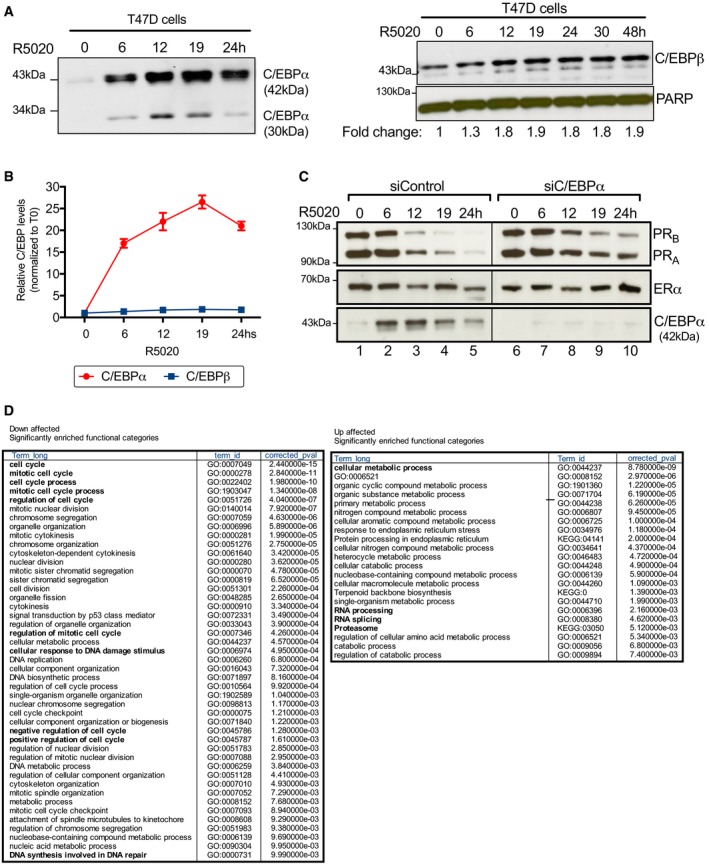
- Time kinetics of C/EBPα expression after hormone induction in T47D cells. T47D cells treated with ethanol or with 10 nM R5020 as indicated were lysed and analyzed by immunoblot with α‐C/EBPα (left panel) or with α‐C/EBPβ (right panel). An antibody to PARP was used as loading control.
- Quantitation of the bands shown in panel (A) using densitometric analysis and ImageJ software. Values were normalized to time zero (T0). Each bar represents the mean ± SD from two experiments.
- T47D cells transfected with control or C/EBPα siRNAs and treated or not with 10 nM R5020 as indicated were lysed and analyzed by immunoblot with specific antibodies for PR, ER, and C/EBPα.
- GO analysis of C/EBPα‐dependent genes in T47D cells. We used g:Profiler (Reimand et al, 2016) to identify Gene Ontology Biological Process (GO:BP) in the differentially expressed genes.
Source data are available online for this figure.
To test whether CEBPα influences the regulation of hormone‐dependent genes, T47D cells were transfected with control siRNAs and with siRNAs against C/EBPα. We found that progestin induction of the Dual Specificity Phosphatase 1 (DUSP1) gene and progestin repression of the PR gene (PGR) were significantly reduced by C/EBPα knockdown (Figs 1B and EV1C). To explore the generality of these observations, we performed RNA‐seq experiments in cells transfected with siRNA against C/EBPα or with control siRNA and exposed to hormone for 6 h. In siControl cells, we found 1,187 up‐regulated and 819 genes down‐regulated by hormone (q < 0.01) (Fig 1C). Of the hormone‐induced genes, 20% were C/EBPα dependent, while for the hormone‐repressed genes the proportion was 33.5%, suggesting that C/EBPα plays a more general role during hormonal gene regulation, particularly in down‐regulated genes (Fig 1C). Analysis of the Gene Ontology (GO) categories revealed that the C/EBPα‐dependent up‐regulated genes were primarily implicated in cellular metabolic process, RNA splicing and processing, while the down‐regulated genes were involved in the regulation of cell cycle, mitotic cell cycle, cellular response to DNA damage and DNA repair (Fig EV1D). Among them, we found 49 genes associated with cell cycle and cell proliferation such as the cell cycle modulators CNTD1, CKS2, and the cyclin‐dependent kinase 4 inhibitor CDKN2D; the G2 mitotic‐specific cyclin B2 (CCNB2), the transcription factor involved in cell cycle E2F1, and genes associated with DNA repair such as XRCC2, PCNA, BRCA1, and its associated RING domain protein BARD1 and BRIP1 (Appendix Fig S1A). Confirming their proliferative function, the expression of these 49 genes increased proportionally with the breast cancer tumor grade (Appendix Fig S1B, left panel) and its expression is associated with a decreased overall survival of the patients (Appendix Fig S1B, right panel). We also found several up‐regulated genes with anti‐proliferative functions such as DUSP1, DUSP2, STAT4, IL12, GAS7, and RASAL2 that could complement the cell growth arrest program triggered by progestins. Expression of these genes tends to be anti‐correlated to tumor grade, though not significantly (Appendix Fig S1C, left panel) and was associated with longer survival (Appendix Fig S1C, right panel).
C/EBPα mediates the inhibitory effect of progestins on cell proliferation
Previous studies using PR‐positive breast cancer cell lines demonstrated a biphasic cellular response to progestins, with an immediate proliferative burst followed by a sustained growth arrest (Groshong et al, 1997; Musgrove et al, 1998; Skildum et al, 2005). We confirmed these findings in T47D cells and found that progestin‐induced cell proliferation was significantly increased by C/EBPα knockdown (Fig 1D). Cells depleted of C/EBPα showed a delayed growth arrest phase, suggesting an anti‐proliferative role for C/EBPα (Gery et al, 2005). To expand this finding, we generated a T47D cell line that expresses recombinant CEBPα under the control of doxycycline (Dox) (T47DindC/EBPα). The increase in C/EBPα (Fig 1E, left and middle panels) correlated with a decrease in cell number (Fig EV2A, upper panel) as well as in the percentage of cells in S phase detected after hormone (Fig 1E, right panel). The effect of overexpressed C/EBPα was also observed in MCF7 and BT474 cells (Fig EV2A, middle and lower panels), although these breast cancer cell lines express different levels of estrogen receptor (ER) and PR (Fig EV2A, inset). Moreover, in MCF7 cells, we confirmed that C/EBPα knockdown increased cell growth as observed in T47D cells (Fig EV2B).
Figure EV2. Overexpression of C/EBPα decreases cell proliferation in BT474, MCF7, and T47D breast cancer cells.
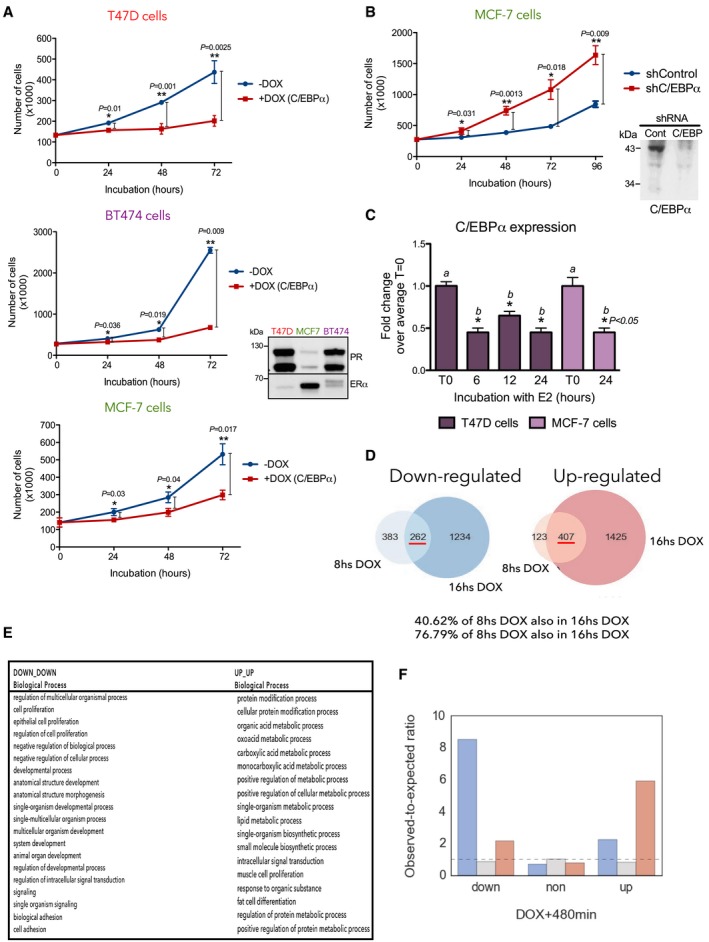
- T47DindC/EBPα (upper panel), BT474indC/EBPα (middle panel), and MCF7indC/EBPα cells (lower panel) and expressing recombinant C/EBPα under the control of doxycycline (DOX) were used in proliferation assays. Data are represented as mean and SD from three experiments performed in duplicate. P‐values were obtained by Student's t‐test. Inset: The levels of expression of PR and ER for each of the three cell lines are shown.
- MCF‐7 cells expressing shControl or shC/EBPα were subjected to proliferation assays. Data are represented as mean ± SD from three experiments performed in duplicate. The P‐values obtained using Student's t‐test are indicated.
- T47D and MCF‐7 cells were treated with 10 nM 17β‐estradiol (+E2) as indicated; cDNA was generated and used as template for real‐time PCR with C/EBPα and GAPDH specific primers. Each bar represents the mean ± SD from two experiments. Different italic letters (a,b) are significantly different from each other (P ≤ 0.05) using Student's t‐test.
- Overlap between down‐ and up‐regulated transcripts. Transcripts are scored as significant (q < 0.01) differentially expressed transcripts (DETs) upon treatment with DOX for either 8 or 16 h (see Materials and Methods). DETs are further partitioned into those showing a decrease (down‐regulated) or increase (up‐regulated) in expression after the treatment.
- Gene ontology enrichment analysis. Gene Ontology:Biological Process (GO:BP) and KEGG significantly enriched (P < 0.01) in DETs inferred in each of the two DOX treatments.
- Overlap between R5020‐ and DOX‐DETs. Observed‐to‐expected ratios for the different transcript categories (see Materials and Methods) are shown. The dashed line denotes a ratio of 1 and, as expected, it matches the ratio seen for transcripts scored as non‐regulated in R5020‐ and DOX‐DETs sets of data.
Source data are available online for this figure.
In contrast to progestins, estrogens induce continuous proliferation of breast cancer cells. We found that estrogens did not increase but rather decreased the levels of CEBPα by 40–50% in T47D and MCF‐7 cells (Fig EV2C). This could in part explain the strong proliferative action of estrogens in breast cancer cells.
To identify the genes responsible for the anti‐proliferative role of CEBPα, we performed RNA‐seq in T47D cells expressing Dox‐inducible CEBPα untreated or treated for 8 and 16 h with Dox. We found 407 and 262 genes up‐ and down‐regulated, respectively, at these time points (Fig EV2D). Gene ontology analysis revealed categories associated with the regulation of cell proliferation, metabolic process, signaling, developmental processes, and cell differentiation (Fig EV2E). We compared the RNA‐seq data from T47D cells exposed to hormone with T47DindC/EBPα cells treated with Dox for 8 h. We found that 13% of the down‐ and 17% of the up‐regulated genes identified after Dox induction—around 8‐ and 6‐fold more than expected by chance, respectively—showed the same trend in cells exposed to R5020 (Fig EV2F).
C/EBPα acts as a cell growth modulator in vivo
During the development of the mammary gland, progestins stimulate the growth of stem and progenitor cells (Joshi et al, 2010). In hormone‐responsive breast cancers, progestins increase the stem cell‐like population by converting ER+/PR+ cells to receptor negative cells that acquire expression of the tumor‐initiating markers CD44 and cytokeratin 5 (CK5; Cittelly et al, 2013). Therefore, we explored whether C/EBPα is involved in the hormone‐dependent dedifferentiation of luminal breast cancer cells. To address this point, we used the cell line expressing Dox‐inducible C/EBPα and measured the proportion of CD44high/CD24low cells by FACS sorting. Upon overexpressing CEBPα, the proportion of cells expressing CD44high/CD24low was reduced by 5‐fold (Fig 2A, right panel). Exposure of T47D cells to R5020 for 24 h increased by approximately 3‐fold the cell population expressing stem cell markers. Upon overexpression of CEBPα, the dedifferentiated cells increased with hormone, but the proportion of these cells remained very low (Fig 2A, left panel). Thus, high levels of C/EBPα compromise the ability of hormones to induce dedifferentiation, suggesting that C/EBPα may function as a growth modulator in breast cancer cells.
Figure 2. C/EBPα inhibits hormone‐induced expression of stem cell markers and acts as a cell growth modulator in vivo .
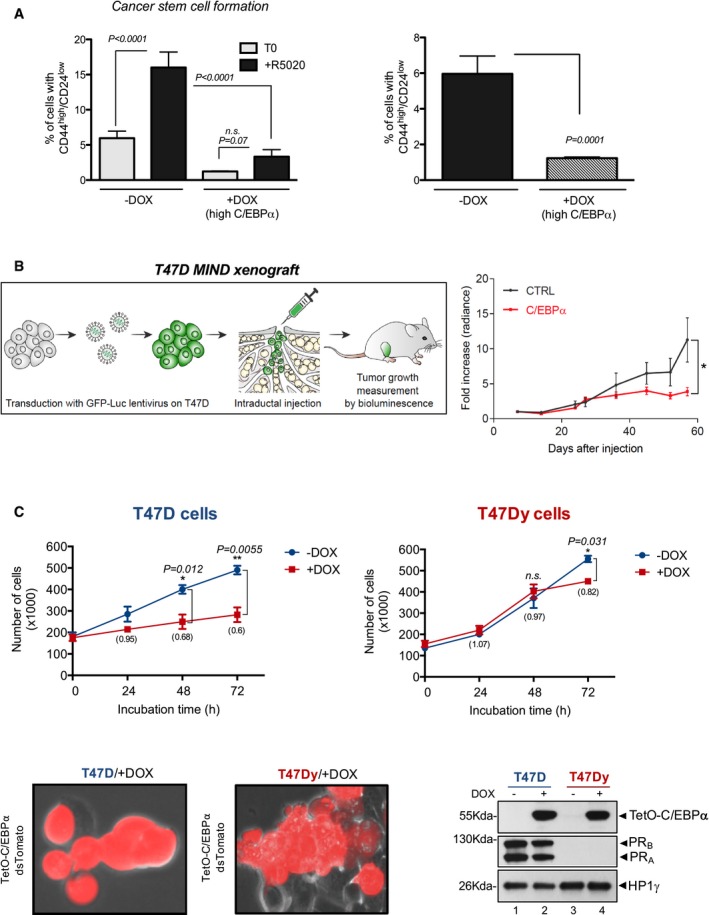
- T47DindC/EBPα cells induced or not with Dox were untreated or exposed to R5020 for 24 h, and the expression of CD44 and CD24 was measured using specific antibodies (CD44‐APC and CD24‐P, respectively) and fluorescence‐activated cell sorting analysis. Data are represented as mean and SD from three experiments performed in duplicate. The P‐values were obtained by ANOVA followed by Tukey test.
- Left panel: Scheme of the Mouse Intra‐Ductal (MIND) xenograft approach: tumor cells expressing luciferase are injected intraductally via the teat (Sflomos et al, 2016). Seven days after injection, CEBPα expression is induced with 2 mg/ml doxycycline in the drinking water maintained throughout the experiment. Tumor growth is assessed by bioluminescence (IVIS). Right panel: Tumor growth of T47DindC/EBPα‐MIND marked with luciferase (Sflomos et al, 2016) treated or not with Dox was assessed by bioluminescence. Results are shown as means ± SEM. Five mice were used for each condition. In total, 17 and 19 glands were analyzed from control and C/EBPα. P‐values were obtained by Student's t‐test and were calculated relative to control animals (*P ≤ 0.05)
- Upper panels: T47DindC/EBPα and T47DyindC/EBPα (PR‐) cells expressing inducible recombinant C/EBPα were exposed to Doxycycline (1 μg/ml) for different times, and then, the number of cells was monitored. Data are represented as mean ± SD from two experiments performed in triplicate. P‐values were obtained by Student's t‐test and were calculated relative to untreated (−DOX). The numbers in parenthesis correspond to the average of the ratio +DOX over −DOX for each time point. Lower panels: T47D‐ and T47Dy‐inducible C/EBPα cells were treated with doxycycline (1 μg/ml Dox) for 24 h, and the expression of C/EBPα was monitored by immune fluorescence microscopy (left) and Western blot (right). The Western blot confirms that T4Dy cells do not express PR. HP1γ was used as loading control. The band with a molecular weight around 55 kDa correspond to the TetO‐C/EBPα ds Tomato fusion protein.
Source data are available online for this figure.
An analysis of a cohort of 21 human invasive breast carcinomas and 90 invasive ductal and lobular carcinomas (Curtis et al, 2012) revealed reduced levels of C/EBPα compared with 144 samples of normal breast (Rhodes et al, 2004), and C/EBPα overexpression correlates with better prognosis and longer survival (Appendix Fig S2A and B). These observations suggest that C/EBPα may behave as a tumor suppressor in breast cancers. To test this hypothesis in a more physiological in vivo model, we injected Control and C/EBPα‐overexpressing T47D cells into the mouse milk duct of immune‐suppressed mature female mice (Sflomos et al, 2016). The intraductal microenvironment enables ER+ breast cancer cells to grow in vivo and form tumors that recapitulate the human disease, with the mouse systemic hormone levels (Fig 2B, left panel) (Sflomos et al, 2016). In vivo monitoring of engrafted mice by luminescence showed that the tumors derived from T47D overexpressing C/EBPα grew significantly slower than control T47D cells (Fig 2B, right panel), supporting a role of C/EBPα as tumor modulator in breast cancer.
We next asked if the suppressive effect of C/EBPα on cell proliferation was directly associated with the presence of PR. To answer this question, we established a C/EBPα‐inducible system in the PR‐negative T47Dy cells (Horwitz et al, 1995). The inhibitory effect of growth upon C/EBPα induction by DOX was significantly delayed in T47Dy cells compared to the wild‐type counterpart (Fig 2C), indicating that C/EBPα requires progestin activity to elicit its full effect as growth regulator in breast cancer cells.
Genome‐wide distribution of PR and C/EBPα
To explore the mechanism of the CEBPα effect on hormone‐induced cell proliferation, we performed ChIP‐seq using specific PR and C/EBPα antibodies (Nacht et al, 2016) and Appendix Fig S3A to analyze the genome‐wide distribution of these proteins in T47D cells. Using a stringent peak calling approach (see Materials and Methods), we found 143 PR peaks before and 13,766 peaks after 6‐h hormone exposure, while for C/EBPα we found 8,468 peaks before and 32,482 after hormone exhibiting considerable (36.3%) overlap (Fig 3A and B). Integration of the ChIP‐seq profiles from both transcription factors identified three unique clusters of PR and C/EBPα‐binding (Fig 3C, left panel):
Figure 3. Genome‐wide binding of PR and C/EBPα in T47D cells.
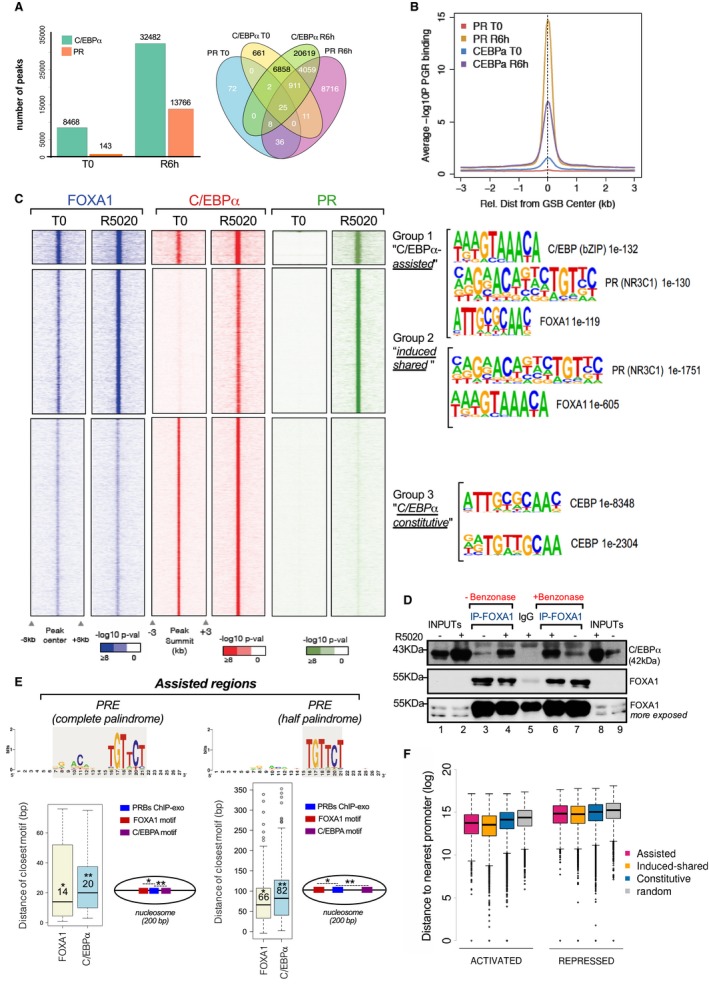
- Left panel: number of peaks identified by ChIP‐seq for PR and C/EBPα. Right panel: Venn diagrams of the overlap between PR and C/EBPα at T0 and 6 h after hormone induction.
- Overlapping of PR and C/EBPα genome binding sites. Based on the analysis of ChIP‐seq experiments, the Sitepro (CEAS package) analysis (see Materials and Methods) was employed to plot PR and C/EBPα (at time T0 and 6 h after hormone exposure). For each factor, the average −logPoisson P‐values were plotted within a window of ± 3 kb.
- Heat maps of the genomic binding of FOXA1, C/EBPα, and PR before and after exposure to R5020 for 60 min. The ChIP reads are shown within a 16 kb window centered on the C/EBPα peaks. Based on the analysis of ChIP‐seq experiments, sequenced reads were aligned to the human genome, the peaks of enrichment were defined, and the P‐values of the ChIP reads were calculated in each window (50 bp). Significant peaks were defined as previously shown (Ferrari et al, 2012). The most abundant binding motifs identified using MEME suite SpaMo tool are shown on the right.
- The interaction between FOXA 1 and C/EBPα is not dependent on DNA binding. T47D cells exposed or not to hormone and treated or not with benzonase were lysed and immunoprecipitated either with α‐FOXA1‐specific antibody or IgG. The immunoprecipitates (IP) were analyzed by immunoblotting with specific antibodies for C/EBPα and FOXA1.
- Neighboring motif analysis on “assisted” regions. The PR ChIP‐exo peaks either bound to a complete PRE (left) or half PRE (right) in assisted regions were mapped, and the average distance with FOXA1 and C/EBPA motifs was calculated by using MEME suite SpaMo tool (Whitington et al, 2011).
- “Assisted” and “Induced‐shared” regions are closer to hormone regulates genes. In each of the three clusters of ChIP‐seq peaks identified in C, the closest differentially expressed gene was determined (DETs, see Materials and Methods). Distribution of distances to the nearest hormone‐regulated promoter is shown for each cluster (bp). The distances corresponding to randomly selected regions were significant in all cases.
Cluster 1 composed of 1,061 C/EBPα‐assisted PR binding sites, which are occupied by C/EBPα but not by PR prior to hormone exposure and exhibit both factors bound after hormone exposure.
Cluster 2 composed of 8,987 induced shared PR‐C/EBPα binding sites, which do not exhibit binding of either factor prior to hormone exposure and are occupied by both PR and C/EBPα upon hormone exposure.
Cluster 3 composed of 27,715 C/EBPα−constitutive sites, which are constitutively occupied by C/EBPα and exhibit very low if any PR binding after hormone exposure.
Motif analysis showed that the NR3C1 group of nuclear receptors (GRE, PRE, ARE), C/EBPA and FKHD (FOXA1) motifs were enriched in group 1, while only GRE and FOXA1 in group 2 (Fig 3C, right panels). Interestingly, although C/EBPα is bound before PR in cluster 1, other additional DNA motifs as FOXA1 and PRE were found significantly enriched at almost the same level as the CEBPA (1e‐132, 1e‐130, and 1e‐119 for CEBPA, PRE, and FOXA1, respectively). Moreover, in “assisted” regions the CEBPA motif is underestimated compared to “C/EBPα constitutive” (55 vs. 79.3%), a cluster exclusively driven by C/EBPA and where only the CEBPA motif is found (Fig 3C, right panels). Therefore, binding of C/EBPα to cluster 1 would occur via a different sequence motif or via protein–protein interaction with another pre‐bound factor, possibly FOXA1.
As the FKHD motif (G/A‐C/T‐AAAC/T‐A) is found in the “assisted” regions at T0 (Fig 3C), we hypothesized that the pioneer factor FOXA1 could be enriched in these regions and could participate in C/EBPα anchoring. To address this point, we performed the ChIP‐seq of FOXA1 in T47D cells and found that before hormone exposure “assisted” regions exhibit higher FOXA1 occupancy, compared with the other clusters (Fig 3C, left panels). Only a slight increase in FOXA1 binding is observed after hormone exposure, supporting its function as a pioneer factor. The “induced shared” cluster showed increased in FOXA1 signal after hormone exposure compared with group 3 (Fig 3C, left panel). To determine whether FOXA1 and C/EBPα can physically interact, we performed co‐immunoprecipitation (co‐IPs) experiments using breast cancer cell extracts. We found a weak interaction in the absence of hormone that increases after 6 h of hormone exposure (Fig 3D, lanes 3–4). As DNA binding proteins can associate during IP due to their adjacent binding on DNA, we repeated the co‐IP experiments in the presence of the nuclease benzonase. We found that the interaction between and FOXA1 is not affected by the presence of benzonase, pointing to a direct protein–protein interaction (Fig 3D, compare lanes 4 and 6).
To identify at near single nucleotide resolution binding locations of PR genome‐wide, we carried out PR ChIP‐exo experiments in T47D cells treated with hormone (Rhee & Pugh, 2011). First, we examine the gap in bps between the PRE and adjacent motifs. In 590 “assisted” regions, PR binds to a half palindrome (5′‐TGTTCT‐3′) around FOXA1 and C/EBPα motifs located in average at 66 and 82 bp, respectively (Fig 3E, right panel). In the remaining 69 regions, PR binds to a complete PRE palindrome and the most frequent gap between PRE and FOXA1 was 14 and 20 bp between PRE and C/EBPα (Fig 3E, left panel). Thus, FOXA1 and C/EBPα could facilitate PR binding to “assisted” regions through a short‐range mechanism, within a nucleosome.
To obtain a global measure of the contribution to gene regulation of C/EBPα‐ and PR binding regions, we calculated the distance of the peaks in each of these three clusters to the closest transcription start sites (TSS) of hormone‐regulated genes (Fig 3F). Regions in clusters 1 and 2 are localized closer to the TSS of hormone‐activated genes compared to the rest (Fig 3F, left panel). Cluster 3 sites are significantly further away from up‐regulated transcripts compared to “assisted” and “induced shared” peaks (P‐value = 1 e‐101). This is particularly remarkable considering the difference in the number of peaks between this cluster (~27,000) and clusters 1 and 2 (~1,000 and ~9,000, respectively). When hormone‐repressed genes were analyzed, “assisted” and “induced shared” regions are found at the same distance from the TSS of regulated genes as cluster 3, and the differences with the random regions are reduced (Fig 3F, right panel).
Of note, in the “C/EBPα‐constitutive” cluster 3 binding of C/EBPα is gained in the presence of the hormone without a direct participation of PR as a tethering factor (Fig 3C). The increase in C/EBPα signal in this category could be due to the global increase in C/EBPα protein levels upon hormone exposure (Fig EV1A, left panel). Analysis of the genomic distribution showed similarities between the different clusters with an over‐representation of introns and of 1–5 kb from the TSS in “assisted” and “induced shared” regions (Appendix Fig S3B).
Next, we use Genomic Regions Enrichment of Annotations Tool (GREAT; McLean et al, 2010) to gain insight into the gene ontology of “assisted” and “induced shared” clusters. “Assisted” regions were enriched for terms associated to genes with the following characteristics: (i) down‐regulated in breast cancer and in basal subtype of breast cancer, (ii) involved in therapy resistance to tamoxifen (breast tumors and xenografts), and (iii) related to breast cancer (Appendix Fig S4, upper panel). On the other hand, categories associated with regulation of genes by estrogens and androgens in MCF7 and LNCAP cells, respectively, as well as genes regulated by Beta Parvin in 3D organoids of the triple‐negative MDA‐MB‐231 cells were overrepresented in the “induced shared” regions (Appendix Fig S4, bottom panel). This data conclude that “assisted” genomic regions might play a more direct role in regulating genes associated with breast cancer.
“Assisted” regions are packaged into H1‐depleted nucleosomes and exhibit characteristics of active enhancers
Taking into account that C/EBPα and FOXA1 could cooperate to allow PR binding in a nucleosomal range (Fig 3E), we next asked if the DNA of the “assisted” regions is packaged into nucleosomes. We performed micrococcal nuclease (MNase)‐seq assays in T47D cells at two concentrations of MNase (30 and 270 units). Our results showed that a nucleosome‐like particle is detected in both “induced shared” and “assisted” regions, while “C/EBPα ‐constitutive” and random regions do not showed protection to MNase (Fig 4A, left panel). The increase in nuclease concentration caused the loss of nucleosomal signal, preferably in the “assisted” regions, which would support the presence of a loose nucleosome (Fig 4A, middle panel).
Figure 4. “Assisted” and “Induced shared” regions exhibit characteristics of active enhancers.
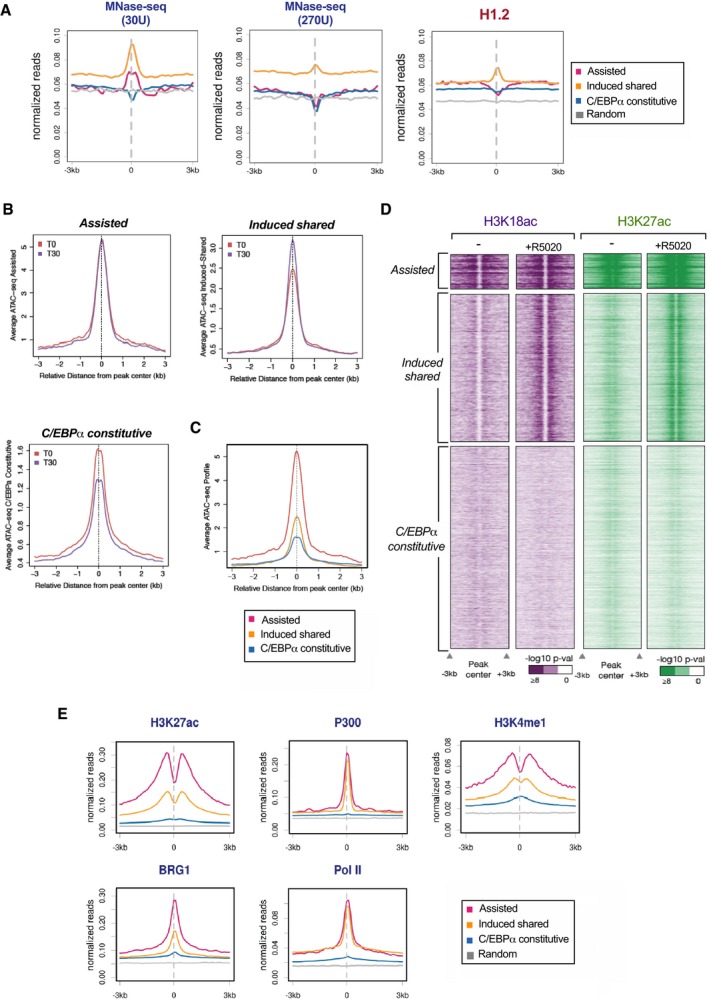
- Nucleosomes occupancy in the different clusters was analyzed using MNase‐seq in untreated T47D cells at 30 and 270 units of MNase as indicated (left panels). ChIP‐seq using H1.2‐specific antibody was performed in untreated T47D cells (right panel). Average profiles of the reads for each group are shown.
- Sitepro (CEAS package) analysis was employed to plot average ATAC‐seq for each cluster of Fig 3C. The average ATAC‐seq values before and after hormone exposure (+R5020) were plotted within a window of ± 3 kb of the C/EBPα peaks.
- Sitepro (CEAS package) analysis was employed to plot average ATAC‐seq for each cluster at time T0 within the same y axis.
- Heat map showing the enrichment (−logP) of H3K18ac and H3K27ac for each cluster of Fig 3C. Increasing color shades represent enrichment as calculated above(see scales under the heat maps).
- Behavior of enhancer marks around C/EBPα peaks as detected using ChIP‐seq experiments with specific antibodies to H3K27ac, H3K4me1, P300, BRG1, and RNA Pol II, in untreated and in cells exposed to hormone. Average profiles of the reads for each group are shown.
The linker histone H1 binds to the entry/exit sites of DNA on the surface of the nucleosomal core particle and completes the nucleosome. H1 has been linked to higher‐order chromatin compaction and global gene silencing (Hergeth & Schneider, 2015). Next, we wonder whether this open nucleosome that is detected in the “assisted” regions lacks histone H1. Analysis of the average profiles of the H1.2 ChIP‐seq corresponding to “assisted” regions showed a significant depletion of H1.2 compared to the other clusters (Fig 4A, right panel). Therefore, the “assisted” regions are assembled in nucleosomes depleted of H1, which constitutes an optimal platform for the cooperation between PR, FOXA1, and C/EBPα.
“Assisted” and to a lesser extent “induced shared” regions exhibit an open chromatin conformation and high degree of histone acetylation compared with “C/EBPα‐constitutive” regions (Fig 4B–D). To explore whether these regions function as enhancers in breast cancer cells, we performed ChIP‐seq in the presence of hormone for the most representative enhancer marks. Average profiles of the reads corresponding to “assisted” as well as “induced shared” regions present significantly more H3K27ac, P300, H3K4me1, BRG1, and RNA polymerase II than “C/EBPα‐constitutive” and random regions (Fig 4E). Thus, these regions exhibit epigenetic features of active enhancers in breast cancer cells.
C/EBPα is involved in 3D chromatin looping
So far, we have described that the “assisted” regions are located in enhancers and closer to genes regulated by hormone. To perform their action at distance, enhancers must interact with the promoter regions of the target genes by establishing chromatin loops. To explore this possibility, we used in situ Hi‐C experiments, a method that probes the 3D architecture of whole genomes by coupling proximity‐based ligation with massively parallel sequencing (Lieberman‐Aiden et al, 2009). We found that prior to hormone exposure “assisted” and “induced shared” regions associated with the promoters of hormone‐regulated genes exhibited more contacts compared with random regions, and that upon hormone exposure the density of contact increases significantly for the “assisted” regions (P < 0.05) (Fig 5A). Moreover, the increase was limited to hormone‐dependent promoters. If the analysis was made for all the promoters, no significant change in contact density was observed upon hormone exposure for any of the analyzed regions (Fig EV3A). These findings support a function of the C/EBPα in “assisted” regions, helping PR to accomplish its function in hormone‐dependent enhancers.
Figure 5. C/EBPα is involved in 3D chromatin looping.
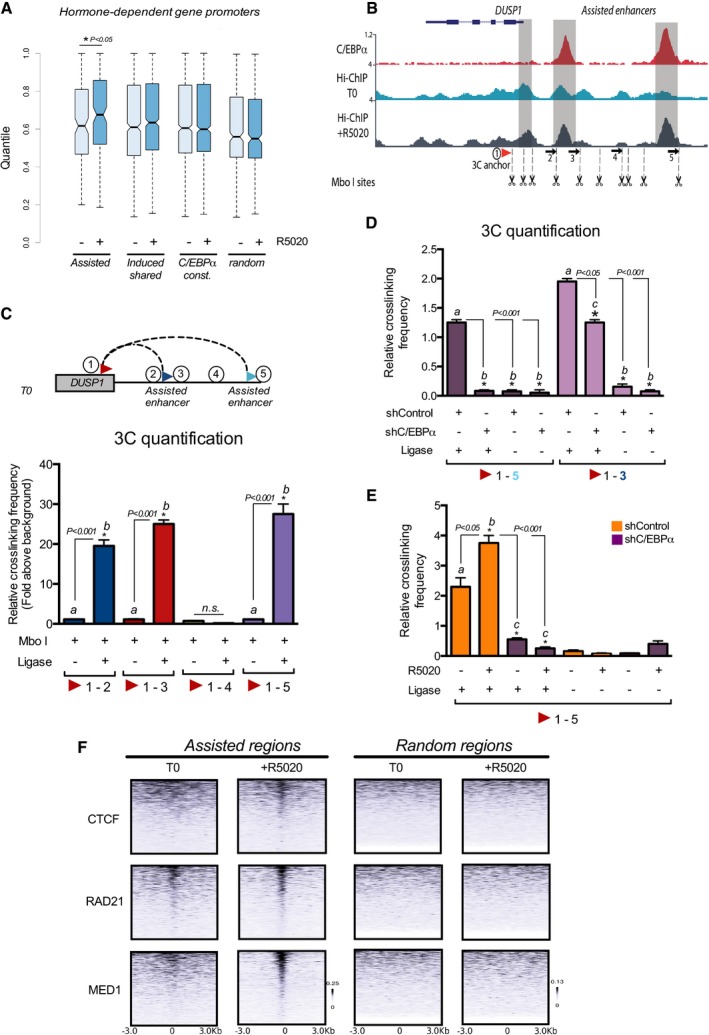
- Hi‐C contacts of the indicated regions with promoters of hormone‐dependent genes. Pair (promoter/region) significance was obtained by contrasting the observed number of contacts against a null distribution composed of 500 random pairs at the same distance. Then, promoter enrichment was estimated as the 99 percentile (a robust maximum) of the nearby selected regions (from 20 to 200 kb) corresponding to the most likely putative regulator. The boxes represent interquartile ranges, horizontal lines the medians, and whiskers extend to the maximum and minimum values.
- Snapshot of the genome browser for the DUSP1 gene (schematically shown at the top). Upper line: C/EBPα ChiP‐seq. Two upstream peaks located at −1.3 and −5 kb from the TSS and showing C/EBPα binding are depicted. Middle line: Hi‐ChIP profile using a PR‐specific antibody and cells prior to hormone exposure. Lower line: similar Hi‐ChIP profile in cells exposed to progestin for 60 min. Underneath the graph, the regions selected for designing the oligonucleotides used for 3C quantification by PCR are indicated.
-
Top panel: Scheme showing the interactions analyzed by 3C.Bottom panel: Quantification of the 3C experiments performed on Mbo I digested and religated or not chromatin from T47D cells as indicated. 3C results were quantified by qPCR using primers spanning within the DUSP1 gene and the two putative enhancers. Primer #4 as well as primers including the PR binding site in the Bcl‐x gene were used as controls. Data are representative of four experiments. Data are represented as mean and SD from four experiments performed in duplicate.
- 3C experiments were performed on chromatin obtained from untreated shControl and shC/EBPα T47D cells. The combination of the anchor primer 1 with primers 5 or 3 indicates the contacts between the DUSP1 gene and the two putative enhancers. Data are representative of four experiments. Data are represented as mean and SD from four experiments performed in duplicate.
- 3C experiments were performed on Mbo I digested and religated or not chromatin from T47D shControl and sh C/EBPα cells treated or not with hormone as indicated. Data are represented as mean and SD from four experiments performed in duplicate.
- Heat maps of the ChIP‐seq profiles of CTCF, RAD21, and Med1 for the “assisted” and random regions in cells prior to hormone exposure (T0) and in cells exposed to progestins for 60 min (+R5020).
Figure EV3. C/EBPα knockdown compromised the hormone‐dependent recruitment of RAD21 to “assisted” regions.
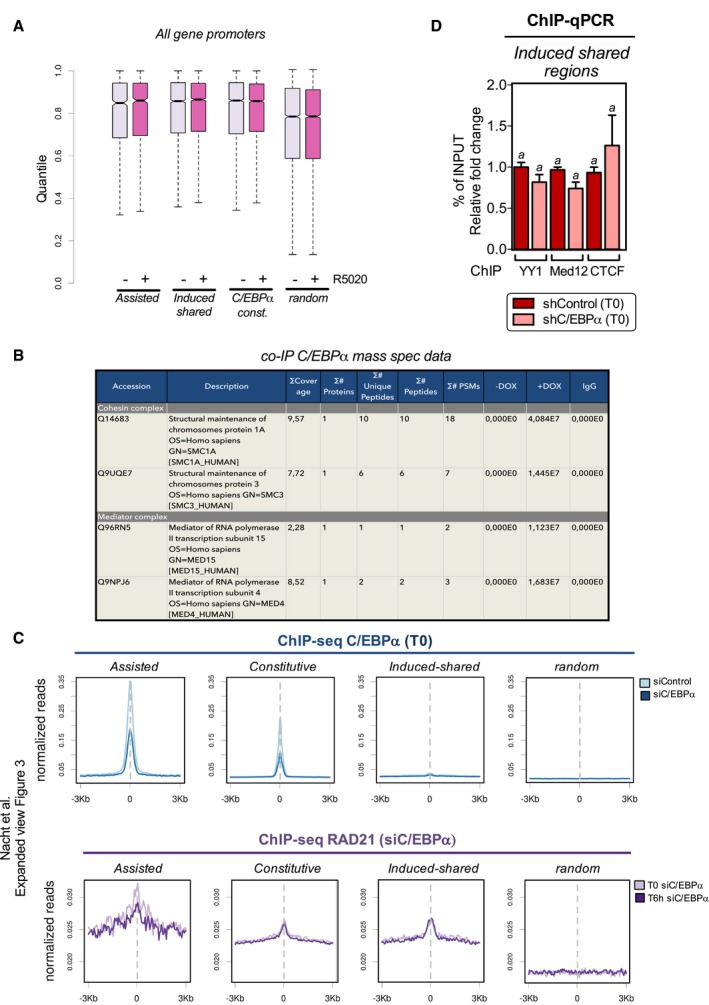
- Hi‐C contacts from promoter of all genes and selected regions were analyzed. Pair (promoter/region) significance was obtained by contrasting the observed number of contacts against a null distribution composed of 500 random pairs at the same distance. Then, promoter enrichment was estimated as the 99 percentile (a robust maximum) of the nearby selected regions (from 20 to 200 kb) corresponding to the most likely putative regulator. The boxes represent interquartile ranges, horizontal lines the medians, and whiskers extend to the maximum and minimum values
- Immunoprecipitation of C/EBPα followed by mass spec in a T47D cell line expressing inducible C/EBPα (see Materials and Methods). This table shows the interactors found associated with the Cohesin and Mediator complexes.
- ChIP‐seq experiments of C/EBPα were performed in cells expressing either shControl or shC/EBPα at uninduced conditions (upper panel). Reads profile for “assisted”, “constitutive”, “induced shared”, and random regions are shown. Lower panel: ChIP‐seq of RAD21 was performed in cells expressing either shControl or shC/EBPα treated or not with hormone for 6 h.
- ChIP experiments of YY1, Med12, and CTCF were performed in cells expressing either shControl or shC/EBPα at T0. Precipitated DNA was analyzed by PCR for the presence of sequences corresponding to the induced shared regions. Data are represented as mean ± SD from three experiments performed in duplicate. (a) Superscript letters are not significantly different from each other using Student's t‐test.
To gain additional insight into the molecular mechanism by which PR and C/EBPα regulate target genes, we selected for further analysis the gene DUSP1, which presents two “assisted” regions located at 5 kb and 1.3 kb upstream of the TSS (Fig 5B). DUSP1 inactivates ERK1/2 by dephosphorylation of both threonine and tyrosine residues (Sun et al, 1993), and its hormone‐dependent induction requires of C/EBPα (Fig 1B). Therefore, to verify the function of C/EBPα we performed 3C assays in cells transfected with shRNA against C/EBPα or shControl. We found that the two “assisted” regions contact the promoter of DUSP1 before hormone exposure (T0) (Fig 5C) and the interaction increases upon hormone exposure (Fig 5E). Both the interactions at T0 and at T30 were dependent on C/EBPα (Fig 5D). These results along with the expression assays (Fig 1B) highlight the role of C/EBPα as an organizer of the genome architecture allowing an appropriate hormonal induction.
To evaluate in more depth the effect of the hormone on enhancer–promoter interactions, we perform Hi‐ChIP of PR (Mumbach et al, 2016). We found that after hormone exposure, the −5 kb PR and C/EBPα binding sites presented more contacts compared with the untreated sample and involved the −1.3 kb “assisted” enhancer and the DUSP1 proximal promoter (Fig 5B). Thus, the C/EBPα found at T0 in the “assisted” enhancers could be mediating the interactions with the promoter that are preformed and detected before hormone, while hormone induction increased these contacts.
C/EBPα interacts with cohesin, CTCF, YY1, and Mediator
Genome‐wide studies showed that architectural proteins CTCF and its frequent binding partner cohesin are important for organization of the genome into topologically associating domains or TADs and in the formation of promoter–enhancer loops (Dixon et al, 2012). In addition, the large Mediator complex that is recruited to enhancers and the transcription factor YY1 are also central players in connecting enhancers to promoters (Weintraub et al, 2017). Therefore, we explored whether these architectural proteins participate in C/EBPα ‐mediated looping by using co‐IP experiments followed by mass spec. We found that C/EBPα interacts with components of cohesin complex SMC1A, SMC3, as well as with Med15 and Med 4, subunits of the Mediator complex (Fig EV3B). In ChIP‐seq experiments of CTCF, RAD21, and Med1 in T47D cells, we found an enrichment of RAD21 at T0 in the “assisted” regions and a further increase after hormone exposure, whereas CTCF showed an increased binding only after hormone exposure (Fig 5F, left top panels). Analysis of Med1 ChIP‐seq data also showed association of Mediator complex at T0 in “assisted” regions and further increased after hormone (Fig 5F, left lower panels). When a similar analysis was performed with random regions, no significant enrichment was detected (Fig 5F, right panels).
Next, we tested whether the presence of CTCF, RAD21, YY1, and Med1 in the “assisted” regions depends on C/EBPα. Knockdown of C/EBPα reduced by 60% C/EBPα binding at “assisted” regions associated with the hormone‐regulated genes AZIN1, SRCAP, ZBED3, PPP1R14D, and DUSP1 (Fig 6A, left panel). We confirm these results genome wide using as control the ChIP‐seq of C/EBPα in C/EBPα knockdown cells (Fig EV3C, upper panel). Concomitant with the low levels of C/EBPα, we found a reduction in the levels of chromatin‐bound YY1, Med12, and CTCF at T0 in the DUSP1 “assisted” enhancer as well as in several other “assisted” regions (Fig 6A, right panel and Fig 6B). This effect was specific for the “assisted” regions, as no change was detected in the “induced shared” exclusive regions (Fig EV3D). In addition, we found that the hormone‐induced RAD21 binding to “assisted” regions was also dependent on C/EBPα, as its knockdown completely abrogated RAD21 recruitment (Fig 6C, left panel). Moreover, ChIP‐seq of RAD21 in C/EBPα depleted cells compromised the hormone‐dependent recruitment of RAD21 to “assisted” regions (Fig EV3C, lower panel). In fact, in the presence of hormone C/EBPα knockdown relocated 52% of RAD21 peaks to new genomic regions (958 out of 2010 RAD21 binding sites). Therefore, several structural proteins known to mediate the 3D contacts between enhancers and promoters, such as YY1, Mediator, and the cohesin complex, depend on C/EBPα to enable the gene structure that ensures proper regulation.
Figure 6. C/EBPα is required for YY1, Med12, and RAD21 loading in “assisted” regions.

- Left panel: ChIP experiments of C/EBPα in cells expressing either shControl or shC/EBPα. The hormone‐regulated genes associated with the “Assisted” regions and tested by RT–PCR were AZIN1, SRCAP, ZBED3, PPP1R14D, and DUSP1 (n = 5; left panel). The inset shows the extent of C/EBPα depletion, using LSD1 as loading control. Right panel: ChIP experiments of YY1, Med12, and CTCF in cells expressing either shControl or shC/EBPα at T0. Precipitated DNA was analyzed by PCR for the presence of sequences corresponding to the assisted regions localized at −1.3 kb from the DUSP1 gene. Data are represented as mean ± SD from three experiments performed in duplicate.
- ChIP experiments of YY1 and Med12 in cells expressing either shControl or shC/EBPα before and after exposure to hormone. Precipitated DNA was analyzed by PCR for the presence of sequences corresponding to the “assisted” regions (n = 5) as shown in panel (A). Data are represented as mean ± SD from three experiments performed in duplicate.
- ChIP experiments of RAD21 in cells expressing either shControl or shC/EBPα before and after exposure to hormone. Precipitated DNA was analyzed by PCR for the presence of sequences corresponding to the “assisted” regions (n = 5) as shown in panel (A) (left panel) and DUSP1 (right panel). Data are represented as mean ± SD from three experiments performed in duplicate.
- ChIP‐seq experiments of PR in cells expressing either shControl or shC/EBPα after exposure to hormone. Reads profile for “assisted”, “constitutive”, “induced shared”, and “random” regions are shown.
- DNase I accessibility assays in cells expressing shControl and shC/EBPα and treated or not with hormone (see Materials and Methods). Accessibility was tested in 5 “assisted” regions as described in panel (A) and in 5 “induced shared” regions associated with ACOTT, WTAP, MUC21, CREBL2, and CAMKK2 genes. Each value corresponds to the mean ± SD of three experiments performed in duplicate. **P < 0.05 using Student's t‐test
C/EBPα maintains the “assisted” regions in an accessible chromatin conformation
To explore whether C/EBPα is required for proper PR binding at “assisted” and “induced shared” regions, we performed ChIP‐seq experiments of PR in cells expressing either shRNA‐Control or shRNA‐C/EBPα before and after exposure to hormone. Hormone‐dependent binding of PR to “assisted” regions was compromised by 50% in C/EBPα‐depleted vs Control cells (Fig 6D, left panel), in line with the extent of chromatin‐bound C/EBPα in knockdown cells (Fig EV3C, upper panel). Binding of PR to “induced shared” regions was partially affected by C/EBPα knockdown (Fig 6D, third panel from the left), suggesting that C/EBPα would have a role as PR co‐activator in these regions.
To gain insight into the molecular mechanism of this effect, we performed quantitative DNAse I accessibility experiments (Di Stefano et al, 2014) on “assisted” regions. Before hormone exposure (T0), depletion of C/EBPα decreased access of the nuclease to DNA in “assisted” regions, compared with shControl cells, while no difference was detected in “induced shared” regions (Fig 6E). Thus, C/EBPα maintains the “assisted” regions in accessible conformation ready for binding of ligand‐activated PR and associated proteins.
A role for Topoisomerase 2α and P300
Transcription factors often cooperate with multiprotein complexes that contribute to their transcriptional output. To identify the proteins that interact with C/EBPα and could mediate its effects, we performed immunoprecipitation of C/EBPα followed by mass spec in a T47D cell line expressing inducible C/EBPα (see Materials and Methods). Analysis of the enriched peptides showed that the interacting proteins function in chromatin remodeling, chromatin modification, mitotic cell cycle, DNA conformation change, DNA repair, and nucleosome organization (Fig EV4A, upper panel). By using the CORUM database (Ruepp et al, 2008), we identify SWI/SNF (BAF), NURD, and CHD complexes as well as histone deacetylases (Fig EV4A, lower panel) as enriched interactors. Moreover, we found five members of the KDM1A/HDAC/REST (HP1γ‐LSD1.com) previously reported as a complex involved in basal and active repression of genes (Vicent et al, 2013; Nacht et al, 2016). Besides BRG1, BAF170 and BAF57, subunits of SWI/SNF (BAF) complex, we also found the acetyltransferase P300 associated with C/EBPα (Fig EV4A, upper panel). We confirmed by ChIP‐seq that P300 as well as BRG1 are both enriched in “assisted” and “induced shared” regions (Fig EV4B and E). Trim 28 (Friedman et al, 1996) and the RSF complex (LeRoy et al, 1998; Orphanides et al, 1998) were also found associated with C/EBPα (Fig 5C). We validated by co‐IP several of the C/EBPα interactors: BRG1, BAF170, SNF2h, CHD4, KDM1, CBX3, and PR (Fig EV4C). Interestingly, other enriched interactors such as DDB1, PARP1, Topo I, Topo2α, and Rad50 are known to be associated with DNA repair (Nacht et al, 2016). We found that BRG1, P300, SNF2h, CHD4, LSD1, HDAC1, Topo2α, and TRIM28 were also associated with endogenous C/EBPα in the presence of hormone (Fig EV4C, right panel).
Figure EV4. The interactome of C/EBPα reveals a preferential interaction with SWI/SNF, NURD, KDM1A/HDAC/REST, and RSF chromatin remodeling complexes.
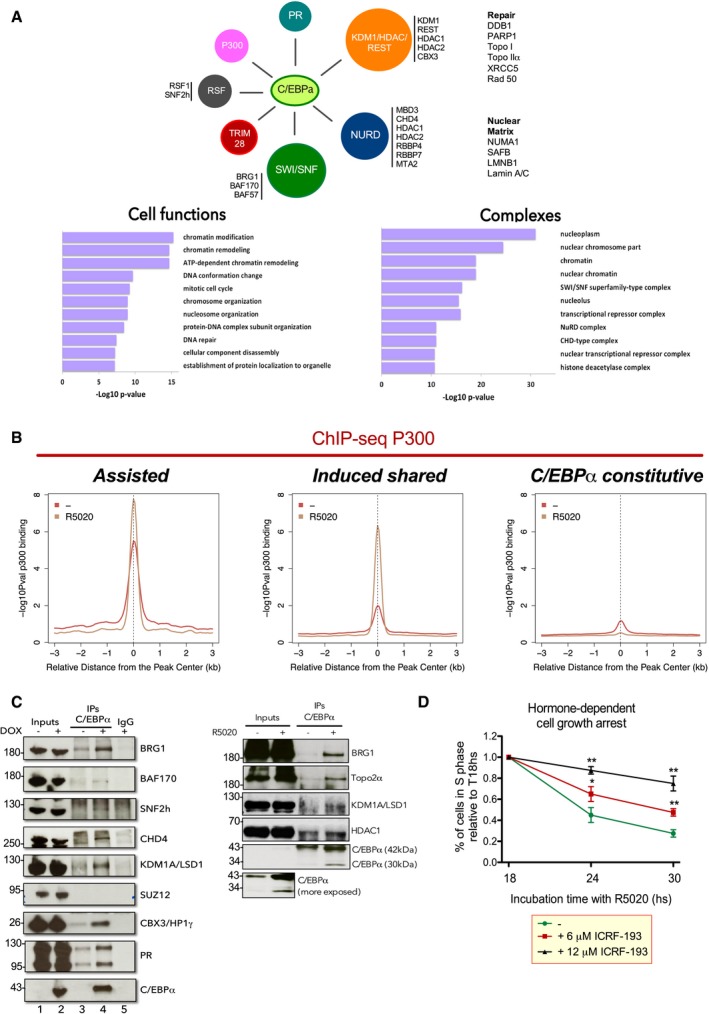
- Immunoprecipitation of C/EBPα followed by mass spec in a T47D cell line expressing inducible C/EBPα (see Materials and Methods). Lower panels: Analysis of the peptides showed that the interacting proteins function in chromatin remodeling, chromatin modification, mitotic cell cycle, DNA conformation change, DNA repair, and nucleosome organization. Right panel: By using the CORUM database (Ruepp et al, 2008), we identify SWI/SNF (BAF), NURD, and CHD complexes as well as histone deacetylases.
- Analysis of the ChIP‐seq of P300 in T47D cells treated or not with hormone in “assisted”, “induced shared”, “PR‐exclusive”, “C/EBPα‐constitutive”, and “C/EBPα‐exclusive” regions.
- Validation of the C/EBPα interactors by co‐IP in T47DindC/EBPα cells −/+ dox (left panel) and T47D cells −/+ hormone (right panel). The immunoprecipitates (IP) were analyzed by immunoblotting with specific antibodies for C/EBPα, Topo 2α, BRG1, BAF170, SNF2h, CHD4, KDM1, CBX3, and PR.
- T47D cells treated or not with 6 and 12 μM of ICRF193 and 10 nM R5020 as indicated were subjected to flow cytometric analysis of cell cycle to quantify the cells in S phase. Each value corresponds to the mean ± SD of three experiments performed in duplicate. **P < 0.01, *P < 0.05 using Student's t‐test.
Source data are available online for this figure.
We were intrigued by the possible role of Topo2 in view of the importance of topoisomerases during gene activation by nuclear receptors in MCF7 breast cancer cells (Ju et al, 2006). Moreover, it has been recently proposed that Topo2α synergizes with BAF (SWI/SNFa) to promote binding of pluripotency factors in embryonic stem cells (Miller et al, 2017). Therefore, we tested whether PR, C/EBPα, and Topo2α physically interact in breast cancer cells. By co‐IP experiments, we found that PR is associated with C/EBPα and Topo2α only after exposure to hormone (Fig 7A, upper panel, compare lanes 2 vs. 3). Conversely, Topo2β and C/EBPβ did not interact with PR (Fig 7A, lower panel). Next, we tested whether Topo2α could be anchored by C/EBPα at the “assisted” regions using (eto)‐ChIP (Miller et al, 2017) and specific antibodies for Topo2α. Using ChIP‐seq, we found that C/EBPα depletion significantly reduced Topo2α in “assisted” regions independently of hormone (Figs 7B and EV5A). Inhibition of Topo2α with the catalytic inhibitor ICRF‐193 makes the chromatin of “assisted” regions unaccessible, similarly to what was observed with C/EBPα knockdown (Fig 7C and compare with Fig 6E). The involvement of Topo2α in DUSP1 gene regulation is supported by the observation that the hormone‐dependent induction was impaired in a dose‐dependent manner by the specific inhibitor ICRF‐193 (Fig 7D). In order to extend our conclusions, we performed RNA‐seq experiments in cells treated or not with hormone and ICRF‐193. We found that 61.3% of the up‐regulated and 50% of the down‐regulated genes (702 and 311 genes, respectively) require Topo2α activity to properly respond to hormone (Fig 7E). The combined analysis with the RNA‐seq performed in C/EBPα depleted cells (Fig 1C) showed that 67% of the down‐regulated and 74% of the up‐regulated genes (321 and 258, respectively) were dependent on both C/EBPα and Topo2α, supporting a functional link required for hormone‐dependent transcriptional activity. As an example, ICRF‐193 reduced the chromatin accessibility of the −1.3 kb DUSP1 region and delayed hormone‐dependent cell growth arrest phase (Fig EV4D), highlighting the role of Topo2α in DUSP1 gene regulation and hence in cell cycle. This observation is in line with previous reports that increasing DUSP1 protein expression suppresses growth of MCF‐7 breast cancer cells (Chen et al, 2005).
Figure 7. C/EBPα anchors P300 and Topo2α maintaining an open chromatin conformation in “assisted” regions.
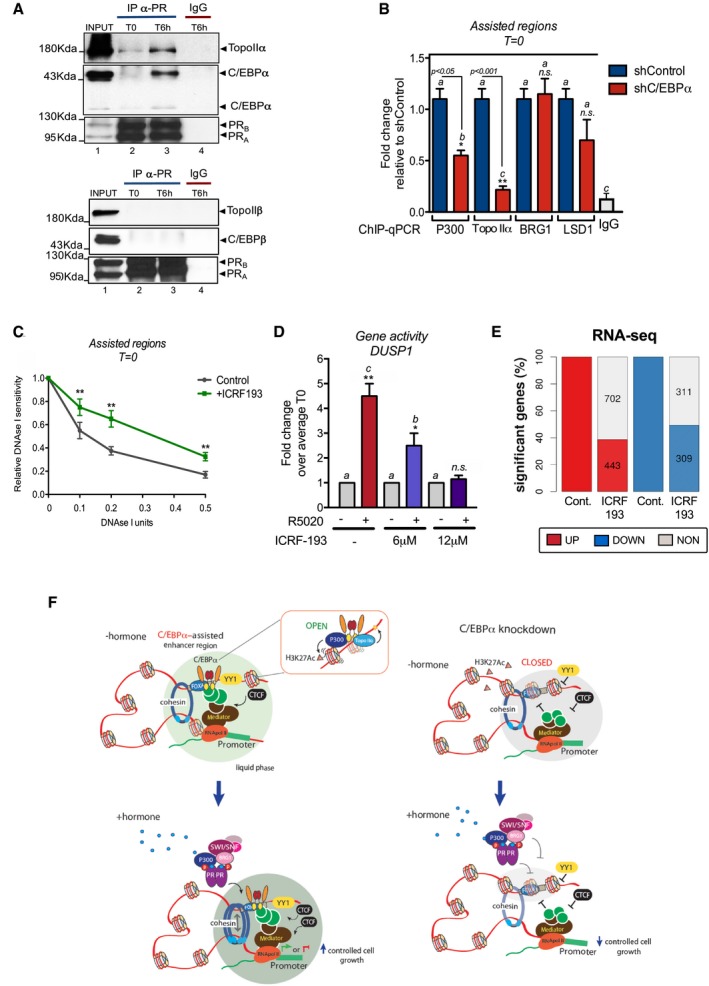
- Immunoprecipitates (IP) of lysates from T47D cells treated or not with hormone using an α‐PR‐specific antibody or IgG were electrophoresed and analyzed by Western blot by immunoblotting with specific antibodies for C/EBPα, Topo2α (top panel), Topo2β (bottom panel), C/EBP β, and PR.
- ChIP experiments of P300, Topo2α, BRG1, and LSD1 were performed in cells expressing either shControl or shC/EBPα and tested in the “assisted” regions described in Fig 6C.
- T47D cells treated or not with 12 μM of the Topo2α inhibitor ICRF193 were subjected to DNase I accessibility assays. The “assisted” regions described in panel (A) were tested by RT–PCR. Each value corresponds to the mean ± SD of three experiments performed in duplicate. **P < 0.05 using Student's t‐test.
- T47D cells were treated or not with 6 and 12 μM of the Topo2α inhibitor ICRF193 and 10 nM R5020 as indicated; cDNA was generated and used as template for real‐time PCR for the assisted region localized at −1.3 kb from the DUSP1.
- T47D cells treated or not with 12 μM of ICRF193 and 10 nM R5020 as indicated were subjected to RNA‐seq experiments performed as described in the Materials and Methods. The number of significant (q < 0.01) differentially expressed genes upon treatment with R5020 for 6 h identified in Control (Cont.) and ICRF‐193 is shown.
- Model for the crosstalk between C/EBPα and PR in breast cancer cells. Upper left panel: In around 1,000 DNA “assisted” enhancer regions, C/EBPα assists PR binding by maintaining the chromatin in an open conformation. To fulfill this function, C/EBPα anchors the acetyltransferase P300 and the topoisomerase 2α (Topo2α). C/EBPα interacts also with components of the Mediator and cohesin complexes to maintain a 3D chromatin structure that assures the development of a gene expression program that enables controlled cell growth in the presence of hormone. Lower left panel: PR and its associated co‐regulators are recruited to assisted regions upon hormone exposure. The structural proteins RAD21 and CTCF are also increased along with enhancer–promoter contacts. Right panels: In the absence of C/EBPα, “assisted” enhancer regions are in a “closed” conformation and the 3D chromatin structure is lost along with the presence of architectural proteins. PR and coactivators cannot longer be properly recruited to these regions, and the gene program associated with controlled cell growth is compromised.
Figure EV5. C/EBPα depletion reduced the presence of Topo2α in “assisted” regions independently of hormone.

- ChIP‐seq experiments of Topo2α were performed in cells expressing either shControl or shC/EBPα in the presence and in the absence of hormone (lower and upper panels, respectively). Reads profile for “assisted”, “constitutive”, “induced shared”, and “random” regions are shown.
- T47D and T47DindC/EBPα cells treated with 10 nM R5020 or doxycycline as indicated were lysed and the increase in PARP cleavage, an indicator of apoptosis was assayed by Western blot. This blot was previously used as loading control in EV1A, right panel. Right panel: T47D cells were incubated with staurosporine for 48 h. The results presented are representative of two independent experiments.
Source data are available online for this figure.
In addition, we found that P300 is enriched in “assisted” regions prior to hormone exposure (Fig EV4B, left panel) and follows a similar trend as Topo2α upon C/EBPα depletion (Fig 7B). As P300 acetylates H3K27 and H3K18, it could contribute to the opening of the “assisted” regions along with Topo2α (Ju et al, 2006; Miller et al, 2017).
Discussion
Though de‐regulation of C/EBPα has been reported in several types of cancer (Tomizawa et al, 2003; Roepman et al, 2005; Tseng et al, 2009; Lourenco & Coffer, 2017), the role of C/EBPα in breast cancer and its regulation by estrogens and progesterone are largely unknown. It is known that exposure of T47D breast cancer cells to progestins induces entry in the cycle and one round of cell division, followed by cell cycle arrest (Groshong et al, 1997; Musgrove et al, 1998; Skildum et al, 2005). In the context of these two phases, our results revealed a complex crosstalk between PR and C/EBPα in controlling hormone‐dependent cell proliferation. Progestins induce C/EBPα expression in breast cancer cells, and the increasing C/EBPα levels turned out to be critical for cell cycle arrest and for the expression of progestin regulated genes. Around one‐third (33.5%) of down‐regulated and almost one‐fifth (20%) of the up‐regulated genes were affected by C/EBPα knockdown, showing that C/EBPα is a main player during hormonal gene regulation. The genes dependent on C/EBPα are involved in cellular metabolic process, RNA processing, cell cycle, mitosis, and DNA damage repair.
After 20–48 h of incubation, progestins exert an inhibitory effect on cell proliferation (Fig 1D); gene analysis indicated that this effect would be mediated either by the inhibition of the pro‐proliferative genes involved in cell cycle (Appendix Fig S1), or by the activation of anti‐proliferative genes as DUSP1, resulting in an arrest in the cell cycle. Notably, both gene expression programs depend on C/EBPα.
Breast cancer cells depleted of C/EBPα proliferate faster, suggesting an anti‐proliferative role for C/EBPα in breast cancer cells. Although C/EBPα is important for tamoxifen‐induced apoptosis (Cheng et al, 2007), in T47D cells exposed to progestins, we see growth arrest but no indication of apoptosis, as detected by cleavage of PARP1 (Fig EV5A). In addition, high levels of C/EBPα compromise the ability of progestins to induce the dedifferentiated cell phenotype in 2D cultures and slow down the growth of cells injected in mouse mammary ducts (Fig 2A and B), supporting a role of C/EBPα as tumor modulator. Thus, C/EBPα is induced by progestins and contributes to limiting the proliferative activity of breast cancer cells by modulating the hormonal regulation of genes involved in cell growth.
Compared to progestins, estrogens are strong inducers of cell proliferation in breast cancer cells and induce multiple rounds of cell division (Groshong et al, 1997; Musgrove et al, 1998; Skildum et al, 2005). In contrast to what is observed with progestins, estrogens decreased C/EBPα expression, in agreement with a negative role in breast cancer cell proliferation. By ChIP‐seq experiments, we found an estrogen receptor binding site localized at 1.2 kb from the TTS of the CEBPA gene, but whether this region is important for estrogen mediated inhibition of C/EBPα is an interesting question that requires further experiments as CRISPR/Cas9‐mediated genome editing.
We used ChIP‐seq to explore the molecular mechanism of the cooperation between the two transcription factors at the level of gene expression, and found various forms of cooperative binding to gene regulatory sequences (Bulger & Groudine, 2011). Although the C/EBPα levels in untreated breast cancer cells are low (see Fig 1), C/EBPα binding is concentrated in thousands of genes and assists binding of ligand‐activated PR by keeping its binding sites in an accessible chromatin conformation prior to hormone exposure (Fig 7F, top panel). These regions present both the consensus DNA binding motif of C/EBPα and also a high chromatin accessibility, which makes them attractive regions to the limited amount of C/EBPα available.
In fact, gene ontology analysis revealed that “assisted” regions might play a role in regulating genes associated with breast cancer (Appendix Fig S4).
Our results point to a cooperation between PR and C/EBPα to enhance a gene expression program that enables controlled cell growth in the presence of hormone. This may be a more general function of C/EBP family proteins, as glucocorticoid receptor (GR)‐binding sites are preoccupied by C/EBPβ in liver, and disruption of C/EBPβ binding results in decreased chromatin accessibility and reduced GR recruitment (Grontved et al, 2013). In the progestin‐induced gene DUSP1, where two “assisted” regions are found at −1.3 and −5 kb from the TSS, the function of C/EBPα is to maintain a preformed loop with the promoter region. In fact, C/EBPα knockdown decreases the contacts between the “assisted” regions and the DUSP1 gene promoter both in the presence and in the absence of hormone (Fig 7F).
Our Hi‐C data showed that “assisted” regions exhibit more long distance interactions with hormone‐regulated promoters compared with the other regions. This suggests that C/EBPα could be involved in maintaining the genomic loops that ensure the regulated expression of PR target genes associated with “assisted” regions.
We could infer from our data that part of the function of C/EBPα in breast cancer is to provide the adequate 3D conformation to the genome that guarantees the hormonal effect. For this, C/EBPα stabilizes binding of structural proteins such as RAD21, YY1, and Mediator in the appropriate arrangement, allowing chromatin looping between enhancers and promoters (Fig 6A–C). Therefore, C/EBPα would participate promoting a nuclear architecture that guarantees the correct hormonal‐mediated gene regulation.
How does C/EBPα maintain “assisted” regions accessible for PR binding? We found that C/EBPα achieves this function by interacting with lysine acetyltransferase P300 and with DNA Topoisomerase 2α, and we cannot exclude a role for the chromatin remodeling complex SWI/SNF. Although C/EBPα depletion did not significantly affect the presence of SWI/SNF in several “assisted” regions (data not shown), genome‐wide analysis showed that BRG1 is enriched in these regions and interacts with PR and C/EBPα. Acetylation catalyzed by P300 could stabilize SWI/SNF binding through the bromo domain of the ATPase BRG1. In fact, SWI/SNF complexes interact directly with Topo2α and both remodelers could synergize to keep the target chromatin in an open conformation, as previously reported for the resolution of facultative heterochromatin (Miller et al, 2017). However, maintaining the “assisted” regions accessible likely requires additional factors. A good candidate is the pioneer factor FOXA1, which interacts with C/EBPα in breast cancer cells, as both are found in “assisted” regions. FOXA1 and C/EBPα could cooperate at the nucleosome level to facilitate PR binding to the “assisted” regions. FOXA1 by promoting the selective displacement of histone H1 from “assisted” regions (Fig 4A), as previously reported in liver (Iwafuchi‐Doi et al, 2016), would enable loading of C/EBPα ensuring chromatin opening and PR binding in the presence of hormone.
It has been reported that topoisomerase 2β (Topo2 β) participates in the activation of the pS2 gene in the presence of estrogens (Ju et al, 2006). One of the mechanisms proposed for Topo2β regulation of gene transcription is by favoring the exchange of histone H1 by HMBG1/2 (Ju et al, 2006). This is compatible with the finding that “assisted” regions are enriched in Topo2α at T0 and depleted of H1 (Figs 4A, 7B and EV5A). In fact, either depletion of C/EBPα or the inhibition of the topoisomerase closed the “assisted” regions as would be expected in the presence of H1.
In this study, we provide a global picture of the cooperation network between PR and C/EBPα in controlling chromatin remodeling and gene expression, but many questions remain to be investigated. In particular, how post‐translational modifications activated by hormones can affect this network is an intriguing one. In the liver, phosphorylation of C/EBPα residue Ser193 has been shown to be crucial for its growth arrest capacity as mutational studies demonstrate that this modification is critical for the association with cyclin‐dependent kinases 2 and 4 (CDK2/CDK4) and E2F–Rb–Brm complexes (Wang et al, 2001, 2006). In contrast, homozygous CebpaΔPHR/ΔPHR (ΔPHR) mice, carrying a modified cebpa allele lacking amino acids 180–194, displayed no apparent adverse phenotypes and failed to show any cell cycle or developmental differences between the ΔPHR mice and their control littermates (Porse et al, 2006). Thus, further experiments performed in well‐defined systems will be necessary to reconcile these conflicting results.
The phosphatase PP2A and the kinase GSK3 were also reported to regulate C/EBPα phosphorylation (Wang et al, 2004; Datta et al, 2007). C/EBPα can also be directly phosphorylated by ERK1/2 on S21, leading to a conformational change that affects the ability of C/EBPα to induce myeloid differentiation (Ross et al, 2004; Radomska et al, 2006). Since progestins rapidly and transiently induced ERK1/2 activity, it is possible that the activated kinases phosphorylate C/EBPα and keep it inactive, enabling the initial activation of the cell cycle in response to hormone. A few hours later, when C/EBPα accumulates and ERK1/2 is no longer activated, dephosphorylation could contribute to the anti‐proliferative action of C/EBPα. We have preliminary evidence for C/EBPα S21 phosphorylation and its sensitivity to ERK1/2 inhibitors, but further studies with appropriate mutants are required to clarify the significance of this signaling pathway.
Acetylation catalyzed by lysine acetyltransferases is another important post‐translational modification that plays a pivotal role in numerous cellular processes. GCN5 can interact with and acetylate C/EBPα on at least lysine K298 and K302 in the basic DNA binding domain (DBD), leading to impaired DNA binding activity and granulocytic differentiation capacity (Bararia et al, 2016). Interestingly, in the presence of hormone PR associated with C/EBPα also interacts with several acetyltransferases and HDACs (Fig EV4C and Vicent et al, 2013, 2009) that could regulate its activity.
The female sex steroids estrogen and progesterone are essential for the proliferation of mammary epithelial cell, and the question arises to what extend the role of C/EBPα we found in cancer cells is relevant for the hormonal control of normal mammary gland development and function. Targeted deletion of the C/EBPβ isoforms results in severe inhibition of lobuloalveolar development, a block of functional differentiation, and subtle changes in ductal morphogenesis (Grimm & Rosen, 2003). The C/EBPα mRNA is expressed throughout murine mammary gland development, but C/EBPα is not essential during this process. C/EBPα mRNA and protein are high during the early stages of lactation, expressed at lower levels during the late stages of lactation/early involution, and only the mRNA increases again during late involution (Raught et al, 1995; Sabatakos et al, 1998). Thus, the development and function of the mammary gland in mice depends more on C/EBPβ. However, the fact that the levels of C/EBPα can change throughout the development, that a small amount of C/EBPα is sufficient to show its effect in the “assisted” regions, and the possibility that development of the human and the mouse mammary gland exhibit differences in C/EBPα expression and distribution, leaves open a possible role of C/EBPα in human normal breast.
The molecular mechanism by which C/EBPα acts in the crosstalk with PR is mainly associated with its effect on transcriptional activation since it is involved in the decompaction of chromatin, the increase of promoter–enhancer contacts, and the recruitment of chromatin remodeling machineries (Figs 3, 5 and 6). However, we must bear in mind that C/EBPα is also directly or indirectly involved in the repression of a significant number of hormone‐regulated genes (Fig 1C). In fact, several mechanisms of gene repression involving chromatin looping such as in the Dlx5‐Dlx6 locus in mice and the expression of the Kit cytokine receptor during terminal erythroid differentiation have been reported to involve C/EBPα (Horike et al, 2005; Jing et al, 2008).
In conclusion, we show that PR and C/EBPα cooperate to enhance a gene expression program that enables controlled cell growth and provides a possible explanation for the tumor “suppressor” role of C/EBPα in hormone‐dependent breast cancer.
Materials and Methods
Cell culture and hormone treatments
T47D breast cancer cells were routinely grown in RPMI 1640 medium supplemented with 10% FBS, 2 mM l‐glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. For the experiments, cells were plated in RPMI medium without phenol red supplemented with 10% dextran‐coated charcoal treated FBS (DCC/FBS), and 48 h later, medium was replaced by fresh medium without serum. After 24 h in serum‐free conditions, cells were incubated with R5020 (10 nM) for different times at 37°C. For the experiments with a short arrest, cells were incubated for 4 h with 10% charcolized serum before hormone treatment.
T47D shControl, T47D C/EBPα knockdown (shC/EBPα), inducible C/EBPα (T47DindC/EBPα), and inducible C/EBPα MCF‐7 (MCF7indC/EBPα) cell lines were established from T47D and MCF‐7 cells (Sancho et al, 2008).
Chromatin immunoprecipitation in cultured cells
Chromatin immunoprecipitation (ChIP) assays were performed as described (Strutt & Paro, 1999) using anti‐PR (Santa Cruz, H190), anti‐C/EBPα (Cell Signaling, 8178S), anti‐BRG1 [EPNCIR111A] (Abcam, ab110641), anti‐LSD1 (Abcam, ab17721), anti‐Topo2α (Santa Cruz, sc‐3659), anti‐P300 (Santa Cruz, sc‐584x and sc‐585x), and anti‐SRC3 (Santa Cruz, sc‐9119x). Quantification of ChIP was performed by real‐time PCR using Roche Lightcycler (Roche). The fold enrichment of target sequence in the immunoprecipitated (IP) compared to input (Ref) fractions was calculated using the comparative Ct (the number of cycles required to reach a threshold concentration) method with the equation 2Ct(IP)‐Ct(Ref). Each of these values were corrected by the human β‐globin gene and referred as relative abundance over time zero. Primer sequences are available on request.
RNA interference experiments
All siRNAs were transfected into the T47D‐MTVL cells using Lipofectamine 2000 (Invitrogen). After 48 h, the medium was replaced by fresh medium without serum. After 16 h in serum‐free conditions, cells were incubated with R5020 (10 nM) or vehicle (ethanol) for different times at 37°C. The down‐regulation of C/EBPα expression was determined by Western blotting. Primer sequences are available on request. C/EBPα siRNAs were purchased from Dharmacon (Thermo Scientific).
RNA extraction and RT–PCR
Total RNA was prepared and cDNA generated as previously described (Vicent et al, 2006). Quantification of LUC and GAPDH gene products was performed by real‐time PCR. Each value calculated using the standard curve method was corrected by the human GAPDH and expressed as relative RNA abundance over time zero. Primer sequences for DUSP1, C/EBPα, and PGR are available on request.
Co‐immunoprecipitation assay
T47D cells as well as shControl and shC/EBPα cells were lysed and cell extracts (1 mg protein) were incubated overnight with protein A/G agarose beads previously coupled with 3 μg of the corresponding antibodies or an unspecific control antibody. The immunoprecipitated proteins (IPs) were eluted by boiling in SDS‐sample buffer. Inputs and IPs were analyzed by Western blot using PR‐, C/EBPα, Topo2α, and BRG1‐specific antibodies.
Cell proliferation
T47D‐shControl, T47D‐C/EBPα knockdown (shC/EBPα), inducible C/EBPα (T47DindC/EBPα), and inducible C/EBPα MCF‐7 (MCF7indC/EBPα) cell lines were plated in triplicate 24‐well plates at a seeding density of 100,000 cells per well, using medium described above in the presence or absence of doxycycline when induction of C/EBPα was required. Cells were counted every 24 h and allowed to grow for up to 4 days.
Flow cytometry
T47D cells were plated into duplicate wells of six‐well plastic dishes and preincubated as described. After 24 h, 10 nM R5020 or ethanol was added. Cells were harvested at the start of treatment (control, zero time) and every 6 or 12 h after hormone addition. The cell suspension was pelleted, stained with propidium iodide, and treated with ribonuclease (RNase). Samples were cooled to 4°C, and 10,000 cells were analyzed on a BD FACSCanto analyzer flow cytometer.
In Fig 2A, T47DindC/EBPα cells untreated or treated with doxycycline were labeled with antibodies CD44‐APC, CD24‐PE (BD Biosciences, Franklin Lakes, NJ, USA) at a concentration of 10 × 106 cells per ml under optimized conditions, and were subjected to fluorescence‐activated cell analysis in a LSRII (Becton and Dickinson, NJ, USA). For cell sorting, the FACSAria II Sorp (Becton and Dickinson, NJ, USA) was used.
Intraductal growth of breast cancer cell lines
T47DindC/EBPα were lentivirally infected to express GFP and Luciferase. 100,000 cells were injected through the test of thoracic and inguinal gland of 10 adult NSG mice (5 per group), creating a MIND (Mouse Intra‐Ductal) xenograft (Sflomos et al, 2016). Seven days after injection, CEBPα expression was induced with doxycycline in the drinking water (2 mg/ml) and maintained throughout the experiment. The number of glands injected was 19 for CEBPα and 17 for the control. Tumor growth was assessed by bioluminescence (IVIS). Five mice were used per condition resulting in 19 glands for CEBPα and 17 glands for the Control.
Animal husbandry
Animals are housed in IVC cages—Green line—from Tecniplast®, type II long in polysulfone, with bottles in polysulfone as well. Cages are in over‐pressure compared to the pressure in the housing room. Housing rooms are in over‐pressure compared to the pressure outside of the barrier unit. Bedding: Aspen Tapvei® (little squares about 4 × 4 × 1 mm). Water: acidified (pH between 2.5 and 3) on resin column (Prominent® CH system). Diet: ref 3242, irradiated, from Provimi‐Kliba®. Enrichment: nesting material (tissues) and cardboard tunnel. Housing room temperature: 22°C ± 2°C Housing room humidity: 55% ± 10% Light cycle: 12 h light start from 7 am, 12 h dark start from 7 pm.
All mouse experiments were performed in accordance with protocols approved by the Service de la Consommation et des Affaires Vétérinaires of Canton de Vaud (VD1865.4).
DNAse I digestion analysis
Chromatin samples obtained as described before from two biological replicas were subjected to DNAse I digestion. Briefly, 2 μg of chromatin was treated with 0.15 and 0.4 units of DNAse I (Roche) for 3 min at 37°C in 1× DNAse incubation buffer. Control samples were incubated in the absence of DNAse I. Reactions were terminated by addition of 40 mM EDTA final concentration, and the cross‐linking was reversed by incubating the samples at 65°C. After 6 h, Proteinase K (40 μg/ml final concentration) was added to each reaction and incubated overnight at 37°C. After careful phenol–chloroform extractions, the DNA was quantified and used as template for real‐time PCR products using specific primers.
Chromatin conformation capture
Chromatin conformation capture (3C) assays were carried out essentially as follows. A total of 107 cells were cross‐linked using 1% (vol/vol) formaldehyde for 10 min at room temperature (RT). The cross‐linking reaction was quenched with 0.125 M glycine at room temperature for 5 min followed by 15 min on ice, and then, the cells were washed twice with cold PBS. Cells were lysed in 0.5 ml of cell lysis buffer containing protease inhibitors on ice for 30 min. The nuclei were resuspended in NEBuffer 2 (New England Biolabs) containing 0.5% SDS and incubated in a thermomixer 10 min at 65°C. Next, 1.8% (vol/vol) Triton X‐100 was added to sequester the SDS and the samples were incubated for an additional 15 min at 37°C. The cross‐linked DNA was digested with 400 units of Mbo I (New England Biolabs) at 37°C overnight. Mbo I was heat inactivated at 65°C for 20 min. For ligation of DNA ends, T4 DNA ligase was added and the samples were incubated for 4 h at 16°C. The ligated material was resuspended in NEBuffer 2 followed by RNAse treatment. Cross‐links were reversed by incubating with Proteinase K (10 mg/ml) at 65°C overnight. Finally, the DNA was phenol/chloroform‐extracted and concentrated by ethanol precipitation. A total of 20 ng of DNA was analyzed by PCR (primers are available upon request).
RNA‐seq
RNA was extracted from T47D‐MTVL cells treated or not for 6 h with R5020 and submitted to massive sequencing using the Solexa Genome Analyzer. The protocol followed to analyze the RNA‐seq data can be found in the Appendix Supplementary Methods section.
ChIP‐seq
ChIP‐DNA was purified and subjected to deep sequencing using the Solexa Genome Analyzer (Illumina, San Diego, CA). The protocol followed to analyze the ChIP‐seq data can be found in the Appendix Supplementary Methods section.
ATAC‐seq
ATAC experiments were performed as described (Buenrostro et al, 2013).
Extended Bioinformatics methods can be found in the Appendix Supplementary Methods section.
Hi‐C experiments
Hi‐C libraries were generated from T47D cells treated or not with R5020 for 60 min according the previously published Hi‐C protocol with minor adaptations (Lieberman‐Aiden et al, 2009). Hi‐C libraries were generated independently in both conditions using HindIII and NcoI restriction enzymes. Hi‐C libraries were controlled for quality and sequenced on an Illumina Hiseq2000 sequencer. Illumina Hi‐seq paired‐end reads were processed by aligning to the reference human genome (GRCh37/hg19) using BWA.
ChIP‐exo experiments
ChIP‐exo experiments were performed in untreated T47D cells using the ChIP‐exo kit from Active Motif and anti‐PR (Santa Cruz, H190). PGR ChIP‐exo‐identified peaks were subject to Exo‐Profiler analysis by using transfect matrices of MEME‐ChIP suit SpaMo tool (PMID: 21486936) identified motifs of FOXA1 (AC: MA0148.3) and CEBP/a (AC: MA0102.3). Only regions containing one unique motif of either FOXA1 and/or CEBPa were considered for the analysis of the distance to PGR ChIP‐exo sites.
Mass spec analysis
T47DindC/EBPα cells were lysed, and cell extracts (2.5 mg) were incubated overnight with protein A magnetic beads (Invitrogen) previously coupled with 30 μg of the C/EBPα antibody or an unspecific control antibody. The immunoprecipitated proteins were digested with trypsin and LysC, and each tryptic digest sample was subjected to LC‐MS/MS analysis using a 1‐h gradient in the LTQ‐Orbitrap Velos Pro with a CID method. As a quality control, BSA controls were run between each of your samples to avoid carryover and assess the instrument performance. The samples were searched against SwissProt Human database, using the search algorithm Mascot v2.5.1 (http://www.matrixscience.com/). Peptides have been filtered based on FDR, and only peptides showing an FDR lower than 5% have been retained.
Statistical analysis
Cell proliferation, ChIPs, DNAse accessibility, Chromatin conformation capture (3C), gene activity, and FACS assays were performed at least three times in duplicates. The values are given as mean ± SD, and P‐values were obtained by Student's t‐test and were calculated relative to control conditions. For the experiments in Figs 1D and E, and 2A, the P‐values were obtained by ANOVA followed by Tukey test.
Author contributions
ASN conducted the majority of the experiments with participation from RZ, VS and FLD; RF, JC‐C, and JQ performed the bioinformatics analysis; AL generated the inducible T47D cell line T47DindC/EBPα; GPV conceived the original ideas, designed the experiments, and wrote the paper with input from MB and CB; GPV and MB drafted and revised the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We wish to thank Thomas Graf and Luciano Di Croce for advice on the manuscript. The experimental work was supported by grants from the Departament d'Innovació Universitat i Empresa (DIUiE), and the Spanish Ministry of Economy and Competitiveness (SAF2016‐75006P), “Centro de Excelencia Severo Ochoa 2013‐2017”, SEV‐2012‐0208 and ERC Synergy Grant “4DGenome” nr: 609989.
The EMBO Journal (2019) 38: e101426
Contributor Information
Miguel Beato, Email: miguel.beato@crg.eu.
Guillermo P Vicent, Email: guillermo.vicent@crg.eu.
Data availability
The raw sequencing data from this study (ChIP‐seq and RNA‐seq) were submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) with the following accession number: GSE132649.
References
- Bararia D, Kwok HS, Welner RS, Numata A, Sarosi MB, Yang H, Wee S, Tschuri S, Ray D, Weigert O et al (2016) Acetylation of C/EBPalpha inhibits its granulopoietic function. Nat Commun 7: 10968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nat Methods 10: 1213–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger M, Groudine M (2011) Functional and mechanistic diversity of distal transcription enhancers. Cell 144: 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Umek RM, McKnight SL (1991) Regulated expression of three C/EBP isoforms during adipose conversion of 3T3‐L1 cells. Genes Dev 5: 1538–1552 [DOI] [PubMed] [Google Scholar]
- Chen YW, Huang SC, Lin‐Shiau SY, Lin JK (2005) Bowman‐Birk inhibitor abates proteasome function and suppresses the proliferation of MCF7 breast cancer cells through accumulation of MAP kinase phosphatase‐1. Carcinogenesis 26: 1296–1306 [DOI] [PubMed] [Google Scholar]
- Cheng J, Yu DV, Zhou JH, Shapiro DJ (2007) Tamoxifen induction of CCAAT enhancer‐binding protein alpha is required for tamoxifen‐induced apoptosis. J Biol Chem 282: 30535–30543 [DOI] [PubMed] [Google Scholar]
- Cittelly DM, Finlay‐Schultz J, Howe EN, Spoelstra NS, Axlund SD, Hendricks P, Jacobsen BM, Sartorius CA, Richer JK (2013) Progestin suppression of miR‐29 potentiates dedifferentiation of breast cancer cells via KLF4. Oncogene 32: 2555–2564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y et al (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486: 346–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta J, Majumder S, Kutay H, Motiwala T, Frankel W, Costa R, Cha HC, MacDougald OA, Jacob ST, Ghoshal K (2007) Metallothionein expression is suppressed in primary human hepatocellular carcinomas and is mediated through inactivation of CCAAT/enhancer binding protein alpha by phosphatidylinositol 3‐kinase signaling cascade. Cancer Res 67: 2736–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stefano B, Sardina JL, van Oevelen C, Collombet S, Kallin EM, Vicent GP, Lu J, Thieffry D, Beato M, Graf T (2014) C/EBPalpha poises B cells for rapid reprogramming into induced pluripotent stem cells. Nature 506: 235–239 [DOI] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson RL, Hemati N, Ross SE, MacDougald OA (2001) p300 coactivates the adipogenic transcription factor CCAAT/enhancer‐binding protein alpha. J Biol Chem 276: 16348–16355 [DOI] [PubMed] [Google Scholar]
- Faure AJ, Schmidt D, Watt S, Schwalie PC, Wilson MD, Xu H, Ramsay RG, Odom DT, Flicek P (2012) Cohesin regulates tissue‐specific expression by stabilizing highly occupied cis‐regulatory modules. Genome Res 22: 2163–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari R, Su T, Li B, Bonora G, Oberai A, Chan Y, Sasidharan R, Berk AJ, Pellegrini M, Kurdistani SK (2012) Reorganization of the host epigenome by a viral oncogene. Genome Res 22: 1212–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flodby P, Barlow C, Kylefjord H, Ahrlund‐Richter L, Xanthopoulos KG (1996) Increased hepatic cell proliferation and lung abnormalities in mice deficient in CCAAT/enhancer binding protein alpha. J Biol Chem 271: 24753–24760 [DOI] [PubMed] [Google Scholar]
- Freytag SO, Paielli DL, Gilbert JD (1994) Ectopic expression of the CCAAT/enhancer‐binding protein alpha promotes the adipogenic program in a variety of mouse fibroblastic cells. Genes Dev 8: 1654–1663 [DOI] [PubMed] [Google Scholar]
- Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, Rauscher FJ III (1996) KAP‐1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev 10: 2067–2078 [DOI] [PubMed] [Google Scholar]
- Gery S, Tanosaki S, Bose S, Bose N, Vadgama J, Koeffler HP (2005) Down‐regulation and growth inhibitory role of C/EBPalpha in breast cancer. Clin Cancer Res 11: 3184–3190 [DOI] [PubMed] [Google Scholar]
- Grimm SL, Rosen JM (2003) The role of C/EBPbeta in mammary gland development and breast cancer. J Mammary Gland Biol Neoplasia 8: 191–204 [DOI] [PubMed] [Google Scholar]
- Grontved L, John S, Baek S, Liu Y, Buckley JR, Vinson C, Aguilera G, Hager GL (2013) C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J 32: 1568–1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groshong SD, Owen GI, Grimison B, Schauer IE, Todd MC, Langan TA, Sclafani RA, Lange CA, Horwitz KB (1997) Biphasic regulation of breast cancer cell growth by progesterone: role of the cyclin‐dependent kinase inhibitors, p21 and p27(Kip1). Mol Endocrinol 11: 1593–1607 [DOI] [PubMed] [Google Scholar]
- Hergeth SP, Schneider R (2015) The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep 16: 1439–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horike S, Cai S, Miyano M, Cheng JF, Kohwi‐Shigematsu T (2005) Loss of silent‐chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet 37: 31–40 [DOI] [PubMed] [Google Scholar]
- Horwitz KB, Tung L, Takimoto GS (1995) Novel mechanisms of antiprogestin action. J Steroid Biochem Mol Biol 53: 9–17 [DOI] [PubMed] [Google Scholar]
- Iwafuchi‐Doi M, Donahue G, Kakumanu A, Watts JA, Mahony S, Pugh BF, Lee D, Kaestner KH, Zaret KS (2016) The pioneer transcription factor FoxA maintains an accessible nucleosome configuration at enhancers for tissue‐specific gene activation. Mol Cell 62: 79–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing H, Vakoc CR, Ying L, Mandat S, Wang H, Zheng X, Blobel GA (2008) Exchange of GATA factors mediates transitions in looped chromatin organization at a developmentally regulated gene locus. Mol Cell 29: 232–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson PF (2005) Molecular stop signs: regulation of cell‐cycle arrest by C/EBP transcription factors. J Cell Sci 118: 2545–2555 [DOI] [PubMed] [Google Scholar]
- Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL, Stingl J, Waterhouse PD, Khokha R (2010) Progesterone induces adult mammary stem cell expansion. Nature 465: 803–807 [DOI] [PubMed] [Google Scholar]
- Ju BG, Lunyak VV, Perissi V, Garcia‐Bassets I, Rose DW, Glass CK, Rosenfeld MG (2006) A topoisomerase IIbeta‐mediated dsDNA break required for regulated transcription. Science 312: 1798–1802 [DOI] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS et al (2010) Mediator and cohesin connect gene expression and chromatin architecture. Nature 467: 430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy G, Orphanides G, Lane WS, Reinberg D (1998) Requirement of RSF and FACT for transcription of chromatin templates in vitro . Science 282: 1900–1904 [DOI] [PubMed] [Google Scholar]
- Lieberman‐Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO et al (2009) Comprehensive mapping of long‐range interactions reveals folding principles of the human genome. Science 326: 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco AR, Coffer PJ (2017) A tumor suppressor role for C/EBPalpha in solid tumors: more than fat and blood. Oncogene 36: 5221–5230 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu GD, Leung CH, Yan B, Tan CM, Low SY, Aung MO, Salto‐Tellez M, Lim SG, Hooi SC (2010) C/EBPalpha is up‐regulated in a subset of hepatocellular carcinomas and plays a role in cell growth and proliferation. Gastroenterology 139, 632–643 [DOI] [PubMed] [Google Scholar]
- Malik S, Roeder RG (2010) The metazoan Mediator co‐activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet 11: 761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G (2010) GREAT improves functional interpretation of cis‐regulatory regions. Nat Biotechnol 28: 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EL, Hargreaves DC, Kadoch C, Chang CY, Calarco JP, Hodges C, Buenrostro JD, Cui K, Greenleaf WJ, Zhao K et al (2017) TOP2 synergizes with BAF chromatin remodeling for both resolution and formation of facultative heterochromatin. Nat Struct Mol Biol 24: 344–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming J, Zhou Y, Du J, Fan S, Pan B, Wang Y, Fan L, Jiang J (2015) miR‐381 suppresses C/EBPα‐dependent Cx43 expression in breast cancer cells. Biosci Rep 35: e00266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, Chang HY (2016) HiChIP: efficient and sensitive analysis of protein‐directed genome architecture. Nat Methods 13: 919–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musgrove EA, Lee CS, Buckley MF, Sutherland RL (1994) Cyclin D1 induction in breast cancer cells shortens G1 and is sufficient for cells arrested in G1 to complete the cell cycle. Proc Natl Acad Sci USA 91: 8022–8026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musgrove EA, Swarbrick A, Lee CS, Cornish AL, Sutherland RL (1998) Mechanisms of cyclin‐dependent kinase inactivation by progestins. Mol Cell Biol 18: 1812–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacht AS, Pohl A, Zaurin R, Soronellas D, Quilez J, Sharma P, Wright RH, Beato M, Vicent GP (2016) Hormone‐induced repression of genes requires BRG1‐mediated H1.2 deposition at target promoters. EMBO J 35: 1822–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J et al (2012) Spatial partitioning of the regulatory landscape of the X‐inactivation centre. Nature 485: 381–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CT, Corces VG (2014) CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 15: 234–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides G, LeRoy G, Chang CH, Luse DS, Reinberg D (1998) FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 92: 105–116 [DOI] [PubMed] [Google Scholar]
- Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB (1998) Progesterone regulates transcription of the p21(WAF1) cyclin‐ dependent kinase inhibitor gene through Sp1 and CBP/p300. J Biol Chem 273: 10696–10701 [DOI] [PubMed] [Google Scholar]
- Pedersen TA, Kowenz‐Leutz E, Leutz A, Nerlov C (2001) Cooperation between C/EBPalpha TBP/TFIIB and SWI/SNF recruiting domains is required for adipocyte differentiation. Genes Dev 15: 3208–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips‐Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y et al (2013) Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153: 1281–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas‐Silva MD, Weinberg RA (1997) The restriction point and control of cell proliferation. Curr Opin Cell Biol 9: 768–772 [DOI] [PubMed] [Google Scholar]
- Planas‐Silva MD, Donaher JL, Weinberg RA (1999) Functional activity of ectopically expressed estrogen receptor is not sufficient for estrogen‐mediated cyclin D1 expression. Cancer Res 59: 4788–4792 [PubMed] [Google Scholar]
- Porse BT, Pedersen TA, Hasemann MS, Schuster MB, Kirstetter P, Luedde T, Damgaard I, Kurz E, Schjerling CK, Nerlov C (2006) The proline‐histidine‐rich CDK2/CDK4 interaction region of C/EBPalpha is dispensable for C/EBPalpha‐mediated growth regulation in vivo . Mol Cell Biol 26: 1028–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prall OW, Sarcevic B, Musgrove EA, Watts CK, Sutherland RL (1997) Estrogen‐induced activation of Cdk4 and Cdk2 during G1‐S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin‐dependent kinase inhibitor association with cyclin E‐Cdk2. J Biol Chem 272: 10882–10894 [DOI] [PubMed] [Google Scholar]
- Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, Tenen DG (1998) CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol 18: 4301–4314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radomska HS, Basseres DS, Zheng R, Zhang P, Dayaram T, Yamamoto Y, Sternberg DW, Lokker N, Giese NA, Bohlander SK et al (2006) Block of C/EBP alpha function by phosphorylation in acute myeloid leukemia with FLT3 activating mutations. J Exp Med 203: 371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raught B, Liao WS, Rosen JM (1995) Developmentally and hormonally regulated CCAAT/enhancer‐binding protein isoforms influence beta‐casein gene expression. Mol Endocrinol 9: 1223–1232 [DOI] [PubMed] [Google Scholar]
- Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, Vilo J (2016) g:Profiler‐a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res 44: W83–W89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF (2011) Comprehensive genome‐wide protein‐DNA interactions detected at single‐nucleotide resolution. Cell 147: 1408–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM (2004) ONCOMINE: a cancer microarray database and integrated data‐mining platform. Neoplasia 6: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepman P, Wessels LF, Kettelarij N, Kemmeren P, Miles AJ, Lijnzaad P, Tilanus MG, Koole R, Hordijk GJ, van der Vliet PC et al (2005) An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nat Genet 37: 182–186 [DOI] [PubMed] [Google Scholar]
- Ross SE, Radomska HS, Wu B, Zhang P, Winnay JN, Bajnok L, Wright WS, Schaufele F, Tenen DG, MacDougald OA (2004) Phosphorylation of C/EBPalpha inhibits granulopoiesis. Mol Cell Biol 24: 675–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp A, Brauner B, Dunger‐Kaltenbach I, Frishman G, Montrone C, Stransky M, Waegele B, Schmidt T, Doudieu ON, Stumpflen V et al (2008) CORUM: the comprehensive resource of mammalian protein complexes. Nucleic Acids Res 36: D646–D650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatakos G, Davies GE, Grosse M, Cryer A, Ramji DP (1998) Expression of the genes encoding CCAAT‐enhancer binding protein isoforms in the mouse mammary gland during lactation and involution. Biochem J 334(Pt 1): 205–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho M, Diani E, Beato M, Jordan A (2008) Depletion of human histone H1 variants uncovers specific roles in gene expression and cell growth. PLoS Genet 4: e1000227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster MB, Porse BT (2006) C/EBPalpha: a tumour suppressor in multiple tissues? Biochim Biophys Acta 1766: 88–103 [DOI] [PubMed] [Google Scholar]
- Screpanti I, Romani L, Musiani P, Modesti A, Fattori E, Lazzaro D, Sellitto C, Scarpa S, Bellavia D, Lattanzio G et al (1995) Lymphoproliferative disorder and imbalanced T‐helper response in C/EBP beta‐deficient mice. EMBO J 14: 1932–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seagroves TN, Krnacik S, Raught B, Gay J, Burgess‐Beusse B, Darlington GJ, Rosen JM (1998) C/EBPbeta, but not C/EBPalpha, is essential for ductal morphogenesis, lobuloalveolar proliferation, and functional differentiation in the mouse mammary gland. Genes Dev 12: 1917–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sflomos G, Dormoy V, Metsalu T, Jeitziner R, Battista L, Scabia V, Raffoul W, Delaloye JF, Treboux A, Fiche M et al (2016) A preclinical model for ERalpha‐positive breast cancer points to the epithelial microenvironment as determinant of luminal phenotype and hormone response. Cancer Cell 29: 407–422 [DOI] [PubMed] [Google Scholar]
- Skildum A, Faivre E, Lange CA (2005) Progesterone receptors induce cell cycle progression via activation of mitogen‐activated protein kinases. Mol Endocrinol 19: 327–339 [DOI] [PubMed] [Google Scholar]
- Strutt H, Paro R (1999) Mapping DNA target sites of chromatin proteins in vivo by formaldehyde crosslinking. Methods Mol Biol 119: 455–467 [DOI] [PubMed] [Google Scholar]
- Sun H, Charles CH, Lau LF, Tonks NK (1993) MKP‐1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo . Cell 75: 487–493 [DOI] [PubMed] [Google Scholar]
- Tomizawa M, Watanabe K, Saisho H, Nakagawara A, Tagawa M (2003) Down‐regulated expression of the CCAAT/enhancer binding protein alpha and beta genes in human hepatocellular carcinoma: a possible prognostic marker. Anticancer Res 23: 351–354 [PubMed] [Google Scholar]
- Tomizawa M, Horie H, Yamamoto H, Matsunaga T, Sasaki F, Hashizume K, Hiyama E, Kaneko M, Suita S, Ando H et al (2007) Reciprocal expression of CCAAT/enhancer binding proteins alpha and beta in hepatoblastomas and its prognostic significance. Oncol Rep 17: 341–344 [PubMed] [Google Scholar]
- Tseng HH, Hwang YH, Yeh KT, Chang JG, Chen YL, Yu HS (2009) Reduced expression of C/EBP alpha protein in hepatocellular carcinoma is associated with advanced tumor stage and shortened patient survival. J Cancer Res Clin Oncol 135: 241–247 [DOI] [PubMed] [Google Scholar]
- Vicent GP, Ballare C, Nacht AS, Clausell J, Subtil‐Rodriguez A, Quiles I, Jordan A, Beato M (2006) Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol Cell 24: 367–381 [DOI] [PubMed] [Google Scholar]
- Vicent GP, Zaurin R, Nacht AS, Li A, Font‐Mateu J, Le Dily F, Vermeulen M, Mann M, Beato M (2009) Two chromatin remodeling activities cooperate during activation of hormone responsive promoters. PLoS Genet 5: e1000567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicent GP, Nacht AS, Zaurin R, Font‐Mateu J, Soronellas D, Le Dily F, Reyes D, Beato M (2013) Unliganded progesterone receptor‐mediated targeting of an RNA‐containing repressive complex silences a subset of hormone‐inducible genes. Genes Dev 27: 1179–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ND, Finegold MJ, Bradley A, Ou CN, Abdelsayed SV, Wilde MD, Taylor LR, Wilson DR, Darlington GJ (1995) Impaired energy homeostasis in C/EBP alpha knockout mice. Science 269: 1108–1112 [DOI] [PubMed] [Google Scholar]
- Wang H, Iakova P, Wilde M, Welm A, Goode T, Roesler WJ, Timchenko NA (2001) C/EBPalpha arrests cell proliferation through direct inhibition of Cdk2 and Cdk4. Mol Cell 8: 817–828 [DOI] [PubMed] [Google Scholar]
- Wang GL, Iakova P, Wilde M, Awad S, Timchenko NA (2004) Liver tumors escape negative control of proliferation via PI3K/Akt‐mediated block of C/EBP alpha growth inhibitory activity. Genes Dev 18: 912–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Shi X, Salisbury E, Sun Y, Albrecht JH, Smith RG, Timchenko NA (2006) Cyclin D3 maintains growth‐inhibitory activity of C/EBPalpha by stabilizing C/EBPalpha‐cdk2 and C/EBPalpha‐Brm complexes. Mol Cell Biol 26: 2570–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub AS, Li CH, Zamudio AV, Sigova AA, Hannett NM, Day DS, Abraham BJ, Cohen MA, Nabet B, Buckley DL et al (2017) YY1 is a structural regulator of enhancer‐promoter loops. Cell 171: 1573–1588.e1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitington T, Frith MC, Johnson J, Bailey TL (2011) Inferring transcription factor complexes from ChIP‐seq data. Nucleic Acids Res 39: e98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Yan M, Patra J, Natrajan R, Yan Y, Swagemakers S, Tomaszewski JM, Verschoor S, Millar EK, van der Spek P et al (2011) Enhanced RAD21 cohesin expression confers poor prognosis and resistance to chemotherapy in high grade luminal, basal and HER2 breast cancers. Breast Cancer Res 13: R9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Enge M, Whitington T, Dave K, Liu J, Sur I, Schmierer B, Jolma A, Kivioja T, Taipale M et al (2013) Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell 154: 801–813 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The raw sequencing data from this study (ChIP‐seq and RNA‐seq) were submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) with the following accession number: GSE132649.