

Abstract
RIG‐I‐MAVS antiviral signaling represents an important pathway to stimulate interferon production and confer innate immunity to the host. Upon binding to viral RNA and Riplet‐mediated polyubiquitination, RIG‐I promotes prion‐like aggregation and activation of MAVS. MAVS subsequently induces interferon production by activating two signaling pathways mediated by TBK1‐IRF3 and IKK‐NF‐κB respectively. However, the mechanism underlying the activation of MAVS downstream pathways remains elusive. Here, we demonstrated that activation of TBK1‐IRF3 by MAVS‐Region III depends on its multimerization state and identified TRAF3IP3 as a critical regulator for the downstream signaling. In response to virus infection, TRAF3IP3 is accumulated on mitochondria and thereby facilitates the recruitment of TRAF3 to MAVS for TBK1‐IRF3 activation. Traf3ip3‐deficient mice demonstrated a severely compromised potential to induce interferon production and were vulnerable to RNA virus infection. Our findings uncover that TRAF3IP3 is an important regulator for RIG‐I‐MAVS signaling, which bridges MAVS and TRAF3 for an effective antiviral innate immune response.
Keywords: innate immunity, interferon, MAVS, RIG‐I, TRAF3IP3
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
Introduction
Innate immunity plays an important role in protecting higher organisms against pathogen infection (Palm & Medzhitov, 2009; Takeuchi & Akira, 2010). In the past two decades, distinct signaling pathways have been identified to mediate proper and efficient immune responses to virus infection (Yoneyama & Fujita, 2010; Aoshi et al, 2011; Tan et al, 2018). RIG‐I, one of the RIG‐I‐like receptors (RLRs), senses viral RNA in the cytoplasm and triggers immune signaling through its downstream adaptor protein MAVS (also known as VISA, IPS1, Cardif; Yoneyama et al, 2004; Kawai et al, 2005; Meylan et al, 2005; Seth et al, 2005; Xu et al, 2005; Chow et al, 2018; Loo et al, 2008), while cyclic GMP–AMP synthase (cGAS) detects viral DNA in the cytoplasm and launches immune response through the adaptor protein STING (also known as MITA, ERIS, MYPS; Chen et al, 2016). RIG‐I‐MAVS transduces antiviral signal to downstream kinases, including TBK1/IKKε and IKKα/β, which further activate transcriptional factors IRF3/IRF7 and NF‐κB, respectively, to stimulate the expression of type I interferon (IFN) and proinflammatory cytokines (tenOever et al, 2004). Type I IFN (α and β) induces the expression of hundreds of interferon‐stimulated genes (ISGs) and eventually an antiviral state to provide innate immunity for host cells.
Upon detection of viral RNA, RIG‐I switches from a “closed” conformation to an “open” one, thereby activating MAVS by inducing its prion‐like aggregation (Hou et al, 2011; Kowalinski et al, 2011; Luo et al, 2011; Cai et al, 2014; Peisley et al, 2014; Wu et al, 2014). It has been shown recently that Riplet‐mediated ubiquitination is crucial for RIG‐I‐mediated MAVS activation and the signal transduction from RIG‐I to MAVS could be elegantly reconstituted in vitro using purified components (Zeng et al, 2010; Shi et al, 2017). In contrast to such an elaborate understanding of the MAVS upstream signaling pathway, its downstream signaling has been unclear. MAVS contains multiple tumor necrosis factor receptor‐associated factor (TRAF)‐binding sites for TRAF proteins, including TRAF2, TRAF3, TRAF5, and TRAF6, which play redundant roles in mediating MAVS downstream signaling (Seth et al, 2005; Xu et al, 2005; Pineda et al, 2007; Liu et al, 2013; Fang et al, 2017). The identification of Ubc5 as an essential player for MAVS downstream signaling indicated the involvement of a ubiquitination event and supported the role of the TRAF proteins, which are E3 ubiquitin ligases (Zeng et al, 2009). In addition, other molecules, including TRADD and TANK, were also indicated in MAVS downstream signaling (Guo & Cheng, 2007; Michallet et al, 2008). A recent report refined two previously identified TRAF‐binding sites of MAVS as Regions I and II, which contribute to IKK‐NF‐κB activation, and further uncovered MAVS‐Region III as a novel site responsible for TBK1‐IRF3 activation (Shi et al, 2015). Phosphorylation of MAVS at a site located in its Region III was demonstrated to be critical for TBK1‐IRF3 activation (Liu et al, 2015). However, how MAVS‐Region III recruits and activates its downstream signaling molecules such as TRAFs is largely unknown.
The recruitment of TRAF3 to MAVS is an essential step in RIG‐I‐MAVS antiviral signaling (Saha et al, 2006; Paz et al, 2011; Belgnaoui et al, 2012). However, the mechanism of TRAF3 recruitment by MAVS remains unknown. Interestingly, TRAF3 interacting protein 3 (TRAF3IP3), also known as TRAF3‐interacting JNK‐activating modulator (T3JAM), has been reported to associate with TRAF3 and express at high levels in a variety of immune organs (Zou et al, 2015; Yu et al, 2018). Previous studies indicated that TRAF3IP3 and TRAF3 synergistically promote JNK but not NF‐κB activation, and TRAF3IP3‐mediated ERK signaling is critical in thymocyte development (Dadgostar et al, 2003). TRAF3IP3 also promotes autophagy and thus plays an important role in marginal zone B‐cell development and survival (Peng et al, 2015). Nevertheless, how TRAF3IP3 might contribute to antiviral innate immune signaling and whether it might be involved in RIG‐I‐MAVS pathway have not been explored.
Prion‐like aggregation of MAVS is required for its activation and presumably releases its autoinhibition on Region III after virus infection (Shi et al, 2015). Activated MAVS‐Region III promotes its downstream TBK1‐IRF3 activation, which is central to RIG‐I signaling. To gain mechanistical insight into how MAVS activates TBK1‐IRF3 signaling, we expressed MAVS‐Region III in vitro and in cells and found that it becomes active after multimerization. We then identified TRAF3IP3 by affinity purification as a binding partner to multimerized active MAVS‐Region III. We further found that TRAF3IP3 is accumulated on the mitochondria and mediates TRAF3 recruitment to MAVS upon virus infection. Remarkably, TRAF3IP3 plays a specific role in regulating TBK1‐IRF3 but not IKK‐NF‐κB activation upon RNA virus infection. Traf3ip3‐deficient mice showed attenuated interferon response and thus severely crippled innate immunity to RNA virus infection. Therefore, our data identify TRAF3IP3 as a critical regulator in MAVS‐mediated TBK1‐IRF3 activation for type I interferon production against RNA virus infection.
Results
The activity of MAVS‐Region III depends on its multimerization state
Upon virus infection, MAVS N‐terminal CARD domain goes through a prion‐like aggregation to release the autoinhibitory effect on Region III by its adjacent regions, which is essential for MAVS to activate downstream signaling (Shi et al, 2015). MAVS‐Region III (aa‐401–450) can specifically activate the TBK1‐IRF3 but not IKK‐NF‐κB signaling (Shi et al, 2015). To investigate the mechanisms that govern Region III in MAVS signaling, we first determined whether the activity of Region III depends on its multimerization state per se. MAVS‐Region III was fused with various GCN4‐derived peptides, which are composed of about 31–32 amino acids and differ from each other by a few amino acids (Fig 1A). These GCN4‐derived peptides were designed to form stable dimer, trimer, and tetramer, respectively (Harbury et al, 1993). Fusion proteins of MAVS‐Region III were expressed in bacteria and purified to be homogeneity. These proteins demonstrated the same mobility pattern on SDS–PAGE, consistent with their similar molecular weights (Fig EV1A). In contrast, they showed differential shifting mobility on native gel electrophoresis, indicating that they have distinct multimerization states as designed (Fig EV1B).
Figure 1. TRAF3IP3 associates with active MAVS‐Region III .
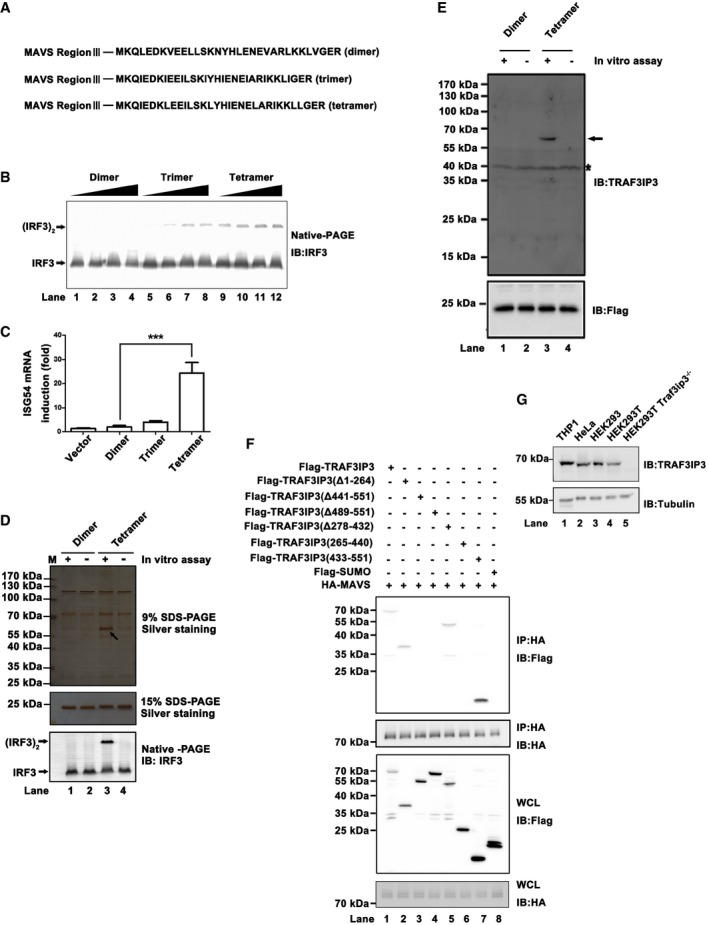
- Diagram for three constructs expressing MAVS‐Region III C terminally tagged with GCN4‐derived peptides, whose sequences are presented in bold.
- Recombinant MAVS‐Region III‐dimer (lanes 1–4), trimer (lanes 5–8), or tetramer (lanes 9–12) were subjected to IRF3 dimerization assay, respectively, in vitro. Increasing doses of recombinant MAVS‐Region III were used and are shown in Fig EV1C. Immunoblotting analysis was performed following native‐PAGE for the reaction mixture.
- pcDNA3‐based constructs expressing MAVS‐Region III‐dimer, trimer, and tetramer were transfected into HEK293T cells, respectively. ISG54 induction was measured 36 h after transfection by quantitative PCR (qPCR). Expression levels of MAVS‐Region III are shown in Fig EV1D.
- Recombinant MAVS‐Region III‐dimer or tetramer was immunoprecipitated with anti‐FLAG M2 beads with or without a preincubation with S100 from HEK293T cells following an experimental procedure outlined in Fig EV1G. The IP products were resolved in SDS–PAGE, and a specific protein band pulled down with MAVS‐Region III‐tetramer is indicated by an arrow. TRAF3IP3 was identified in the specific band by mass spectrometry.
- Protein samples as described in (D) were subjected to immunoblotting analysis. The TRAF3IP3 band is indicated by an arrow and the asterisk indicated nonspecific bands.
- pcDNA3‐FLAG‐TRAF3IP3 (full‐length and truncated forms) and pcDNA3‐HA‐MAVS were transfected into HEK293T cells, which were harvested 36 h post‐transfection. Co‐IP experiments were performed with anti‐HA antibody, followed by immunoblotting. A diagram depicting various TRAF3IP3 deletions used in the co‐IP experiments is shown in Fig EV1H.
- Human cells, including THP1, HeLa, HEK293, HEK293T as well as Traf3ip3 −/− HEK293T, were lysed and subjected to immunoblotting to examine TRAF3IP3 expression.
Figure EV1. TRAF3IP3 binds to multimerized and active MAVS‐Region III (related to Fig 1).
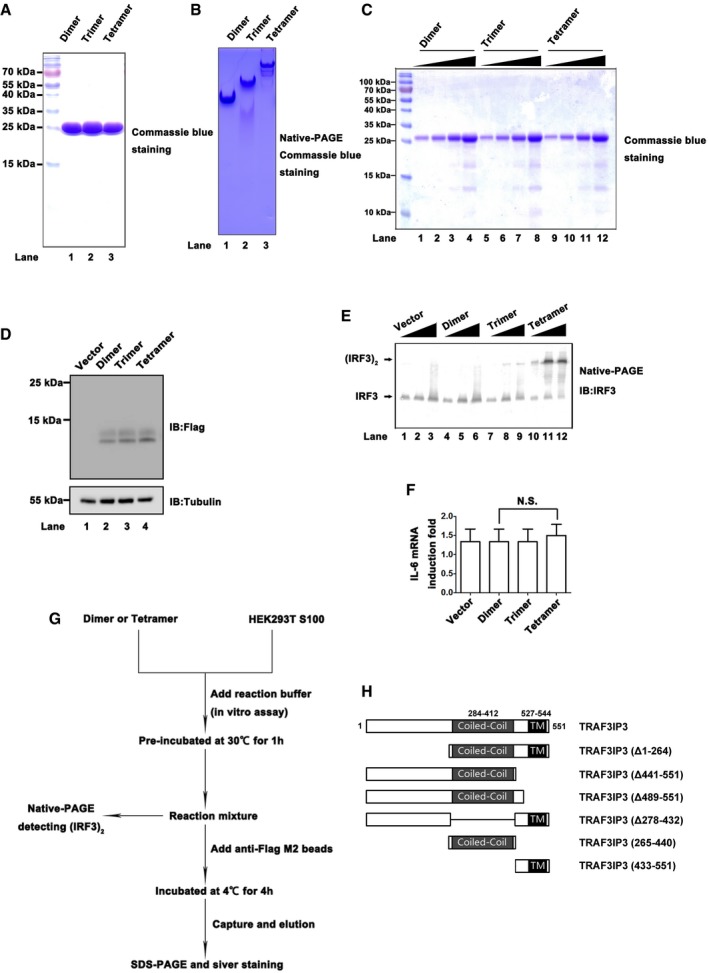
- Recombinant MAVS‐Region III‐dimer, trimer, or tetramer was expressed in E. coli and purified as described in methods. Proteins were separated by SDS–PAGE and visualized by Coomassie blue staining.
- Recombinant MAVS‐Region III‐dimer, trimer, or tetramer was separated by native‐PAGE and visualized by Commassie blue staining.
- SDS–PAGE and Coomassie blue staining of recombinant MAVS‐Region III‐dimer, trimer, or tetramer‐FLAG as described in Fig 1B.
- Immunoblotting analysis indicates expression levels of pcDNA3‐MAVS‐Region III‐dimer, trimer, or tetramer‐FLAG as described in Fig 1C.
- Increasing amounts of pcDNA3‐MAVS‐Region III‐dimer, trimer, and tetramer were transfected into HEK293T cells, respectively, and IRF3 dimerizations were examined by Western immunoblotting.
- pcDNA3‐based constructs expressing MAVS‐Region III‐dimer, trimer, and tetramer were transfected into HEK293T cells, respectively. IL6 induction was measured 36 h after transfection by qPCR. All data are presented as the mean values based on three independent experiments, and error bars indicate s.d. P values were determined by unpaired two‐tailed Student's t‐test. NS indicates no statistically significant difference.
- A cell‐free system for identification of MAVS‐interacting factors facilitate downstream IRF3 activation. In vitro assay was initiated by incubation of HEK293T cell S100 fractions with recombinant MAVS‐Region III‐dimer or tetramer under the reaction buffer. One portion of the reaction mixture was subjected to native‐PAGE for detecting (IRF3)2, while the others were incubated with anti‐FLAG M2 beads. Subsequently, FLAG‐tagged MAVS‐(Region III) and MAVS‐(Region III‐L439A), as well as their associated proteins, were captured by M2 beads and eluted with FLAG peptides.
- A diagram illustrating domain architectures of wild‐type and mutant TRAF3IP3 as described in Fig 1F.
The recombinant MAVS‐Region III was subjected to an activity assay in vitro, in which IRF3 dimerization was examined to monitor TBK1‐IRF3 activation. As shown in Fig 1B, while MAVS‐Region III‐dimer could not trigger IRF3 dimerization, MAVS‐Region III‐tetramer was highly potent in stimulating IRF3 dimerization. These results indicate that a higher‐order multimerization state is indispensable for the activity of MAVS‐Region III. Moreover, we expressed various forms of MAVS‐Region III in cells and examined ISG54 and IL6 induction, as indicators for TBK1‐IRF3 and IKK‐NF‐κB activation, respectively. Consistent with the results above, MAVS‐Region III‐tetramer but not Region III‐dimer induced the expression of ISG54 dramatically in Mavs knockout HEK293T cells (Fig 1C). IRF3 dimerization could be readily detected in cells expressing MAVS‐Region III‐tetramer (Fig EV1E). On the other hand, MAVS‐Region III‐tetramer did not induce the expression of IL6, confirming our previous findings that Region III can only activate TBK1‐IRF3 but not IKK‐NF‐κB (Shi et al, 2015; Fig EV1F). These data suggested that high‐order multimerization of MAVS‐Region III is a prerequisite for its activation and function in RIG‐I‐MAVS signaling.
TRAF3IP3 binds to multimerized and active MAVS‐Region III
We next used MAVS‐Region III‐tetramer as a bait to identify its binding partners involved in MAVS downstream signaling. Recombinant MAVS‐Region III‐tetramer or ‐dimer were preincubated with lysates from HEK293T cells and subsequently pulled down with anti‐FLAG M2 beads (Fig EV1G). In the products coimmunoprecipitated with MAVS‐Region III‐tetramer, a specific protein band with an apparent molecular weight of ~60 k Dalton was revealed by silver staining following SDS–PAGE (Fig 1D). This band was further identified by mass spectrometry as TRAF3IP3, a TRAF3‐binding protein that was previously known to be involved in multiple adaptive immune signaling (Table EV1). Immunoblotting analysis confirmed the presence of TRAF3IP3 in the pulled‐down products by MAVS‐Region III‐tetramer (Fig 1E). Strikingly, TRAF3IP3 was not coimmunoprecipitated with MAVS‐Region III‐dimer or MAVS‐Region III‐tetramer without preincubation in vitro, suggesting that TRAF3IP3 was recruited to the active MAVS‐Region III‐tetramer during the preincubation process. In support of these data, the association of TRAF3IP3 with full‐length MAVS was revealed in the co‐immunoprecipitation assay following transient expression of both proteins in HEK293T cells (Fig 1F). Protein structural predictions indicate that the C‐terminal region (residues 491–507) of TRAF3IP3 constitutes a transmembrane helix domain (TM). Our results showed that the TM domain of TRAF3IP3 was critical for its interaction with MAVS (Fig 1F). Additionally, we examined a panel of human cells and observed a broad spectrum of endogenous TRAF3IP3 expression (Fig 1G), in contrast to previous reports indicating that mouse TRAF3IP3 expression is limited to immune organs and tissues (Zou et al, 2015).
TRAF3IP3 regulates IFN response to virus infection in human cells
To investigate the role of TRAF3IP3 in MAVS signaling, we used small interfering RNA to downregulate TRAF3IP3 expression and analyzed the effects on virus‐stimulated IFNβ induction (Figs 2A). When TRAF3IP3 was knocked down in HEK293 cells, IFNβ production in response to Sendai virus (SeV) infection was reduced to an extent that was correlated with the knockdown efficiency (Fig 2B). Similarly, IFNB induction in response to vesicular stomatitis virus (VSV) infection or poly(I:C) treatment was also greatly attenuated when TRAF3IP3 was knocked down (Fig 2C). In contrast, IFNB induction by a DNA virus (herpes simplex virus, HSV‐1) in HEK293 cells was not affected by reduced expression of TRAF3IP3. These data together demonstrated that TRAF3IP3 is specifically involved in RNA virus‐triggered IFNβ induction.
Figure 2. TRAF3IP3 regulates interferon production in response to RNA virus infection.
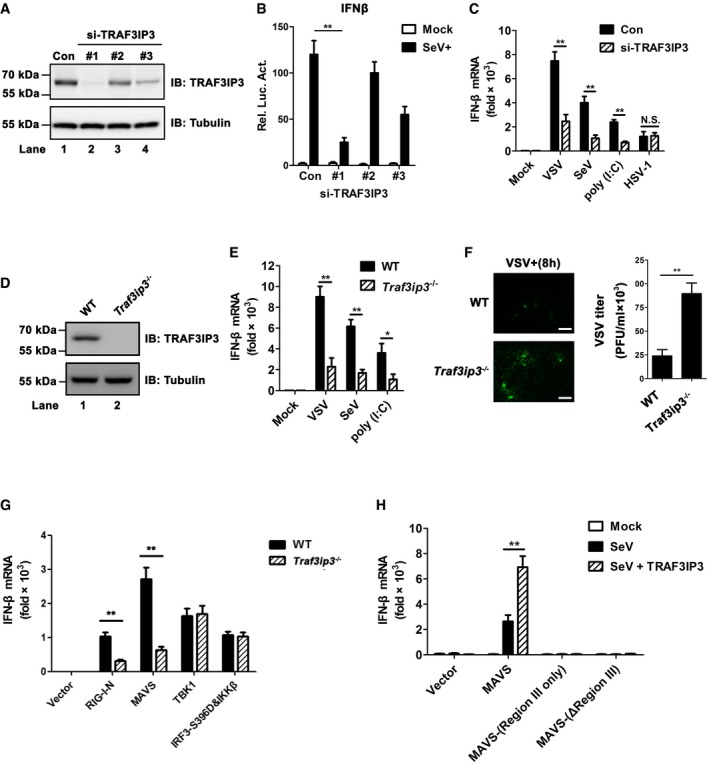
- Three pairs of small interfering RNA against TRAF3IP3 were transfected into HEK293T cells, respectively, and immunoblotting was then employed to examine the knockdown efficiency. A pair of scrambled RNA oligos was transfected and signified as con.
- Three pairs of si‐TRAF3IP3 together with IFNβ‐luciferase reporter were transfected into HEK 293T cells. Twenty‐four hours after transfection, the cells were infected with (SeV) or without (mock) Sendai virus, which were harvested 6 h post‐infection to measure the luciferase activity.
- Si‐TRAF3IP3 (oligo #1) was transfected into HEK 293 cells, which were then treated with mock, VSV, SeV, poly(I:C), or HSV‐1. After various treatments, IFNB inductions were measured by qPCR.
- Immunoblotting to detect TRAF3IP3 in wild‐type and Traf3ip3 −/− HEK293T cells.
- Wild‐type and Traf3ip3 −/− HEK293T cells were treated with or without VSV, SeV, or poly (I:C) as indicated for 12 h. IFNB induction was measured by qPCR.
- Wild‐type and Traf3ip3 −/− HEK293T cells were infected with GFP‐VSV. Fluorescent images were taken to examine GFP‐VSV proliferation at 8 h post‐infection (left). Scale bar represents 10 micrometers. GFP‐VSV titers were quantitated by plaque assay (right).
- Various expression constructs were transfected into WT and Traf3ip3 −/− HEK293T cells. Thirty‐six hours after transfection, IFNB induction was measured by qPCR. Protein expression levels are shown in Fig EV2A.
- Mavs −/− HEK293T cells were transfected with empty vector, pcDNA‐FLAG‐MAVS, pcDNA‐FLAG‐MAVS‐(Region III only), or pcDNA‐FLAG‐MAVS‐(ΔRegion III), together with pcDNA3‐FLAG‐TRAF3IP3 or not as indicated. Twenty‐four hours after transfection, cells were infected with SeV for 12 h. IFNB induction was measured by qPCR. Protein expression levels are shown in Fig EV2B.
Figure EV2. Expression levels of overexpressed proteins as indicated in Fig 2G and H (related to Fig 2).
- Immunoblotting to show the expression levels of FLAG‐RIG‐I(N), FLAG‐MAVS, FLAG‐TBK1, and FLAG‐IRF3 S396D as indicated in Fig 2G.
- Immunoblotting to show the expression levels of pcDNA‐FLAG‐MAVS, pcDNA‐FLAG‐MAVS‐(Region III only), pcDNA‐FLAG‐MAVS‐(ΔRegion III), or pcDNA3‐FLAG‐TRAF3IP3 as indicated in Fig 2H.
We then made Traf3ip3‐deficient HEK293T cells by CRISPR Cas9‐mediated gene editing (Fig 2D). Consistent with the data above, IFNB production in response to infections by VSV or SeV and treatment with poly(I:C) was markedly impaired in Traf3ip3 −/− cells (Fig 2E). As a result, Traf3ip3 −/− cells showed compromised antiviral immunity and were more permissible to VSV proliferation than wild‐type cells (Fig 2F). To dissect the mechanism by which TRAF3IP3 regulates the RIG‐I‐MAVS pathway, we compared IFN induction in HEK293T wild type with the Traf3ip3 −/− line that ectopically expressed RIG‐I, MAVS or TBK1. We found that Traf3ip3 deficiency crippled RIG‐I‐ and MAVS‐triggered IFNB production (Fig 2G). In contrast, TBK1‐ or IRF3 (S396D)‐triggered IFNB induction was not affected in Traf3ip3‐deficient cells (Fig 2G). Collectively, these results showed that TRAF3IP3 plays a critical role downstream of MAVS and upstream of TBK1 in RIG‐I‐mediated antiviral signaling.
We then examined whether TRAF3IP3 could trigger downstream signaling and IFNs production independently of MAVS by using two MAVS mutants, i.e., MAVS‐Region III and MAVS‐(ΔRegion III). It is known that MAVS downstream signaling bifurcates into two pathways that involve TBK1‐IRF3 and IKK‐NF‐κB, respectively. MAVS‐Region III can only activate the TBK1‐IRF3 pathway, while MAVS‐(ΔRegion III) can solely stimulate the IKK‐NF‐κB pathway. As expected, IFNB production was induced upon SeV infection in the presence of full‐length MAVS, which was enhanced dramatically by transient expression of TRAF3IP3 (Fig 2H). In contrast, neither MAVS‐Region III nor MAVS‐(ΔRegion III) alone could induce IFNB expression upon virus infection. Ectopic co‐expression of TRAF3IP3 could not restore IFNB induction in cells expressing either MAVS‐Region III or MAVS‐(ΔRegion III) (Fig 2H). These data demonstrated that TRAF3IP3 promotes IFN induction in a MAVS‐dependent manner.
TRAF3IP3 is involved in TBK1‐IRF3 activation downstream of MAVS
We then found that ISG54 but not IL6 induction was severely attenuated upon SeV infection in cells with TRAF3IP3 knocked down (Fig EV3A and B). This result suggested that TRAF3IP3 is required for TBK1‐IRF3 activation but not IKK‐NF‐κB activation in virus‐triggered IFNB induction. Indeed, the depletion of TRAF3IP3 dampened ISG54 induction pronouncedly upon VSV infection or poly(I:C) treatment (Fig 3A left). In contrast, depletion of TRAF3IP3 conferred marginal effect on IL6 induction under the same stimulations (Fig 3A right), suggesting that TRAF3IP3 might not be involved in IKK‐NF‐κB activation. As expected, the depletion of TRAF3IP3 showed little effect on either TBK1‐IRF3 or IKK‐NF‐κB activation upon HSV‐1 infection, confirming that TRAF3IP3 is not involved in DNA virus‐triggered IFNβ signaling (Fig 3A). In support of these data, we found that IRF3 dimerization was inhibited dramatically in response to RNA virus infection when TRAF3IP3 was knocked down, further demonstrating that IRF3 activation was impaired in the absence of TRAF3IP3 (Fig 3B). Consistently, ISG54 but not IL6 induction was significantly crippled in Traf3ip3 −/− cells upon infection by VSV or SeV, or poly(I:C) treatment (Fig 3C). Moreover, we examined the effects of TRAF3IP3 on signaling in Mavs −/− cells expressing MAVS‐Region III or MAVS‐(ΔRegion III). As mentioned above, both MAVS‐Region III‐ and MAVS‐(ΔRegion III)‐expressing cells lack the ability to produce IFNβ in response to virus infection. Strikingly, viral infection of MAVS‐Region III‐expressing cells induced ISG54 expression, which was further enhanced by co‐expression of TRAF3IP3 (Fig EV3C). In contrast, TRAF3IP3 had no effect on virus‐induced IL6 production in cells expressing MAVS‐(ΔRegion III) (Fig EV3D). These data further confirmed that TRAF3IP3 is specifically involved in TBK1‐IRF3 activation but not in IKK‐NF‐κB activation.
Figure EV3. TRAF3IP3 is required for ISG54 induction but not for IL6 induction upon RNA virus infection (related to Fig 3).
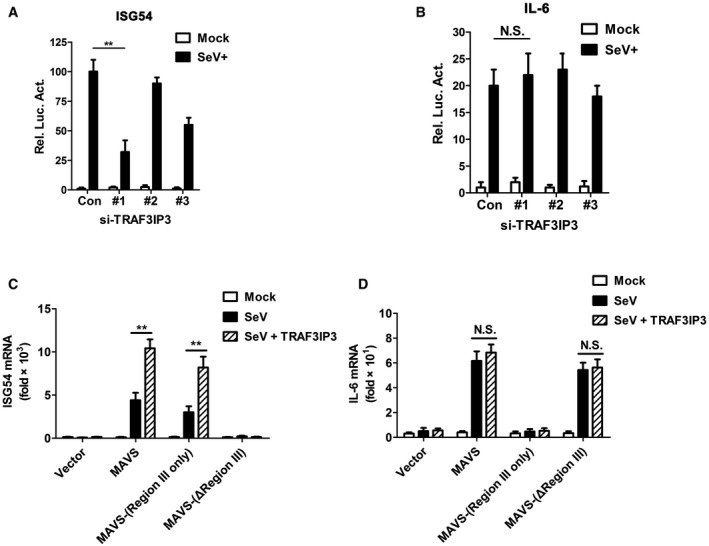
Figure 3. TRAF3IP3 is specifically required for TBK1‐IRF3 activation.
- The experiment was performed as described in Fig 2C. Instead of IFNB, ISG54 and IL6 inductions were measured by qPCR.
- HEK293 cells were transfected with si‐TRAF3IP3 oligos and then treated with various stimuli. Following subcellular fractionation, S5 fractions were obtained and subjected to native‐PAGE for detecting IRF3 dimer by immunoblotting. The whole‐cell lysates were also subjected to SDS–PAGE for anti‐IRF3 and tubulin immunoblotting.
- The experiment was performed as described in Fig 2E. ISG54 and IL6 induction was measured by qPCR.
- Wild‐type and Traf3ip3 −/− HEK293T cells were infected with or without VSV. At indicated time points, cells were collected and analyzed by immunoblotting. Following subcellular fractionation, S5 fractions were subjected to native‐PAGE to examine IRF3 dimer, and P5 fractions contained mitochondria were subjected to SDD‐AGE to examine MAVS aggregation. The mitochondrial outer membrane protein prohibitin was immunoblotted as an internal control.
We also examined whether TRAF3IP3 regulates the prion‐like aggregation of MAVS, a critical step in its activation. The results showed that viral infection of cells lacking TRAF3IP3 induced robust prion‐like aggregation of MAVS, comparable to those of wild‐type cells, indicating that MAVS aggregation and activation is independent of TRAF3IP3 (Fig 3D). Once again, we observed decreased IRF3 dimerization and TBK1 phosphorylation in the absence of TRAF3IP3, confirming that TRAF3IP3 is required for the activation of TBK1 and IRF3.
TRAF3IP3 mediates TRAF3 recruitment to MAVS upon virus infection
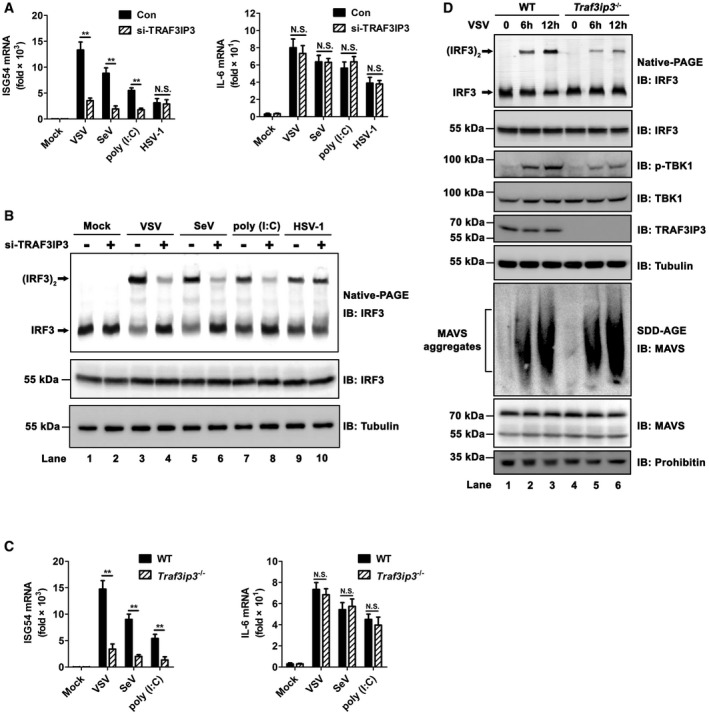
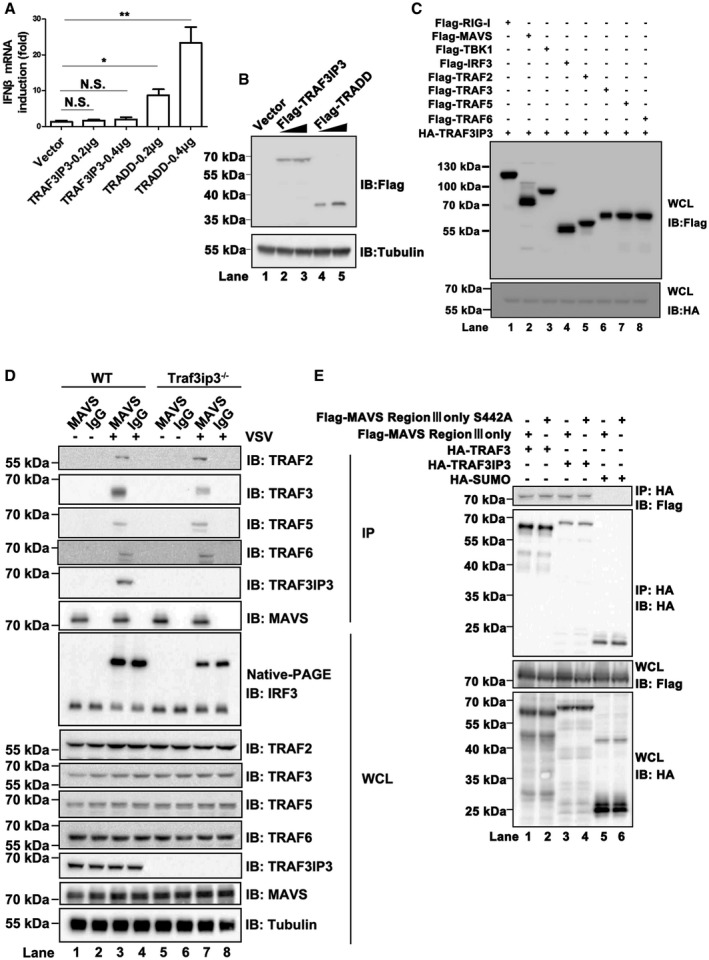
In spite of extensive investigations, the molecular mechanism by which MAVS activates its downstream kinases and transcriptional factors upon virus infection remains obscure. Overexpression of TRAF3IP3 alone did not lead to activation of either TBK1‐IRF3 or IKK‐NF‐κB (Figs 4A and EV4A). In contrast, overexpression of TRADD, a protein involved in MAVS downstream signaling, triggered the activation of TBK1‐IRF3, IKK‐NF‐κB, and ultimately interferon (Figs 4A and EV4A). Therefore, we reasoned that TRAF3IP3 might function in a manner different from TRADD and postulated that TRAF3IP3 might act as an adaptor between MAVS and its downstream signaling molecules. In particular, TRAF3IP3 as a potential TRAF‐binding protein may mediate the interaction between MAVS and TRAF proteins, which are known downstream signaling molecules in the MAVS pathway. We therefore examined whether TRAF3IP3 associates with TRAF proteins by co‐immunoprecipitation from HEK293T cells transfected with TRAF3IP3 and various TRAF proteins. We observed that TRAF3IP3 specifically associated with TRAF3 but not TRAF2/5/6. On the other hand, TRAF3IP3 interacted with MAVS but not RIG‐I, TBK1, or IRF3 (Fig 4B). We showed that TRAF3IP3 binds to MAVS‐Region III (Fig 1D and E), which does not contain any TRAF‐binding motif. Based on these data, we hypothesized that TRAF3IP3 mediates the recruitment of TRAF3 to MAVS‐Region III upon virus infection. Indeed, upon virus infection, MAVS‐Region III recruited significant amount of both TRAF3 and TRAF3IP3 (Fig 4C). Remarkably, the amount of TRAF3 recruited to MAVS‐Region III was severely compromised in cells with TRAF3IP3 knocked down. In contrast, the recruitment of TRAF5 to MAVS‐Region III was not affected by TRAF3IP3 knockdown. These data suggested that TRAF3IP3 plays a preferential role in the recruitment of TRAF3 to MAVS upon virus infection. We also carried out similar experiments by using Traf3ip3 −/− cells, which showed the same results (Fig 4D). Furthermore, we investigated the association of endogenous MAVS with various TRAFs in primary bone marrow‐derived macrophages (BMDMs). As shown in Fig EV4D, upon virus infection, endogenous MAVS was associated with TRAF2/3/5/6. Strikingly, the association between MAVS and TRAF3, but not other TRAFs, was dramatically compromised in the absence of TRAF3IP3. It has been shown that phosphorylation S442 in Region III of MAVS plays a role in its downstream signaling (Liu et al, 2015). Our data showed that the S442 mutation did not affect the interaction between MAVS‐Region III and TRAF3IP3, indicating that the interaction between TRAF3IP3 and MAVS‐Region III is independent of the phosphorylation (Fig EV4E).
Figure 4. TRAF3IP3 mediates TRAF3 recruitment to MAVS upon virus infection.
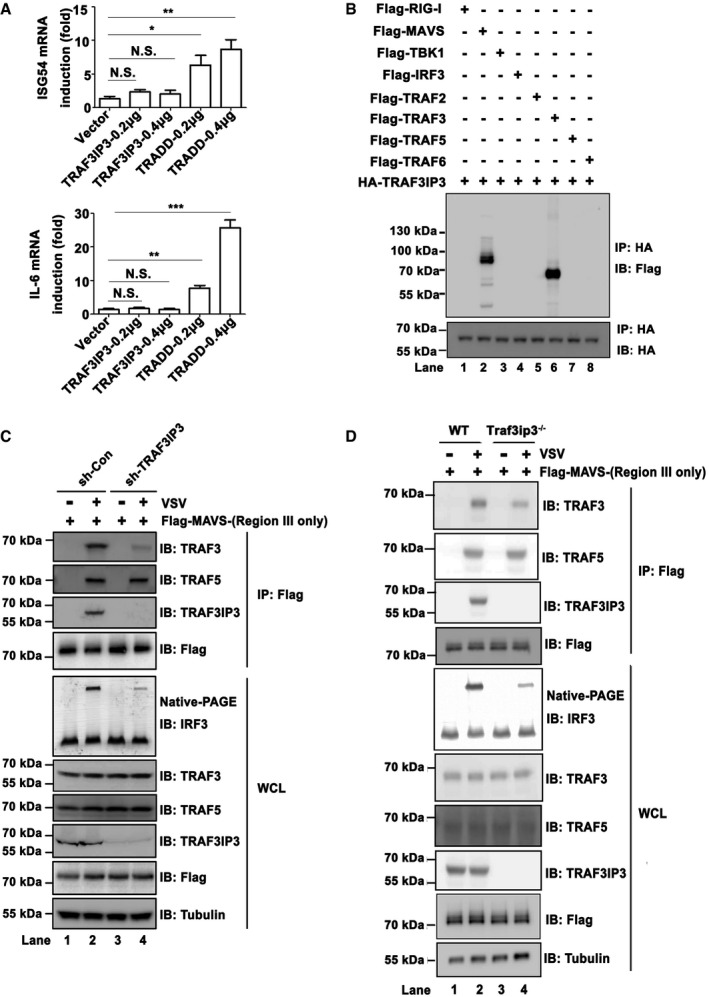
- pcDNA‐FLAG‐TRADD and pcDNA‐FLAG‐TRAF3IP3 were transfected into HEK293T cells at increasing amount, respectively. ISG54 and IL6 induction were measured 36 h post‐transfection by qPCR. Expression levels are shown in Fig EV4B. All data are presented as the mean values based on three independent experiments, and error bars indicate s.d. P values were determined by unpaired two‐tailed Student's t‐test. *P < 0.05, **P < 0.01, and ***P < 0.001. NS indicates no statistically significant difference.
- HEK293T cells were transfected with pcDNA3‐HA‐TRAF3IP3, in combination with pcDNA3‐FLAG‐ RIG‐I, MAVS, TBK1, IRF3, TRAF2, TRAF3, TRAF5, or TRAF6 as indicated. Thirty‐six hours after transfection, cells were collected and anti‐HA immunoprecipitations were performed. IP products were subjected to immunoblotting. Protein expression levels are shown in Fig EV4C.
- sh‐RNA targeting TRAF3IP3 (sh‐TRAF3IP3) was transduced into Mavs −/− HEK293T cells. Twenty‐four hours after transduction, cells were treated with puromycin (2 μg/ml) for 48 h and then transfected with pcDNA3‐FLAG‐MAVS‐(Region III only). Twenty‐four hours after transfection, cells were infected with or without VSV for 12 h. Cells were then collected and subjected to immunoprecipitation assay and immunoblotting.
- Wild‐type and Traf3ip3 −/− HEK293T cells were transfected with pcDNA3‐FLAG‐MAVS‐(Region III only). Twenty‐four hours post‐transfection, cells were infected with or without VSV. Twelve hours post‐infection, cells were collected and subjected to immunoprecipitation assay and immunoblotting.
Source data are available online for this figure.
Figure EV4. TRAF3IP3 associates with both MAVS and TRAF3 (related to Fig 4).
- pcDNA‐FLAG‐TRADD and pcDNA‐FLAG‐TRAF3IP3 were transfected into HEK293T cells, respectively. IFNB induction was measured 36 h after transfection by qPCR. All data are presented as the mean values based on three independent experiments, and error bars indicate s.d. P values were determined by unpaired two‐tailed Student's t‐test. *P < 0.05 and **P < 0.01. NS indicates no statistically significant difference.
- Immunoblotting to show the expression levels of FLAG‐TRADD and FLAG‐TRAF3IP3 as indicated in (A) and Fig 4A.
- Immunoblot analysis of whole‐cell lysates of HEK293T cells to show the expression of various constructs as indicated in Fig 4B.
- Primary BMDMs were isolated from wild‐type and Traf3ip3 −/− mice and infected with or without VSV. Twelve hours post‐infection, cells were collected and subjected to immunoprecipitation assay and following immunoblotting as indicated.
- Various expression constructs were transfected into HEK293T cells as indicated. Thirty‐six hours after transfection, Co‐IP experiments were performed with anti‐HA antibodies, followed by Western immunoblotting.
TRAF3IP3 accumulates on the Mitochondria after virus infection
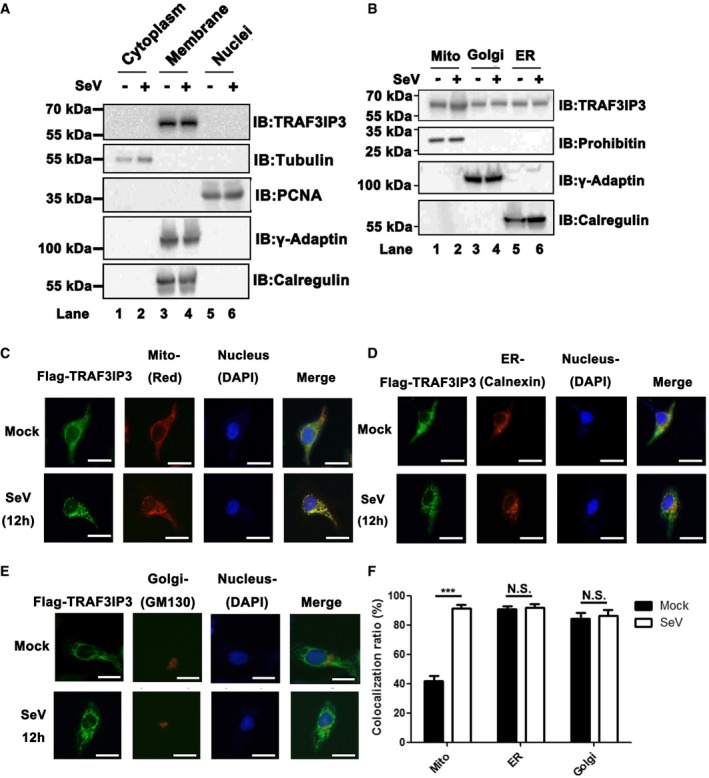
Having established that TRAF3IP3 mediates the recruitment of TRAF3 to MAVS for IFN induction, we next investigated how TRAF3IP3 might regulate antiviral signaling on the subcellular level. A previous study has shown that TRAF3IP3 mediates MEK‐BRAF interaction to facilitate ERK signaling at Golgi apparatus (Zou et al, 2015). The interaction with MAVS, however, suggests that TRAF3IP3 can localize to the mitochondrion because MAVS is a mitochondrial outer membrane protein. We first performed subcellular fractionation analyses and found that endogenous TRAF3IP3 indeed localized predominantly to the membranous fraction with or without virus infection (Fig 5A, lanes 3 and 4). We further separated the membranous fractions into three distinct organelle‐containing fractions, i.e., the mitochondrion (Mito), Golgi apparatus, and endoplasmic reticulum (ER), each of which could be identified by a specific marker, respectively (Fig 5B). To our surprise, TRAF3IP3 was distributed among all three organelles examined. Remarkably, more TRAF3IP3 was detected on the mitochondrion upon virus infection (Fig 5B, lane 2). In contrast, we did not observe any increase of TRAF3IP3 on ER or Golgi in response to virus infection. These data suggested that TRAF3IP3 is preferentially recruited or accumulated on mitochondria upon virus infection. We next performed immunostaining assays, in which ectopically expressed TRAF3IP3 was imaged as our antibody for endogenous TRAF3IP3 did not work for immunofluorescent staining. As shown in Fig 5C, FLAG‐TRAF3IP3 was distributed evenly outside the nuclei without viral stimulation. Upon virus infection, FLAG‐TRAF3IP3 was accumulated on the mitochondrion pronouncedly, as shown by overlapping signal of FLAG‐TRAF3IP3 with MitoTracker, an indicator for mitochondria (Fig 5C). In contrast, no accumulation signal of TRAF3IP3 could be observed on either ER or Golgi upon virus infection (Fig 5D–F). Collectively, these results demonstrated that in response to virus infection, TRAF3IP3 is accumulated specifically on the mitochondrion, consistent with its role as an adaptor between MAVS and TRAF3 in antiviral signaling.
Figure 5. TRAF3IP3 is accumulated on the mitochondria after virus infection.
-
ASubcellular fractionation was performed for HEK293T cells infected with or without SeV to get three fractions, i.e., cytoplasm, membrane, and nuclei extracts, which were then subjected to immunoblotting analysis as indicated.
-
BMembranous fraction obtained in Fig 5A were further separated into mitochondria, Golgi, and ER extracts as described in methods, followed by immunoblotting analysis.
-
C–EImmunofluorescent microscopic imaging for TRAF3IP3. HeLa cells were transfected with pcDNA3‐FLAG‐TRAF3IP3. Twenty‐four hours after transfection, cells were infected with or without SeV for 12 h. Anti‐FLAG M2 (FITC) was used for immunofluorescence staining of FLAG‐TRAF3IP3. Nuclei were stained with DAPI. Mitochondria were stained with MitoTracker Red (C). Calnexin (an ER protein) was stained for ER (D). GM130 (a Golgi protein) was stained for Golgi (E). Scale bar represents 5 μm.
-
FQuantitative analysis of yellow color as shown in (C–E), indicating TRAF3IP3 localization on various organelles with or without virus infection. The quantification was performed with ImageJ. All data are presented as the mean values based on three independent experiments, and error bars indicate s.d. P values were determined by unpaired two‐tailed Student's t‐test. ***P < 0.001. NS indicates no statistically significant difference.
Source data are available online for this figure.
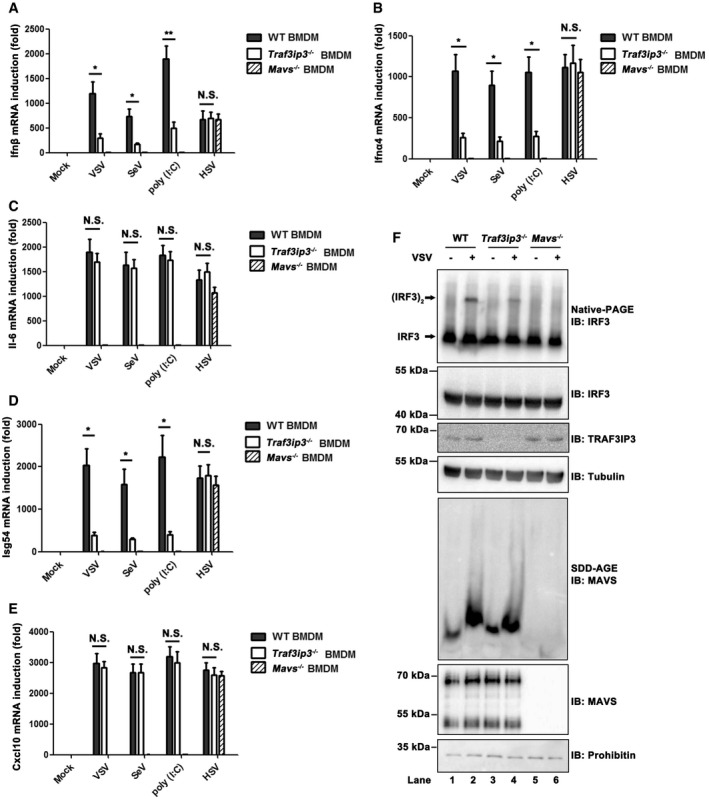
TRAF3IP3 mediates interferon response to RNA virus infection in mouse primary cells
To confirm the antiviral function of TRAF3IP3 in a more physiological context, we generated Traf3ip3 knockout mice and isolated primary cells including BMDMs and peritoneal exudate macrophages (PEMs). These cells were then treated with diverse stimuli, and their expression of IFNs and proinflammatory cytokines were measured. As shown in Figs 6A and EV5A, Ifnb induction in Traf3ip3 −/− BMDMs in response to SeV or VSV infection was drastically attenuated when compared with wild‐type cells. Similarly, Ifna4 production was also impaired in Traf3ip3 −/− BMDMs upon RNA virus infection or poly(I:C) stimulation (Fig 6B). These results indicate that TRAF3IP3 is indeed involved in RNA virus‐triggered immune signaling in BMDMs. In contrast, Ifnb induction in BMDMs upon HSV‐1 infection was not affected in the absence of TRAF3IP3 or MAVS, which is expected as TRAF3IP3 and MAVS are not required for DNA virus‐triggered immune signaling in BMDMs.
Figure 6. TRAF3IP3 mediates interferon response to virus infection in BMDMs and PEMs from mice.
-
A–EBMDMs from wild‐type, Traf3ip3 −/−, and Mavs −/− mice were treated with or without VSV, SeV, poly (I:C), or HSV‐1, respectively, as indicated for 6 h. Ifnb (A), Ifna4 (B), Il6 (C), Isg54 (D), and Cxcl10 (E) induction was measured, respectively, by qPCR. All data are presented as the mean values based on three independent experiments, and error bars indicate s.d. P values were determined by unpaired two‐tailed Student's t‐test. *P < 0.05 and **P < 0.01. NS indicates no statistically significant difference.
-
FBMDMs from wild‐type, Traf3ip3 −/−, and Mavs −/− mice were infected with or without VSV for 6 h. Cells were then collected and following subcellular fractionation, S5 fractions were subjected to native‐PAGE to examine IRF3 dimer, and P5 fractions contained mitochondria were subjected to SDD‐AGE to examine MAVS aggregation.
Source data are available online for this figure.
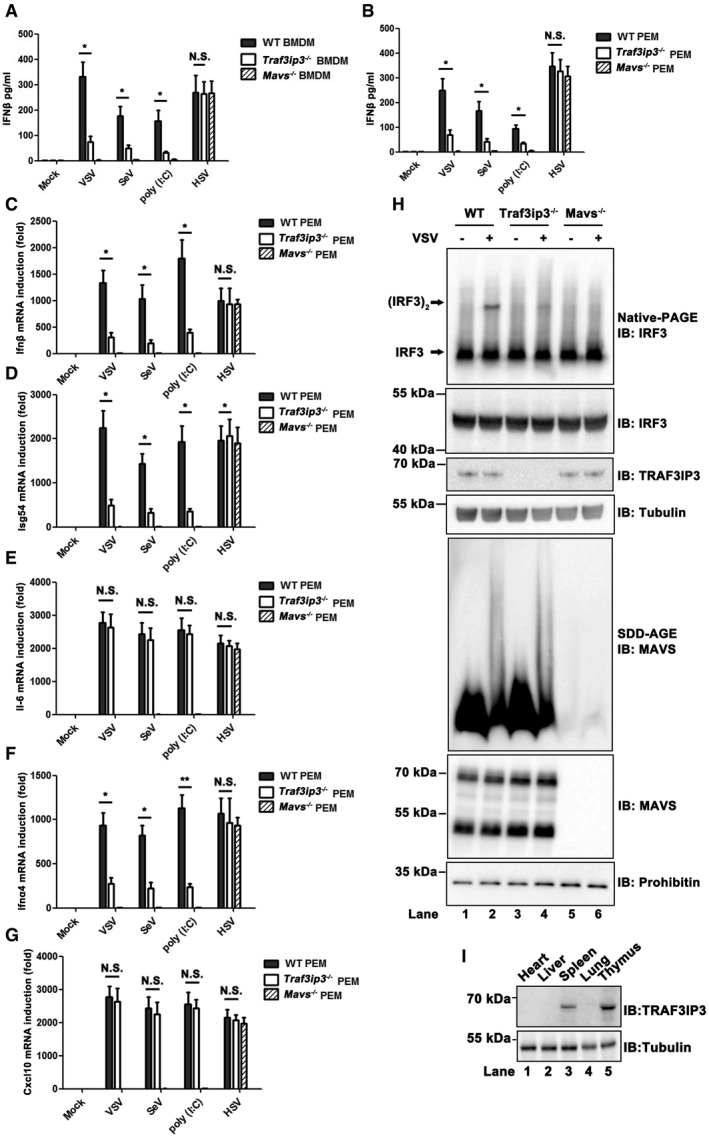
Figure EV5. TRAF3IP3 is involved in antiviral innate immune signaling in mice (related to Fig 6).
-
A, BBMDMs (A) and PEMs (B) from wild‐type, Traf3ip3 −/−, and Mavs −/− mice were treated with or without VSV, SeV, poly (I:C), or HSV‐1, respectively, as indicated for 6 h. The culture supernatants were then collected, and IFNβ production was measured by ELISA.
-
C–GPEMs from wild‐type, Traf3ip3 −/−, and Mavs −/− mice were treated with or without VSV, SeV, poly (I:C), or HSV‐1 as indicated for 6 h. Ifnb (C), Isg54 (D), Il6 (E), Ifna4 (F), or Cxcl10 (G) induction was measured by qPCR.
-
HPEMs from wild‐type, Traf3ip3 −/−, and Mavs −/− mice were infected with or without VSV for 6 h. Cells were collected and analyzed by immunoblotting. Following subcellular fractionation, S5 fractions were subjected to native‐PAGE to examine IRF3 dimer and P5 fractions contained mitochondria were subjected to SDD‐AGE to examine MAVS aggregation.
-
IImmunoblot analysis of TRAF3IP3 expression in heart, liver, spleen, lung, or thymus from mice.
To investigate how TRAF3IP3 might regulate IFN induction mechanistically in primary cells upon virus infection, productions of Isg54 and Il6 were both measured. While Traf3ip3 deficiency did not affect Il6 induction (Fig 6C), it attenuated Isg54 induction pronouncedly upon RNA virus infection (Fig 6D). These results confirm the findings in human cells that TRAF3IP3 is specifically involved in IRF3 but not NF‐κB activation in RIG‐I‐MAVS‐mediated IFN induction. In line with these results, induction of Cxcl10, a proinflammatory cytokine, was not affected in the absence of TRAF3IP3 upon RNA virus infection (Fig 6E). Similar results were observed in Traf3ip3 −/− PEMs (Fig EV5B–G). Furthermore, we examined the status of key molecules implicated in RIG‐I‐MAVS pathway to provide further mechanistic insights underlying antiviral function of TRAF3IP3. In Traf3ip3 −/− BMDMs or PEMs, MAVS aggregation was not affected upon VSV infection, while IRF3 dimerization or activation was severely impaired, confirming that TRAF3IP3 indeed plays a critical role in transducing antiviral signaling between MAVS and TBK1 (Figs 6F and EV5H).
Traf3ip3 deficiency attenuates antiviral immunity in mice
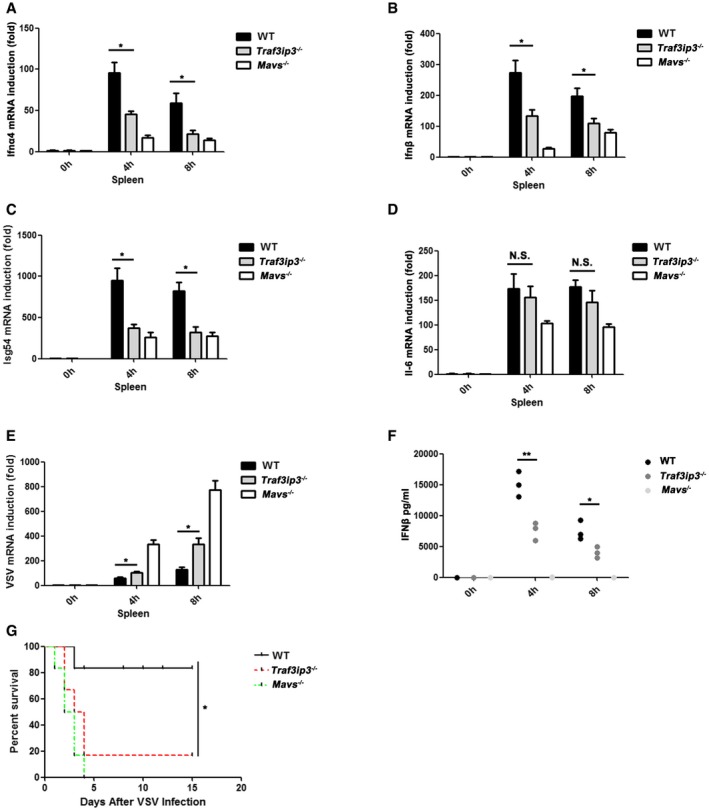
To investigate the role of TRAF3IP3 in regulating antiviral immunity in mice, we injected VSV into Traf3ip3 −/− and wild‐type mice intravenously. TRAF3IP3 is preferentially expressed at high level in the spleen and thymus in mice (Fig EV5I). We therefore isolated spleens from virus‐treated mice and measured the production of IFNs and proinflammatory cytokines. Notably, the absence of TRAF3IP3 resulted in a severely compromised induction of both Ifna4 and Ifnb upon VSV infection (Fig 7A and B). As expected, induction of Isg54 was attenuated in Traf3ip3 −/− mice upon VSV infection, whereas Il6 induction was not affected with the same stimulation (Fig 7C and D). As a result, spleens from Traf3ip3 −/− mice were more permissive to VSV amplification than those from wild‐type mice (Fig 7E). We also examined the sera from Traf3ip3 −/− and wild‐type mice after VSV infection. Traf3ip3 −/− mice produced much less Ifnb than wild‐type mice in their sera at 4 and 8 h after VSV infection (Fig 7F). Similarly, Ifnb induction was compromised severely in Traf3ip3 −/− mice upon infection with another RNA virus, influenza A virus (IAV). Once again, we observed the downregulation of Isg54 but not Il6 induction upon IAV infection in Traf3ip3 −/− mice (Fig EV6A–D). In contrast, Ifnb induction was not affected in the absence of TRAF3IP3 upon infection with HSV‐1 (Fig EV6E–H). Furthermore, the majority of wild‐type mice survived VSV infection, in contrast to that more than eighty percent of Traf3ip3 −/− mice succumbed to VSV infection within 5 days. These results demonstrate that Traf3ip3 −/− mice were more vulnerable to VSV killing than wild‐type mice, confirming the critical role of TRAF3IP3 in immediate innate antiviral defense in vivo (Fig 7G). Traf3ip3 −/− mice surviving VSV infection for 5 days recovered and stayed alive beyond the duration of our experimental observation (over 2 weeks), probably due to the adaptive immunity developed subsequently. Collectively, we concluded that TRAF3IP3 played an important role in IFN‐mediated antiviral innate immunity in vivo.
Figure 7. Traf3ip3 deficiency attenuates antiviral immunity in mice.
-
A–FWild‐type, Traf3ip3 −/−, and Mavs −/− mice (three littermates per group) were infected intravenously with VSV at a MOI of 2 × 108. The spleens were collected 4 h or 8 h after infection, and Ifna4 (A), Ifnb (B), Isg54 (C), and Il6 (D) inductions were measured, respectively, by qPCR. VSV RNA levels were measured by qPCR (E). The sera were collected from the treated mice and used for the measurement of IFNβ production by ELISA (F). All data are presented as the mean values based on three independent experiments, and error bars indicate s.d. P values were determined by unpaired two‐tailed Student's t‐test. *P < 0.05 and **P < 0.01. NS indicates no statistically significant difference.
-
GWild‐type, Traf3ip3 −/−, and Mavs −/− mice (n = 6 each) were injected with VSV via tail vein injection at 5 × 107 pfu per mouse, and the survival rates were monitored for 15 days. P values were determined by unpaired two‐tailed Student's t‐test. *P < 0.05.
Figure EV6. Traf3ip3 deficiency attenuates antiviral immunity against IAV but not HSV‐1 infection (related to Fig 7).
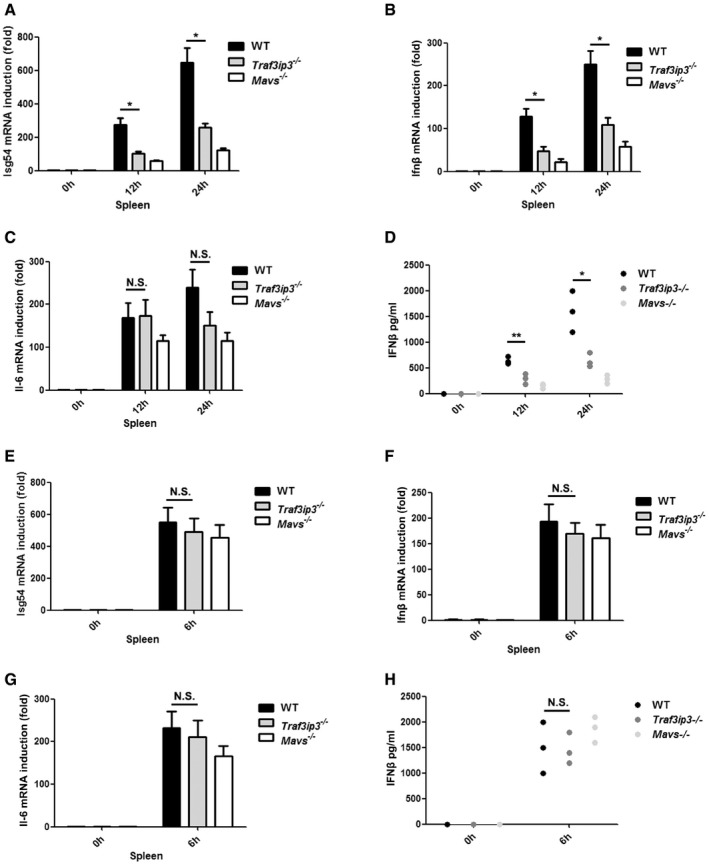
-
A–DWild‐type, Traf3ip3 −/−, and Mavs −/− mice (three littermates per group) were infected intranasally with IAV at a pfu of 3 × 108. The spleens were collected 12 h or 24 h after infection and Isg54 (A), Ifnb (B), and Il6 (C) inductions were measured, respectively, by qPCR. The sera were collected from the treated mice and used for the measurement of IFNβ production by ELISA (D).
-
E–HWild‐type, Traf3ip3 −/−, and Mavs −/− mice (three littermates per group) were infected intravenously with HSV‐1 at a dose of 5 × 107 pfu per mouse. The spleens were collected 6 h after infection and Isg54 (E), Ifnb (F), and Il6 (G) inductions were measured, respectively, by qPCR. The sera were collected from the treated mice and used for the measurement of IFNβ production by ELISA (H).
Discussion
MAVS was identified as an essential player in RIG‐I signaling more than a decade ago and Mavs‐deficient mice were shortly generated to provide evidence substantiating the pivotal role of MAVS in interferon induction by virus infection (Sun et al, 2006). In contrast, dissecting the molecular mechanisms underlying MAVS activation and how MAVS activates its downstream signaling has been arduous. As such, a number of proteins have been identified to modulate MAVS antiviral activity (Moore et al, 2008; Lei et al, 2012; Liu et al, 2012, 2017; Zhang et al, 2014; Yan et al, 2017). Among them, TRAF proteins were found to play redundant roles downstream of MAVS. Despite these studies, there are remaining gaps in our understanding of signaling from MAVS to its downstream molecules. In this study, we reported an important regulator TRAF3IP3, which mediates the recruitment of TRAF3 to MAVS to promote MAVS downstream signaling and specifically regulates TBK1‐IRF3 but not IKK‐ NF‐κB activation (Fig 8).
Figure 8. A schematic model illustrating functional involvement of TRAF3IP3 in RIG‐I‐MAVS‐mediated antiviral signaling.
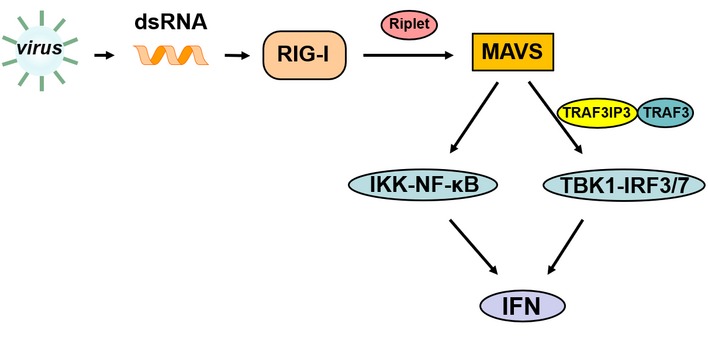
Upon virus infection, RIG‐I binds to viral dsRNA and activates MAVS with the help of Riplet. Furthermore, MAVS recruits TRAF3 through TRAF3IP3 to activate downstream TKB1‐IRF3 and MAVS activates IKK‐NF‐κB probably by directly binding to other TRAFs. IRF3 and NF‐κB enter the nucleus to turn on type I IFN expression.
Multimerization or aggregation has been demonstrated as a key feature of MAVS antiviral function. MAVS C‐terminal transmembrane domain mediates a homotypic interaction between full‐length MAVS and its truncated isoforms to prevent its accidental spontaneous aggregation in cells, which represents one multimerization element of MAVS (Qi et al, 2017). Moreover, N‐terminal CARD domain of MAVS mediates its prion‐like aggregation upon virus infection, which unleashes the autoinhibitory effect of MAVS on its Region III. MAVS‐Region III can then activate downstream TBK1‐IRF3 signaling. Following these studies, there is an intriguing question as to whether the activity of MAVS‐Region III depends on its multimerization state on its own. To address this question, we enforced the multimerization of MAVS‐Region III by artificially tagging it with GCN4‐derived peptides, which are able to mediate multimerization independently. Based on both in vitro and in cellulo evidence, we concluded that a high‐order multimerization of MAVS‐Region III is indispensable for its activity. As a matter of fact, our previous report showed that without N‐terminal CARD domain, overexpressed MAVS‐Region III‐TM could also activate TBK1‐IRF3 in cells, which is conceivable as MAVS transmembrane domain could mediate homotypic interaction and thus multimerization (Shi et al, 2015). Collectively, we reasoned that prion‐like aggregation of MAVS can not only release its autoinhibition on Region III, but also result in the multimerization and activation of Region III, highlighting an unappreciated layer of consequence and significance of MAVS prion‐like aggregation. Following structural study on MAVS‐Region III in both autoinhibitory and high‐order multimerized states should provide more mechanistic insight into its antiviral function.
In the effort investigating how MAVS‐Region III activates its downstream signaling, we identified TRAF3IP3 as a binding partner to MAVS‐Region III and an important regulator in mediating TBK1‐IRF3 activation downstream of MAVS. Interestingly, TRAF3IP3 specifically associated with TRAF3 but not other TRAF proteins. Therefore, we postulated a mechanism underlying TRAF3IP3‐mediated TRAF3 recruitment to MAVS for antiviral signaling. In support of this mechanism, TRAF3IP3 only binds to multimerized and active MAVS‐Region III but not inactive MAVS‐Region III, indicating it is a signaling‐dependent interaction. Furthermore, TRAF3 recruitment to MAVS was severely impaired in the absence of TRAF3IP3 in response to virus infection. Our findings thus shed light into an unidentified mechanism suggesting that TRAF3IP3 mediates the recruitment of TRAF3 to MAVS upon virus infection, highlighting the involvement of TRAF3 in antiviral signaling in a sense. We further investigated the antiviral role of TRAF3IP3 in vivo. In the absence of TRAF3IP3, type I IFN induction was severely compromised upon RNA virus infection in mouse primary cells. Mechanistic studies revealed that ISG54 but not IL‐6 induction was impaired in Traf3ip3 −/− cells, indicating that TRAF3IP3 was indeed required for TBK1‐IRF3 but not IKK‐NF‐κB activation and nevertheless essential for IFNs induction. As a result, Traf3ip3 −/− mice were defective in antiviral innate immunity and more vulnerable to VSV‐mediated killing than wild‐type mice, providing supportive evidence demonstrating the antiviral function of TRAF3IP3 in vivo.
In addition to Region III, MAVS also harbors Region I and II. MAVS‐Region I and II contain putative TRAF protein‐binding sites, which presumably mediate TRAF2/3/5/6 recruitment directly upon virus infection. In support of this view, point mutations of these TRAF‐binding sites completely disrupt the function of Region I and II in activating downstream IKK‐NF‐κB (Liu et al, 2013; Shi et al, 2015). These studies emphasized the importance of putative TRAF‐binding sites of MAVS in antiviral signaling. Intriguingly, MAVS‐Region III could also recruit TRAF3 and TRAF5 for TBK1‐IRF3 activation in response to virus infection, regardless of that it does not harbor any putative TRAF‐binding sites (Shi et al, 2015). Our identification of TRAF3IP3 involved in RIG‐I‐MAVS signaling revealed a mechanism underlying TRAF3 recruitment to MAVS through the adaptor protein TRAF3IP3 upon virus infection. In light of that MAVS‐Region III could also recruit TRAF5 for TBK1‐IRF3 activation upon virus infection, it is possible that there might be unknown molecules mediating the recruitment of TRAF5 to MAVS‐Region III for antiviral signaling. Furthermore, considering that TRAF proteins play redundant roles in MAVS downstream signaling, it will be interesting to further investigate the remaining question as to how or whether these TRAF proteins might promote the activation of downstream signaling molecules including TBK1 and IKK by different mechanisms.
Materials and Methods
Mice
Traf3ip3 fl/fl mice in C57BL/6 background were from Dr. Qiang Zou (Shanghai Jiao Tong University School of Medicine) as described (Yu et al, 2018). EIIA‐cre (E2a‐cre) transgenic mice expressing Cre recombinase were generously provided by Dr. Weiguo Zou (Shanghai Institute of Biochemistry and Cell Biology). Mavs knockout mice were generated with the CRISPR–Cas9 technique by Shanghai Model Organisms Center, Inc. (SMOC). Homozygous Traf3ip3 fl/fl mice were mated with EIIA‐cre mice to generate mice carrying both the cre gene and the heterozygous loxP‐flanked Traf3ip3 (Traf3ip3 fl/+ EIIA‐cre). The filial generations were further crossed to Traf3ip3 fl/fl mice to generate Traf3ip3‐deficient mice (Traf3ip3 fl/fl EIIA‐cre). Littermates who lacked the Cre gene (Traf3ip3 fl/fl EIIA‐cre−/−) were used as controls. The genotyping was performed by standard PCR. Mice were maintained in a specific pathogen‐free (SPF) conditions, and all animal experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) at the Shanghai Institute of Biochemistry and Cell Biology.
Plasmids and antibodies
Human complementary DNA was synthesized from total RNA extracted from HEK293T cells, and pMD‐18T‐TRAF3IP3 was a gift from Dr. Jiahuai Han (Xiamen University). cDNAs of TRAF3IP3, MAVS, TRAF3/5/6, RIG‐I(N), IRF3 S396D, TBK1, and TRADD were subcloned into pcDNA3‐FLAG or pcDNA3‐HA expression vector and primers used for the amplification can be found in Table EV2. All mutations and deletions were constructed with the Fast‐mutagenesis Kit (TransGen Biotech, Beijing, China), QuickChange Lightning Multi Site‐Directed Mutagenesis Kit (Stratagene), or overlapping PCR strategy, respectively. All constructs were confirmed by DNA sequencing. Antibody against human MAVS and mouse MAVS were raised by immunizing rabbits with recombinant proteins His‐sumo‐hMAVS‐(aa‐301–460) and His‐sumo‐mMAVS‐(aa‐394–473). Commercial antibodies included anti‐FLAG (Sigma, F3165, F7425, dilution 1:5,000), anti‐tubulin (Sigma, T5168, dilution 1:7,500), anti‐HA (Cell Signaling Technology, 3724S, dilution 1:2,000), anti‐GM130 (BD, 610823, dilution 1:800), anti‐calnexin (Sigma, C4731, dilution 1:800), anti‐TBK1 (Cell Signaling Technology, 3504, dilution 1:1,000), anti‐pTBK1 S172 (Cell Signaling Technology, 5483S, dilution 1:1,000), anti‐IRF3 (proteintech, 11312‐1‐AP, dilution 1:2,000), anti‐TRAF3IP3 (Santa Cruz, SC‐366384, dilution 1:2,000), anti‐TRAF3 (Santa Cruz, SC‐1828, dilution 1:1,000), anti‐TRAF5 (Santa Cruz, SC‐74502, dilution 1:1,000), anti‐TRAF6 (Santa Cruz, SC7221, dilution 1:1,000), anti‐PCNA (Santa Cruz, SC56, dilution 1:1,000), anti‐γ‐adaptin (Santa Cruz, SC‐10763, dilution 1:1,000), and anti‐calregulin (Santa Cruz, SC‐6468, dilution 1:1,000); antibodies against TRAF5 (ab12123, dilution 1: 2,000), IRF3 (Abcam, 2241‐1, dilution 1:3,000), and prohibitin (ab75766, dilution 1:10,000) were from Abcam.
Cells and viruses
HeLa cells, HEK293 cells, PEMs, and BMDMs were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS, ExCell Bio, FSP500), penicillin (100 U/ml), and streptomycin (100 μg/ml). THP1 cells were cultured in RPMI Medium 1640 basic medium with 10% FBS and antibiotics. HEK293T cells were cultured in DMEM supplemented with 10% calf serum and antibiotics. HeLa and THP‐1 cells were from the Cell Resource Center (Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences). Recombinant virus VSV‐∆M51‐GFP was amplified in Vero cells and used with a multiplicity of infection (MOI) of 1. Sendai virus, herpesvirus herpes simplex virus‐1 (HSV‐1), and IAV (influenza A/Puerto Rico/8/1934 [H1N1] PR8 strain) were used at a concentration of 50 HA units/ml.
Generation of knockout cell lines based on HEK293T
Traf3ip3‐deficient HEK293T cells were established by CRISPR/Cas9 technique, and the guide RNA sequence can be found in Table EV2. Briefly, guide RNA sequences for each gene were cloned into a CRISPR/Cas9‐based vector pX330 with a puromycin‐resistant selection marker. These vectors were transfected into HEK293T cells by Lipofectamine 2000 (Invitrogen). After puromycin (0.5 μg/ml) selection, single colonies were picked and verified by genome sequencing and Western blotting. Mavs‐deficient HEK293T cells were described (Shi et al, 2015).
Generation of stable cell lines based on HEK293T
To cells stably expressing TRAF3IP3 and its truncation mutants, HEK293T cells were infected with lentivirus as indicated, respectively, for 24 h. Puromycin selection was performed for 48 h post‐infection. After immunoblotting analysis, cells were seeded in 96‐well dishes. Single colonies were then picked and verified by Western blotting.
Immunoprecipitations
HEK293T cells were transfected with indicated constructs by Lipofectamine 2000 (Invitrogen). Thirty‐six hours after transfection, cells were harvested and resuspended with lysis buffer (HEPES 20 mM pH 7.5, MgCl2 5 mM, KCl 10 mM, EGTA 0.5 mM, Triton X‐100 1%, and protease inhibitor cocktail (Roche)). Following a brief centrifugation, anti‐FLAG M2 affinity gel (Sigma A2220) or anti‐HA agarose beads (Thermo 26181) were added to cell lysate for 4 h at 4°C. After incubation, M2 or HA beads were spun down and washed for three times with lysis buffer followed by immunoblotting analysis. For immunoprecipitation of endogenous MAVS, BMDM cellular lysates were incubated with antibody against mMAVS or IgG overnight, and then, protein G beads were added. Four hours later, protein G beads were spun down and washed for three times with lysis buffer followed by immunoblotting analysis.
Luciferase reporter assay
HEK293T cells were seeded in 12‐well plates at a density of 1 × 105 cells per well and 6 h later co‐transfected with 20 ng of reporter gene (IFN‐luciferase, ISRE‐luciferase, or NF‐κB‐luciferase), 20 ng of pCMV‐LacZ as internal control, and indicated expression vectors by calcium phosphate method. Twenty‐four hours post‐transfection, cells were stimulated with or without VSV or SeV. Thirty‐six hours after transfection, cells were harvested and lysed in passive lysis buffer (Promega). Firefly luciferase activities were measured with luminometer using the Luciferase Reporter Kit (Promega), and LacZ activities were measured by o‐nitrophenyl‐b‐D‐galactopyranoside (ONPG) assay in buffer (Na2HPO4/NaH2PO4 200 mM pH 7.3, MgCl2 2 mM, b‐mercaptoethanol 100 mM, ONPG 1.33 mg/ml) following a protocol provided by Sigma Technical Bulletin (GALA‐1KT). Fold induction of firefly luciferase was normalized to LacZ activity. Data were showed as fold induction over empty vector‐transfected controls.
Real‐time PCR
In brief, total RNA was prepared using the RNA simple total RNA kit (Tiangen, Shanghai, China) from cells, and the reverse transcription was carried out using the GoScript Reverse Transcription system (Promega). cDNAs were then used as templates for qPCR assay by the use of SuperReal Premix Plus (Tiangen). And induction fold was determined with the ∆∆Cq method, and qPCR primers used to amplify specific genes are shown in Table EV2.
ELISA
Concentrations of the cytokine in cell culture supernatants and mouse serum were measured, respectively, by IFN‐BETA Mouse ELISA (High Sensitivity, PBL Assay Science) according to the manufacturer's instructions.
RNA interfering
All siRNA duplexes were obtained from Gene‐Pharma, and the sequence targeting TRAF3IP3 can be found in Table EV2. HEK293T cells were seeded at a confluence of 10%, and 6 h later, si‐TRAF3IP3 was transfected into HEK293T cells by calcium phosphate method. Thirty‐six hours post‐transfection, si‐TRAF3IP3 transfection was performed again. Thirty‐six hours after second transfection, cells were harvested and following experiments were carried out. We used PLKO.1‐based lenti‐viral transduction and the sequence of sh‐RNA can be found in Table EV2. Lenti‐viral vector and viral packaging vectors were transfected into HEK293T cells using Lipofectamine 2000 (Invitrogen). Forty‐eight hours later, culture medium containing packaged lenti‐viral particles were collected and filtered through 0.45 μm low protein‐binding durapore membrane (Millipore). HEK293T cells were incubated with lentivirus in the presence of 8 μg/ml polybrene for 24 h before fresh medium was served. Twenty‐four hours post‐infection, puromycin (2 μg/ml) was added to the culture medium to select cells stably expressing the sh‐RNA transduced. After another 48 h, the cells were used for following experiments.
Recombinant protein preparation
MAVS‐Region III‐GCN4‐dimer‐FLAG and MAVS‐Region III‐GCN4‐dimer‐FLAG were cloned into pET28a with an N‐terminal 6X His tag and sumo tag and then expressed in BL21 strain (PJY2), which were induced by 0.2 mM isopropyl‐b‐D‐thiogalactoside (IPTG) for 9 h at 18°C. Recombinant proteins were purified with Ni‐NTA agarose beads according to the manufacturer's protocol (Qiagen, 30210).
IRF3 activation assay in vitro
Cytosolic extracts (S100) and mitochondrial fractions (P5) were isolated from HEK293T cells. Briefly, HEK293T cellular homogenates in hypotonic buffer (Tris–Cl 10 mM pH 7.5, KCl 10 mM, EGTA 0.5 mM, and MgCl2 1.5 mM) were centrifuged at 1,000 g for 5 min, and the pellet was discarded. The supernatant was further fractionated by centrifugation at 10,000 g, and the pellet was P5 while the supernatant was S5. S5 was further centrifuged at 100,000 g for 30 min to get supernatant S100. P5 and S5 were used for IRF3 dimerization assay in vitro followed by native‐PAGE and Western blot analysis as described (Shi et al, 2015).
Semidenaturing detergent agarose gel electrophoresis
Crude mitochondria (P5) were isolated from HEK293T cells and resuspended in 1 × sample buffer (0.5 × TBE, 10% glycerol, 2% SDS, and 0.0025% bromophenol blue) and loaded onto a vertical 1.5% agarose gel. After electrophoresis in the running buffer (1 × TBE and 0.1% SDS) for 30 min with a constant voltage of 100V at 4°C, Western immunoblotting was performed.
Fluorescence microscopy
Traf3ip3 −/− HEK293T cells and wild‐type HEK293T cells were seeded in 6‐well dish. Ten hours post‐infection by VSV, fluorescent images were taken from live cells using Olympus IX71 inverted fluorescence microscope. For immunofluorescence microscopy of the subcellular localization of TRAF3IP3, HeLa cells were transfected with pcDNA3‐FLAG‐TRAF3IP3 by Lipofectamine 2000. Twenty‐four hours after transfection, cells were uninfected or infected with SeV for 12 h. Cells were then incubated with 150 nM MitoTracker Red CMXRos (Thermo Fisher Scientific) for 15 min at room temperature. After staining, cells were washed with pre‐warmed 1 × PBS and fixed with 4% formaldehyde for 15 min. Cells were then permeabilized with 0.5% Triton X‐100 for 15 min and incubated with anti‐FLAG M2 (FITC), anti‐calnexin, or anti‐GM130, respectively overnight. After incubation, cells were washed with pre‐warmed 1 × PBS for three times. Before fluorescent imaging, cells were incubated with appropriate secondary antibodies and DAPI (Beyotime, C1002). All the images were taken with Olympus IX71 inverted fluorescence microscope.
Plaque assay
Plaque assay was performed as recently described (Qi et al, 2017). In brief, HEK293T cells in 6‐well plates were infected with serial dilutions of the recovered VSV for 1 h. The infected cells were overlaid with 1% soft agar dissolved in DMEM and incubated for 48 h. Plates were stained with 0.1% crystal violet in DMEM to display plaques, which were then quantitated.
Subcellular fractionation
To isolate mitochondria, Golgi, and ER, cells (4 × 108) infected with or without SeV were collected. After homogenization, cells went through a series of centrifugation steps as described (Wieckowski et al, 2009; von Blume et al, 2012; Galen & Blume, 2013). Then, immunoblotting was performed.
Statistical analysis
All data are showed as mean values ± s.d. based on data from three independent experiments. Statistics significance between two groups was determined by two‐tailed Student's t‐test, and a P value of less than 0.05 was recognized as a statistically significance.
Author contributions
WZ, NQ, JL, RZ, YC, and CW conducted experiments. SC contributed to the mass spectrometric analysis and XL helped with viruses. FH and NQ organized the study and prepared the manuscript with the help of SQ. All authors discussed the results and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (91754117 and 31671471 to Fajian Hou) and National Science Foundation for Young Scientists of China (31600700 to Nan Qi) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB19000000). We thank Dr. Qiang Zou (Shanghai Jiao Tong University School of Medicine) for providing traf3ip3 fl/fl mice and Dr. Weiguo Zou (Shanghai Institute of Biochemistry and Cell Biology) for providing EIIA‐cre (E2a‐cre) transgenic mice.
The EMBO Journal (2019) 38: e102075
Contributor Information
Nan Qi, Email: qinan@zjut.edu.cn.
Fajian Hou, Email: fhou@sibcb.ac.cn.
References
- Aoshi T, Koyama S, Kobiyama K, Akira S, Ishii KJ (2011) Innate and adaptive immune responses to viral infection and vaccination. Curr Opin Virol 1: 226–232 [DOI] [PubMed] [Google Scholar]
- Belgnaoui SM, Paz S, Samuel S, Goulet ML, Sun Q, Kikkert M, Iwai K, Dikic I, Hiscott J, Lin R (2012) Linear ubiquitination of NEMO negatively regulates the interferon antiviral response through disruption of the MAVS‐TRAF3 complex. Cell Host Microbe 12: 211–222 [DOI] [PubMed] [Google Scholar]
- von Blume J, Alleaume AM, Kienzle C, Carreras‐Sureda A, Valverde M, Malhotra V (2012) Cab45 is required for Ca(2 + )‐dependent secretory cargo sorting at the trans‐Golgi network. J Cell Biol 199: 1057–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, Chen ZJ (2014) Prion‐like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 156: 1207–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sun L, Chen ZJ (2016) Regulation and function of the cGAS‐STING pathway of cytosolic DNA sensing. Nat Immunol 17: 1142–1149 [DOI] [PubMed] [Google Scholar]
- Chow KT, Gale M Jr, Loo YM (2018) RIG‐I and other RNA sensors in antiviral immunity. Annu Rev Immunol 36: 667–694 [DOI] [PubMed] [Google Scholar]
- Dadgostar H, Doyle SE, Shahangian A, Garcia DE, Cheng G (2003) T3JAM, a novel protein that specifically interacts with TRAF3 and promotes the activation of JNK11 Nucleotide sequence data reported are available in the DDB/EMBL/GenBank databases under accession number AY383616. FEBS Lett 553: 403–407 [DOI] [PubMed] [Google Scholar]
- Fang R, Jiang Q, Zhou X, Wang C, Guan Y, Tao J, Xi J, Feng JM, Jiang Z (2017) MAVS activates TBK1 and IKKepsilon through TRAFs in NEMO dependent and independent manner. PLoS Pathog 13: e1006720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galen JV, Blume JV (2013) Enrichment of Golgi membranes from HeLa cells by sucrose gradient ultracentrifugation. Bio Protoc 3: e906 [Google Scholar]
- Guo B, Cheng G (2007) Modulation of the interferon antiviral response by the TBK1/IKKi adaptor protein TANK. J Biol Chem 282: 11817–11826 [DOI] [PubMed] [Google Scholar]
- Harbury PB, Zhang T, Kim PS, Alber T (1993) A switch between two‐, three‐, and four‐stranded coiled coils in GCN4 leucine zipper mutants. Science 262: 1401–1407 [DOI] [PubMed] [Google Scholar]
- Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ (2011) MAVS forms functional prion‐like aggregates to activate and propagate antiviral innate immune response. Cell 146: 448–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S (2005) IPS‐1, an adaptor triggering RIG‐I‐ and Mda5‐mediated type I interferon induction. Nat Immunol 6: 981–988 [DOI] [PubMed] [Google Scholar]
- Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, Gerlier D, Cusack S (2011) Structural basis for the activation of innate immune pattern‐recognition receptor RIG‐I by viral RNA. Cell 147: 423–435 [DOI] [PubMed] [Google Scholar]
- Lei Y, Wen H, Yu Y, Taxman DJ, Zhang L, Widman DG, Swanson KV, Wen KW, Damania B, Moore CB et al (2012) The mitochondrial proteins NLRX1 and TUFM form a complex that regulates type I interferon and autophagy. Immunity 36: 933–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HM, Loo YM, Horner SM, Zornetzer GA, Katze MG, Gale M Jr (2012) The mitochondrial targeting chaperone 14‐3‐3epsilon regulates a RIG‐I translocon that mediates membrane association and innate antiviral immunity. Cell Host Microbe 11: 528–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Chen J, Cai X, Wu J, Chen X, Wu YT, Sun L, Chen ZJ (2013) MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLife 2: e00785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV et al (2015) Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347: aaa2630 [DOI] [PubMed] [Google Scholar]
- Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, Wang P, Zhao K, Hou J, Wang X et al (2017) The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63‐linked polyubiquitination. Nat Immunol 18: 214–224 [DOI] [PubMed] [Google Scholar]
- Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez‐Sobrido L, Akira S, Gill MA, Garcia‐Sastre A, Katze MG et al (2008) Distinct RIG‐I and MDA5 signaling by RNA viruses in innate immunity. J Virol 82: 335–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo D, Ding SC, Vela A, Kohlway A, Lindenbach BD, Pyle AM (2011) Structural insights into RNA recognition by RIG‐I. Cell 147: 409–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J (2005) Cardif is an adaptor protein in the RIG‐I antiviral pathway and is targeted by hepatitis C virus. Nature 437: 1167–1172 [DOI] [PubMed] [Google Scholar]
- Michallet MC, Meylan E, Ermolaeva MA, Vazquez J, Rebsamen M, Curran J, Poeck H, Bscheider M, Hartmann G, Konig M et al (2008) TRADD protein is an essential component of the RIG‐like helicase antiviral pathway. Immunity 28: 651–661 [DOI] [PubMed] [Google Scholar]
- Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti‐Loper MA, Madden VJ, Sun L, Ye Z et al (2008) NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451: 573–577 [DOI] [PubMed] [Google Scholar]
- tenOever BR, Sharma S, Zou W, Sun Q, Grandvaux N, Julkunen I, Hemmi H, Yamamoto M, Akira S, Yeh WC et al (2004) Activation of TBK1 and IKKvarepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J Virol 78: 10636–10649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm NW, Medzhitov R (2009) Pattern recognition receptors and control of adaptive immunity. Immunol Rev 227: 221–233 [DOI] [PubMed] [Google Scholar]
- Paz S, Vilasco M, Werden SJ, Arguello M, Joseph‐Pillai D, Zhao T, Nguyen TL, Sun Q, Meurs EF, Lin R et al (2011) A functional C‐terminal TRAF3‐binding site in MAVS participates in positive and negative regulation of the IFN antiviral response. Cell Res 21: 895–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisley A, Wu B, Xu H, Chen ZJ, Hur S (2014) Structural basis for ubiquitin‐mediated antiviral signal activation by RIG‐I. Nature 509: 110–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S, Wang K, Gu Y, Chen Y, Nan X, Xing J, Cui Q, Chen Y, Ge Q, Zhao H (2015) TRAF3IP3, a novel autophagy up‐regulated gene, is involved in marginal zone B lymphocyte development and survival. Clin Exp Immunol 182: 57–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda G, Ea CK, Chen ZJ (2007) Ubiquitination and TRAF signaling. Adv Exp Med Biol 597: 80–92 [DOI] [PubMed] [Google Scholar]
- Qi N, Shi Y, Zhang R, Zhu W, Yuan B, Li X, Wang C, Zhang X, Hou F (2017) Multiple truncated isoforms of MAVS prevent its spontaneous aggregation in antiviral innate immune signalling. Nat Commun 8: 15676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha SK, Pietras EM, He JQ, Kang JR, Liu SY, Oganesyan G, Shahangian A, Zarnegar B, Shiba TL, Wang Y et al (2006) Regulation of antiviral responses by a direct and specific interaction between TRAF3 and Cardif. EMBO J 25: 3257–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF‐kappaB and IRF 3. Cell 122: 669–682 [DOI] [PubMed] [Google Scholar]
- Shi Y, Yuan B, Qi N, Zhu W, Su J, Li X, Qi P, Zhang D, Hou F (2015) An autoinhibitory mechanism modulates MAVS activity in antiviral innate immune response. Nat Commun 6: 7811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Yuan B, Zhu W, Zhang R, Li L, Hao X, Chen S, Hou F (2017) Ube2D3 and Ube2N are essential for RIG‐I‐mediated MAVS aggregation in antiviral innate immunity. Nat Commun 8: 15138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, Chen ZJ (2006) The specific and essential role of MAVS in antiviral innate immune responses. Immunity 24: 633–642 [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140: 805–820 [DOI] [PubMed] [Google Scholar]
- Tan X, Sun L, Chen J, Chen ZJ (2018) Detection of microbial infections through innate immune sensing of nucleic acids. Annu Rev Microbiol 72: 447–478 [DOI] [PubMed] [Google Scholar]
- Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P (2009) Isolation of mitochondria‐associated membranes and mitochondria from animal tissues and cells. Nat Protoc 4: 1582–1590 [DOI] [PubMed] [Google Scholar]
- Wu B, Peisley A, Tetrault D, Li Z, Egelman EH, Magor KE, Walz T, Penczek PA, Hur S (2014) Molecular imprinting as a signal‐activation mechanism of the viral RNA sensor RIG‐I. Mol Cell 55: 511–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB (2005) VISA is an adapter protein required for virus‐triggered IFN‐beta signaling. Mol Cell 19: 727–740 [DOI] [PubMed] [Google Scholar]
- Yan BR, Zhou L, Hu MM, Li M, Lin H, Yang Y, Wang YY, Shu HB (2017) PKACs attenuate innate antiviral response by phosphorylating VISA and priming it for MARCH5‐mediated degradation. PLoS Pathog 13: e1006648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG‐I has an essential function in double‐stranded RNA‐induced innate antiviral responses. Nat Immunol 5: 730–737 [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Fujita T (2010) Recognition of viral nucleic acids in innate immunity. Rev Med Virol 20: 4–22 [DOI] [PubMed] [Google Scholar]
- Yu X, Teng XL, Wang F, Zheng Y, Qu G, Zhou Y, Hu Z, Wu Z, Chang Y, Chen L et al (2018) Metabolic control of regulatory T cell stability and function by TRAF3IP3 at the lysosome. J Exp Med 215: 2463–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Xu M, Liu S, Sun L, Chen ZJ (2009) Key role of Ubc5 and lysine‐63 polyubiquitination in viral activation of IRF3. Mol Cell 36: 315–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ (2010) Reconstitution of the RIG‐I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 141: 315–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhu L, Feng P (2014) Dissecting innate immune signaling in viral evasion of cytokine production. J Vis Exp 85: e51078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Q, Jin J, Xiao Y, Hu H, Zhou X, Jie Z, Xie X, Li JY, Cheng X, Sun SC (2015) T cell development involves TRAF3IP3‐mediated ERK signaling in the Golgi. J Exp Med 212: 1323–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6