

Abstract
From bacteria to mammalian cells, damaged DNA is sensed and targeted by DNA repair pathways. In eukaryotes, kinases play a central role in coordinating the DNA damage response. DNA damage signaling kinases were identified over two decades ago and linked to the cell cycle checkpoint concept proposed by Weinert and Hartwell in 1988. Connections between the DNA damage signaling kinases and DNA repair were scant at first, and the initial perception was that the importance of these kinases for genome integrity was largely an indirect effect of their roles in checkpoints, DNA replication, and transcription. As more substrates of DNA damage signaling kinases were identified, it became clear that they directly regulate a wide range of DNA repair factors. Here, we review our current understanding of DNA damage signaling kinases, delineating the key substrates in budding yeast and humans. We trace the progress of the field in the last 30 years and discuss our current understanding of the major substrate regulatory mechanisms involved in checkpoint responses and DNA repair.
Keywords: checkpoint, DNA damage response, DNA repair, kinase, mass spectrometry
Subject Categories: DNA Replication, Repair & Recombination
Glossary
- BER
Base excision repair
- BIR
Break‐induced replication
- CDK
Cyclin‐dependent kinase
- DDK
Dbf4‐dependent kinase
- dNTP
deoxyribonucleotide
- GCR
Gross chromosomal rearrangement
- HR
Homologous recombination
- ICL
Interstrand crosslink
- IR
Ionizing radiation
- MMR
Mismatch repair
- NER
Nucleotide excision repair
- NHEJ
Non‐homologous end joining
- PIKK
PI3 kinase‐related kinase
- RNR
Ribonucleotide reductase
- SAC
Spindle assembly checkpoint
- SDSA
Synthesis‐dependent strand annealing
- SSA
Single‐strand annealing
Introduction
From pathway to network
In eukaryotes, kinases play a central role in the DNA damage response, from sensing DNA damage to regulating cellular processes. Work in yeast and mammalian systems in the 1990s identified an evolutionarily conserved set of DNA damage signaling kinases, including phosphatidylinositol 3′ kinase (PI3K)‐related kinases (PIKKs) and PIKK‐regulated downstream kinases. These kinases were found to be involved in cell cycle control (Allen et al, 1994; Carr, 1995; Morrow et al, 1995; Savitsky et al, 1995; Cimprich et al, 1996; Furnari et al, 1997; Peng et al, 1997; Sanchez et al, 1997) and were linked to the “cell cycle checkpoint” concept proposed by Weinert and Hartwell (Weinert & Hartwell, 1988; Hartwell & Weinert, 1989). Subsequent work in the late 1990s and early 2000s revealed how these kinases establish the checkpoint and control processes beyond the cell cycle, such as apoptosis, transcription, and DNA replication (Santocanale & Diffley, 1998; Sun et al, 1998; Zhou & Elledge, 2000). In 2007, the use of mass spectrometry (MS)‐based proteomics allowed a more systematic analysis of the network of phosphorylation events triggered by DNA damage signaling kinases (Matsuoka et al, 2007; Smolka et al, 2007). As a result, the perception that DNA damage signaling kinases operate within a simple signaling pathway (Fig 1; the classical “linear” depiction of DNA damage signaling) evolved to a more comprehensive view in which DNA damage signaling kinases function in an elaborate signaling network comprised of hundreds of substrates.
Figure 1. DNA damage signaling via PIKKs and checkpoint kinases in budding yeast and humans.
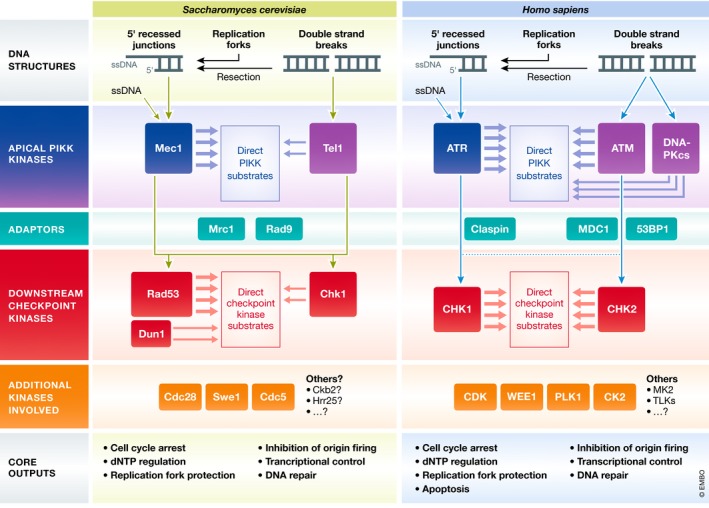
DNA damage signaling is initiated at DNA structures that form during DNA damage or replication stress, including single‐strand DNA (ssDNA) and broken DNA ends. The apical PIKKs are recruited to these structures and become activated to initiate downstream signaling. Mec1/ATR is recruited to RPA‐coated ssDNA, while Tel1/ATM and DNA‐PKcs initially associate with DNA ends formed by double‐strand breaks. Adaptor proteins are often required to mediate the transfer of phosphorylation from apical to downstream checkpoint kinases. Apical and downstream checkpoint kinases function coordinately to mediate cellular responses to DNA damage, either directly or through the regulation of additional downstream kinases. PIKKs also target an extensive network of substrates independently of downstream checkpoint kinases.
From checkpoint to DNA repair
After the discovery of DNA damage signaling kinases, mechanistic links between these kinases and the DNA repair machinery were virtually non‐existent. It was not immediately appreciated that these kinases directly target and regulate the DNA repair machinery. Now that dozens of DNA repair proteins have been shown to be phosphorylated by DNA damage signaling kinases, there is little doubt that active and direct control of the DNA repair machinery is a core function of DNA damage signaling kinases. However, the precise mechanisms by which these kinases control the action of these substrates remain incompletely understood and represent a significant knowledge gap in the field. Here, we review our current understanding of the integrated action of DNA damage signaling kinases. We delineate the key substrates for checkpoint and DNA repair in budding yeast and highlight the potential parallels in humans. Based on the accumulated knowledge of the mechanisms of substrate regulation, we discuss how our understanding of the action of DNA damage signaling kinases in genome maintenance has evolved over the last 30 years.
DNA damage signaling kinases
DNA damage signaling kinases have been traditionally categorized as either apical or effector kinases. The apical PIKKs, or ATR, ATM, and DNA‐PKcs in mammals and Mec1 and Tel1 in budding yeast (Fig 1; see Table 1 for gene name overview in model organisms), associate with DNA structures that form as byproducts of DNA damage or replication stress, including single‐strand DNA (ssDNA) and broken DNA ends (Fig 1). In the canonical mode of action, Mec1/ATR is recruited to ssDNA, whereas Tel1/ATM and DNA‐PKcs associate with the ends of double‐strand DNA (dsDNA) breaks. The mechanism of activation for each of these kinases is different, but recruitment to damaged DNA is often a requirement (for details on the mechanism of how kinases associate with DNA structures, as well as co‐factors involved, refer to the following reviews: Blackford & Jackson, 2017; Di Domenico et al, 2014; Saldivar et al, 2017; Zou, 2013). Activated apical kinases transfer stimulatory phosphorylation to the downstream checkpoint kinases (Rad53, Chk1, and Dun1 in yeast; CHK2 and CHK1 in mammals), which catalyze phosphorylation events that mediate cellular responses to DNA damage as part of the canonical DNA damage checkpoint (Fig 1). Whereas Rad53 mediates nearly all checkpoint‐related functions in budding yeast, with Chk1 playing only a minor role (Sanchez et al, 1999), a more balanced division of labor exists for CHK1 and CHK2 in humans. Human CHK2 shares sequence and structural similarities with yeast Rad53, including an FHA domain (Matsuoka et al, 1998), but the functional similarities are limited to the DSB signaling response. Human CHK1 and yeast Rad53 play a crucial role in the replication stress response, but they share little or no sequence/structural similarity.
Table 1.
Key protein names in budding yeast, fission yeast, and humans
| Budding yeast | Fission yeast | Humans | |
|---|---|---|---|
| Apical PIKK kinase | Mec1 | Rad3 | ATR |
| Tel1 | Tel1 | ATM | |
| DNA‐PKcs | |||
| Downstream checkpoint kinase | Rad53 | Cds1 | CHK2/CHK1a |
| Chk1 | Chk1 | CHK1? | |
| MRN complex | Mre11 | Mre11 | MRE11 |
| Rad50 | Rad50 | RAD50 | |
| Xrs2 | Nbs1 | NBS1 | |
| ATR cofactor | Lcd1 (Ddc2) | Rad26 | ATRIP |
| 9‐1‐1 complex | Ddc1 | Rad9 | RAD9 |
| Mec3 | Hus1 | HUS1 | |
| Rad17 | Rad1 | RAD1 | |
| ATR activators | Dpb11 | Rad4/Cut5 | TOPBP1 |
| Dna2 | |||
| Ddc1 | |||
| ETAA1 | |||
| Adaptors | Rad9 | Crb2 | MDC1/53BP1 |
| Mrc1 | Mrc1 | CLASPIN |
Human CHK1 is a functional analog of yeast Rad53.
Adaptor proteins as substrates for downstream signaling activation
In budding yeast, the transfer of phosphorylation from apical to downstream checkpoint kinases requires the checkpoint adaptor proteins Rad9 and Mrc1, which recruit the downstream checkpoint kinases in proximity to the apical kinase. Mec1 and Tel1 promote the recruitment of the Rad9 adaptor by phosphorylating lesion‐proximal substrates, such as histone H2A (Downs et al, 2000) and the 9‐1‐1 complex (Paciotti et al, 1998), which are then recognized directly or indirectly by Rad9 (Fig 2; Toh et al, 2006; Hammet et al, 2007; Pfander & Diffley, 2011). Once recruited, Rad9 is phosphorylated by Mec1 or Tel1 (Emili, 1998; Vialard et al, 1998), which promotes its oligomerization (Soulier & Lowndes, 1999; Usui et al, 2009) and further stabilization on DNA (Naiki et al, 2004). Mec1‐ and Tel1‐mediated phosphorylation of Rad9 creates docking sites for the recruitment of the downstream effector kinase Rad53 (Fig 2; Gilbert et al, 2001; Schwartz et al, 2002; Sun et al, 1998), which, upon recruitment to Rad9, is phosphorylated and activated by Mec1 or Tel1 (Sanchez et al, 1996; Sun et al, 1996). Chk1 also relies on Rad9 for its activation by Mec1 (Blankley & Lydall, 2004); however, unlike Rad53, the phosphorylation events that facilitate the recruitment of Chk1 to Rad9 are catalyzed by cyclin‐dependent kinase (CDK; Fig 2; Abreu et al, 2013). Rad53 can also be activated via the Mrc1 adaptor (Alcasabas et al, 2001; Osborn & Elledge, 2003). Mrc1, being an intrinsic component of the replisome, is already “on‐site” for mediating activation, obviating the need for a devoted recruitment mechanism, as is the case with Rad9. In fact, Mrc1 mediates a more rapid response to replication stress than Rad9‐dependent DNA damage signaling (Pardo et al, 2017; Bacal et al, 2018). Similar to Rad9 and Mrc1, Sgs1 has been proposed to mediate Rad53 recruitment in a manner that depends upon its phosphorylation by Mec1 (Hegnauer et al, 2012). Mec1 and Tel1 also facilitate DNA damage signaling activation and propagation by recruiting chromatin modifiers and remodelers near sites of DNA damage (van Attikum et al, 2004; Downs et al, 2004; Morrison et al, 2004, 2007), which may help de‐condense chromatin in a way that permits adaptor assembly.
Figure 2. Recruitment of DNA damage signaling kinases and adaptor proteins to DNA lesions: conserved features between budding yeast and humans.
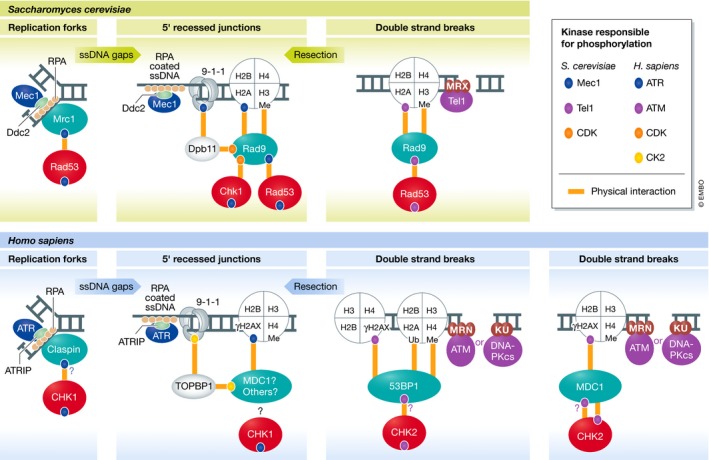
Phosphorylation and adaptor proteins play a key role in the recruitment of downstream checkpoint kinases. The colored ovals indicate phosphorylation events mediated by DNA damage signaling kinases (see kinase key). The orange lines indicate protein–protein interactions promoted by the indicated phosphorylation events (also methylation (me) or ubiquitylation (Ub)). Activation of the downstream checkpoint kinases by the apical PIKK kinases requires adaptor proteins (outlined in green). In most cases, these adaptor proteins act as scaffolds to directly bind to and recruit the downstream checkpoint kinase. The model, mostly based on extensive work in yeast, posits that the recruitment of the downstream checkpoint kinase to the proximity of the apical PIKK kinase enables the phosphorylation and activation of the downstream checkpoint kinase. In addition to activating the downstream checkpoint kinase, phosphorylation events mediated by the apical PIKK kinases are critical for scaffold assembly, often promoting protein–protein interactions. Accordingly, a conserved feature of several adaptor proteins in budding yeast and humans is the presence of protein domains responsible for binding phosphorylated proteins (FHA and BRCT domains). Notably, other kinases such as CDK and CK2 also catalyze phosphorylation events involved in adaptor recruitment, although these events are often not induced by DNA damage. For DNA‐PKcs, while this kinase has been implicated in the phosphorylation of H2AX and 53BP1, it does not seem to be involved in CHK2 phosphorylation.
Similar as in yeast, vertebrate ATM and ATR utilize checkpoint adaptor proteins to mediate their transfer of phosphorylation to the checkpoint effector kinases. ATR primarily relies on Claspin, the homolog of yeast Mrc1, to mediate the activation of CHK1 (Kumagai & Dunphy, 2000). Like Mrc1, Claspin associates with the replisome (Lee et al, 2003) and recruits CHK1 upon exposure to replication stress. Also similar to yeast, the Claspin–CHK1 interaction depends on ATR activity (Kumagai & Dunphy, 2003; Lindsey‐Boltz et al, 2009). While it is currently unknown whether mammalian ATR directly phosphorylates Claspin, Xenopus ATR has been shown to directly phosphorylate Claspin at threonines 817 and 819, which are critical for CHK1 recruitment (Fig 2; the uncertainty of ATR's direct phosphorylation of Claspin in humans is denoted by “?”; Yoo et al, 2006; in‐depth discussion of the similarities and differences between Claspin orthologs can be found here: Smits et al, 2019). Once bound to CHK1, Claspin is thought to both stabilize CHK1 and tether it in proximity to ATR, allowing for extensive phosphorylation and full activation of the effector kinase (Liu et al, 2006). While ATR utilizes Claspin to facilitate its phosphorylation of CHK1, the phosphorylation of many other ATR substrates does not require Claspin, suggesting that, like in yeast, the adaptor proteins are primarily responsible for facilitating Mec1/ATR's phosphorylation of the effector kinases and are not required for the phosphorylation of other substrates (like many of those depicted below in Fig 4).
Two mammalian adaptors have been linked to the ATM–CHK2 signaling axis: MDC1 and 53BP1. Despite extensive research on these proteins, it remains unclear precisely how they function in transducing ATM signaling toward CHK2 activation. While 53BP1 is the functional ortholog of yeast Rad9, MDC1 also shares functional similarities with Rad9 in the context of the DNA damage signaling response. Similar to Rad9, MDC1 possesses BRCT domains that directly bind to phosphorylated histone H2AX (Fig 2; γH2AX; analogous to histone H2A phosphorylated at the C‐terminal SQ site in yeast; Stucki et al, 2005). Once associated with γH2AX at DNA breaks, MDC1 is phosphorylated by ATM and contributes to CHK2 activation (Goldberg et al, 2003; Peng & Chen, 2003; Stewart et al, 2003; Wu et al, 2008). MDC1 has also been shown to be important for CHK1 activation through its interaction with the TOPBP1 scaffold (Wang et al, 2011; Leung et al, 2013), which may functionally resemble the Rad9–Dpb11 interaction in budding yeast (Fig 2).
ATM promotes the recruitment of 53BP1 through two distinct mechanisms. Similar to yeast Rad9, 53BP1 recognizes γH2AX through its C‐terminal pair of BRCT domains (Fig 2), an interaction which has been controversial, but has recently gained additional support (Baldock et al, 2015; Kleiner et al, 2015). Nonetheless, the prominent mechanism of 53BP1 recruitment to DNA breaks involves ATM‐ and γH2AX‐mediated recruitment of MDC1, which becomes phosphorylated by ATM and recruits the E3 ubiquitin ligases RNF8 and RNF168, leading to ubiquitylation of H2A that is recognized directly by 53BP1 (Fig 2; for a detailed review, Hustedt & Durocher, 2016). Notably, this ubiquitylation‐dependent recruiting mechanism is absent in budding yeast. Consistent with an adaptor function for 53BP1, it interacts with CHK2, and the loss of 53BP1 results in reduced ATM‐mediated phosphorylation of CHK2 in response to low doses of ionizing radiation (IR; Wang et al, 2002). However, 53BP1 appears to regulate activation of the checkpoint through a more complex mechanism, as the physical interaction between CHK2 and 53BP1 rapidly decreases upon IR radiation rather than becoming stabilized (Wang et al, 2002), in contrast to the Rad9–Rad53 interaction, which increases after DNA damage in budding yeast. Both 53BP1 and Rad9 play a key role in the control of DNA end resection, highlighting the connection and coordination between checkpoint signaling and the regulation of DNA repair (discussed in detail later in this review; Lazzaro et al, 2008; Bunting et al, 2010; Chapman et al, 2013; Zimmermann et al, 2013; Ferrari et al, 2015; Liu et al, 2017). In both cases, phosphorylation of Rad9/53BP1 by Mec1 or Tel1 in yeast or ATM in humans is essential for suppressing DNA end resection (Bothmer et al, 2011; Ferrari et al, 2015), and it is possible that 53BP1's function in preventing resection contributes to the stabilization of ATM at breaks, which may indirectly promote ATM–CHK2 signaling.
Post‐recruitment events in downstream checkpoint kinase activation
Recruitment to sites of DNA lesions via checkpoint adaptors enables the downstream checkpoint kinases to be directly phosphorylated by the upstream PIKKs, triggering initial kinase activation and subsequent autophosphorylation for further kinase activation. In the case of Rad53, for example, initial phosphorylation by Mec1 or Tel1 promotes its kinase activity by interfering with a kinase auto‐inhibitory domain (Fiorani et al, 2008), which then stimulates Rad53 to phosphorylate other Rad9‐bound Rad53 molecules (Jia‐Lin Ma & Stern, 2008). Such trans‐autophosphorylation events contribute to dissociate Rad53 from Rad9 and prevent further Rad9 oligomerization (Usui et al, 2009). Notably, overexpression of Rad53 in bacteria cells, which lack PIKKs and checkpoint adaptors, results in hyper‐activated Rad53 (Gilbert et al, 2001). This finding supports a model whereby increased local concentration of Rad53 is enough for activation, with adaptors building increased local concentration at the site of lesions and PIKKs facilitating the initial trigger by reducing the minimal concentration threshold required for activation. In mammals, the apical kinases ATR and ATM drive the key events leading to activation of CHK1 and CHK2, respectively. ATR‐ and ATM‐mediated phosphorylation not only recruits CHK1 and CHK2 to sites of DNA lesions, but also directly phosphorylates these downstream kinases to promote their activation. Like in yeast, these priming phosphorylation events are mainly required to relieve inhibitory domains or to drive monomer‐to‐oligomer kinase transition (reviewed in Bartek & Lukas, 2003). ATM‐mediated phosphorylation of CHK2 at threonine 68, an established marker of CHK2 activation, allows for the dimerization of two inactive CHK2 monomers and for their subsequent trans‐ and cis‐phosphorylation (Ahn et al, 2000; Xu et al, 2002; Schwarz et al, 2003). CHK2 dimerization is a transient state, since the multiple trans‐ and cis‐autophosphorylation events promote rapid dimer dissociation, leading to full active monomers (Ahn & Prives, 2002; Xu et al, 2002; Cai et al, 2009). Similar to CHK2, CHK1 activation is a multistep process that requires ATR phosphorylation at serine 317 and serine 345; however, unlike Rad53 or CHK2, CHK1 activation does not appear to involve dimerization or oligomerization (Liu et al, 2000; Zhao & Piwnica‐Worms, 2001).
Substrates mediating the core DNA damage signaling responses
Once activated, DNA damage signaling kinases mediate hallmark responses that include the arrest of the cell cycle, inhibition of origin firing, protection and restart of stalled replication forks, induction of a transcriptional response, initiation of apoptosis, and control of dNTP levels. More recent work has demonstrated that the DNA damage signaling kinases also regulate a range of other processes, such as autophagy, gene gating, chromosome mobility, transcription–replication conflicts, and many more whose mechanistic connections to DNA damage signaling and degrees of conservation across eukaryotes remain less clear. Here, we focus on a select set of core conserved functions of the DNA damage signaling kinases with defined substrates, delineating the parallels between budding yeast and humans (Fig 3).
Figure 3. Yeast‐to‐human parallels in core checkpoint responses mediated by DNA damage signaling.
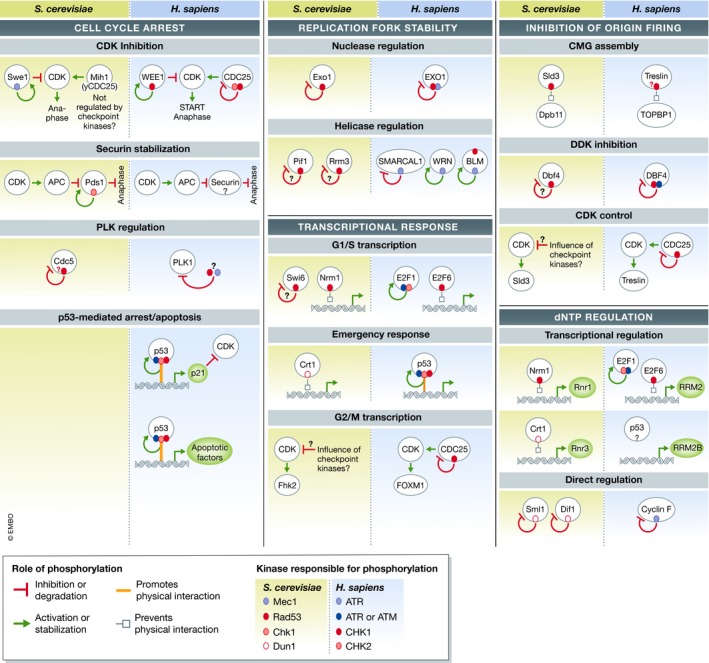
Substrate map highlighting the phosphorylation events involved in core DNA damage signaling responses in yeast and humans (see text for detailed discussion of each substrate). Conserved or functionally analogous phosphorylation events are positioned parallel to each another. The colored ovals indicate phosphorylation events mediated by DNA damage signaling kinases (see “kinase‐dependency” key). The arrows or lines that emanate from the colored ovals represent the role phosphorylation plays in regulating that protein (see “role of phosphorylation” key). Question marks indicate uncertainty, either in the functionality of the phosphorylation event or in the identity of the kinase or substrate. Arrows that impinge on CDK demonstrate how DNA damage signaling can indirectly inhibit CDK activity.
Cell cycle control
The most classical and widely known function of DNA damage signaling kinases is the imposition of a cell cycle arrest that prevents entry into mitosis. Exactly how this arrest is imposed in budding yeast remains elusive. The paradigm for how cell cycle arrest occurs comes from a series of works conducted in fission yeast and metazoans in 1997 (Furnari et al, 1997; Peng et al, 1997; Sanchez et al, 1997; Weinert, 1997), which revealed that DNA damage signaling inhibits CDC25, a phosphatase that removes inhibitory phosphorylation at a key tyrosine residue in mitotic‐CDK (M‐CDK; Fig 3). In addition, DNA damage signaling kinases stimulate WEE1 (O'Connell et al, 1997; Boddy et al, 1998; Lee et al, 2001), a kinase responsible for phosphorylating the same inhibitory tyrosine site (Fig 3; Mueller et al, 1995). While DNA damage signaling in budding yeast does not appear to impinge upon the Cdc25 phosphatase homolog Mih1, the budding yeast kinase Swe1 (the WEE1 homolog) is regulated similar to its counterpart in higher eukaryotes and fission yeast. Swe1 is likely phosphorylated and activated by DNA damage signaling, which results in inhibition of M‐CDK (Fig 3; Edenberg et al, 2015; Palou et al, 2015). DNA damage signaling in budding yeast is also able to suppress M‐CDK activity through additional redundant mechanisms that remain unclear (Palou et al, 2015). In addition to inhibiting M‐CDK‐dependent activation of the anaphase‐promoting complex (APC/C), DNA damage signaling in budding yeast more directly inhibits the onset of anaphase through Chk1, which phosphorylates and stabilizes the anaphase‐inhibiting Pds1/securin protein (Fig 3; Sanchez et al, 1999; Wang et al, 2001), preventing the separation of sister chromatids. Moreover, Rad53 can influence the mitotic spindle assembly checkpoint (SAC) through inhibition of the polo‐like kinase Cdc5 (Fig 3; Sanchez et al, 1999; Valerio‐Santiago et al, 2013; Zhang et al, 2009). Rad53‐dependent inactivation of Cdc5, particularly in response to telomere damage, prevents Cdc5's phosphorylation of the spindle assembly checkpoint protein Bfa1 (Valerio‐Santiago et al, 2013); however, it is unclear whether Rad53 phosphorylates Cdc5 directly (Fig 3). The inhibition of Cdc5 by DNA damage signaling also indirectly influences the resolution of joint molecules prior to M phase, as Cdc5 activity is important for promoting the activity of the Mus81 resolvase (Szakal & Branzei, 2013). Cdc5 has also been implicated in the down‐regulation of the DNA damage response (Donnianni et al, 2010; Vidanes et al, 2010). A more in‐depth review exploring Cdc5's complex relationship with the DNA damage response in yeast can be found in Botchkarev and Haber (2018). Similar to its budding yeast counterpart, the mammalian polo‐like kinase PLK1 is also inhibited by DNA damage signaling, albeit in an indirect manner (Fig 3; Bruinsma et al, 2017; Qin et al, 2013).
A critical mediator of cell cycle arrest in humans is the p53 transcription factor, whose classical function is to trigger the apoptotic program (reviewed in Chen, 2016). p53 is directly phosphorylated and stabilized by all the DNA damage signaling kinases, eliciting a p53‐dependent transcriptional response that impacts the DNA damage response (reviewed in Kruse & Gu, 2009). p53‐mediated expression of the CDK inhibitor protein p21 represents the primary mechanism by which p53 blocks progression through the cell cycle (Fig 3; Harper et al, 1993, 1995). Apical kinases in mammalian DNA damage signaling also phosphorylate the p53 inhibitor Mdm2, impairing its ability to promote p53 nuclear export and its subsequent degradation (Mayo et al, 1997; Maya et al, 2001; Shinozaki et al, 2003). Unlike most factors covered in Fig 3, Saccharomyces cerevisiae does not have clear p53 or Mdm2 homologs. That said, for reasons discussed in the section on dNTP regulation, the DNA damage signaling‐mediated control of p53 might functionally resemble the control of the Crt1 transcription regulator by Rad53 and Dun1 (Huang et al, 1998).
Fork stability and protection
During DNA replication, stalled replication forks may be targeted and degraded by nucleases. Since fork degradation pathways impair replication fork restart after stalling, leading to persistent DNA lesions, they are considered a major driving force of genomic instability (recently reviewed in Pasero & Vindigni, 2017; Patel & Weiss, 2018). In S. cerevisiae, Rad53 is believed to play a major role in fork stability. It is worth mentioning that Rad53 is essential, and that the lethality of rad53 cells can be rescued by deletion of SML1, an inhibitor of the ribonucleotide reductase enzyme responsible for catalyzing the rate‐limiting step in DNA precursor synthesis (Zhao et al, 1998). Nonetheless, even in the absence of SML1, Rad53 mutants are exquisitely sensitive to chemical agents that damage DNA or stall DNA replication forks (Allen et al, 1994; Sanchez et al, 1999; Gunjan & Verreault, 2003). This sensitivity to DNA damaging agents has been primarily attributed to Rad53's role in stabilizing and restarting stalled replication forks (reviewed in detail here: Segurado & Tercero, 2009). Rad53 protects stalled replication forks from nucleolytic processing by phosphorylating and inhibiting the Exo1 exonuclease (Fig 3; Cotta‐Ramusino et al, 2005; Morin et al, 2008; Segurado & Diffley, 2008). In addition, Rad53 regulates the Pif1 and Rrm3 helicases, potentially limiting replication fork reversal and subsequent fork degradation (Fig 3; Rossi et al, 2015). However, precisely how DNA damage signaling maintains replication fork integrity is unknown and represents a fundamental knowledge gap in the field. Recent work has revealed that Rad53 phosphorylates the replicative helicase component Cdc45, which in turn recruits and stabilizes Rad53 at replication complexes (Can et al, 2018). Identification of additional key substrates and recruiting mechanisms will be necessary to deconstruct Rad53's functions in maintaining fork stability, which may involve a range of redundant phosphorylation events in more than one essential replisome protein.
Mammalian DNA damage signaling kinases also play key roles in protecting stalled replication forks (Fig 3). Human EXO1 is phosphorylated by ATR, which promotes ubiquitylation‐dependent degradation of EXO1 and prevents chromosome fragmentation due to unrestrained EXO1 processing activity (El‐Shemerly et al, 2008; Tomimatsu et al, 2017). CHK1 also phosphorylates EXO1 directly on serine 746, creating a docking site for binding to 14‐3‐3 proteins, which prevent recruitment of EXO1 to chromatin and limit EXO1 action at stalled forks (Engels et al, 2011; Li et al, 2019). In addition, ATR governs the recruitment and/or stability of several helicases important for remodeling the stalled fork and promoting fork restart. For example, ATR phosphorylates the Werner syndrome helicase WRN at multiple S/T‐Q sites, promoting WRN‐RPA co‐localization at replication stress sites (Ammazzalorso et al, 2010). Mutation of these ATR sites causes stalled fork breakage and severely impacts the resumption of DNA replication. Several studies have demonstrated that the Bloom syndrome helicase, BLM, is directly phosphorylated by ATR and that this phosphorylation is important for both promoting replication fork restart after HU‐mediated arrest and suppressing dormant origin firing (Davies et al, 2004, 2007). CHK1 may also constitutively phosphorylate BLM to prevent proteasome‐dependent BLM degradation, suppress chromatin bridge formation, and promote an interaction with 53BP1 (Sengupta et al, 2004; Tripathi et al, 2007; Kaur et al, 2010; Petsalaki et al, 2014). Interestingly, in yeast, Rad53 and Sgs1 (a BLM ortholog) physically interact (Hegnauer et al, 2012), and Sgs1 has been reported to display a strong genetic interaction with Rad9, 53BP1's yeast homolog (Nielsen et al, 2013). Finally, David Cortez's group described an additional mechanism of fork stability regulation through the ATR's phosphorylation of the SMARCAL1 helicase (Couch et al, 2013). ATR‐dependent phosphorylation of SMARCAL1 inhibits fork remodeling activity, preventing subsequent pathological fork degradation by active nucleases (Couch et al, 2013; Kolinjivadi et al, 2017). Collectively, these examples highlight how yeast and human DNA damage signaling kinases protect replication fork integrity via the direct regulation of nucleases and helicases.
Inhibition of origin firing
One mechanism for preventing genome instability during replication stress is to inhibit the firing of late origins (reviewed in McIntosh & Blow, 2012; Yekezare et al, 2013). DNA damage signaling kinases control origin firing through two main pathways, which are conserved from yeast to higher eukaryotes (Fig 3). During genotoxic stress in S phase, Rad53 phosphorylates Dbf4, a subunit of the Dbf4‐dependent kinase (DDK) complex, and the Sld3 component of pre‐RCs (pre‐replication complexes) to inhibit the firing of late‐replicating origins (Lopez‐Mosqueda et al, 2010; Zegerman & Diffley, 2010), thereby slowing the progression of DNA synthesis and preventing the exhaustion of RPA (Toledo et al, 2013, 2017). While the mechanism by which Rad53 phosphorylation regulates DDK is unknown, the phosphorylation of Sld3 by Rad53 is thought to prevent the recruitment of Dpb11 to primed replication origins (Lopez‐Mosqueda et al, 2010; Zegerman & Diffley, 2010). In vertebrates, the Dpb11 ortholog TOPBP1 docks to CDK2‐phosphorylated Treslin (the Sld3 ortholog) to mediate origin firing (Kumagai et al, 2010; Boos et al, 2011). Conditions that perturb DNA replication and trigger CHK1 activation have been shown to disrupt the Treslin–TOPBP1 interaction (Boos et al, 2011) in a mechanism analogous to Rad53‐dependent control of the Sld3–Dpb11 interaction in S. cerevisiae. CHK1 has also been shown to directly phosphorylate Treslin in Xenopus egg extracts, inhibiting DNA replication (Guo et al, 2015). Metazoan ATR/ATM and CHK1 phosphorylate the DBF4 subunit of DDK, inhibiting pre‐RC assembly and origin firing (Costanzo et al, 2003; Heffernan et al, 2007; Lee et al, 2012). CHK1 is also assumed to inhibit origin firing indirectly by suppressing CDK activity via the inhibition of CDC25 (Shechter et al, 2004; Sorensen & Syljuasen, 2012). Consistent with this assumption, inhibition of CDK rescues replication stress sensitivity of cells lacking ATR or CHK1 signaling, likely by reducing origin firing and preventing RPA exhaustion (Toledo et al, 2013; Dungrawala et al, 2015). Paradoxically, Mec1 (and potentially ATR/ATM) also promotes DNA replication by phosphorylating core components of the replisome, such as the MCM helicase (Cortez et al, 2004; Yoo et al, 2004; Randell et al, 2010).
Transcriptional control
In yeast, Rad53 plays a central role in the transcriptional response to DNA damage and replication stress. In G1, Rad53 was proposed to influence the timing of START by phosphorylating Swi6, a component of the MBF/SBF transcription factor (analog of human E2F; Sidorova & Breeden, 1997, 2003). Rad53 can also regulate the transcription of MBF/SBF targets at the G1/S transition and during S phase through phosphorylation and inhibition of the Nrm1 transcriptional repressor protein, promoting transcription of the largest set of co‐regulated genes in the DNA damage response (Fig 3; Bastos de Oliveira et al, 2012; de Bruin et al, 2008; Travesa et al, 2012). In addition, Dun1 up‐regulates the transcription of a specific set of DNA damage‐induced genes, including subunits of ribonucleotide reductase (RNR), by inducing the phosphorylation and inactivation of the transcriptional repressor protein, Crt1 (Huang et al, 1998; Fig 3). In‐depth transcriptome analyses performed in budding yeast suggest that DNA damage signaling kinases may impinge upon other, as yet unknown, transcription factors (Jaehnig et al, 2013).
DNA damage signaling kinases profoundly impact multiple transcriptional programs in mammals (Fig 3). The E2F pathway (analogous to yeast SBF/MBF) represents a major transcriptional system regulated by DNA damage signaling kinases. ATM‐, ATR‐, and CHK2‐dependent phosphorylation activates E2F1 (Lin et al, 2001; Stevens et al, 2003), whereas CHK1 inhibits the transcriptional repressor E2F6 through direct phosphorylation (Bertoli et al, 2013a). Interestingly, CHK1's inhibition of E2F6 resembles the Rad53‐dependent inhibition of the S. cerevisiae transcriptional repressor Nrm1, as discussed above (more details can be found here: Bertoli et al, 2013b). Similar to yeast, E2F‐dependent transcription in humans includes a large set of genes that play a role in DNA replication, cell cycle, and DNA repair (reviewed here: Bracken et al, 2004; Poppy Roworth et al, 2015). Not surprisingly, inhibition of the ATR–CHK1 pathway causes major remodeling of the proteome via control of the E2F transcriptional circuit and strongly impacts the DNA repair machinery (Kim et al, 2018).
Finally, recent work in mammalian cells has revealed a role for ATR in enforcing a checkpoint between the S and G2 phases of the cell cycle. ATR suppresses CDK‐dependent phosphorylation and activation of the FOXM1 transcription factor by limiting CDK activity during S phase (Fig 3), thereby preventing a premature expression of G2‐specific genes (Saldivar et al, 2018). While the function of the FOXM1 transcription factor is conserved in yeast (Pic‐Taylor et al, 2004; Murakami et al, 2010), it is unclear whether the budding yeast checkpoint kinases also restrict the expression of G2/M‐specific transcripts through an analogous mechanism (Fig 3).
Regulation of dNTP production
In budding yeast, the regulation of dNTP production is considered an essential function of DNA damage signaling. This is supported by the fact that the lethality caused by deletion of MEC1 or RAD53 can be suppressed by deletion of SML1, an inhibitor of the ribonucleotide reductase enzyme complex, which catalyzes the limiting step in dNTP production (Zhao et al, 1998). The Rad53–Dun1 signaling axis induces the activity of the RNR complex by controlling several of its subunits via distinct mechanisms (Allen et al, 1994; Bashkirov et al, 2003; Fig 3). Once activated by Rad53, Dun1 directly phosphorylates Sml1, resulting in its degradation and the activation of the Rnr1 subunit (Zhao et al, 2001; Zhao & Rothstein, 2002; Lee et al, 2008). In addition, Dun1‐mediated phosphorylation of Dif1, an SML1 paralog, results in Dif1's degradation and the subsequent export of the Rnr2–Rnr4 subunits from the nucleus to the cytosol, which enables the formation of the active RNR complex (Lee et al, 2008). Dun1 also up‐regulates the transcription of RNR3 by inducing the phosphorylation and inactivation of the transcriptional repressor protein, Crt1 (Huang et al, 1998; Fig 3). Rad53 also plays a Dun1‐independent role in RNR control. For example, Rad53's phosphorylation of the transcriptional repressor protein Nrm1, upon exposure to replication stress, results in the MBF‐mediated induction of RNR1 transcription (Bastos de Oliveira et al, 2012; Huang et al, 1998; Travesa et al, 2012; Fig 3). Since cells lacking Dun1 are viable, in contrast to rad53Δ cells, it is possible that the essential function of Rad53 is due to both its Dun1‐dependent and Dun1‐independent roles in modulating RNR or, alternatively, a combination of Rad53's function in dNTP regulation (via Dun1) and other roles in, for example, replication fork stability.
Despite the critical importance of dNTP regulation in yeast, no clear Sml1, Dif1, or Dun1 orthologs or functional analogs have been identified so far in mammalian cells. Nevertheless, it is intriguing that an extra genomic copy of the RRM2 gene (RNR regulatory subunit) can extend the lifespan of ATR‐deficient mice (Lopez‐Contreras et al, 2015). Whether ATR directly phosphorylates RRM2 is to date not clear, although ATR may promote RRM2 stabilization by regulating the degradation of the Cyclin F subunit of the SCF ubiquitin ligase complex (Fig 3), which prevents SCF‐mediated degradation of RRM2 (D'Angiolella et al, 2012). RRM2 transcription is also induced by E2F and thus can be indirectly up‐regulated by the ATR–CHK1 kinases (as discussed in the section “transcriptional control”; Ishida et al, 2001). Interestingly, p53 promotes transcription of the RRM2B subunit of human RNR (Tanaka et al, 2000), and it is tempting to speculate on a potential analogy to the role of yeast Crt1 in inducing RNR3 transcription (Huang et al, 1998). In response to hypoxia, this p53‐dependent induction of RRM2B is important for maintaining the fidelity of DNA replication in low oxygen conditions (Foskolou et al, 2017). However, a direct link to the DNA damage signaling kinases has yet to be made.
Control of DNA repair by DNA damage signaling kinases
The control of DNA repair is a core function of DNA damage signaling kinases. Mounting genetic evidence from budding yeast illustrates the central role of DNA damage signaling to maintaining genome integrity. For example, using a genetic assay to assess chromosome instability in yeast, known as the gross chromosomal rearrangement (GCR) assay, the Kolodner Lab showed that cells lacking Mec1 and Tel1 exhibit extremely high rates of GCRs (Myung et al, 2001). In fact, mec1Δ tel1Δ cells are some of the most genetically unstable strains isolated to date. These cells undergo GCRs at a rate of five orders of magnitude higher than wild‐type cells (Myung et al, 2001). As discussed below, current evidence supports a model whereby the ability of Mec1 and Tel1 to suppress GCRs and related forms of genetic instabilities is associated with the role these kinases play in directly targeting and regulating components of the DNA repair machinery. In mammals, ATM and ATR are also responsible for maintaining chromosome stability (Xu et al, 1996; Brown & Baltimore, 2000); as shown in Fig 4, ATM and ATR phosphorylate and regulate a large set of DNA repair factors. Importantly, the high levels of genetic instability in yeast or mammalian cells lacking Mec1/ATR and Tel1/ATM have not yet been recapitulated by any combination of mutations of phosphorylation sites, suggesting that the key substrates through which these kinases suppress genomic instabilities remain unclear.
Figure 4. Yeast‐to‐human parallels in the regulation of DNA repair proteins by DNA damage signaling: substrates and mechanisms.
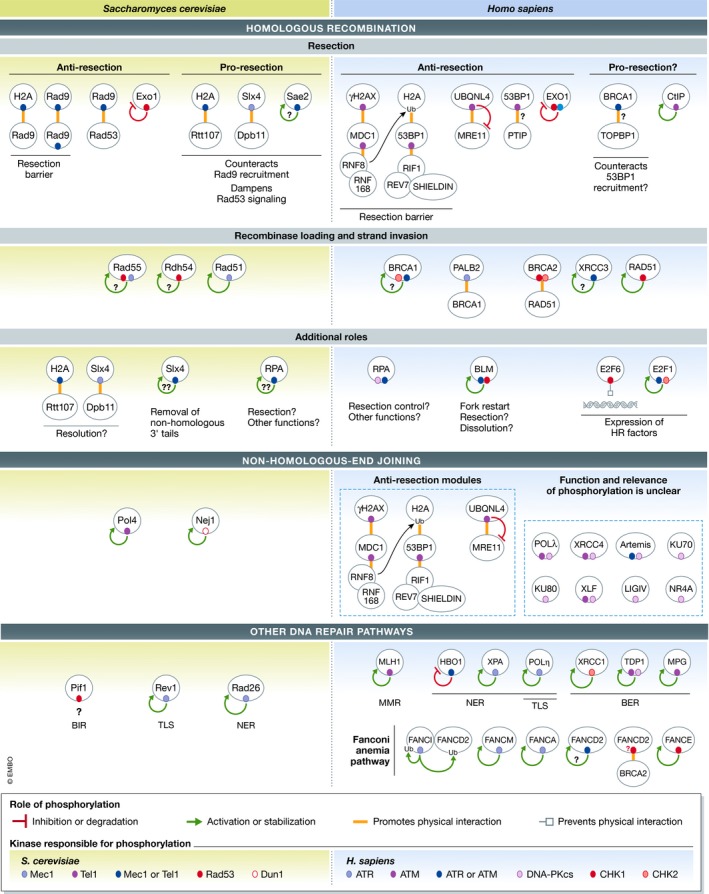
Substrate map cataloging the DNA damage signaling events regulating DNA repair proteins (see text for detailed discussion of each substrate). Conserved or analogous substrates involved in related DNA repair pathways are positioned parallel to each other. The colored ovals indicate phosphorylation events mediated by DNA damage signaling kinases (see “kinase‐dependency” key). The arrows or lines that emanate from the colored nodes represent the role phosphorylation plays in regulating that protein (see “role of phosphorylation” key). Question marks indicate uncertainty, either in the functionality of the phosphorylation event or in the identity of the kinase or substrate.
Phosphorylation control of the homologous recombination machinery: substrates and mechanisms
In both yeast and humans, DNA damage signaling kinases phosphorylate proteins in homologous recombination (HR)‐directed repair and have been shown to play an important regulatory role. For example, Mec1 phosphorylates Rtt107, Slx4, and Sgs1, all of which are known to play multiple roles in HR‐mediated repair, from the control of resection to the dissolution or resolution of joint chromosome structures (Sarbajna & West, 2014; Hang & Zhao, 2016; Cussiol et al, 2017; Guervilly & Gaillard, 2018). Recent work from our laboratory has revealed that phosphorylation of these proteins correlates with the ability of cells to suppress GCRs, suggesting that proper control of HR repair is essential for preventing chromosomal rearrangements (Lanz et al, 2018). Phosphorylation of Slx4 by Mec1 promotes DNA end resection, the first step in the HR process (Dibitetto et al, 2016; Liu et al, 2017). Mechanistically, Mec1 phosphorylation mediates the interaction between Slx4 and Dpb11, a multi‐BRCT domain scaffold that bridges Slx4 to the 9‐1‐1 clamp loaded at 5′ recessed junctions (Ohouo et al, 2010; Cussiol et al, 2015; Fig 4). Such stabilization of Slx4 at DNA lesions, which is also dependent on Rtt107's interaction with phosphorylated H2A (Fig 4), is believed to counteract a resection block promoted by Rad9, thereby allowing long‐range resection to occur (Dibitetto et al, 2016; Liu et al, 2017). Importantly, the role of Rad9 in counteracting resection relies on its oligomerization and Rad53 signaling (Clerici et al, 2014; Ferrari et al, 2015; Gobbini et al, 2015), which are both dependent on Mec1‐mediated phosphorylation events. Therefore, Mec1 plays opposing roles in HR, inhibiting resection via the Rad9–Rad53 signaling axis or promoting resection by mediating the Slx4–Dpb11 interaction (Fig 4).
Mec1's phosphorylation of Slx4 has also been linked to a role in single‐strand annealing (SSA), specifically in regulating cleavage of 3′ non‐homologous DNA tails by Rad1–Rad10 (Toh et al, 2010). In another example, which supports a key role for Mec1 in the control of HR, phosphorylation of multiple S/T‐Q residues in Sae2, a pro‐resection factor homologous to human CtIP, contributes to Mec1‐mediated GCR suppression (Liang et al, 2015). Furthermore, Mec1 phosphorylates the strand exchange factor Rad55, and Rad53 promotes the phosphorylation and DNA binding of the Tid1/Rdh54 translocase (Fig 4; Bashkirov et al, 2006; Ferrari et al, 2013; Herzberg et al, 2006). Mec1 also phosphorylates Rad51 at serine 192, likely supporting its ATPase activity and capacity for strand invasion (Fig 4; Flott et al, 2011).
The ssDNA binding protein RPA, which plays a critical role in coating resected DNA ends during the homologous recombination process, is one of the most established targets of the PIKKs. In both yeast and humans, RPA, through an interaction with Rad52, is thought to promote the loading of Rad51 on ssDNA (Park et al, 1996; Sugiyama et al, 1998; Sugiyama & Kowalczykowski, 2002). How PIKK phosphorylation regulates RPA function is not well understood, although studies point to a role in homologous recombination. RPA phosphorylation was initially proposed to promote the interaction between RPA and Rad52 (Deng et al, 2009); however, recent work using single‐molecule imaging reports that RPA phosphorylation inhibits DNA end resection via inhibition of the BLM helicase (Soniat et al, 2019). RPA phosphorylation in human cells has also been reported to impact DNA synthesis under stress (Vassin et al, 2009), the recruitment of factors to stalled replication forks (Murphy et al, 2014), and the imposition of the cell cycle checkpoint (Vassin et al, 2009). In budding yeast, mutation of the residues in RPA targeted by Mec1 and Tel1 has yet to yield an observable phenotype. Overall, the accumulated evidence supports a model whereby DNA damage signaling plays a key role in the control of the HR machinery by targeting multiple components that participate in discrete steps of the HR process (Fig 4).
ATR is a key regulator of homologous recombination‐mediated repair
ATR's primary action occurs in S phase, a period during the cell cycle where a sister DNA template is available for homology‐directed repair, and it functions mainly as a pro‐HR kinase. Depletion or inhibition of ATR impairs the ability of cells to utilize HR and leads to synergistic cell death with replication stress‐inducing drugs (Wang et al, 2004a; Vriend et al, 2016; Yazinski et al, 2017; Kim et al, 2018). One model proposes that during HR‐mediated repair, an ATM‐to‐ATR transition occurs, where ATM initiates resection and triggers the ATR activation that governs the later steps of homologous recombination (Cuadrado et al, 2006; Shiotani & Zou, 2009). Our laboratory proposed that ATR drives HR by promoting the stabilization of the pro‐resection factor BRCA1 at DNA lesions via interaction with TOPBP1 (human ortholog of Dpb11), which may counteract recruitment of the anti‐resection factor 53BP1 to sites of DNA lesions (Liu et al, 2017). It is tempting to speculate that this mechanism could be analogous to the Mec1‐mediated Dpb11–Slx4 interaction in yeast, which is also important for DSB resection (Dibitetto et al, 2016; Liu et al, 2017; Ohouo et al, 2010; Fig 4). A recent study from the Zou Lab provided additional insights into how ATR promotes HR‐mediated repair. They showed that while initial DSB resection requires CDK activity, later steps in HR require a “CDK‐to‐ATR switch” to promote proper recruitment of the key HR factors PALB2 and BRCA2, which are required for strand invasion. Mechanistically, ATR activated at resected ends recruits PALB2–BRCA2 to DNA damage sites by phosphorylating PALB2 on serine 59 to promote its interaction with BRCA1 (Buisson et al, 2017). At the same time, ATR inhibits CDK to prevent a CDK‐dependent phosphorylation at PALB2's serine 64 that inhibits the BRCA1–PALB2 interaction (Buisson et al, 2017). Finally, the control of E2F transcription via ATR–CHK1 signaling strongly impacts the ability of cells to utilize HR‐mediated repair by ensuring proper expression of key components of the HR machinery (Kim et al, 2018). Similar to yeast, RAD51 deposition and RAD51‐mediated strand invasion are regulated by DNA damage signaling kinases. CHK1 has been shown to drive the formation of RAD51 foci at stalled replication forks, likely through direct phosphorylation (Sorensen et al, 2005). CHK1 and CHK2 may also be important to regulate the association of RAD51 with BRCA2 (Bahassi et al, 2008). Moreover, RAD51C, a putative homolog of yeast Rad55, associates with XRCC3, whose phosphorylation by ATM and ATR in response to IR is important for HR regulation (Somyajit et al, 2013).
ATM and DNA‐PKcs in resection control
ATM signaling blocks DNA end resection via recruitment of an anti‐resection complex formed by 53BP1‐RIF1‐REV7‐SHIELDIN (Setiaputra & Durocher, 2019). This process is achieved through a defined order of events: phosphorylation of H2AX and MDC1, followed by recruitment of the RNF8 and RNF168 ubiquitin ligases, followed by ubiquitylation of H2A and recruitment of 53BP1, whose phosphorylation by ATM serves as a docking platform for RIF1 and, indirectly, the REV7–SHIELDIN complex (Fig 4). In addition, ATM limits resection by phosphorylating the proteasomal shuttle factor UBQLN4, which leads to degradation of MRE11, a component of the MRN complex required for initial steps in resection (Jachimowicz et al, 2019). Paradoxically, ATM may also promote resection by phosphorylating CtIP, a key factor required for resection initiation (Shibata et al, 2011). DNA‐PKcs also plays opposing roles in resection control. Whereas binding of DNA‐PKcs to DNA ends has a major anti‐resection function by preventing recruitment of the EXO1 nuclease, DNA‐PKcs’ autophosphorylation is required to promote dissociation from DNA ends, thereby allowing EXO1 binding and resection (Zhou & Paull, 2013). More work is needed to better understand and define the extent to which the actions of ATM and DNA‐PKcs impact overall resection in cells.
ATM and DNA‐PKcs promote NHEJ
ATM and DNA‐PKcs are key players in the control of non‐homologous end joining (NHEJ), a pathway for DSB repair in which DNA ends are ligated without the need for a homologous template. ATM and DNA‐PKcs play partially redundant roles in promoting NHEJ, as impairment of both kinases results in a stronger NHEJ defect compared with impairment of one of the kinases (Gapud & Sleckman, 2011; Gapud et al, 2011; Zha et al, 2011). In addition, combined ATM and DNA‐PKcs deficiency leads to embryonic lethality in mice and a more severe DSB repair defect during immunoglobulin class‐switch recombination than loss of only one of the kinases (Callen et al, 2009). The ability of ATM and DNA‐PKcs to limit DNA end resection is a key part of their pro‐NHEJ function; however, these kinases likely regulate additional processes to promote NHEJ, since chemical inhibition of both kinases severely inhibits NHEJ without inducing resection due to persistent DNA‐PKcs at break ends. ATM and DNA‐PKcs redundantly phosphorylate several NHEJ repair components, such as XLF, Artemis, and POLλ (Zhang et al, 2004; Goodarzi et al, 2006; Yu et al, 2008; Sastre‐Moreno et al, 2017) (Fig 4). In addition, DNA‐PKcs solely phosphorylates the XRCC4, KU70, KU80, and LIG‐IV core components (Chan et al, 1999; Lee et al, 2004; Wang et al, 2004b; Douglas et al, 2005; Amiri Moghani et al, 2018). However, mutation of these phosphorylation sites, which are dependent on ATM and/or DNA‐PKcs, does not impair NHEJ or phenocopy the loss or inhibition of ATM and DNA‐PKcs function. Notably, phosphorylation of a non‐conventional DNA‐PKcs substrate, the transcription factor NR4A, was proposed to be important for NHEJ, as demonstrated by a phosphosite mutation, but the impact of the mutation in NHEJ remains unclear (Malewicz et al, 2011).
In S. cerevisiae, little is known about the crosstalk between DNA damage signaling kinases and NHEJ repair factors. Tel1 phosphorylates Pol4, stimulating its gap‐filling activity and preventing the appearance of deleterious chromosome translocations (Ruiz et al, 2013). Another report showed that the Dun1 kinase is also important for NHEJ by phosphorylating Nej1, a key activator of the NHEJ ligase Dnl4 (Ahnesorg & Jackson, 2007). DNA damage signaling kinases in budding yeast do not appear to impinge upon the NHEJ pathway as extensively as they do in humans and may reflect budding yeast's strong preference for homologous recombination‐mediated DNA repair (Jasin & Rothstein, 2013).
Other DNA repair pathways
While DNA damage signaling kinases appear to extensively impinge upon multiple factors and steps of recombinational DNA repair, they also target and regulate other repair mechanisms (Fig 4), suggesting that their action is context‐dependent and, in some cases, important in repair pathway choice. In budding yeast, Mec1 phosphorylates the nucleotide excision repair (NER) protein Rad26 and regulates the transcription‐coupled NER mode of repair (Taschner et al, 2010). In humans, ATR also controls NER by phosphorylating XPA (Wu et al, 2006) and, in conjunction with ATM, the histone acetyltransferase HBO1. HBO1 phosphorylation stabilizes its interaction with DDB2, promoting its proteasome‐mediated degradation (Matsunuma et al, 2016). In addition, ATR phosphorylates POLη at serine 601, facilitating post‐replication repair and promoting checkpoint activation at UV damage sites (Gohler et al, 2011). In yeast, Mec1 regulates the translesion synthesis polymerase Rev1 potentially through direct phosphorylation (Pages et al, 2009). This complements earlier studies, where it was shown that Mec1 is essential for Rev1 binding to chromosomes (Hirano & Sugimoto, 2006; Sabbioneda et al, 2007). DNA damage signaling kinases are also important for the execution of mismatch repair (MMR) and base excision repair (BER). ATM phosphorylates the MMR protein MLH1, contributing to its stabilization after DNA damage (Romeo et al, 2011). Also, ATM directly phosphorylates the BER proteins TDP1 (Das et al, 2009) and MPG (Agnihotri et al, 2014), enhancing their DNA repair activity and promoting cell survival after exposure to alkylating agents. DNA‐PKcs is also able to phosphorylate TDP1 at serine 81 (Das et al, 2009), allowing interaction with the DNA repair protein XRCC1, which is itself a CHK2 target for BER regulation (Chou et al, 2008). Finally, Rad53 was shown to be important in break‐induced replication (BIR) through the phosphorylation of the Pif1 helicase (Vasianovich et al, 2014).
Fanconi anemia pathway
The Fanconi anemia pathway is a genetic network involved in the repair of DNA interstrand crosslinks (ICLs) in the genome (Niraj et al, 2019). It combines the action of nucleotide excision repair and homologous recombination factors, several of which are regulated by ATR (Fig 4). ATR plays a crucial role at an early step in the Fanconi anemia pathway by phosphorylating the FANCI protein and inducing ubiquitylation of the FANCI–FANCD2 complex (Ishiai et al, 2008; Shigechi et al, 2012). The ubiquitylated FANCI–FANCD2 complex serves as a key scaffold for the recruitment of several DNA repair proteins, including structure‐specific nucleases and TLS polymerases involved in ICL repair (Knipscheer et al, 2009). In addition to promoting repair and replication fork start, phosphorylation of FANCI by ATR modulates DNA replication by inhibiting dormant origin firing (Chen et al, 2015). FANCD2 is also phosphorylated by ATM and ATR (Ho et al, 2006), whereas CHK1 phosphorylation is important for the FANCD2–BRCA2 interaction (Zhi et al, 2009). CHK1 phosphorylates FANCE at threonine 346 and serine 374 to promote resistance to mitomycin C (Wang et al, 2007). Additionally, ICL resistance has been linked to the ATR‐mediated phosphorylation of FANCA at serine 1449 and FANCM at serine 1045 (Collins et al, 2009; Singh et al, 2013; Fig 4).
Coordination of checkpoint and DNA repair: where are we 30 years later?
Following the checkpoint paper by Weinert and Hartwell in 1988 (Weinert & Hartwell, 1988), it was not at all clear how eukaryotic cells sense DNA damage and couple lesion detection to the control of the cell cycle. At that time, Weinert and Hartwell defined checkpoint as “control mechanisms enforcing dependency in the cell cycle”, implying the existence of some undefined regulatory mechanism. While the 1988 paper marks the identification of the first checkpoint factor in budding yeast (Rad9), the checkpoint concept largely preceded the identification of most key components mediating lesion detection and signaling. It was not even clear whether kinases were involved; in fact, the word “kinase” is absent from both the checkpoint papers of 1988 and 1989 (Weinert & Hartwell, 1988; Hartwell & Weinert, 1989). Identification of the apical PIKK kinases and downstream checkpoint kinases, as well as their involvement in mediating DNA damage responses, would not become evident until the mid‐1990s. Decades later, our understanding of both the mechanisms of lesion detection and the signaling circuitry connecting lesion detection to regulation of biological processes has radically changed. The field has defined many of the key proteins required for lesion detection and uncovered an extensive network of kinase substrates. The concept that checkpoint is a simple mechanism coupling lesion detection to cell cycle arrest also dramatically changed to incorporate both a range of biological outputs and a network of functional and physical connections. Nonetheless, at its essence, the central concept that DNA damage checkpoints coordinate cell cycle with DNA repair continues to hold true.
A unified model for the coordination of checkpoint and repair in the DSB response
When Weinert and Hartwell first proposed the checkpoint concept, it would be considered improbable that the circuitry responsible for promoting cell cycle arrest also directly and actively controls the DNA repair machinery. However, over the last 30 years, accumulated knowledge from work in yeast and mammals points to a key role for the apical PIKKs and downstream checkpoint kinases in the spatiotemporal coordination of checkpoint responses and DNA repair transactions. The response to DSBs epitomizes such intricate coordination. Mounting evidence supports a model whereby early response to DSBs involves rapid activation of apical PIKK kinases and the establishment of an emergency response that inhibits improper nuclease action (and resection) to prevent improper processing of DNA ends. These early response actions include cell cycle arrest, general inhibition of origin firing (if in S phase), and extensive transcriptional re‐programming. Following establishment of a robust checkpoint response and early resection block, cells must decide when to initiate DNA repair, including which repair pathway to utilize and when to resume the cell cycle. This decision is simpler in G1 cells, as the engagement of NHEJ for the repair of DSBs is highly preferred and can occur as a natural consequence of the resection block. However, for many DNA lesions in S phase and G2, the use of HR‐mediated repair is the pathway of choice for error‐free repair. Cells must, therefore, oppose the initial anti‐resection effects of DNA damage signaling to initiate resection, the first step in HR. Given the importance of the decision to initiate (or not to initiate) resection for repair pathway choice, this step is under intense regulation by phosphorylation. In yeast, Mec1 promotes resection by phosphorylating Slx4 and likely Sae2, opposing its own anti‐resection function via Rad53 activation. Interestingly, such dual and opposite functions for Mec1 could be key to providing a highly controllable system in which resection is spatiotemporally fine‐tuned. This is particularly important, as the extent of resection impacts which HR‐mediated mechanisms will follow [e.g., canonical HR, single‐strand annealing, synthesis‐dependent strand annealing (SDSA), and break‐induced replication (BIR)]. In humans, current evidence points to ATR, ATM, and DNA‐PKcs playing an important role in the regulation of resection, although much less is understood compared with yeast. In both yeast and humans, phosphatases are expected to play a key role in counter‐balancing the phosphorylation events that impose the early resection block and influence the DNA repair process. The balance between DNA damage signaling kinases and the phosphatases that oppose them is better appreciated for the more canonical aspects of the checkpoint, but more work is needed to understand how this balance affects choices made during the DNA repair process.
In sum, apical kinases perform highly elaborate actions in the spatiotemporal control of checkpoint responses and DNA repair. Since checkpoint functions and early resection blockades can counteract HR‐mediated DNA repair mechanisms, the ability of cells to modulate DNA damage signaling is essential for repair pathway control, and, in particular, proper execution of HR‐mediated repair. More work is needed to better understand how DNA damage signaling kinases monitor and control subsequent steps in HR (including strand invasion, homology search, and resolution/dissolution), as well as how such regulation ensures the fidelity of HR and prevents the erroneous recombination that can give rise to genetic instability.
What's next? Toward a holistic view of the DNA damage signaling network
In budding yeast and humans, DNA damage signaling kinases target dozens, if not hundreds, of DNA repair proteins, thereby modulating repair pathways. As described above, our understanding of the mechanisms by which these kinases coordinate DNA repair machineries is incomplete. When considering a function associated with the action of a kinase, the gold standard is to demonstrate that the function is impaired by mutation of a discrete set of phosphorylatable residues within a substrate protein(s). In almost all the cases discussed above, the DNA repair‐related phenotypes associated with the disruption of kinase function are far stronger than the phenotypes associated with the introduction of phosphosite mutations on the kinase substrates. Thus, either the proper combination of phosphosite mutations has not yet been uncovered, or there are still many more undiscovered phosphorylation events mediated by the DNA damage signaling kinases that are critical for the control of DNA repair. In some cases, fundamental phenotypes associated with the loss of DNA damage signaling still lack defined substrates and underlying molecular mechanisms. For example, a mechanistic understanding of the genetic instability (mostly monitored as accumulation of GCR) observed upon loss of Mec1 and Tel1 is unknown and represents a significant gap in our understanding of DNA damage signaling in budding yeast. In humans, the lethality and chromosomal fragmentation observed upon inhibition of ATR also lack a clear mechanistic underpinning. Furthermore, while it is established that inhibition of ATM and DNA‐PKcs strongly impair NHEJ, it is unclear which phosphorylation events are disrupted upon inhibition and in what ways they are necessary for NHEJ.
Mass spectrometry as a tool to study DNA damage signaling kinases: strengths, weaknesses, and future directions
Mass spectrometry has been instrumental for discovering in vivo substrates of the DNA damage signaling kinases (Matsuoka et al, 2007; Smolka et al, 2007; Bastos de Oliveira et al, 2015; Wagner et al, 2016; Lanz et al, 2018). The ability to quantitatively assess the phosphoproteome of an organism opened the door for unbiased identification of kinase substrates. Phosphoproteomic screens, performed in budding yeast and humans, have identified hundreds of phosphorylation events catalyzed by the DNA damage signaling kinases. Reassuringly, many of these phosphorylation events map to previously known substrate proteins. Excitingly, most of these events occur in substrate proteins not yet studied, many of which are associated with a broad range of nuclear processes, including DNA repair. However, the scope of the DNA damage signaling network revealed by phosphoproteomics raises the question of how many phosphorylation events have tangible biological significance. The ability to distinguish functional phosphorylation from kinase promiscuity represents the primary challenge of large‐scale phosphoproteomic datasets. For such an inquiry, it is imperative to generate mutant strains that either lack or constitutively mimic the phosphorylated residues in the substrate protein, with the ultimate goal to phenocopy the effects of the kinase's action or inaction. However, this is equivalent to finding a needle in a haystack, and better strategies are needed to enhance our ability to efficiently and systematically predict functional sites and, when necessary, to dissect functional redundancy and combinatorial effects.
Identification of in vivo kinase substrates using mass spectrometry is often performed using quantitative MS‐based approaches, primary SILAC, which provide a highly quantitative comparison between the phosphoproteomic profiles of two different cell populations (Bastos de Oliveira et al, 2015, 2018). Budding yeast represents an ideal system for mapping DNA damage signaling using mass spectrometry due to its relative simplicity compared with the mammalian system. A range of genetic tools and mutants enable powerful genetic–proteomic strategies to deconstruct the action of apical and downstream kinases. For example, loss of Mec1/ATR results in lethality in most eukaryotic systems, but suppressor mutations that rescue the lethality of mec1Δ cells (such as SML1 deletion) are available in yeast and are vital for the identification of proteins containing Mec1/Tel1‐ and Rad53‐dependent phosphorylation using phosphoproteomics (Smolka et al, 2007; Bastos de Oliveira et al, 2015; Lanz et al, 2018).
Phosphoproteomics has also been used to identify targets of the DNA damage signaling kinases in mammalian cell lines (Matsuoka et al, 2007; Stokes et al, 2007; Bennetzen et al, 2010; Pines et al, 2011; Beli et al, 2012; Kirkpatrick et al, 2013; Wagner et al, 2016). The sheer complexity of the human phosphoproteome makes in‐depth phosphoproteomic analyses more challenging than in yeast. Nonetheless, phosphoproteomic studies, such as the 2007 landmark paper by the Elledge group (Matsuoka et al, 2007), have revealed hundreds of potential substrates of DNA damage signaling kinases. One limitation of this work is that it relied on the use of phospho‐specific antibodies for the SQ/TQ phosphosite motif, so it did not identify substrates of the downstream DDC kinases CHK1 and CHK2, nor did it identify potentially direct ATR or ATM substrates phosphorylated at non‐canonical motifs (non‐SQ/TQ). Unlike in yeast, matching a substrate to a specific DNA damage signaling kinase is still a challenge, although the recent development of specific chemical inhibitors of ATR, ATM, and CHK1 has allowed more efficient assignment of kinase dependency. In fact, recent studies have made use of these chemical inhibitors to find ATR‐, ATM‐, and CHK1‐dependent phosphorylation events in vivo (Blasius et al, 2011; Wagner et al, 2016). Of note, the substrates of DNA‐PKcs, a third, human‐specific PI3 kinase‐like kinase, have yet to be extensively profiled by MS, although low‐throughput studies have uncovered substrate proteins associated with NHEJ (DNA‐PKcs is reviewed in detail here: Blackford & Jackson, 2017).
An alternative approach to screening for substrates of a kinase‐of‐interest involves the mutation of gate‐keeping residues in the kinase's ATP‐binding pocket, which enables the accommodation and subsequent utilization of bulky ATP analogs (Hertz et al, 2010). The addition of an analog‐compatible kinase to human cell extracts results in the in vitro modification of its substrate proteins, which are then isolated and identified based on the covalent attachment of analog phosphate groups. This chemical‐genetics approach (commonly referred to as the “Shokat method”) has been used to map the signaling network of several CDKs (Blethrow et al, 2008; Chi et al, 2008) and, more recently, to identify CHK1 substrate proteins in human cell extracts (Blasius et al, 2011). However, these chemical‐genetic strategies require the generation of mutant kinase alleles that are able to utilize bulky ATP analogs, and the kinase labeling reactions normally take place in cell lysates rather than in vivo. Also, efforts to apply the Shokat method to the upstream PIKKs have thus far been unsuccessful, as ATM and ATR appear to be less amenable to mutations in their ATP‐binding pockets.
Overall, phosphoproteomic studies have revealed that the human DNA damage signaling network is extensive, consisting of many nuclear proteins, but also non‐nuclear proteins. In addition to the established DNA repair‐related substrates previously described in low‐throughput studies, phosphoproteomic analyses have uncovered a large proportion of substrates involved in transcription and RNA processing, although, like in yeast, the functional significance of these phosphorylation events remains unclear. While the work done thus far in mammalian systems has yielded many candidate substrates, the DNA damage signaling kinases may operate differently in different cell types. Thus, it will be important for future investigations to thoroughly assess how the DNA damage signaling network varies in different cell types and pathological statuses. Attaining full coverage of the phosphoproteome is also a concern, as available phosphoproteome datasets do not cover all phosphorylation events in a cell due to technical limitations with mass spectrometry (Engholm‐Keller & Larsen, 2013). Currently, however, instruments have become more sensitive, and the ability to use mass spectrometry to cover the full phosphoproteome may become a reality in the near future. Looking ahead, it will be crucial to develop more efficient approaches for identifying functional phosphorylation events within the large phosphoproteomic datasets. One possibility is to use structural prediction analysis to identify phosphorylation events that are likely to impair protein function (such as disrupting protein–protein or protein–DNA interactions) (preprint: Lanz et al, 2019). In conclusion, a holistic understanding of the role and action of DNA damage signaling kinases will require powerful technologies capable of quantitatively monitoring the full extent of the phosphoproteome in combination with systematic approaches for functional and mechanistic analyses.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We thank Ethan Sanford and Robert Weiss for careful reading of the article and suggestions. This work was supported by M.B.S. grants from the National Institutes of Health (R01GM097272 and R01HD095296).
The EMBO Journal (2019) 38: e101801
See the Glossary for abbreviations used in this article.
References
- Abreu CM, Kumar R, Hamilton D, Dawdy AW, Creavin K, Eivers S, Finn K, Balsbaugh JL, O'Connor R, Kiely PA et al (2013) Site‐specific phosphorylation of the DNA damage response mediator rad9 by cyclin‐dependent kinases regulates activation of checkpoint kinase 1. PLoS Genet 9: e1003310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnihotri S, Burrell K, Buczkowicz P, Remke M, Golbourn B, Chornenkyy Y, Gajadhar A, Fernandez NA, Clarke ID, Barszczyk MS et al (2014) ATM regulates 3‐methylpurine‐DNA glycosylase and promotes therapeutic resistance to alkylating agents. Cancer Discov 4: 1198–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn JY, Schwarz JK, Piwnica‐Worms H, Canman CE (2000) Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res 60: 5934–5936 [PubMed] [Google Scholar]
- Ahn J, Prives C (2002) Checkpoint kinase 2 (Chk2) monomers or dimers phosphorylate Cdc25C after DNA damage regardless of threonine 68 phosphorylation. J Biol Chem 277: 48418–48426 [DOI] [PubMed] [Google Scholar]
- Ahnesorg P, Jackson SP (2007) The non‐homologous end‐joining protein Nej1p is a target of the DNA damage checkpoint. DNA Repair (Amst) 6: 190–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, Furuya K, Diffley JF, Carr AM, Elledge SJ (2001) Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol 3: 958–965 [DOI] [PubMed] [Google Scholar]
- Allen JB, Zhou Z, Siede W, Friedberg EC, Elledge SJ (1994) The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage‐induced transcription in yeast. Genes Dev 8: 2401–2415 [DOI] [PubMed] [Google Scholar]
- Amiri Moghani AR, Sharma MK, Matsumoto Y (2018) In cellulo phosphorylation of DNA double‐strand break repair protein XRCC4 on Ser260 by DNA‐PK. J Radiat Res 59: 700–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammazzalorso F, Pirzio LM, Bignami M, Franchitto A, Pichierri P (2010) ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J 29: 3156–3169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Attikum H, Fritsch O, Hohn B, Gasser SM (2004) Recruitment of the INO80 complex by H2A phosphorylation links ATP‐dependent chromatin remodeling with DNA double‐strand break repair. Cell 119: 777–788 [DOI] [PubMed] [Google Scholar]
- Bacal J, Moriel‐Carretero M, Pardo B, Barthe A, Sharma S, Chabes A, Lengronne A, Pasero P (2018) Mrc1 and Rad9 cooperate to regulate initiation and elongation of DNA replication in response to DNA damage. EMBO J 37: e99319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahassi EM, Ovesen JL, Riesenberg AL, Bernstein WZ, Hasty PE, Stambrook PJ (2008) The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene 27: 3977–3985 [DOI] [PubMed] [Google Scholar]
- Baldock RA, Day M, Wilkinson OJ, Cloney R, Jeggo PA, Oliver AW, Watts FZ, Pearl LH (2015) ATM localization and heterochromatin repair depend on direct interaction of the 53BP1‐BRCT2 domain with gammaH2AX. Cell Rep 13: 2081–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3: 421–429 [DOI] [PubMed] [Google Scholar]
- Bashkirov VI, Bashkirova EV, Haghnazari E, Heyer WD (2003) Direct kinase‐to‐kinase signaling mediated by the FHA phosphoprotein recognition domain of the Dun1 DNA damage checkpoint kinase. Mol Cell Biol 23: 1441–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashkirov VI, Herzberg K, Haghnazari E, Vlasenko AS, Heyer WD (2006) DNA damage‐induced phosphorylation of Rad55 protein as a sentinel for DNA damage checkpoint activation in S. cerevisiae . Methods Enzymol 409: 166–182 [DOI] [PubMed] [Google Scholar]
- Bastos de Oliveira FM, Harris MR, Brazauskas P, de Bruin RA, Smolka MB (2012) Linking DNA replication checkpoint to MBF cell‐cycle transcription reveals a distinct class of G1/S genes. EMBO J 31: 1798–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos de Oliveira FM, Kim D, Cussiol JR, Das J, Jeong MC, Doerfler L, Schmidt KH, Yu H, Smolka MB (2015) Phosphoproteomics reveals distinct modes of Mec1/ATR signaling during DNA replication. Mol Cell 57: 1124–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos de Oliveira FM, Kim D, Lanz M, Smolka MB (2018) Quantitative analysis of DNA damage signaling responses to chemical and genetic perturbations. Methods Mol Biol 1672: 645–660 [DOI] [PubMed] [Google Scholar]
- Beli P, Lukashchuk N, Wagner SA, Weinert BT, Olsen JV, Baskcomb L, Mann M, Jackson SP, Choudhary C (2012) Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol Cell 46: 212–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen MV, Larsen DH, Bunkenborg J, Bartek J, Lukas J, Andersen JS (2010) Site‐specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol Cell Proteomics 9: 1314–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli C, Klier S, McGowan C, Wittenberg C, de Bruin RA (2013a) Chk1 inhibits E2F6 repressor function in response to replication stress to maintain cell‐cycle transcription. Curr Biol 23: 1629–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli C, Skotheim JM, de Bruin RA (2013b) Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 14: 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford AN, Jackson SP (2017) ATM, ATR, and DNA‐PK: the trinity at the heart of the DNA damage response. Mol Cell 66: 801–817 [DOI] [PubMed] [Google Scholar]
- Blankley RT, Lydall D (2004) A domain of Rad9 specifically required for activation of Chk1 in budding yeast. J Cell Sci 117: 601–608 [DOI] [PubMed] [Google Scholar]
- Blasius M, Forment JV, Thakkar N, Wagner SA, Choudhary C, Jackson SP (2011) A phospho‐proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol 12: R78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blethrow JD, Glavy JS, Morgan DO, Shokat KM (2008) Covalent capture of kinase‐specific phosphopeptides reveals Cdk1‐cyclin B substrates. Proc Natl Acad Sci USA 105: 1442–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy MN, Furnari B, Mondesert O, Russell P (1998) Replication checkpoint enforced by kinases Cds1 and Chk1. Science 280: 909–912 [DOI] [PubMed] [Google Scholar]
- Boos D, Sanchez‐Pulido L, Rappas M, Pearl LH, Oliver AW, Ponting CP, Diffley JF (2011) Regulation of DNA replication through Sld3‐Dpb11 interaction is conserved from yeast to humans. Curr Biol 21: 1152–1157 [DOI] [PubMed] [Google Scholar]
- Botchkarev VV Jr, Haber JE (2018) Functions and regulation of the Polo‐like kinase Cdc5 in the absence and presence of DNA damage. Curr Genet 64: 87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E et al (2011) Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell 42: 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken AP, Ciro M, Cocito A, Helin K (2004) E2F target genes: unraveling the biology. Trends Biochem Sci 29: 409–417 [DOI] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D (2000) ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 14: 397–402 [PMC free article] [PubMed] [Google Scholar]
- de Bruin RA, Kalashnikova TI, Aslanian A, Wohlschlegel J, Chahwan C, Yates JR III, Russell P, Wittenberg C (2008) DNA replication checkpoint promotes G1‐S transcription by inactivating the MBF repressor Nrm1. Proc Natl Acad Sci USA 105: 11230–11235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruinsma W, Aprelia M, Garcia‐Santisteban I, Kool J, Xu YJ, Medema RH (2017) Inhibition of Polo‐like kinase 1 during the DNA damage response is mediated through loss of Aurora A recruitment by Bora. Oncogene 36: 1840–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R, Niraj J, Rodrigue A, Ho CK, Kreuzer J, Foo TK, Hardy EJ, Dellaire G, Haas W, Xia B et al (2017) Coupling of homologous recombination and the checkpoint by ATR. Mol Cell 65: 336–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez‐Capetillo O, Cao L et al (2010) 53BP1 inhibits homologous recombination in Brca1‐deficient cells by blocking resection of DNA breaks. Cell 141: 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Chehab NH, Pavletich NP (2009) Structure and activation mechanism of the CHK2 DNA damage checkpoint kinase. Mol Cell 35: 818–829 [DOI] [PubMed] [Google Scholar]
- Callen E, Jankovic M, Wong N, Zha S, Chen HT, Difilippantonio S, Di Virgilio M, Heidkamp G, Alt FW, Nussenzweig A et al (2009) Essential role for DNA‐PKcs in DNA double‐strand break repair and apoptosis in ATM‐deficient lymphocytes. Mol Cell 34: 285–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Can G, Kauerhof AC, Macak D, Zegerman P (2018) Helicase subunit Cdc45 targets the checkpoint kinase Rad53 to both replication initiation and elongation complexes after fork stalling. Mol Cell 73: 562–573.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr AM (1995) DNA structure checkpoints in fission yeast. Semin Cell Biol 6: 65–72 [DOI] [PubMed] [Google Scholar]
- Chan DW, Ye R, Veillette CJ, Lees‐Miller SP (1999) DNA‐dependent protein kinase phosphorylation sites in Ku 70/80 heterodimer. Biochemistry 38: 1819–1828 [DOI] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas‐Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ (2013) RIF1 is essential for 53BP1‐dependent nonhomologous end joining and suppression of DNA double‐strand break resection. Mol Cell 49: 858–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Jones MJ, Yin Y, Crist SB, Colnaghi L, Sims RJ III, Rothenberg E, Jallepalli PV, Huang TT (2015) ATR‐mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol Cell 58: 323–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J (2016) The cell‐cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med 6: a026104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Y, Welcker M, Hizli AA, Posakony JJ, Aebersold R, Clurman BE (2008) Identification of CDK2 substrates in human cell lysates. Genome Biol 9: R149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou WC, Wang HC, Wong FH, Ding SL, Wu PE, Shieh SY, Shen CY (2008) Chk2‐dependent phosphorylation of XRCC1 in the DNA damage response promotes base excision repair. EMBO J 27: 3140–3150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, Shin TB, Keith CT, Schreiber SL (1996) cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc Natl Acad Sci USA 93: 2850–2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Trovesi C, Galbiati A, Lucchini G, Longhese MP (2014) Mec1/ATR regulates the generation of single‐stranded DNA that attenuates Tel1/ATM signaling at DNA ends. EMBO J 33: 198–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins NB, Wilson JB, Bush T, Thomashevski A, Roberts KJ, Jones NJ, Kupfer GM (2009) ATR‐dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood 113: 2181–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Glick G, Elledge SJ (2004) Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc Natl Acad Sci USA 101: 10078–10083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J (2003) An ATR‐ and Cdc7‐dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell 11: 203–213 [DOI] [PubMed] [Google Scholar]
- Cotta‐Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, Foiani M (2005) Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint‐defective cells. Mol Cell 17: 153–159 [DOI] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA et al (2013) ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27: 1610–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado M, Martinez‐Pastor B, Murga M, Toledo LI, Gutierrez‐Martinez P, Lopez E, Fernandez‐Capetillo O (2006) ATM regulates ATR chromatin loading in response to DNA double‐strand breaks. J Exp Med 203: 297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cussiol JR, Jablonowski CM, Yimit A, Brown GW, Smolka MB (2015) Dampening DNA damage checkpoint signalling via coordinated BRCT domain interactions. EMBO J 34: 1704–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cussiol JR, Dibitetto D, Pellicioli A, Smolka MB (2017) Slx4 scaffolding in homologous recombination and checkpoint control: lessons from yeast. Chromosoma 126: 45–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, Saraf A, Florens L, Washburn MP, Pagano M (2012) Cyclin F‐mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 149: 1023–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das BB, Antony S, Gupta S, Dexheimer TS, Redon CE, Garfield S, Shiloh Y, Pommier Y (2009) Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA‐PK. EMBO J 28: 3667–3680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SL, North PS, Dart A, Lakin ND, Hickson ID (2004) Phosphorylation of the Bloom's syndrome helicase and its role in recovery from S‐phase arrest. Mol Cell Biol 24: 1279–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SL, North PS, Hickson ID (2007) Role for BLM in replication‐fork restart and suppression of origin firing after replicative stress. Nat Struct Mol Biol 14: 677–679 [DOI] [PubMed] [Google Scholar]
- Deng X, Prakash A, Dhar K, Baia GS, Kolar C, Oakley GG, Borgstahl GE (2009) Human replication protein A‐Rad52‐single‐stranded DNA complex: stoichiometry and evidence for strand transfer regulation by phosphorylation. Biochemistry 48: 6633–6643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico EG, Romano E, Del Porto P, Ascenzioni F (2014) Multifunctional role of ATM/Tel1 kinase in genome stability: from the DNA damage response to telomere maintenance. Biomed Res Int 2014: 787404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibitetto D, Ferrari M, Rawal CC, Balint A, Kim T, Zhang Z, Smolka MB, Brown GW, Marini F, Pellicioli A (2016) Slx4 and Rtt107 control checkpoint signalling and DNA resection at double‐strand breaks. Nucleic Acids Res 44: 669–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnianni RA, Ferrari M, Lazzaro F, Clerici M, Tamilselvan Nachimuthu B, Plevani P, Muzi‐Falconi M, Pellicioli A (2010) Elevated levels of the polo kinase Cdc5 override the Mec1/ATR checkpoint in budding yeast by acting at different steps of the signaling pathway. PLoS Genet 6: e1000763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P, Gupta S, Morrice N, Meek K, Lees‐Miller SP (2005) DNA‐PK‐dependent phosphorylation of Ku70/80 is not required for non‐homologous end joining. DNA Repair (Amst) 4: 1006–1018 [DOI] [PubMed] [Google Scholar]
- Downs JA, Lowndes NF, Jackson SP (2000) A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 408: 1001–1004 [DOI] [PubMed] [Google Scholar]
- Downs JA, Allard S, Jobin‐Robitaille O, Javaheri A, Auger A, Bouchard N, Kron SJ, Jackson SP, Cote J (2004) Binding of chromatin‐modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell 16: 979–990 [DOI] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, Cortez D (2015) The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol Cell 59: 998–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg ER, Mark KG, Toczyski DP (2015) Ndd1 turnover by SCF(Grr1) is inhibited by the DNA damage checkpoint in Saccharomyces cerevisiae . PLoS Genet 11: e1005162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Shemerly M, Hess D, Pyakurel AK, Moselhy S, Ferrari S (2008) ATR‐dependent pathways control hEXO1 stability in response to stalled forks. Nucleic Acids Res 36: 511–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emili A (1998) MEC1‐dependent phosphorylation of Rad9p in response to DNA damage. Mol Cell 2: 183–189 [DOI] [PubMed] [Google Scholar]
- Engels K, Giannattasio M, Muzi‐Falconi M, Lopes M, Ferrari S (2011) 14‐3‐3 proteins regulate exonuclease 1‐dependent processing of stalled replication forks. PLoS Genet 7: e1001367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engholm‐Keller K, Larsen MR (2013) Technologies and challenges in large‐scale phosphoproteomics. Proteomics 13: 910–931 [DOI] [PubMed] [Google Scholar]
- Ferrari M, Nachimuthu BT, Donnianni RA, Klein H, Pellicioli A (2013) Tid1/Rdh54 translocase is phosphorylated through a Mec1‐ and Rad53‐dependent manner in the presence of DSB lesions in budding yeast. DNA Repair (Amst) 12: 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari M, Dibitetto D, De Gregorio G, Eapen VV, Rawal CC, Lazzaro F, Tsabar M, Marini F, Haber JE, Pellicioli A (2015) Functional interplay between the 53BP1‐ortholog Rad9 and the Mre11 complex regulates resection, end‐tethering and repair of a double‐strand break. PLoS Genet 11: e1004928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorani S, Mimun G, Caleca L, Piccini D, Pellicioli A (2008) Characterization of the activation domain of the Rad53 checkpoint kinase. Cell Cycle 7: 493–499 [DOI] [PubMed] [Google Scholar]
- Flott S, Kwon Y, Pigli YZ, Rice PA, Sung P, Jackson SP (2011) Regulation of Rad51 function by phosphorylation. EMBO Rep 12: 833–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskolou IP, Jorgensen C, Leszczynska KB, Olcina MM, Tarhonskaya H, Haisma B, D'Angiolella V, Myers WK, Domene C, Flashman E et al (2017) Ribonucleotide reductase requires subunit switching in hypoxia to maintain DNA replication. Mol Cell 66: 206–220 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari B, Rhind N, Russell P (1997) Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science 277: 1495–1497 [DOI] [PubMed] [Google Scholar]
- Gapud EJ, Dorsett Y, Yin B, Callen E, Bredemeyer A, Mahowald GK, Omi KQ, Walker LM, Bednarski JJ, McKinnon PJ et al (2011) Ataxia telangiectasia mutated (Atm) and DNA‐PKcs kinases have overlapping activities during chromosomal signal joint formation. Proc Natl Acad Sci USA 108: 2022–2027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gapud EJ, Sleckman BP (2011) Unique and redundant functions of ATM and DNA‐PKcs during V(D)J recombination. Cell Cycle 10: 1928–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert CS, Green CM, Lowndes NF (2001) Budding yeast Rad9 is an ATP‐dependent Rad53 activating machine. Mol Cell 8: 129–136 [DOI] [PubMed] [Google Scholar]
- Gobbini E, Villa M, Gnugnoli M, Menin L, Clerici M, Longhese MP (2015) Sae2 function at DNA double‐strand breaks is bypassed by dampening Tel1 or Rad53 activity. PLoS Genet 11: e1005685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohler T, Sabbioneda S, Green CM, Lehmann AR (2011) ATR‐mediated phosphorylation of DNA polymerase eta is needed for efficient recovery from UV damage. J Cell Biol 192: 219–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J, Jackson SP (2003) MDC1 is required for the intra‐S‐phase DNA damage checkpoint. Nature 421: 952–956 [DOI] [PubMed] [Google Scholar]
- Goodarzi AA, Yu Y, Riballo E, Douglas P, Walker SA, Ye R, Harer C, Marchetti C, Morrice N, Jeggo PA et al (2006) DNA‐PK autophosphorylation facilitates Artemis endonuclease activity. EMBO J 25: 3880–3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guervilly JH, Gaillard PH (2018) SLX4: multitasking to maintain genome stability. Crit Rev Biochem Mol Biol 53: 475–514 [DOI] [PubMed] [Google Scholar]
- Gunjan A, Verreault A (2003) A Rad53 kinase‐dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae . Cell 115: 537–549 [DOI] [PubMed] [Google Scholar]
- Guo C, Kumagai A, Schlacher K, Shevchenko A, Shevchenko A, Dunphy WG (2015) Interaction of Chk1 with Treslin negatively regulates the initiation of chromosomal DNA replication. Mol Cell 57: 492–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammet A, Magill C, Heierhorst J, Jackson SP (2007) Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep 8: 851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang L, Zhao X (2016) The Rtt107 BRCT scaffold and its partner modification enzymes collaborate to promote replication. Nucleus 7: 346–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ (1993) The p21 Cdk‐interacting protein Cip1 is a potent inhibitor of G1 cyclin‐dependent kinases. Cell 75: 805–816 [DOI] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ, Keyomarsi K, Dynlacht B, Tsai LH, Zhang P, Dobrowolski S, Bai C, Connell‐Crowley L, Swindell E et al (1995) Inhibition of cyclin‐dependent kinases by p21. Mol Biol Cell 6: 387–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA (1989) Checkpoints: controls that ensure the order of cell cycle events. Science 246: 629–634 [DOI] [PubMed] [Google Scholar]
- Heffernan TP, Unsal‐Kacmaz K, Heinloth AN, Simpson DA, Paules RS, Sancar A, Cordeiro‐Stone M, Kaufmann WK (2007) Cdc7‐Dbf4 and the human S checkpoint response to UVC. J Biol Chem 282: 9458–9468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegnauer AM, Hustedt N, Shimada K, Pike BL, Vogel M, Amsler P, Rubin SM, van Leeuwen F, Guenole A, van Attikum H et al (2012) An N‐terminal acidic region of Sgs1 interacts with Rpa70 and recruits Rad53 kinase to stalled forks. EMBO J 31: 3768–3783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz NT, Wang BT, Allen JJ, Zhang C, Dar AC, Burlingame AL, Shokat KM (2010) Chemical genetic approach for kinase‐substrate mapping by covalent capture of thiophosphopeptides and analysis by mass spectrometry. Curr Protoc Chem Biol 2: 15–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzberg K, Bashkirov VI, Rolfsmeier M, Haghnazari E, McDonald WH, Anderson S, Bashkirova EV, Yates JR III, Heyer WD (2006) Phosphorylation of Rad55 on serines 2, 8, and 14 is required for efficient homologous recombination in the recovery of stalled replication forks. Mol Cell Biol 26: 8396–8409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Sugimoto K (2006) ATR homolog Mec1 controls association of DNA polymerase zeta‐Rev1 complex with regions near a double‐strand break. Curr Biol 16: 586–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho GP, Margossian S, Taniguchi T, D'Andrea AD (2006) Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Mol Cell Biol 26: 7005–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Zhou Z, Elledge SJ (1998) The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 94: 595–605 [DOI] [PubMed] [Google Scholar]
- Hustedt N, Durocher D (2016) The control of DNA repair by the cell cycle. Nat Cell Biol 19: 1–9 [DOI] [PubMed] [Google Scholar]
- Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita‐Kikuta E, Koike T et al (2008) FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat Struct Mol Biol 15: 1138–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, Nevins JR (2001) Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol 21: 4684–4699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jachimowicz RD, Beleggia F, Isensee J, Velpula BB, Goergens J, Bustos MA, Doll MA, Shenoy A, Checa‐Rodriguez C, Wiederstein JL et al (2019) UBQLN4 represses homologous recombination and is overexpressed in aggressive tumors. Cell 176: 505–519 e22 [DOI] [PubMed] [Google Scholar]
- Jaehnig EJ, Kuo D, Hombauer H, Ideker TG, Kolodner RD (2013) Checkpoint kinases regulate a global network of transcription factors in response to DNA damage. Cell Rep 4: 174–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M, Rothstein R (2013) Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5: a012740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia‐Lin Ma N, Stern DF (2008) Regulation of the Rad53 protein kinase in signal amplification by oligomer assembly and disassembly. Cell Cycle 7: 808–817 [DOI] [PubMed] [Google Scholar]
- Kaur S, Modi P, Srivastava V, Mudgal R, Tikoo S, Arora P, Mohanty D, Sengupta S (2010) Chk1‐dependent constitutive phosphorylation of BLM helicase at serine 646 decreases after DNA damage. Mol Cancer Res 8: 1234–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Liu Y, Oberly S, Freire R, Smolka MB (2018) ATR‐mediated proteome remodeling is a major determinant of homologous recombination capacity in cancer cells. Nucleic Acids Res 46: 8311–8325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick DS, Bustos DJ, Dogan T, Chan J, Phu L, Young A, Friedman LS, Belvin M, Song Q, Bakalarski CE et al (2013) Phosphoproteomic characterization of DNA damage response in melanoma cells following MEK/PI3K dual inhibition. Proc Natl Acad Sci USA 110: 19426–19431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner RE, Verma P, Molloy KR, Chait BT, Kapoor TM (2015) Chemical proteomics reveals a gammaH2AX‐53BP1 interaction in the DNA damage response. Nat Chem Biol 11: 807–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, Walter JC (2009) The Fanconi anemia pathway promotes replication‐dependent DNA interstrand cross‐link repair. Science 326: 1698–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L et al (2017) Smarcal1‐mediated fork reversal triggers Mre11‐dependent degradation of nascent DNA in the absence of Brca2 and Stable Rad51 nucleofilaments. Mol Cell 67: 867–881 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse JP, Gu W (2009) Modes of p53 regulation. Cell 137: 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG (2000) Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol Cell 6: 839–849 [DOI] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG (2003) Repeated phosphopeptide motifs in Claspin mediate the regulated binding of Chk1. Nat Cell Biol 5: 161–165 [DOI] [PubMed] [Google Scholar]
- Kumagai A, Shevchenko A, Shevchenko A, Dunphy WG (2010) Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell 140: 349–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz MC, Oberly S, Sanford EJ, Sharma S, Chabes A, Smolka MB (2018) Separable roles for Mec1/ATR in genome maintenance, DNA replication, and checkpoint signaling. Genes Dev 32: 822–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz MC, Yugandhar K, Gupta S, Sanford E, Faça V, Vega S, Joiner A, Fromme C, Yu H (2019) In‐depth and 3‐dimensional exploration of the budding yeast phosphoproteome. bioRxiv 10.1101/700070 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro F, Sapountzi V, Granata M, Pellicioli A, Vaze M, Haber JE, Plevani P, Lydall D, Muzi‐Falconi M (2008) Histone methyltransferase Dot1 and Rad9 inhibit single‐stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J 27: 1502–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kumagai A, Dunphy WG (2001) Positive regulation of Wee1 by Chk1 and 14‐3‐3 proteins. Mol Biol Cell 12: 551–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kumagai A, Dunphy WG (2003) Claspin, a Chk1‐regulatory protein, monitors DNA replication on chromatin independently of RPA, ATR, and Rad17. Mol Cell 11: 329–340 [DOI] [PubMed] [Google Scholar]
- Lee KJ, Jovanovic M, Udayakumar D, Bladen CL, Dynan WS (2004) Identification of DNA‐PKcs phosphorylation sites in XRCC4 and effects of mutations at these sites on DNA end joining in a cell‐free system. DNA Repair (Amst) 3: 267–276 [DOI] [PubMed] [Google Scholar]
- Lee YD, Wang J, Stubbe J, Elledge SJ (2008) Dif1 is a DNA‐damage‐regulated facilitator of nuclear import for ribonucleotide reductase. Mol Cell 32: 70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AY, Chiba T, Truong LN, Cheng AN, Do J, Cho MJ, Chen L, Wu X (2012) Dbf4 is direct downstream target of ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3‐related (ATR) protein to regulate intra‐S‐phase checkpoint. J Biol Chem 287: 2531–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung CC, Sun L, Gong Z, Burkat M, Edwards R, Assmus M, Chen J, Glover JN (2013) Structural insights into recognition of MDC1 by TopBP1 in DNA replication checkpoint control. Structure 21: 1450–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Lavagnino Z, Lemacon D, Kong L, Ustione A, Ng X, Zhang Y, Wang Y, Zheng B, Piwnica‐Worms H et al (2019) Ca(2+)‐stimulated AMPK‐dependent phosphorylation of Exo1 protects stressed replication forks from aberrant resection. Mol Cell 74: 1123–1137 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Suhandynata RT, Zhou H (2015) Phosphorylation of Sae2 mediates forkhead‐associated (FHA) domain‐specific interaction and regulates its DNA repair function. J Biol Chem 290: 10751–10763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin WC, Lin FT, Nevins JR (2001) Selective induction of E2F1 in response to DNA damage, mediated by ATM‐dependent phosphorylation. Genes Dev 15: 1833–1844 [PMC free article] [PubMed] [Google Scholar]
- Lindsey‐Boltz LA, Sercin O, Choi JH, Sancar A (2009) Reconstitution of human claspin‐mediated phosphorylation of Chk1 by the ATR (ataxia telangiectasia‐mutated and rad3‐related) checkpoint kinase. J Biol Chem 284: 33107–33114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini‐Rivera S, DeMayo F, Bradley A et al (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14: 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- Liu S, Bekker‐Jensen S, Mailand N, Lukas C, Bartek J, Lukas J (2006) Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol Cell Biol 26: 6056–6064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Cussiol JR, Dibitetto D, Sims JR, Twayana S, Weiss RS, Freire R, Marini F, Pellicioli A, Smolka MB (2017) TOPBP1Dpb11 plays a conserved role in homologous recombination DNA repair through the coordinated recruitment of 53BP1Rad9. J Cell Biol 216: 623–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Contreras AJ, Specks J, Barlow JH, Ambrogio C, Desler C, Vikingsson S, Rodrigo‐Perez S, Green H, Rasmussen LJ, Murga M et al (2015) Increased Rrm2 gene dosage reduces fragile site breakage and prolongs survival of ATR mutant mice. Genes Dev 29: 690–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Mosqueda J, Maas NL, Jonsson ZO, Defazio‐Eli LG, Wohlschlegel J, Toczyski DP (2010) Damage‐induced phosphorylation of Sld3 is important to block late origin firing. Nature 467: 479–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malewicz M, Kadkhodaei B, Kee N, Volakakis N, Hellman U, Viktorsson K, Leung CY, Chen B, Lewensohn R, van Gent DC et al (2011) Essential role for DNA‐PK‐mediated phosphorylation of NR4A nuclear orphan receptors in DNA double‐strand break repair. Genes Dev 25: 2031–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunuma R, Niida H, Ohhata T, Kitagawa K, Sakai S, Uchida C, Shiotani B, Matsumoto M, Nakayama KI, Ogura H et al (2016) UV damage‐induced phosphorylation of HBO1 triggers CRL4DDB2‐mediated degradation to regulate cell proliferation. Mol Cell Biol 36: 394–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge SJ (1998) Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282: 1893–1897 [DOI] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y et al (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316: 1160–1166 [DOI] [PubMed] [Google Scholar]
- Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, Moas M, Buschmann T, Ronai Z, Shiloh Y et al (2001) ATM‐dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev 15: 1067–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo MW, Wang CY, Cogswell PC, Rogers‐Graham KS, Lowe SW, Der CJ, Baldwin AS Jr (1997) Requirement of NF‐kappaB activation to suppress p53‐independent apoptosis induced by oncogenic Ras. Science 278: 1812–1815 [DOI] [PubMed] [Google Scholar]
- McIntosh D, Blow JJ (2012) Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb Perspect Biol 4: a012955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin I, Ngo HP, Greenall A, Zubko MK, Morrice N, Lydall D (2008) Checkpoint‐dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J 27: 2400–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison AJ, Highland J, Krogan NJ, Arbel‐Eden A, Greenblatt JF, Haber JE, Shen X (2004) INO80 and gamma‐H2AX interaction links ATP‐dependent chromatin remodeling to DNA damage repair. Cell 119: 767–775 [DOI] [PubMed] [Google Scholar]
- Morrison AJ, Kim JA, Person MD, Highland J, Xiao J, Wehr TS, Hensley S, Bao Y, Shen J, Collins SR et al (2007) Mec1/Tel1 phosphorylation of the INO80 chromatin remodeling complex influences DNA damage checkpoint responses. Cell 130: 499–511 [DOI] [PubMed] [Google Scholar]
- Morrow DM, Tagle DA, Shiloh Y, Collins FS, Hieter P (1995) TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell 82: 831–840 [DOI] [PubMed] [Google Scholar]
- Mueller PR, Coleman TR, Dunphy WG (1995) Cell cycle regulation of a Xenopus Wee1‐like kinase. Mol Biol Cell 6: 119–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami H, Aiba H, Nakanishi M, Murakami‐Tonami Y (2010) Regulation of yeast forkhead transcription factors and FoxM1 by cyclin‐dependent and polo‐like kinases. Cell Cycle 9: 3233–3242 [DOI] [PubMed] [Google Scholar]
- Murphy AK, Fitzgerald M, Ro T, Kim JH, Rabinowitsch AI, Chowdhury D, Schildkraut CL, Borowiec JA (2014) Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol 206: 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung K, Datta A, Kolodner RD (2001) Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae . Cell 104: 397–408 [DOI] [PubMed] [Google Scholar]
- Naiki T, Wakayama T, Nakada D, Matsumoto K, Sugimoto K (2004) Association of Rad9 with double‐strand breaks through a Mec1‐dependent mechanism. Mol Cell Biol 24: 3277–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen I, Bentsen IB, Andersen AH, Gasser SM, Bjergbaek L (2013) A Rad53 independent function of Rad9 becomes crucial for genome maintenance in the absence of the Recq helicase Sgs1. PLoS One 8: e81015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niraj J, Farkkila A, D'Andrea AD (2019) The fanconi anemia pathway in cancer. Annu Rev Cancer Biol 3: 457–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell MJ, Raleigh JM, Verkade HM, Nurse P (1997) Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J 16: 545–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohouo PY, Bastos de Oliveira FM, Almeida BS, Smolka MB (2010) DNA damage signaling recruits the Rtt107‐Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell 39: 300–306 [DOI] [PubMed] [Google Scholar]
- Osborn AJ, Elledge SJ (2003) Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev 17: 1755–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paciotti V, Lucchini G, Plevani P, Longhese MP (1998) Mec1p is essential for phosphorylation of the yeast DNA damage checkpoint protein Ddc1p, which physically interacts with Mec3p. EMBO J 17: 4199–4209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages V, Santa Maria SR, Prakash L, Prakash S (2009) Role of DNA damage‐induced replication checkpoint in promoting lesion bypass by translesion synthesis in yeast. Genes Dev 23: 1438–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palou G, Palou R, Zeng F, Vashisht AA, Wohlschlegel JA, Quintana DG (2015) Three different pathways prevent chromosome segregation in the presence of DNA damage or replication stress in budding yeast. PLoS Genet 11: e1005468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo B, Crabbe L, Pasero P (2017) Signaling pathways of replication stress in yeast. FEMS Yeast Res 17: fow101 [DOI] [PubMed] [Google Scholar]
- Park MS, Ludwig DL, Stigger E, Lee SH (1996) Physical interaction between human RAD52 and RPA is required for homologous recombination in mammalian cells. J Biol Chem 271: 18996–19000 [DOI] [PubMed] [Google Scholar]
- Pasero P, Vindigni A (2017) Nucleases acting at stalled forks: how to reboot the replication program with a few shortcuts. Annu Rev Genet 51: 477–499 [DOI] [PubMed] [Google Scholar]
- Patel DR, Weiss RS (2018) A tough row to hoe: when replication forks encounter DNA damage. Biochem Soc Trans 46: 1643–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica‐Worms H (1997) Mitotic and G2 checkpoint control: regulation of 14‐3‐3 protein binding by phosphorylation of Cdc25C on serine‐216. Science 277: 1501–1505 [DOI] [PubMed] [Google Scholar]
- Peng A, Chen PL (2003) NFBD1, like 53BP1, is an early and redundant transducer mediating Chk2 phosphorylation in response to DNA damage. J Biol Chem 278: 8873–8876 [DOI] [PubMed] [Google Scholar]
- Petsalaki E, Dandoulaki M, Morrice N, Zachos G (2014) Chk1 protects against chromatin bridges by constitutively phosphorylating BLM serine 502 to inhibit BLM degradation. J Cell Sci 127: 3902–3908 [DOI] [PubMed] [Google Scholar]
- Pfander B, Diffley JF (2011) Dpb11 coordinates Mec1 kinase activation with cell cycle‐regulated Rad9 recruitment. EMBO J 30: 4897–4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pic‐Taylor A, Darieva Z, Morgan BA, Sharrocks AD (2004) Regulation of cell cycle‐specific gene expression through cyclin‐dependent kinase‐mediated phosphorylation of the forkhead transcription factor Fkh2p. Mol Cell Biol 24: 10036–10046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines A, Kelstrup CD, Vrouwe MG, Puigvert JC, Typas D, Misovic B, de Groot A, von Stechow L, van de Water B, Danen EH et al (2011) Global phosphoproteome profiling reveals unanticipated networks responsive to cisplatin treatment of embryonic stem cells. Mol Cell Biol 31: 4964–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppy Roworth A, Ghari F, La Thangue NB (2015) To live or let die – complexity within the E2F1 pathway. Mol Cell Oncol 2: e970480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin B, Gao B, Yu J, Yuan J, Lou Z (2013) Ataxia telangiectasia‐mutated‐ and Rad3‐related protein regulates the DNA damage‐induced G2/M checkpoint through the Aurora A cofactor Bora protein. J Biol Chem 288: 16139–16144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randell JC, Fan A, Chan C, Francis LI, Heller RC, Galani K, Bell SP (2010) Mec1 is one of multiple kinases that prime the Mcm2‐7 helicase for phosphorylation by Cdc7. Mol Cell 40: 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo F, Falbo L, Di Sanzo M, Misaggi R, Faniello MC, Viglietto G, Cuda G, Costanzo F, Quaresima B (2011) BRCA1 is required for hMLH1 stabilization following doxorubicin‐induced DNA damage. Int J Biochem Cell Biol 43: 1754–1763 [DOI] [PubMed] [Google Scholar]
- Rossi SE, Ajazi A, Carotenuto W, Foiani M, Giannattasio M (2015) Rad53‐mediated regulation of Rrm3 and Pif1 DNA helicases contributes to prevention of aberrant fork transitions under replication stress. Cell Rep 13: 80–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz JF, Pardo B, Sastre‐Moreno G, Aguilera A, Blanco L (2013) Yeast pol4 promotes tel1‐regulated chromosomal translocations. PLoS Genet 9: e1003656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbioneda S, Bortolomai I, Giannattasio M, Plevani P, Muzi‐Falconi M (2007) Yeast Rev1 is cell cycle regulated, phosphorylated in response to DNA damage and its binding to chromosomes is dependent upon MEC1. DNA Repair (Amst) 6: 121–127 [DOI] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, Cimprich KA (2017) The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18: 622–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Hamperl S, Bocek MJ, Chung M, Bass TE, Cisneros‐Soberanis F, Samejima K, Xie L, Paulson JR, Earnshaw WC et al (2018) An intrinsic S/G2 checkpoint enforced by ATR. Science 361: 806–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ (1996) Regulation of RAD53 by the ATM‐like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271: 357–360 [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica‐Worms H, Elledge SJ (1997) Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, Elledge SJ (1999) Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 286: 1166–1171 [DOI] [PubMed] [Google Scholar]
- Santocanale C, Diffley JF (1998) A Mec1‐ and Rad53‐dependent checkpoint controls late‐firing origins of DNA replication. Nature 395: 615–618 [DOI] [PubMed] [Google Scholar]
- Sarbajna S, West SC (2014) Holliday junction processing enzymes as guardians of genome stability. Trends Biochem Sci 39: 409–419 [DOI] [PubMed] [Google Scholar]
- Sastre‐Moreno G, Pryor JM, Moreno‐Onate M, Herrero‐Ruiz AM, Cortes‐Ledesma F, Blanco L, Ramsden DA, Ruiz JF (2017) Regulation of human pollambda by ATM‐mediated phosphorylation during non‐homologous end joining. DNA Repair (Amst) 51: 31–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky K, Bar‐Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S et al (1995) A single ataxia telangiectasia gene with a product similar to PI‐3 kinase. Science 268: 1749–1753 [DOI] [PubMed] [Google Scholar]
- Schwartz MF, Duong JK, Sun Z, Morrow JS, Pradhan D, Stern DF (2002) Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol Cell 9: 1055–1065 [DOI] [PubMed] [Google Scholar]
- Schwarz JK, Lovly CM, Piwnica‐Worms H (2003) Regulation of the Chk2 protein kinase by oligomerization‐mediated cis‐ and trans‐phosphorylation. Mol Cancer Res 1: 598–609 [PubMed] [Google Scholar]
- Segurado M, Diffley JF (2008) Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev 22: 1816–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segurado M, Tercero JA (2009) The S‐phase checkpoint: targeting the replication fork. Biol Cell 101: 617–627 [DOI] [PubMed] [Google Scholar]
- Sengupta S, Robles AI, Linke SP, Sinogeeva NI, Zhang R, Pedeux R, Ward IM, Celeste A, Nussenzweig A, Chen J et al (2004) Functional interaction between BLM helicase and 53BP1 in a Chk1‐mediated pathway during S‐phase arrest. J Cell Biol 166: 801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setiaputra D, Durocher D (2019) Shieldin – the protector of DNA ends. EMBO Rep 20: e47560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechter D, Costanzo V, Gautier J (2004) ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol 6: 648–655 [DOI] [PubMed] [Google Scholar]
- Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher‐Scholz G, Lobrich M et al (2011) Factors determining DNA double‐strand break repair pathway choice in G2 phase. EMBO J 30: 1079–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigechi T, Tomida J, Sato K, Kobayashi M, Eykelenboom JK, Pessina F, Zhang Y, Uchida E, Ishiai M, Lowndes NF et al (2012) ATR‐ATRIP kinase complex triggers activation of the Fanconi anemia DNA repair pathway. Cancer Res 72: 1149–1156 [DOI] [PubMed] [Google Scholar]
- Shinozaki T, Nota A, Taya Y, Okamoto K (2003) Functional role of Mdm2 phosphorylation by ATR in attenuation of p53 nuclear export. Oncogene 22: 8870–8880 [DOI] [PubMed] [Google Scholar]
- Shiotani B, Zou L (2009) Single‐stranded DNA orchestrates an ATM‐to‐ATR switch at DNA breaks. Mol Cell 33: 547–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova JM, Breeden LL (1997) Rad53‐dependent phosphorylation of Swi6 and down‐regulation of CLN1 and CLN2 transcription occur in response to DNA damage in Saccharomyces cerevisiae . Genes Dev 11: 3032–3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova JM, Breeden LL (2003) Rad53 checkpoint kinase phosphorylation site preference identified in the Swi6 protein of Saccharomyces cerevisiae . Mol Cell Biol 23: 3405–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh TR, Ali AM, Paramasivam M, Pradhan A, Wahengbam K, Seidman MM, Meetei AR (2013) ATR‐dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions. Cancer Res 73: 4300–4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits VAJ, Cabrera E, Freire R, Gillespie DA (2019) Claspin – checkpoint adaptor and DNA replication factor. FEBS J 286: 441–455 [DOI] [PubMed] [Google Scholar]
- Smolka MB, Albuquerque CP, Chen SH, Zhou H (2007) Proteome‐wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci USA 104: 10364–10369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somyajit K, Basavaraju S, Scully R, Nagaraju G (2013) ATM‐ and ATR‐mediated phosphorylation of XRCC3 regulates DNA double‐strand break‐induced checkpoint activation and repair. Mol Cell Biol 33: 1830–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soniat MM, Myler LR, Kuo HC, Paull TT, Finkelstein IJ (2019) RPA phosphorylation inhibits DNA resection. Mol Cell 75: 145–153.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T (2005) The cell‐cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol 7: 195–201 [DOI] [PubMed] [Google Scholar]
- Sorensen CS, Syljuasen RG (2012) Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res 40: 477–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulier J, Lowndes NF (1999) The BRCT domain of the S. cerevisiae checkpoint protein Rad9 mediates a Rad9‐Rad9 interaction after DNA damage. Curr Biol 9: 551–554 [DOI] [PubMed] [Google Scholar]
- Stevens C, Smith L, La Thangue NB (2003) Chk2 activates E2F‐1 in response to DNA damage. Nat Cell Biol 5: 401–409 [DOI] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ (2003) MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421: 961–966 [DOI] [PubMed] [Google Scholar]
- Stokes MP, Rush J, Macneill J, Ren JM, Sprott K, Nardone J, Yang V, Beausoleil SA, Gygi SP, Livingstone M et al (2007) Profiling of UV‐induced ATM/ATR signaling pathways. Proc Natl Acad Sci USA 104: 19855–19860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP (2005) MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double‐strand breaks. Cell 123: 1213–1226 [DOI] [PubMed] [Google Scholar]
- Sugiyama T, New JH, Kowalczykowski SC (1998) DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single‐stranded DNA. Proc Natl Acad Sci USA 95: 6049–6054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, Kowalczykowski SC (2002) Rad52 protein associates with replication protein A (RPA)‐single‐stranded DNA to accelerate Rad51‐mediated displacement of RPA and presynaptic complex formation. J Biol Chem 277: 31663–31672 [DOI] [PubMed] [Google Scholar]
- Sun Z, Fay DS, Marini F, Foiani M, Stern DF (1996) Spk1/Rad53 is regulated by Mec1‐dependent protein phosphorylation in DNA replication and damage checkpoint pathways. Genes Dev 10: 395–406 [DOI] [PubMed] [Google Scholar]
- Sun Z, Hsiao J, Fay DS, Stern DF (1998) Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 281: 272–274 [DOI] [PubMed] [Google Scholar]
- Szakal B, Branzei D (2013) Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J 32: 1155–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, Takei Y, Nakamura Y (2000) A ribonucleotide reductase gene involved in a p53‐dependent cell‐cycle checkpoint for DNA damage. Nature 404: 42–49 [DOI] [PubMed] [Google Scholar]
- Taschner M, Harreman M, Teng Y, Gill H, Anindya R, Maslen SL, Skehel JM, Waters R, Svejstrup JQ (2010) A role for checkpoint kinase‐dependent Rad26 phosphorylation in transcription‐coupled DNA repair in Saccharomyces cerevisiae . Mol Cell Biol 30: 436–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh GW, O'Shaughnessy AM, Jimeno S, Dobbie IM, Grenon M, Maffini S, O'Rorke A, Lowndes NF (2006) Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair (Amst) 5: 693–703 [DOI] [PubMed] [Google Scholar]
- Toh GW, Sugawara N, Dong J, Toth R, Lee SE, Haber JE, Rouse J (2010) Mec1/Tel1‐dependent phosphorylation of Slx4 stimulates Rad1‐Rad10‐dependent cleavage of non‐homologous DNA tails. DNA Repair (Amst) 9: 718–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker‐Jensen S, Mailand N, Bartek J, Lukas J (2013) ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155: 1088–1103 [DOI] [PubMed] [Google Scholar]
- Toledo L, Neelsen KJ, Lukas J (2017) Replication catastrophe: when a checkpoint fails because of exhaustion. Mol Cell 66: 735–749 [DOI] [PubMed] [Google Scholar]
- Tomimatsu N, Mukherjee B, Harris JL, Boffo FL, Hardebeck MC, Potts PR, Khanna KK, Burma S (2017) DNA‐damage‐induced degradation of EXO1 exonuclease limits DNA end resection to ensure accurate DNA repair. J Biol Chem 292: 10779–10790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travesa A, Kuo D, de Bruin RA, Kalashnikova TI, Guaderrama M, Thai K, Aslanian A, Smolka MB, Yates JR III, Ideker T et al (2012) DNA replication stress differentially regulates G1/S genes via Rad53‐dependent inactivation of Nrm1. EMBO J 31: 1811–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi V, Nagarjuna T, Sengupta S (2007) BLM helicase‐dependent and ‐independent roles of 53BP1 during replication stress‐mediated homologous recombination. J Cell Biol 178: 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T, Foster SS, Petrini JH (2009) Maintenance of the DNA‐damage checkpoint requires DNA‐damage‐induced mediator protein oligomerization. Mol Cell 33: 147–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerio‐Santiago M, de Los Santos‐Velazquez AI, Monje‐Casas F (2013) Inhibition of the mitotic exit network in response to damaged telomeres. PLoS Genet 9: e1003859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasianovich Y, Harrington LA, Makovets S (2014) Break‐induced replication requires DNA damage‐induced phosphorylation of Pif1 and leads to telomere lengthening. PLoS Genet 10: e1004679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassin VM, Anantha RW, Sokolova E, Kanner S, Borowiec JA (2009) Human RPA phosphorylation by ATR stimulates DNA synthesis and prevents ssDNA accumulation during DNA‐replication stress. J Cell Sci 122: 4070–4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialard JE, Gilbert CS, Green CM, Lowndes NF (1998) The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1‐dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J 17: 5679–5688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidanes GM, Sweeney FD, Galicia S, Cheung S, Doyle JP, Durocher D, Toczyski DP (2010) CDC5 inhibits the hyperphosphorylation of the checkpoint kinase Rad53, leading to checkpoint adaptation. PLoS Biol 8: e1000286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriend LE, Prakash R, Chen CC, Vanoli F, Cavallo F, Zhang Y, Jasin M, Krawczyk PM (2016) Distinct genetic control of homologous recombination repair of Cas9‐induced double‐strand breaks, nicks and paired nicks. Nucleic Acids Res 44: 5204–5217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner SA, Oehler H, Voigt A, Dalic D, Freiwald A, Serve H, Beli P (2016) ATR inhibition rewires cellular signaling networks induced by replication stress. Proteomics 16: 402–416 [DOI] [PubMed] [Google Scholar]
- Wang H, Liu D, Wang Y, Qin J, Elledge SJ (2001) Pds1 phosphorylation in response to DNA damage is essential for its DNA damage checkpoint function. Genes Dev 15: 1361–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Carpenter PB, Elledge SJ (2002) 53BP1, a mediator of the DNA damage checkpoint. Science 298: 1435–1438 [DOI] [PubMed] [Google Scholar]
- Wang H, Powell SN, Iliakis G, Wang Y (2004a) ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res 64: 7139–7143 [DOI] [PubMed] [Google Scholar]
- Wang YG, Nnakwe C, Lane WS, Modesti M, Frank KM (2004b) Phosphorylation and regulation of DNA ligase IV stability by DNA‐dependent protein kinase. J Biol Chem 279: 37282–37290 [DOI] [PubMed] [Google Scholar]
- Wang X, Kennedy RD, Ray K, Stuckert P, Ellenberger T, D'Andrea AD (2007) Chk1‐mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Mol Cell Biol 27: 3098–3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Gong Z, Chen J (2011) MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J Cell Biol 193: 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert TA, Hartwell LH (1988) The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae . Science 241: 317–322 [DOI] [PubMed] [Google Scholar]
- Weinert T (1997) A DNA damage checkpoint meets the cell cycle engine. Science 277: 1450–1451 [DOI] [PubMed] [Google Scholar]
- Wu X, Shell SM, Yang Z, Zou Y (2006) Phosphorylation of nucleotide excision repair factor xeroderma pigmentosum group A by ataxia telangiectasia mutated and Rad3‐related‐dependent checkpoint pathway promotes cell survival in response to UV irradiation. Cancer Res 66: 2997–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Luo K, Lou Z, Chen J (2008) MDC1 regulates intra‐S‐phase checkpoint by targeting NBS1 to DNA double‐strand breaks. Proc Natl Acad Sci USA 105: 11200–11205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D (1996) Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev 10: 2411–2422 [DOI] [PubMed] [Google Scholar]
- Xu X, Tsvetkov LM, Stern DF (2002) Chk2 activation and phosphorylation‐dependent oligomerization. Mol Cell Biol 22: 4419–4432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A et al (2017) ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor‐resistant BRCA‐deficient cancer cells. Genes Dev 31: 318–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekezare M, Gomez‐Gonzalez B, Diffley JF (2013) Controlling DNA replication origins in response to DNA damage – inhibit globally, activate locally. J Cell Sci 126: 1297–1306 [DOI] [PubMed] [Google Scholar]
- Yoo HY, Shevchenko A, Dunphy WG (2004) Mcm2 is a direct substrate of ATM and ATR during DNA damage and DNA replication checkpoint responses. J Biol Chem 279: 53353–53364 [DOI] [PubMed] [Google Scholar]
- Yoo HY, Jeong SY, Dunphy WG (2006) Site‐specific phosphorylation of a checkpoint mediator protein controls its responses to different DNA structures. Genes Dev 20: 772–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Mahaney BL, Yano K, Ye R, Fang S, Douglas P, Chen DJ, Lees‐Miller SP (2008) DNA‐PK and ATM phosphorylation sites in XLF/Cernunnos are not required for repair of DNA double strand breaks. DNA Repair (Amst) 7: 1680–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegerman P, Diffley JF (2010) Checkpoint‐dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 467: 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha S, Jiang W, Fujiwara Y, Patel H, Goff PH, Brush JW, Dubois RL, Alt FW (2011) Ataxia telangiectasia‐mutated protein and DNA‐dependent protein kinase have complementary V(D)J recombination functions. Proc Natl Acad Sci USA 108: 2028–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Succi J, Feng Z, Prithivirajsingh S, Story MD, Legerski RJ (2004) Artemis is a phosphorylation target of ATM and ATR and is involved in the G2/M DNA damage checkpoint response. Mol Cell Biol 24: 9207–9220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Nirantar S, Lim HH, Sinha I, Surana U (2009) DNA damage checkpoint maintains CDH1 in an active state to inhibit anaphase progression. Dev Cell 17: 541–551 [DOI] [PubMed] [Google Scholar]
- Zhao X, Muller EG, Rothstein R (1998) A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2: 329–340 [DOI] [PubMed] [Google Scholar]
- Zhao H, Piwnica‐Worms H (2001) ATR‐mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21: 4129–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Chabes A, Domkin V, Thelander L, Rothstein R (2001) The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J 20: 3544–3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Rothstein R (2002) The Dun1 checkpoint kinase phosphorylates and regulates the ribonucleotide reductase inhibitor Sml1. Proc Natl Acad Sci USA 99: 3746–3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi G, Wilson JB, Chen X, Krause DS, Xiao Y, Jones NJ, Kupfer GM (2009) Fanconi anemia complementation group FANCD2 protein serine 331 phosphorylation is important for fanconi anemia pathway function and BRCA2 interaction. Cancer Res 69: 8775–8783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408: 433–439 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Paull TT (2013) DNA‐dependent protein kinase regulates DNA end resection in concert with Mre11‐Rad50‐Nbs1 (MRN) and ataxia telangiectasia‐mutated (ATM). J Biol Chem 288: 37112–37125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T (2013) 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science 339: 700–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L (2013) Four pillars of the S‐phase checkpoint. Genes Dev 27: 227–233 [DOI] [PMC free article] [PubMed] [Google Scholar]