

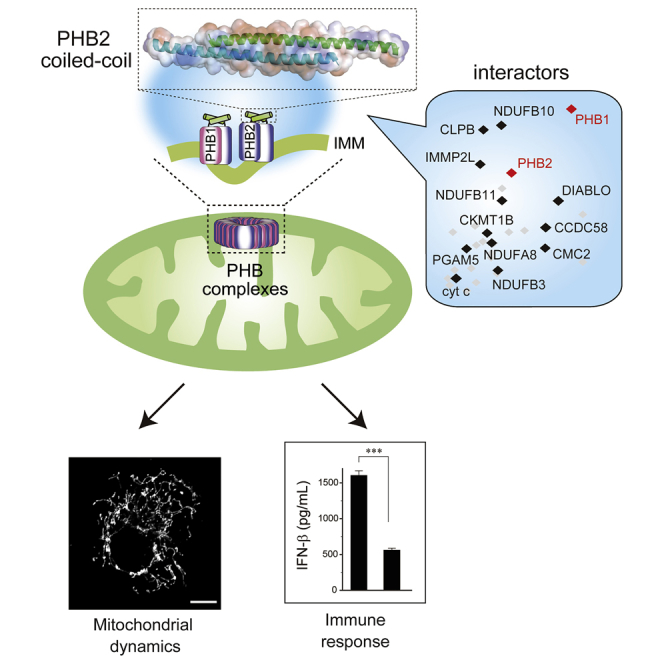
Summary
The coiled-coil motif mediates subunit oligomerization and scaffolding and underlies several fundamental biologic processes. Prohibitins (PHBs), mitochondrial inner membrane proteins involved in mitochondrial homeostasis and signal transduction, are predicted to have a coiled-coil motif, but their structural features are poorly understood. Here we solved the crystal structure of the heptad repeat (HR) region of PHB2 at 1.7-Å resolution, showing that it assembles into a dimeric, antiparallel coiled-coil with a unique negatively charged area essential for the PHB interactome in mitochondria. Disruption of the HR coiled-coil abolishes well-ordered PHB complexes and the mitochondrial tubular networks accompanying PHB-dependent signaling. Using a proximity-dependent biotin identification (BioID) technique in live cells, we mapped a number of mitochondrial intermembrane space proteins whose association with PHB2 relies on the HR coiled-coil region. Elucidation of the PHB complex structure in mitochondria provides insight into essential PHB interactomes required for mitochondrial dynamics as well as signal transduction.
Subject Areas: Molecular Biology, Cell Biology, Structural Biology
Graphical Abstract
Highlights
-
•
Heptad repeat (HR) region of PHB2 is essential for PHB complexes in mitochondria
-
•
The HR region of PHB2 assembles into a dimeric, anti-parallel coiled-coil
-
•
Disruption of the PHB2 coiled-coil abolishes mitochondrial dynamics
-
•
The coiled-coil associates with mitochondrial proteins, invoking an immune response
Molecular Biology; Cell Biology; Structural Biology
Introduction
Mitochondria are dynamic organelles that undergo continual cycles of fusion and fission. The balance of these two processes controls the overall morphology of the mitochondrial population, which contains individual organelles ranging from small isolated spheres to interconnected tubular networks (Westermann, 2010, Chan, 2012, Wai and Langer, 2016). Beyond controlling mitochondrial morphology, mitochondrial fusion also enables cooperation between mitochondria and protects the function of the mitochondrial population. Without adequate mitochondrial fusion, mitochondria exist as autonomous organelles, a state that results in greater heterogeneity and dysfunction of the mitochondrial population (Chen et al., 2005). In mammals, mitochondrial fusion is controlled by several conserved large guanosine triphosphatases of the dynamin family that orchestrate mitofusins (MFN1 and MFN2) at the outer mitochondrial membrane (OMM) and optic atrophy 1 (OPA1), having eight isoforms resulting by transcriptional and post-translational processes, in the inner mitochondrial membrane (IMM) (Chen and Chan, 2010).
Prohibitins (PHBs) are ubiquitously expressed IMM proteins that form ring-like PHB complexes comprising multiple PHB1 and PHB2 subunits (Tatsuta et al., 2005) to maintain the cristae structure and mitochondrial integrity (Artal-Sanz and Tavernarakis, 2009, Osman et al., 2009b). Several lines of evidence indicate that PHB complexes act as a functional platform to sequester mitochondrial proteases from the substrate, OPA1, and control proteolytic processing of the OPA1 balance (coexistence with long and short isoforms) to regulate mitochondrial fusion (Merkwirth et al., 2008 & 2012; Ehses et al., 2009, Richter-Dennerlein et al., 2014, Korwitz et al., 2016). In mice, the absence of PHB complexes is embryonic lethal (Merkwirth et al., 2008), and postnatal neuronal-specific ablation of Phb2 causes neurodegeneration (Merkwirth et al., 2012), defects thought to be linked to the destabilization of fusogenic OPA1. One of the PHB complexes, PHB2, was also recently revealed to act as a crucial receptor for the removal of damaged mitochondria by mitophagy (Wei et al., 2017). Although the embryologic and cellular functions of PHB complexes involved in both mitochondrial morphology and integrity are beginning to be understood, little is known about how these molecules act mechanistically. PHBs have a unique structural motif, a 4,3 hydrophobic heptad repeat ([HR] region: Figure S1A) that is strongly associated with the coiled-coil motif (Figure S1B), which has central roles in protein-protein interactions (Grigoryan and Keating, 2008). The presence of the HR region in mitochondrial proteins is reminiscent of the structure of MFN1, in which the formation of anti-parallel coiled-coil structures mediates mitochondrial tethering (Koshiba et al., 2004). In addition, similar anti-parallel coiled-coils are formed during mitochondrial fission events mediated by dimeric states of mitochondrial division 1 and mitochondrial fission 1 complexes at the OMM (Koirala et al., 2010, Zhang et al., 2012). Understanding how PHB molecules are involved in mitochondrial membrane dynamics via the HR region in their putative coiled-coil motifs will fill a critical gap in our knowledge concerning its actions.
In the present study, we performed a biophysical and cell biologic analysis of the HR region of PHB2. We found that the HR region of PHB2 forms a dimeric antiparallel coiled-coil with a highly charged surface. Surprisingly, in addition to its self-dimerization, we revealed that this coiled-coil motif mediates the tight assembly of the PHB interactome in mitochondria. Mutations that disrupt the HR coiled-coil or change the HR surface charge properties severely disrupt the PHB interactome assembly, as well as mitochondrial morphology and its functions, indicating that PHB complexes are important for mitochondrial integrity. These structural insights contribute to elucidate the mechanistic roles of PHB complexes essential for mitochondrial functions.
Results
Heptad Repeat Region of PHB2 Is Essential for PHB Complexes in Mitochondria
To understand the structural basis of PHB complexes in mitochondria through its HR region, we sought to determine interactors that rely on this region. We first established HEK293 cell lines stably expressing either variant 1 (V1: used as the standard variant in this study) or variant 3 (V3: an alternative splicing variant lacking most of the HR sequence) of PHB2 (Figures S1A and S1B), which harbor a C-terminal biotin ligase (BirA) with a hemagglutinin tag, and screened live cells for proximate and interacting proteins. Validating these BirA-fused PHB2 variants biochemically, both recombinants behaved almost identically to endogenous PHB2, showing its potential localization in the mitochondria (Figure S1C) and association with the membrane (Figure S1D), although the affinity of PHB2(V3)-BirA with the membrane was slightly less than that of the V1 construct. The assembly formation of PHB2(V1)-BirA was also similar to that of endogenous PHB2, and it formed large PHB complexes <1 MDa, as revealed by blue native polyacrylamide gel electrophoresis (BN-PAGE) (Figure S1E). The complex formation of PHB2(V3)-BirA, however, was largely disrupted and exhibited an almost disordered complex state in the mitochondria.
To further characterize these structural differences of the PHB complexes, we used a proximity-based biotin labeling method in live cells. Using the improved proximity-dependent biotin identification screen (BioID2) (Kim et al., 2016) in our established HEK293 cell lines, we followed large-scale pull-downs of biotinylated mitochondrial proteins by mass spectrometric (MS) analysis (Figures 1A and 1B, and Table S1). We detected at least seven overlapping hits (PHB1, PHB2, AFG3L2, YME1L1, TIMM50, NDUFA5, and ATP5B) with previously identified PHB2(V1) interactors (Richter-Dennerlein et al., 2014) (Table S1). Remarkably, comparative analysis between these two variants revealed that a selective group of mitochondrial proteins, including PHB1; several subunits of electron transport chains (ETCs, mostly mitochondrial complex I); caseinolytic peptidase B protein homolog (CLPB), which is also known as mitochondrial ATPases associated with diverse cellular activities (AAA) ATPase chaperonin; and other functional proteins, including proteases, were significantly enriched in the PHB2(V1)-BirA fraction (Figure 1C). These results clearly demonstrated that the assembly of PHB2 with such interactors (an interactome) in mitochondria depended on the HR region.
Figure 1.

Identification of PHB2 Interactors that Rely on the HR Region
(A–C) Volcano plot showing PHB2(V1)-BirA versus control (A), PHB2(V3)-BirA versus control (B), and PHB2(V1)-BirA versus PHB2(V3)-BirA (C) plotted against the p value of quadruplicate (n = 4) experiments. The dashed boxes in (A) and (B) show significantly increased proteins (abundance ratio >4, p < 0.01), and the enrichment proteins (110 for A and 169 for B) were analyzed by gene ontology annotations defining the concepts of the biologic process mapping shown in the pie charts (%) on the right. In (C), proteins annotated with known IMS proteins are indicated in black and the positions of PHB1 and PHB2 are indicated in red. The control cells were the original HEK293 cell lines.
(D) Schematic view of the chemical cross-linking coupled with mass spectrometry approach. Mitochondria (Mt) from HEK293 cells stably expressing PHB2(V1)-Myc were chemically cross-linked with MS-cleavable cross-linker disuccinimidyl dibutyric urea (DSBU) (maximal distance constraint of 12.5 Å) for 30 min, followed by stringent extraction of the protein lysates. The extracted cross-linked proteins were affinity purified for subsequent analysis and identification by liquid chromatography-tandem MS.
(E) Cross-link map showing the sequence position of all observed Lys-Lys cross-linked pairs between PHB2 and other mitochondrial proteins. Cross-links within PHB2 could not be distinguished either intramolecularly (one monomer) or intermolecularly (monomers) in this analysis. The cross-linking experiments were performed under 50 μM (red dashed line) or 100 μM (blue line) DSBU concentrations, and the red solid lines show cross-linked pairs observed in both conditions.
Structural Profiling of PHB Complexes by Cross-Linking MS
Having identified that well-ordered PHB complexes in mitochondria are responsible for the presence of the HR region, we next applied a chemical cross-linking approach coupled with MS (Leitner et al., 2016) to elucidate the spatial arrangement of the complexes at the subunit level. Isolated mitochondria from cells expressing a Myc-tagged version of PHB2(V1) were chemically cross-linked using an MS-cleavable cross-linker, disuccinimidyl dibutyric urea, the resultant extracts were purified by affinity beads, and the digested peptides were analyzed by liquid chromatography-tandem MS (Figure 1D). We identified a total of 33 unique Lys-Lys cross-links in the PHB complexes (Table S2), and the vast majority of identified peptide pairs were between its heterotypic PHB1-PHB2 (11 cross-links) or homotypic PHB2-PHB2 (8 cross-links) (Figure 1E), suggesting that these molecules tightly interact, consistent with a previous report (Tatsuta et al., 2005). Three pairs of heterotypic peptides in particular (PHB1K202-PHB2K216, PHB1K202-PHB2K262, and PHB1K208-PHB2K218) were substantiated by the results of a reverse cross-linking experiment (using a Myc-tagged version of PHB1 construct, Table S2). We also found an intriguing linkage between PHB2 (K216) and DIABLO (K203), or between PHB2 (K89) and cytochrome c (K100), both of which are involved in apoptosis, suggesting a substantial role of PHB2 in cell death (Merkwirth et al., 2008). Of note, the site-specific interactions in the PHB complexes converged around the HR region (Figure 1E), providing us novel structural insight into the complex formation at the subunit level.
The HR Region of PHB2 Assembles into a Helical, Anti-parallel Coiled-Coil Structure
We investigated the biophysical properties of the HR region of PHB2. Based on the coiled-coil prediction program COILS (Lupas et al., 1991), we expressed a histidine-tagged recombinant protein spanning residues 188–247 (denoted PHB2188−247) in bacterial cells. The purified PHB2188−247 appeared to be well folded, because circular dichroism analysis indicated that it was highly helical (>60%) and unfolded cooperatively in neutral pH conditions (Figure S2A). As we expected, introducing a proline residue into the polypeptide (I225P) drastically reduced its helicity (Figure 2A).
Figure 2.

Biophysical Properties of the HR Region of PHB2
(A) Circular dichroism spectra of PHB2188−247 (filled square) and its I225P variant (open triangle) at 4°C in 1× PBS (pH 7.4).
(B) The overall structure of PHB2188−265. The left panel shows the side view of the dimeric, anti-parallel coiled-coil with the N and C termini labeled and the molecular surface in shadow. The right panel shows an end-on view looking down the 2-fold axis of the dimer structure.
(C) The left panel shows a helical wheel diagram with the residues indicated, and the interface residues from the a–d layers are shown in the right panel. In the left panel, positively and negatively charged residues are shown in blue and red, respectively, and four residues (I225, E229, E231, and E233) that were mutated in this study are indicated with an asterisk.
(D) An electrostatic potential map of PHB2188−265. Acidic potential is shown in red, and basic regions are shown in blue. The right panel shows an enlarged view from the central region of the coiled-coil, and the glutamates are projected. The structures in the figure were depicted using PyMOL.
We obtained crystals of PHB2188−265, slightly extended at the C termini, and solved its atomic structure at 1.7 Å by molecular replacement (Table S3). The PHB2188−265 polypeptide folded into a dimeric anti-parallel coiled-coil 113 Å long (Figure 2B). Each of the two helices can be depicted in a helical wheel diagram (Figure 2C, left) showing that the a and d residues of the HR constitute a predominantly hydrophobic interaction interface (Figure 2C, right) with an accessible surface area of 1,430 Å2. The PHB2 coiled-coil shows striking structural similarity (folding) to the coiled-coil region of p-stomatin PH1511p (Yokoyama et al., 2008), which is a member of the stomatin/prohibitin/flotillin/HflKC (SPFH) family (Figure S2B).
Role of the HR Region in PHBs Involved in Complex Formation
Our crystal structure revealed that the PHB2 homodimer folds into an elongated shape providing a highly charged surface (Figure S2C, top). A remarkable feature of the coiled-coil is the presence of a negatively charged patch comprising three glutamates (E229, E231, and E233) with a glutamine (Q227) residue at the center of the structure (Figure 2D). To test whether these acidic residues are essential for its coiled-coil folding and/or the PHB complexes in mitochondria, we designed a mutant in which these residues were substituted with lysine (K229, K231, and K233: denoted PHB2KKK) (Figure S2C, middle).
The substitution had no effect on the folding properties based on the indistinguishable molar ellipticity at 222 nm between the wild-type (WT) and KKK variant (Figure S2D, orange), indicating similar levels of helicity at the polypeptide level. Moreover, the PHB2KKK substitution in the full-length molecule also co-immunoprecipitated with the same level of endogenous PHBs in HEK293 cells as the WT, whereas the coiled-coil-disrupted variant PHB2I225P mostly lost this ability (Figure S2E). Consistent with these findings, we confirmed using the BioID2 assay (Table S4) that the PHB2KKK variant retained the abilities of heterotypic interaction with PHB1 (abundance ratio; WT/KKK = 1.28, Figure S2F, top). These findings indicate that the negatively charged residues on the HR surface do not affect the physical hydrophobic interaction interface between PHBs and consequently its overall coiled-coil folding. The presence of acidic residues, however, is absolutely required for the formation of the PHB interactome in mitochondria. Monitored by BN-PAGE, the PHB complexes comprising the PHB2KKK variant seemed to be destabilized (Figure S2G), and the BioID2 assay further revealed a much lower association with previously identified mitochondrial intermembrane space (IMS) proteins, such as CLPB and subunits of mitochondrial complex I (NDUFA8, NDUFB3, NDUFB10, and NDUFB11), in the PHB2KKK variant than in the WT (Table S4 and Figure S2F).
Role of the PHBs/HR Region in Mitochondrial Morphology and Function in Yeast
We thus studied the role of the HR region of PHB2 involved in mitochondrial morphology using PHB2-depleted cells. Conditional gene targeting of Phb2 in mouse embryonic fibroblast (MEF) cells via Cre recombinase expression had severe effects on the mitochondrial morphology and led to fragmented mitochondria in more than 60% of the cells (Figures 3A and 3B, flox/flox + Cre), as reported previously (Merkwirth et al., 2008). The morphologic defects were restored when the PHB2WT gene, but not the coiled-coil-disrupted variant, PHB2I225P, was re-introduced into the null cells. Importantly, the PHB2KKK variant also had little activity for regulating mitochondrial dynamics (Figures 3A and 3B) and a collapsed cristae morphology, as seen in the PHB2I225P variant, monitored by electron microscopy (Figure 3C).
Figure 3.

Disruption of PHB2 Coiled-Coil Abolishes Mitochondrial Dynamics
(A and B) Phb2flox/flox and its variant MEFs were infected with or without a retrovirus encoding GFP-fused Cre recombinase (+Cre) for 72 h to deplete the endogenous Phb2 gene, and the mitochondrial morphology of each cell was monitored by immunofluorescence microscopy (A). Mitochondria in cells were visualized using an antibody against mtHsp70. Insets depict the magnified images of each yellow boxed area. Scale bar, 10 μm. In (B) quantification of data in (A) is shown. Cells were scored as one of four morphologic categories as depicted on the right. In these experiments, at least 100 cells were scored. Each value represents the mean ± SEM (n = 3 experiments).
(C) Phb2flox/flox and its variant MEFs were infected with or without a retrovirus encoding Cre recombinase (+Cre) for 72 h to deplete the endogenous Phb2 gene, and the mitochondrial ultrastructure of each cell was analyzed by transmission electron microscopy. Bottom images: higher magnification images of regions outlined in the middle panels. Scale bar, 1 μm.
See also Figure S3.
Sequential alignments between PHB2 homologs in different organisms revealed conservation of the glutamate cluster, except for Phb2 of the yeast Saccharomyces cerevisiae (Figure 4A). In yeast, however, Phb1 possesses the conserved glutamates and shows a high probability of the coiled-coil formation (Figure 4B, left), leading us to explore the phenotypic significance of Phb1/HR in vivo. Although single PHB1 deletion (phb1Δ) mutant cells grow at a similar rate to WT cells (Figure 4C, + HygB), deletions of both PHB1 and its genetic interactor PSD1 (phb1Δpsd1Δ) cause lethality (Birner et al., 2003, Osman et al., 2009a) (Figure 4C, + G-418/HygB). The growth defect of the phb1Δpsd1Δ cells was completely rescued by introducing a plasmid encoding WT yeast Phb1 with a C-terminal Myc tag (Phb1WT). Most critically, rescue experiments involving the proline (Phb1V202P, Phb1V213P, and Phb1V202/213P) or lysine (Phb1KKK) variants showed no detectable restoration of cell growth even with abundant protein expression levels (Figure 4C). Taken together, these comparative in vitro and vivo studies highlight the importance of the coiled-coil and the charged residues within the structure involved in PHB function.
Figure 4.

Highly Conserved Glutamic Acid Residues in the HR Coiled-Coil Are Essential for the Function of PHB2 Homologs in Yeast
(A) Alignment of prohibitin homologs (Homo sapiens, Mus musculus, Drosophila melanogaster, Caenorhabditis elegans, Xenopus tropicalis, and Saccharomyces cerevisiae) around the HR region. Colons (:) indicate conserved residues among all species shown in the figure, and dots (·) indicate residues with similar properties. Asterisks at the bottom of the alignment sequences indicate mutated amino acids in the study.
(B) Prediction results of coiled-coil probability of yeast Phb1 (left) and Phb2 (right) using the program COILS (Lupas et al., 1991). The yPhb1 coiled-coil is predicted to be highly probable relative to that of yPhb2.
(C) Heterozygous diploid strains (PSD1/psd1Δ::kanMX4 PHB1/phb1Δ::hphNT1 CAN1/can1Δ::STE2pr-Sp_his5) carrying pRS416 (Empty), pRS416/yPhb1WT-Myc (WT), pRS416/yPhb1V202P-Myc (V202P), pRS416/yPhb1V213P-Myc (V213P), pRS416/yPhb1V202/213P-Myc (V202/213P), and pRS416/yPhb1KKK-Myc (KKK) were allowed to sporulate, plated on the various selection media indicated, and incubated at 30°C for 3 days. All the media contained canavanine without histidine and uracil for the selection of MATα haploid strains carrying pRS416 or its derivatives. + G-418, selection for psd1Δ strains; + HygB, selection for phb1Δ strains. The right panels show protein levels in the heterozygous diploid cells analyzed by western blot with antibodies against the Myc epitope and 3-phosphoglycerate kinase (Pgk1).
PHB2 Is Involved in Mitochondrial-Mediated Antiviral Innate Immunity
The aforementioned experiments indicated that the HR region of PHBs had potential roles in mitochondrial morphology and growth of yeast. To evaluate the physiologic relevance of PHB complexes in mammalian cells, we investigated the role of PHB2 via its HR region on a mitochondrial-mediated antiviral innate immune response, the retinoic acid-inducible gene I (RIG-I) signaling pathway, which is a vital function of mitochondria (Seth et al., 2005, Koshiba et al., 2011, Koshiba, 2013). We verified that Phb2flox/flox MEFs challenged with Sendai virus (SeV), a negative-stranded RNA virus of the Paramyxoviridae family, potently activated RIG-I-mediated signal transduction and the cells produced interferon β (IFN-β) and a proinflammatory cytokine interleukin-6, but the immune responses were attenuated when the Cre recombinase was expressed in the cells (Figure S3A). In contrast, the Phb2-depleted MEFs were rescued by introduction of the PHB2WT gene, but neither PHB2I225P nor PHB2KKK substantially recovered the antiviral responses. The functionality of the Phb2-depleted MEFs, however, was unaffected when the cells were infected with a double-stranded DNA virus, Herpes simplex virus 1, which activates the cGAS/STING cytosolic DNA sensing pathway (Figure S3B). Moreover, cytochrome c oxidase (COX, complex IV) activity was not affected in the Phb2-depleted MEFs (Figure S3C, brown stain). These observations indicate that the HR region contributes to the PHB2-mediated execution of mitochondrial-dependent antiviral innate immunity (RIG-I pathway).
To understand how PHB2 regulates RIG-I-mediated antiviral innate immunity, we focused on the functional role of the HR-dependent PHB2-interacting proteins involved in signal transduction using an RNA interference approach. HEK293 cells treated with each of the 12 gene candidates identified by BioID2 (Table S4) with its gene-specific small interfering RNAs (siRNAs), and activation of the IFN-β reporter was triggered by the delivery of poly(I:C), a synthetic analog of viral double-stranded RNA, into the siRNA-treated cells by transient transfection. Among these candidates, we confirmed that specific targeting of the subunits of mitochondrial complex I (NDUFA8, NDUFB3, and NDUFB11) potently suppressed activation of the IFN-β luciferase reporter (Figure 5A), consistent with our previous findings (Yoshizumi et al., 2017). Knockdown of PHB1 was also sufficient to inhibit RIG-I-mediated antiviral signaling, as seen with depletion of homolog Phb2 in MEFs. In particular, CLPB preferentially impaired the RIG-I-mediated antiviral innate immunity among these candidates (Figure 5A).
Figure 5.

Involvement of PHB Complexes in Antiviral Innate Immunity via CLPB
(A) Activation of the IFN-β reporter in siRNA-treated HEK293 cells (each target RNAi; #1–#3) transfected without (Mock) or with poly(I:C).
(B and C) HEK293 cells treated with control or CLPB siRNAs were transfected with IFN-β luciferase reporter plasmid either with RIG-I1−250 (B) or MAVS (C) expression plasmids (Mock; empty vector). The transfected cells were analyzed 24 h later for IFN-β-dependent luciferase activity.
(D) The siRNA-treated HEK293 cells were mock infected (−) or infected with SeV (4 hemagglutinin (HA) units/mL) for 20 h, and the activation of endogenous IRF-3 was analyzed by immunoblotting with its specific antibody against the phosphorylation of Ser386 site (pIRF-3). Anti-IRF-3 was used as the loading control.
(E and F) (E) Experiments were performed similarly to those in (D), except that the amounts of IFN-β produced by the infected cells were measured by ELISA. In (F), A549 cells were used instead of HEK293 cells. N.D., not detected.
(G) The mitochondrial fraction (Mt) isolated from HEK293 cells was treated with proteinase K (50 μg/mL) under regular (−) or hypotonic swelling (+) conditions for 15 min on ice. The reactants were developed by immunoblotting with antibodies against CLPB or against several mitochondrial markers as indicated. OMM proteins: MAVS and MFN1. IMS proteins: AIF and PHB2. IMM proteins: TIM23 and OPA1. Matrix protein: mtHSP70.
(H) Isolated mitochondria from HEK293 cells were treated with either 1 M NaCl (plus sonication) or 0.1 M Na2CO3 (pH 11.0) for 30 min on ice. After centrifugation, the supernatant (S) and pellets (P) were analyzed by immunoblotting with antibodies against CLPB or against several mitochondrial proteins as indicated.
(I) Interaction of CLPB and MAVS. HEK293 cells stably expressing Myc-tagged CLPB were mock infected (−) or infected with SeV (4 HAU/mL) for 20 h, and postnuclear cell lysates were subjected to immunoprecipitation (IP) with the antibody against Myc followed by analysis of western blots with an antibody against MAVS. The asterisk indicates Myc-tagged CLPB. The graph on the right shows the quantification of MAVS bands from the IP fraction analyzed by densitometry.
(J) Experiments were performed similarly to those in (I), except that the cells used in the study were pre-treated with control or PHB1 siRNAs before SeV infection.
(K) Experiments were performed similarly to those in (E), except that the cells were pre-treated with siRNA against AKAP1 or ATAD3A.
Each value represents the mean ± SEM (n = 3 experiments). Statistical analysis was performed by the two-tailed Student's t test. ***p < 0.001, **p < 0.01, and *p < 0.05. See also Figures S4 and S5 and Tables S4 and S5.
CLPB Bridges PHB2 to the Mitochondrial Adaptor, Mitochondrial Antiviral Signaling Protein
We therefore evaluated the functional role of the PHB2-CLPB axis involved in mitochondrial-mediated antiviral innate immunity. Overexpression of N-terminal CARD domains of RIG-I (designated as RIG-I1−250) in HEK293 cells induced a robust intracellular antiviral response, as reported previously (Seth et al., 2005, Yasukawa et al., 2009), and the activation was attenuated when the cells were treated with the CLPB-specific siRNA (Figure 5B). Knockdown of endogenous CLPB in cells similarly reduced the activation of the IFN-β reporter in response to overexpression of the mitochondrial antiviral signaling protein (MAVS) (Figure 5C), an OMM protein that acts downstream of RIG-I (Seth et al., 2005). Affirmatively, virus-induced phosphorylation of endogenous interferon regulatory factor-3 (IRF-3), the hallmark of IRF-3 activation (Mori et al., 2004), was severely impaired in the CLPB-depleted cells relative to control cells (Figure 5D), and the production of endogenous IFN-β protein was similarly reduced (Figure 5E). We also corroborated the observed effects of CLPB using another type of cell, A549 (Figure 5F). In contrast, loss of endogenous CLPB did not impair the IRF-3 phosphorylation in response to extracellular poly(I:C), which activates the mitochondrial-independent toll-like receptor 3 (TLR-3) pathway (Figure S4A); furthermore, mitochondria in the CLPB-depleted cells were healthy with a mitochondrial membrane potential (Figure S4B) and COX activity (Figure S4C) indistinguishable from those control cells. Taken together, these results demonstrated that CLPB likely acts as a modulator downstream of MAVS in the RIG-I signaling pathway.
We performed a structure-function analysis to fill the topologic gap between CLPB and the mitochondrial adaptor MAVS. Biochemical experiments revealed that sub-mitochondrial localization of CLPB was the IMS because protease accessibility in isolated mitochondria from cells increased only when the OMM was disrupted by hypotonic swelling (Figure 5G). Sonication of mitochondria with high-salt conditions (1 M NaCl) or treatment of mitochondria with an alkaline solution (pH 11.0) allowed us to extract CLPB from the exclusively soluble fractions (Figure 5H), similar to unanchored IMS proteins, such as apoptosis-inducing factor or HtrA serine peptidase 2. Consistent with this fact, we confirmed that the CLPB associated with many kinds of IMS proteins (Table S5) and physically interacted with PHB2 via its putative coiled-coil motif at the central region (Figure S5A). Together, these behaviors indicate that CLPB is a soluble IMS protein that does not embed into the IMM.
Mechanistic Investigation of the Mitochondrial Signalosome
We attempted to clarify how CLPB, as a part of PHB complexes, is involved in the RIG-I signaling pathway. To address this issue, we analyzed immunoprecipitates derived from HEK293 cells to determine whether CLPB could functionally and physically interact with MAVS. Despite the fact that CLPB, which was tightly associated with PHB complexes, barely interacted with endogenous MAVS under physiologic conditions, its interactions were markedly enhanced by SeV infection (Figure 5I). The observed role of CLPB-PHB complexes in antiviral immunity was also verified by PHB2 immunoprecipitation (Figure S5B). Using a co-immunoprecipitation approach, we mapped the region of MAVS that interacts with CLPB through its transmembrane region (Figures S5C and S5D), assuming that their interfaces would present at the IMS. Importantly, the virus-induced MAVS-CLPB interactions were abolished when the PHB proteins were depleted by siRNA (Figure 5J), suggesting that the scaffold function of the PHB complexes is required for the phenomenon.
Finally, we explored additional molecules that might tether CLPB and MAVS at the IMS. By performing BioID2 experiments in HEK293 cells stably expressing either CLPB or MAVS harboring BirA tags, we identified several molecules that were highly enriched in both fractions (also filtered by PHB complexes, Figure S5E and Table S5). Of these, we were interested in two mitochondrial proteins, A-kinase-anchoring protein 1 (AKAP1) in which the N terminus is localized to the IMS (Jun et al., 2016) and ATPase family AAA-domain containing protein 3 A (ATAD3A) that spans the IMM and OMM (Baudier, 2018) (Figure S5F). Consistent with the findings, AKAP1 and ATAD3A both co-immunoprecipitated with endogenous MAVS in the HEK293 cells (Figure S5G), and depletion of these molecules in the cells exhibited strikingly defective antiviral innate immune responses against SeV infection (Figure 5K). We assume that both AKAP1 and ATAD3A tether MAVS throughout the infection process because the BioID2 analysis revealed that these interactions were similar following SeV infection (Figure S5H). Taken together, these results demonstrated that CLPB bridges PHB complexes (IMM) and MAVS (OMM) with the assistance of AKAP1 and ATAD3A at the IMS and invokes an immune response from the mitochondrial signalosome (Figure S5I).
Discussion
PHBs are key components of mitochondrial scaffold proteins that localize in the IMM from yeast to mammals. PHB belongs to the SPFH family of proteins that also function as membrane scaffolds by forming highly ordered oligomers in many species (Browman et al., 2007). Although several biochemical analyses suggest that coiled-coil motifs at their C terminus are required for PHB oligomerization and its functions (Joshi et al., 2003, Tatsuta et al., 2005, He et al., 2008), direct evidence for their roles in mitochondrial functions via the structural motif is limited. Here we show that the HR region at the C terminus of PHB2 forms a dimeric, anti-parallel coiled-coil revealed by X-ray crystallography and demonstrated their involvement in mitochondrial morphology and especially, in antiviral innate immunity (RIG-I pathway).
Our biochemical analysis indicates that the HR region of PHB2, in addition to serving as a homotypic dimerization, mediates a strong interaction with their same region in PHB1 (Table S2 and Figure 1E), which is assumed to fold into a heterotypic dimer (Merkwirth and Langer, 2009). In fact, our simulation of the heterotypic dimerization model predicts proper folding without any structural constraints, and notably, the coiled-coil also showed a negatively charged patch (Figure S2C, bottom), an essential feature for PHB2 in vitro and vivo functions. Although our results demonstrate that the formation of PHB complexes in mitochondria is HR dependent, it should be noted that the interactions between PHB proteins seem to be quite complex, because some weak interactions are still observed in variants without the coiled-coils (Table S1 and Figure S2E). Indeed, a previous study demonstrated that an SPFH domain protein from archaea Pyrococcus horikoshii implied its successive self-oligomerization without the coiled-coil region (Kuwahara et al., 2009). In future work, it will be important to unravel the relative contributions of these multiple interactions in the PHB complexes and how they affect the interactome assembly.
Besides the role of PHBs in mitochondrial integrity, several studies have pointed to the function of these proteins in viral infection, especially an association with viral proteins (Kuadkitkan et al., 2010, Wintachai et al., 2012, Liu et al., 2015). In this context, we found that PHB complexes are involved in RIG-I-mediated antiviral innate immunity via CLPB (Figure 5), an interactor of the PHB2 coiled-coil motif. Our previous study revealed that RIG-I-mediated antiviral innate immunity requires oxidative phosphorylation activity, a prominent physiologic function of mitochondria, and OPA1 ensures the activity and regulates the immune response (Yoshizumi et al., 2017). Consistent with the fact, the interactome assembly of mitochondrial PHB complexes includes several subunits of ETC (Tables S1, S4, and S5), and ablation of these specific genes in cells attenuates RIG-I-mediated signal transduction (Figure 5A). Based on these findings together, we propose a mechanistic model of the role of PHB complexes in mediating virus-triggered innate immune signal transduction (Figure S5I). In this model, a topologic gap between MAVS and the PHB complexes is filled by CLPB, with the assistance of AKAP1 and ATAD3A, that builds a bridge between the OMM and IMM upon viral infection, forming an MAVS signalosome that leads to the recruitment of various downstream effectors, such as tumor necrosis factor receptor-associated factor family members on the OMM (West et al., 2011, Koshiba, 2013). Our biochemical and structural insights might help to clarify how PHB complexes mediate mitochondrial-mediated innate immunity.
Finally, CLPB variants are reported in humans with autosomal recessive mitochondrial disorders (Saunders et al., 2015, Wortmann et al., 2015, Pronicka et al., 2017), although the exact physiologic functions of the protein remain elusive. On the other hand, the bacterial homolog clpB acts as a chaperone involved in the disaggregation of misfolded proteins (Doyle and Wickner, 2009). The findings of the present study may pave the way for understanding the unexpected role of CLPB in structural rearrangements of MAVS, ultimately leading to an activated conformation, and mutations of CLPB may affect to fulfill this role.
Limitations of the Study
We demonstrated that CLPB bridges PHB complexes (IMM) and MAVS (OMM) with the assistance of AKAP1 and ATAD3A at the IMS and invokes mitochondrial-mediated antiviral innate immunity. In the present study, however, we could not determine whether these interactions (MAVS-CLPB-AKAP1-ATAD3A) are direct or indirect. Because MAVS is a C-terminal-tail-anchored OMM protein with few amino acids facing into the IMS, we cannot exclude the possibility that additional molecules, such as mitochondrial contact site and cristae-organizing system (MICOS) complex (Rampelt et al., 2017), mediate accessible contact sites between the IMM and OMM and assist in establishing the mitochondrial signalosome. Indeed, our BioID2 data revealed that both MAVS and CLPB substantially associate with many MICOS components (Table S5). Further studies to elucidate the mechanistic and functional roles of these MICOS components will improve our understanding of how PHB interactomes are involved in MAVS signaling, particularly the linkage between mitochondrial physiology and innate immunity. Finally, because the prominent function of MAVS signaling is precisely regulated upon viral infection, it is also crucial to clarify how the mitochondrial signalosome is inactivated or terminated by inhibitors such as MFN2 (Yasukawa et al., 2009, Sasaki et al., 2013). Our study did not capture the whole mitochondrial signalosome during the infection process, and additional BioID studies of the kinetics under viral infection will reveal the transient structural rearrangement and provide clues regarding the communication between PHB interactomes and MFNs.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We are grateful to Thomas Langer (University of Cologne) and Jacques Baudier (French Institute of Health and Medical Research) for providing the Phb2flox/flox MEFs and the anti-ATAD3A (C-term) antibody, respectively. We also thank Koji Okamoto (Osaka University) and Katsuyoshi Mihara (Kyushu University) for their valuable comments on the study. We appreciate Non Miyata (Kyushu University) for his critical suggestions on western blotting analysis, Yuko Fuchigami (Kyushu University) for her technical assistance with DNA cloning and sequencing, and Mitsugu Yamada (JAXA) for early stage of crystallization and diffraction experiments. We thank the PX beamline staff of SPring-8 and Photon Factory (proposal No. BINDS0459). This work was supported by the JSPS KAKENHI Grants (No. 17H03667, 17K19561, and 18H04863), the Takeda Science Foundation, The Novartis Foundation (Japan) for the Promotion of Science, and Joint Usage and Joint Research Programs, the Institute of Advanced Medical Sciences, Tokushima University to T.K. This study was also partly supported by the Platform Project for Supporting Drug Discovery and Life Science Research [Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)] from AMED Grant (JP18am0101071 to T.T) and by the JSPS KAKENHI Grant (19H04966 to H.K.).
Author Contributions
T. Yoshinaka, T. Yoshizumi, and T.K. performed most of the mammalian cell culture experiments. R.F., Y.H., and T.T. performed bacterial expression, purification of recombinant proteins, and CD experiments. Y.H. and T.T. crystalized the recombinant protein and determined its crystal structure. O.K. performed the yeast study and H.K. performed the BioID2 and XL-MS analyses. All the authors analyzed the data and interpreted all the experimental results. T.K. designed experiments and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: September 27, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.08.056.
Supplemental Information
Mass spectrometry analysis of proximity interactors of the indicated *BirA-hemagglutinin (HA) fusion proteins. Note that the *BirA-HA is fused to the C terminus of PHB2 proteins. Proximity interactors are ranked in the order of the abundance ratios (log2) of PHB2(V1)/PHB2(V3) (average of four independent experiments).
The cross-linking experiments were performed under disuccinimidyl dibutyric urea (DSBU) concentrations of 50 or 100 μM. The resultant peptides (after digestion by trypsin/Lys-C mix) were divided into two halves and subjected to liquid chromatography tandem MS analysis using both Q Exactive Plus (QEplus) and Orbitrap Fusion Lumos (Lumos) mass spectrometers. The cross-linking experiment of PHB1 was performed under DSBU concentration of 100 μM.
Mass spectrometry analysis of proximity interactors of the indicated *BirA- hemagglutinin (HA) fusion proteins. Note that the *BirA-HA is fused to the C terminus of PHB2 proteins. Proteins are ranked in the order of the abundance ratios (log2) of PHB2WT/PHB2KKK.
Mass spectrometry analysis of proximity interactors of the indicated Myc-BirA* or *BirA- hemagglutinin (HA) fusion proteins. Note that Myc-BirA* is fused to the N terminus of MAVS and *BirA-HA is fused to the C terminus of CLPB or PHB1. Proteins are ranked in the order of the abundance ratios of CLPB/control.
References
- Artal-Sanz M., Tavernarakis N. Prohibitin and mitochondrial biology. Trends Endocrinol. Metab. 2009;20:394–401. doi: 10.1016/j.tem.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Baudier J. ATAD3 proteins: brokers of a mitochondria-endoplasmic reticulum connection in mammalian cells. Biol. Rev. Camb. Philos. Soc. 2018;93:827–844. doi: 10.1111/brv.12373. [DOI] [PubMed] [Google Scholar]
- Birner R., Nebauer R., Schneiter R., Daum G. Synthetic lethal interaction of the mitochondrial phosphatidylethanolamine biosynthetic machinery with the prohibitin complex of Saccharomyces cerevisiae. Mol. Biol. Cell. 2003;14:370–383. doi: 10.1091/mbc.E02-05-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browman D.T., Hoegg M.B., Robbins S.M. The SPFH domain-containing proteins: more than lipid raft markers. Trends Cell Biol. 2007;17:394–402. doi: 10.1016/j.tcb.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Chan D.C. Fusion and fission: interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- Chen H., Chan D.C. Physiological functions of mitochondrial fusion. Ann. N. Y. Acad. Sci. 2010;1201:21–25. doi: 10.1111/j.1749-6632.2010.05615.x. [DOI] [PubMed] [Google Scholar]
- Chen H., Chomyn A., Chan D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- Doyle S.M., Wickner S. Hsp104 and ClpB: protein disaggregating machines. Trends Biochem. Sci. 2009;34:40–48. doi: 10.1016/j.tibs.2008.09.010. [DOI] [PubMed] [Google Scholar]
- Ehses S., Raschke I., Mancuso G., Bernacchia A., Geimer S., Tondera D., Martinou J.C., Westermann B., Rugarli E.I., Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009;187:1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan G., Keating A.E. Structural specificity in coiled-coil interactions. Curr. Opin. Struct. Biol. 2008;18:477–483. doi: 10.1016/j.sbi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B., Feng Q., Mukherjee A., Lonard D.M., DeMayo F.J., Katzenellenbogen B.S., Lydon J.P., O'Malley B.W. A repressive role for prohibitin in estrogen signaling. Mol. Endocrinol. 2008;22:344–360. doi: 10.1210/me.2007-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi B., Ko D., Ordonez-Ercan D., Chellappan S.P. A putative coiled-coil domain of prohibitin is sufficient to repress E2F1-mediated transcription and induce apoptosis. Biochem. Biophys. Res. Commun. 2003;312:459–466. doi: 10.1016/j.bbrc.2003.10.148. [DOI] [PubMed] [Google Scholar]
- Jun Y.W., Park H., Lee Y.K., Kaang B.K., Lee J.A., Jang D.J. D-AKAP1a is a signal-anchored protein in the mitochondrial outer membrane. FEBS Lett. 2016;590:954–961. doi: 10.1002/1873-3468.12123. [DOI] [PubMed] [Google Scholar]
- Kim D.I., Jensen S.C., Noble K.A., Kc B., Roux K.H., Motamedchaboki K., Roux K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell. 2016;27:1188–1196. doi: 10.1091/mbc.E15-12-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koirala S., Bui H.T., Schubert H.L., Eckert D.M., Hill C.P., Kay M.S., Shaw J.M. Molecular architecture of a dynamin adaptor: implications for assembly of mitochondrial fission complexes. J. Cell Biol. 2010;191:1127–1139. doi: 10.1083/jcb.201005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korwitz A., Merkwirth C., Richter-Dennerlein R., Tröder S.E., Sprenger H.G., Quirós P.M., López-Otín C., Rugarli E.I., Langer T. Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria. J. Cell Biol. 2016;212:157–166. doi: 10.1083/jcb.201507022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T. Mitochondrial-mediated antiviral immunity. Biochim. Biophys. Acta. 2013;1833:225–232. doi: 10.1016/j.bbamcr.2012.03.005. [DOI] [PubMed] [Google Scholar]
- Koshiba T., Detmer S.A., Kaiser J.T., Chen H., McCaffery J.M., Chan D.C. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- Koshiba T., Yasukawa K., Yanagi Y., Kawabata S. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci. Signal. 2011;4:ra7. doi: 10.1126/scisignal.2001147. [DOI] [PubMed] [Google Scholar]
- Kuadkitkan A., Wikan N., Fongsaran C., Smith D.R. Identification and characterization of prohibitin as a receptor protein mediating DENV-2 entry into insect cells. Virology. 2010;406:149–161. doi: 10.1016/j.virol.2010.07.015. [DOI] [PubMed] [Google Scholar]
- Kuwahara Y., Unzai S., Nagata T., Hiroaki Y., Yokoyama H., Matsui I., Ikegami T., Fujiyoshi Y., Hiroaki H. Unusual thermal disassembly of the SPFH domain oligomer from Pyrococcus horikoshii. Biophys. J. 2009;97:2034–2043. doi: 10.1016/j.bpj.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A., Faini M., Stengel F., Aebersold R. Crosslinking and mass spectrometry: an integrated technology to understand the structure and function of molecular machines. Trends Biochem. Sci. 2016;41:20–32. doi: 10.1016/j.tibs.2015.10.008. [DOI] [PubMed] [Google Scholar]
- Liu S., Wang W., Brown L.E., Qiu C., Lajkiewicz N., Zhao T., Zhou J., Porco J.A., Jr., Wang T.T. A novel class of small molecule compounds that inhibit hepatitis C virus infection by targeting the prohibitin-CRaf pathway. EBioMedicine. 2015;2:1600–1606. doi: 10.1016/j.ebiom.2015.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A., Van Dyke M., Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Merkwirth C., Langer T. Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochim. Biophys. Acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- Merkwirth C., Dargazanli S., Tatsuta T., Geimer S., Löwer B., Wunderlich F.T., von Kleist-Retzow J.C., Waisman A., Westermann B., Langer T. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008;22:476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C., Martinelli P., Korwitz A., Morbin M., Brönneke H.S., Jordan S.D., Rugarli E.I., Langer T. Loss of prohibitin membrane scaffolds impairs mitochondrial architecture and leads to tau hyperphosphorylation and neurodegeneration. PLoS Genet. 2012;8:e1003021. doi: 10.1371/journal.pgen.1003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M., Yoneyama M., Ito T., Takahashi K., Inagaki F., Fujita T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J. Biol. Chem. 2004;279:9698–9702. doi: 10.1074/jbc.M310616200. [DOI] [PubMed] [Google Scholar]
- Osman C., Haag M., Potting C., Rodenfels J., Dip P.V., Wieland F.T., Brügger B., Westermann B., Langer T. The genetic interactome of prohibitins: coordinated control of cardiolipin and phosphatidylethanolamine by conserved regulators in mitochondria. J. Cell Biol. 2009;184:583–596. doi: 10.1083/jcb.200810189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C., Merkwirth C., Langer T. Prohibitins and the functional compartmentalization of mitochondrial membranes. J. Cell Sci. 2009;122:3823–3830. doi: 10.1242/jcs.037655. [DOI] [PubMed] [Google Scholar]
- Pronicka E., Ropacka-Lesiak M., Trubicka J., Pajdowska M., Linke M., Ostergaard E., Saunders C., Horsch S., van Karnebeek C., Yaplito-Lee J. A scoring system predicting the clinical course of CLPB defect based on the foetal and neonatal presentation of 31 patients. J. Inherit. Metab. Dis. 2017;40:853–860. doi: 10.1007/s10545-017-0057-z. [DOI] [PubMed] [Google Scholar]
- Rampelt H., Zerbes R.M., van der Laan M., Pfanner N. Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochim. Biophys. Acta. 2017;1864:737–746. doi: 10.1016/j.bbamcr.2016.05.020. [DOI] [PubMed] [Google Scholar]
- Richter-Dennerlein R., Korwitz A., Haag M., Tatsuta T., Dargazanli S., Baker M., Decker T., Lamkemeyer T., Rugarli E.I., Langer T. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014;20:158–171. doi: 10.1016/j.cmet.2014.04.016. [DOI] [PubMed] [Google Scholar]
- Sasaki O., Yoshizumi T., Kuboyama M., Ishihara T., Suzuki E., Kawabata S., Koshiba T. A structural perspective of the MAVS-regulatory mechanism on the mitochondrial outer membrane using bioluminescence resonance energy transfer. Biochim. Biophys. Acta. 2013;1833:1017–1027. doi: 10.1016/j.bbamcr.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Saunders C., Smith L., Wibrand F., Ravn K., Bross P., Thiffault I., Christensen M., Atherton A., Farrow E., Miller N. CLPB variants associated with autosomal-recessive mitochondrial disorder with cataract, neutropenia, epilepsy, and methylglutaconic aciduria. Am. J. Hum. Genet. 2015;96:258–265. doi: 10.1016/j.ajhg.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth R.B., Sun L., Ea C.K., Chen Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Tatsuta T., Model K., Langer T. Formation of membrane-bound ring complexes by prohibitins in mitochondria. Mol. Biol. Cell. 2005;16:248–259. doi: 10.1091/mbc.E04-09-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T., Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- Wei Y., Chiang W.C., Sumpter R., Jr., Mishra P., Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell. 2017;168:224–238. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A.P., Shadel G.S., Ghosh S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- Wintachai P., Wikan N., Kuadkitkan A., Jaimipuk T., Ubol S., Pulmanausahakul R., Auewarakul P., Kasinrerk W., Weng W.Y., Panyasrivanit M. Identification of prohibitin as a Chikungunya virus receptor protein. J. Med. Virol. 2012;84:1757–1770. doi: 10.1002/jmv.23403. [DOI] [PubMed] [Google Scholar]
- Wortmann S.B., Ziętkiewicz S., Kousi M., Szklarczyk R., Haack T.B., Gersting S.W., Muntau A.C., Rakovic A., Renkema G.H., Rodenburg R.J. CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am. J. Hum. Genet. 2015;96:245–257. doi: 10.1016/j.ajhg.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa K., Oshiumi H., Takeda M., Ishihara N., Yanagi Y., Seya T., Kawabata S., Koshiba T. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci. Signal. 2009;2:ra47. doi: 10.1126/scisignal.2000287. [DOI] [PubMed] [Google Scholar]
- Yokoyama H., Fujii S., Matsui I. Crystal structure of a core domain of stomatin from Pyrococcus horikoshii illustrates a novel trimeric and coiled-coil fold. J. Mol. Biol. 2008;376:868–878. doi: 10.1016/j.jmb.2007.12.024. [DOI] [PubMed] [Google Scholar]
- Yoshizumi T., Imamura H., Taku T., Kuroki T., Kawaguchi A., Ishikawa K., Nakada K., Koshiba T. RLR-mediated antiviral innate immunity requires oxidative phosphorylation activity. Sci. Rep. 2017;7:5379. doi: 10.1038/s41598-017-05808-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Chan N.C., Ngo H.B., Gristick H., Chan D.C. Crystal structure of mitochondrial fission complex reveals scaffolding function for mitochondrial division 1 (Mdv1) coiled coil. J. Biol. Chem. 2012;287:9855–9861. doi: 10.1074/jbc.M111.329359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mass spectrometry analysis of proximity interactors of the indicated *BirA-hemagglutinin (HA) fusion proteins. Note that the *BirA-HA is fused to the C terminus of PHB2 proteins. Proximity interactors are ranked in the order of the abundance ratios (log2) of PHB2(V1)/PHB2(V3) (average of four independent experiments).
The cross-linking experiments were performed under disuccinimidyl dibutyric urea (DSBU) concentrations of 50 or 100 μM. The resultant peptides (after digestion by trypsin/Lys-C mix) were divided into two halves and subjected to liquid chromatography tandem MS analysis using both Q Exactive Plus (QEplus) and Orbitrap Fusion Lumos (Lumos) mass spectrometers. The cross-linking experiment of PHB1 was performed under DSBU concentration of 100 μM.
Mass spectrometry analysis of proximity interactors of the indicated *BirA- hemagglutinin (HA) fusion proteins. Note that the *BirA-HA is fused to the C terminus of PHB2 proteins. Proteins are ranked in the order of the abundance ratios (log2) of PHB2WT/PHB2KKK.
Mass spectrometry analysis of proximity interactors of the indicated Myc-BirA* or *BirA- hemagglutinin (HA) fusion proteins. Note that Myc-BirA* is fused to the N terminus of MAVS and *BirA-HA is fused to the C terminus of CLPB or PHB1. Proteins are ranked in the order of the abundance ratios of CLPB/control.