

Abstract
Over their lifetime, long-term haematopoietic stem cells (HSC) are exposed to a variety of stress conditions that they must endure. Many stresses, such as infection/inflammation, reactive oxygen species, nutritional deprivation and hypoxia, activate unfolded protein response signalling, which induces either adaptive changes to resolve the stress or apoptosis to clear the damaged cell. Whether unfolded-protein-response signalling plays any role in HSC regulation remains to be established. Here, we report that the adaptive signalling of the unfolded protein response, IRE1α-XBP1, protects HSCs from endoplasmic reticulum stress-induced apoptosis. IRE1α knockout leads to reduced reconstitution of HSCs. Furthermore, we show that oncogenic N-RasG12D activates IRE1α-XBP1, through MEK-GSK3β, to promote HSC survival under endoplasmic reticulum stress. Inhibiting IRE1β-XBP1 abolished N-RasG12D-mediated survival under endoplasmic reticulum stress and diminished the competitive advantage of NrasG12D HSCs in transplant recipients. Our studies illuminate how the adaptive endoplasmic reticulum stress response is advantageous in sustaining self-renewal of HSCs and promoting pre-leukaemic clonal dominance.
The longevity of long-term haematopoietic stem cells (HSCs) exposes them to a wide range of stresses in the bone marrow environment, many of which lead to a perturbation of protein homeostasis and activation of the unfolded protein response (UPR)1,2. Three branches of UPR have been identified in mammalian cells: inositol-requiring enzyme 1α (IRE1α, encoded by ERN1), pancreatic eIF2α kinase (PERK) and activating transcription factor 6 (ATF6). Activation of this endoplasmic reticulum (ER) stress response either restores the balance of protein homeostasis, by constraining protein synthesis and increasing protein folding and degradation, or triggers apoptosis if a cell is deemed unrepairable. The cell context and the intensity and duration of the stress determine the fate of the cell following ER stress1,2.
The ER stress-response pathways have been extensively studied in many mammalian cell types including insulin-secreting P cells and B lymphocytes/plasma cells, where there is a persistent demand for protein quality control due to high levels of protein synthesis and secretion. Although HSCs are quiescent, exhibiting profoundly restricted protein synthesis3, and probably low demand for protein folding, recent studies suggest an important role for protein chaperones in HSC regulation4–7. Whether, and how, UPR signalling plays any role in maintaining HSC functions remains unknown. A recent report showed that human HSCs preferentially activate the pro-apoptotic PERK-eIF2α branch of UPR, thus sensitizing them to ER stress-induced apoptosis to maintain HSC integrity7. However, the same report also demonstrated that HSCs have higher basal PERK-eIF2α activation, which raises the question of how HSCs survive repetitive stress conditions and persist long term if they possess high steady-state levels of pro-apoptotic PERK signalling.
Results
IRE1α-XBP1 promotes HSC survival under ER stress.
To assess the response of long-term HSCs to ER stress, CD48−Lineage−Sca1+cKit+ cells8 (CD48−LSK; containing SLAM HSCs and multi-potent progenitor (MPP) cells; the gating strategy is shown in Supplementary Fig. 1a) were sorted and treated with thapsigargin and tunicamycin9. The robust induction of the key targets of UPR, Grp78, Chop and Xbp1s, was observed after treatment with either thapsigargin or tunicamycin, indicating an increase in ER stress and the activation of UPR (Supplementary Fig. 1b). Following ER stress, IREIα induces Xbp1 splicing and increases XBP1 levels10. In contrast to a previous report7, we found that murine HSCs (CD150+CD48−LSK) exhibited increased Xbp1 splicing demonstrated by the XBP1 splicing assay and quantitative PCR with reverse transcription (qRT–PCR) of Xbp1s/Xbp1 (Fig. 1a,b). To validate the activation of IRE1α–XBP1, we exploited the ER stress-activated indicator (ERAI) mouse strain11. In this model, IRE1α-mediated Xbp1 splicing is monitored by fluorescent protein expression, which can be easily detected by flow cytometry. Consistent with a previous report12, the highest ERAI signal was detected in Mac-1+Gr1+ myeloid cells when compared with B (B220+) and T (CD3+) cells (Supplementary Fig. 1d). After 18 h of treatment with either tunicamycin or thapsigargin, HSCs showed a robust increase of ERAI signal (Fig. 1c), indicating the activation of IRE1α in murine HSCs. This induction was completely blocked by Kira613, an IRE1α kinase inhibitor (Supplementary Fig. 1c), or the polyinosine:polycytosine (pIpC)-mediated deletion of IRE1α in Mx1-cre+; Ire1αfl/fl mice14 (Supplementary Fig. 1e), confirming that ERAI signal faithfully represents IRE1α activity. Thus, long-term murine HSCs activate IRE1α-XBP1 under ER stress. Notably, a significant decrease in ERAI signal was observed following prolonged, in vitro culture of HSCs (Supplementary Fig. 1f), which may explain the difference between our data and a previous study that reported attenuated IRE1α activation in human HSCs after treatment with tunicamycin or thapsigargin7.
Fig. 1 |. IRE1α-XBP1 signalling promotes the survival of HSCs under ER stress in vitro and in vivo.
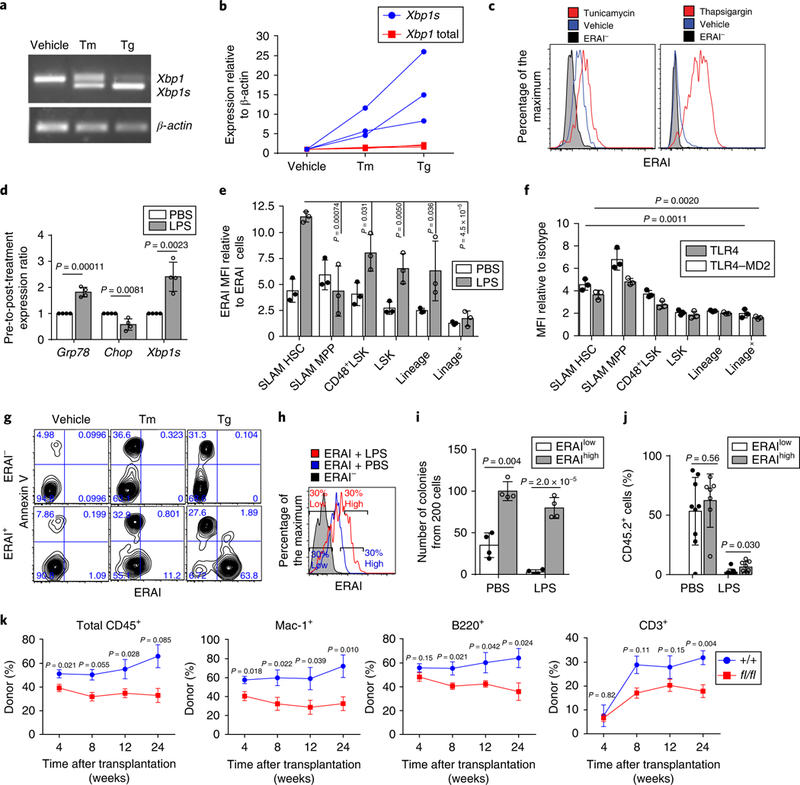
a,b, Representative PCR of Xbp1 splicing (a) and qRT-PCR of Xbp1 and Xbp1s (b) in HSCs treated with either 0.6 μgml−1 tunicamycin (Tm) or 0.2 μM thapsigargin (Tg) for 12h (three independent experiments). The original DNA gel is shown in Supplementary Fig. 7. Each line in b represents data from the same mouse. c, Fluorescence-activated cell sorting (FACS) plot of the ERAI levels in HSCs after treatment with 0.6 μgml−1 Tm (left) or 0.2 μM Tg (right) for 18h (n = 4 biological replicates from 2 independent experiments). d,e, Wild-type mice were treated with either PBS or LPS (2 mgkg−1) for 24h. d, qRT-PCR of UPR targets (n = 4 independent experiments). e, ERAI activation (normalized to ERAI− cells) in bone marrow populations (n = 3 biological replicates from 3 independent experiments). f, TLR4 and TLR4-MD2 levels detected by flow cytometry (n = 3 biological replicates from 3 independent experiments). g, Representative FACS plot of annexin V staining and the ERAI signal in HSCs after 18 h of treatment with 0.6 μgml−1 Tm or 0.2 μM Tg (n = 3 biological replicates from 3 independent experiments). Percentage of cells in each quadrant is shown on FACS plots. h, Gating strategy of ERAIhigh or ERAIlow HSCs. i, Colony formation from 200 ERAIhigh or ERAIlow HSCs that were purified 24h after injection with either PBS or LPS (n = 4 biological replicates from 4 independent experiments). j, ERAIhigh and ERAIlow (CD45.2) HSCs (100 cells each) were purified and transplanted with radioprotectors (CD45.1; 0.3 × 106) into lethally irradiated CD45.1 mice. The percentage of CD45.2 cells in bone marrow HSCs was analysed four weeks after transplantation. n = 9 transplants for the ERAIlow-PBS and ERAIlow-LPS groups, n = 7 transplants for ERAIhigh-PBS and n = 8 transplants for ERAIhigh-LPS, pooled from 2 independent experiments. k, Whole bone marrow cells (0.5×106) from CD45.2 Mx1-cre+; IRE1αfl/fl (fl/fl) or Mx1-cre−; Ire1αfl/fl (+/+) mice were transplanted into lethally irradiated CD45.1 mice, together with CD45.1 competitor cells (0.5×106) and the percentages of CD45.2 cells in total CD45+, myeloid (Mac-1+), B (B220+) and T (CD3+) cells were analysed from peripheral blood. Cells from two donor mice were transplanted into n = 7 (+/+) and 10 (fl/fl) recipients from 2 independent experiments. Data represent the mean ± s.d. for all panels except k, where data represent the mean ± s.e.m. Two-sided Student t-tests were used for statistical analyses. MFI, mean fluorescence intensity.
Lipopolysaccharide (LPS), a bacterial endotoxin and lipoglycan, has been reported to induce HSC hyperproliferation and exhaustion through the binding of its receptor, toll-like receptor 4 (TLR4)15–17. LPS was recently shown to activate ER stress in alveolar epithelial cells18 and macrophages19. To assess whether LPS induced ER stress in HSCs, we treated wild-type mice with LPS (2 mg kg−1 body weight, intraperitoneal injection) and examined the response of UPR signalling. This dose of LPS induced a haematopoietic response (Supplementary Fig. 1g) as previously reported17 and increased the expression of Grp78 and Xbp1s, but not Chop, in HSCs (Fig. 1d). In addition, HSC protein aggregates increased following LPS treatment (Fig. 2d). Together, these data provide evidence that LPS induces ER stress in murine HSCs in vivo. We next compared IRE1α activity in murine HSCs with other haematopoietic populations (Fig. 1e). Among the examined groups, MPPs exhibited the highest basal level of ERAI. Although HSCs showed a lower basal ERAI signal than MPPs, they demonstrated higher basal IRE1α activity than either myeloid progenitors or lineage cells. Importantly, HSCs had the highest ERAI induction after LPS (2 mg kg−1 body mass) was administered. HSCs and MPPs also exhibited higher expression of the LPS receptor TLR4/TLR4–MD2 compared with more differentiated populations (Fig. 1f). These findings indicate that IRE1α signalling is active under steady-state, non-stress conditions and can be further activated following ER stress in vivo in HSCs.
Fig. 2 |. NrasG12D pre-LSCs are resistant to ER stress in vitro and in vivo.
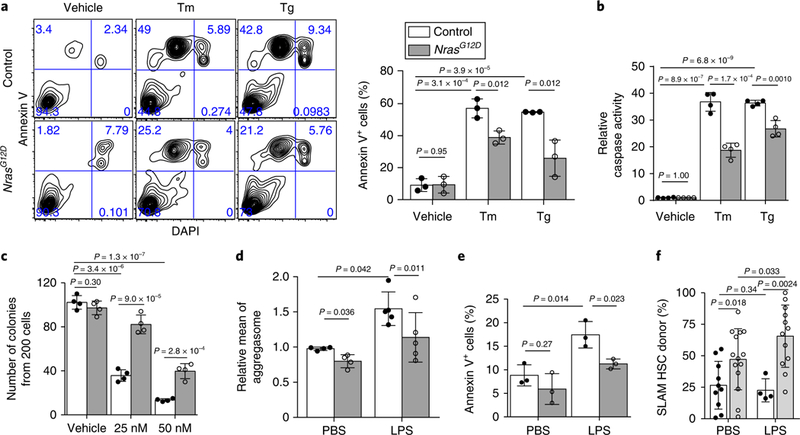
a, Annexin V staining of HSC purified from NrasG12D and control mice at two weeks after pIpC injection, following 18 h of treatment with either 0.6 μg ml−1 Tm or 0.2 μg. Representative FACS plots (left) and a summary from n = 3 biological replicates from 3 independent experiments (right) are shown. Percentage of cells in each quadrant is indicated in FACS plots. b, HSCs (200 cells per test) were purified from NrasG12D and control mice two weeks after pIpC injection, and caspase 3/7 activity was measured after 18h of treatment with either 0.6 μgml−1 Tm or 0.2 μM Tg (n = 4 biological replicates from 4 independent experiments). c, Number of colonies obtained from HSCs (200 cells per test) purified from NrasG12D and control mice two weeks after pIpC injection; the cells were cultured in methycult M3434 (n = 4 biological replicates from 4 independent experiments). The colony numbers were counted after ten days of treatment with 25nM or 50 nM Tg. d, Levels of protein aggregates measured by ProteoStat (see Methods) in HSCs 24h after intraperitoneal injections with LPS (2 mgkg−1 body mass; n = 4 and 5 biological replicates for the PBS- and LPS-treated groups, respectively, from 4 independent experiments). e, Percentage of annexin V+ HSCs 24h after intraperitoneal LPS injection (2 mgkg−1 body mass; n = 3 independent experiments). f, LPS injection increased donor reconstitution from NrasG12D HSCs in transplants engrafted with mixed control and NrasG12D bone marrow cells. Bone marrow cells (0.25×106 cells) from NrasG12D and control CD45.2 mice (two weeks after pIpC injection) were transplanted with 0.5×106 competitor (CD45.1) bone marrow cells into recipient mice. Six weeks after transplantation, transplants were injected with 2 mgkg−1 LPS (three doses) every other day and bone marrow HSCs chimaerism was analysed 2–4 weeks after the LPS injections. n = 10 PBS-treated control, n = 14 PBS-treated NrasG12D, n = 4 LPS-treated control and n = 12 LPS-treated NrasG12D recipients pooled from 3 independent experiments. Data represent the mean ± s.d. for all panels. Two-sided Student’s t-tests were used for statistical analyses, except in f where one-sided f-tests were performed. The legend in a also applies to b-f.
IRE1α activation can be either pro-survival or pro-apoptotic in a context-dependent manner20. To better define the function of IRE1α activation in HSCs, we assessed the correlation between IRE1α-XBP1 activation and apoptosis in HSCs by co-staining with annexin V when measuring the ERAI signal following ER stress (Fig. 1g). Strikingly, the ERAI+ and annexin V+ populations are mutually exclusive following stress. The low level of the ERAI signal in apoptotic cells was not due to ERAI protein degradation, as the proteasome inhibitor MG132 did not accumulate an annexin V+ERAI+ population, and the apoptotic annexin V+ERAI− population expanded, whereas the annexin V−ERAI+ cells persisted, after thapsigargin treatment (Supplementary Fig. 1h,i). Thus, ERAI represents a reliable tool to interrogate IRE1α activation and IRE1α–XBP1 promotes HSC survival following ER stress.
A previous study showed that human HSCs exhibit higher levels of PERK-eIF2α activation than haematopoietic progenitors after treatment with tunicamycin or thapsigargin7, but it was not clear how this could be extrapolated to the fate of HSCs. To understand the effect of PERK-eIF2α on cell survival, a series of reporter constructs for the eIF2α target ATF4 were generated and transduced into the acute myeloid leukaemia (AML) cell lines OCI-AML2 and OCI-AML3 (Supplementary Fig. 2a,b). In this system, PERK-eIF2α activity can be measured by upstream open reading frame (uORF)-mediated stress translation of the ATF4 reporter (PE A4RP1.2, fused with DsRed-Express2) in transduced cells (ZsGreen+). The expression of this reporter was only induced by thapsigargin or Sal003, a compound that increases eIF2α phosphorylation status (Supplementary Fig. 2c). Mutation of uORF1 or uORF2 leads to constitutively inactive (PE A4RP2.2) or active (PE A4RP3.2) expression of the reporter (Supplementary Fig. 2d,e). Thapsigargin treatment induced populations with high ATF4 activation in both OCI-AML2 and OCI-AML3 cell lines that did not show annexin V positivity (Supplementary Fig. 2f), suggesting that PERK-eIF2α activation is not associated with increased apoptosis.
We next assessed whether IRE1α-mediated protection is important to HSC function. We treated ERAI mice with LPS to induce ER stress in vivo, then purified HSCs (Fig. 1h) as either ERAIhigh (top 30% of ERAI intensity) or ERAIlow (bottom 30% intensity) and assessed HSC function (Fig. 1i,j). ERAIhigh HSCs formed significantly more colonies than ERAIlow HSCs (Fig. 1i), indicating that IRE1α activation preserves HSC clonogenicity. To assess HSC function in vivo, ERAIhigh and ERAIlow HSCs (100 cells each; Fig. 1h) were purified from PBS- or LPS-treated CD45.2 mice, combined with 0.3 × 106 radioprotector CD45.1 whole bone marrow cells and transplanted into lethally irradiated CD45.1 recipients (Fig. 1j). Donor reconstitution was assessed by measuring the percentage of donor-derived cells (CD45.2+) in the peripheral blood of transplant recipients. HSCs with higher ERAI levels (that is, higher IRE1α activity) exhibited higher donor engraftment in transplant recipients (Fig. 1j), suggesting that IRE1α activation preserves HSC reconstitution potential in vivo.
Next, we crossed Mx1-cre mice with conditional Ire1α knockout mice14 and injected pIpC to delete IRE1α in haematopoietic tissues. We observed no significant change in steady-state haematopoiesis in Mx1-cre+; Ire1αfl/fl (Ire1α knockout) mice, except for a mild reduction in the number of long-term HSCs (Supplementary Fig. 3a–g). We next combined 5 × l05 whole bone marrow cells from Ire1α knockout or control CD45.2+ mice with 5 × l05 CD45.1+ wild-type bone marrow cells and transplanted them into irradiated CD45.1+ wild-type recipients. The deletion of Ire1α resulted in significantly lower levels of reconstitution in the transplant recipients (Fig. 1k). Combined, our data support that IRE1α-XBP1 activation in HSCs both ameliorates apoptosis following ER stress and preserves HSC clonogenicity and reconstitution capacity.
NrasGI2D mutant HSCs are resistant to ER stress.
Given the role of UPR signalling in normal HSCs, we next wanted to understand whether the ER stress response is dysregulated following leukaemic transformation of HSCs. The transformation of normal haematopoietic cells into leukaemia cells requires a series of mutations that accumulate over years21–23. These mutations initially give rise to pre-leukaemic stem cells (pre-LSCs), which, after gaining additional mutations, develop into fully transformed leukaemic stem cells (LSCs)24. To assess the effect of ER stress on pre-LSCs, we generated Mx1-Cre+; NrasG12D/+ (referred to as NrasG12D or Nras mutant) mice using a previously described conditional knock-in NrasG12D allele25,26 (Supplementary Fig. 4a). Control HSCs exhibited significant increases in apoptosis after treatment with tunicamycin or thapsigargin, as measured by annexin V positivity and caspase activation (Fig. 2a,b). NrasG12D HSCs showed significant protection from tunicamycin-or thapsigargin-induced apoptosis (Fig. 2a,b). In addition, NrasG12D HSCs formed significantly more colonies than controls in the presence of thapsigargin (Fig. 2c). Together, our results show that NrasG12D HSCs are protected from ER stress-induced apoptosis in vitro. Notably, HSCs purified from FLT3-ITD knock-in or Mx1-cre+; Tet2fl/+ mice (two weeks after pIpC injection) demonstrated similar survival rates to controls after treatment with either tunicamycin or thapsigargin (Supplementary Fig. 4b,c), suggesting that the protection from ER stress may be an NrasG12D-specific effect.
We next determined the response of NrasG12D HSCs to ER stress induced by LPS in vivo (Supplementary Fig. 4a). NrasG12D HSCs showed lower levels of protein aggregates and significant protection from apoptosis compared with control HSCs (Fig. 2d,e), demonstrating that hyperactive N-Ras signalling protects pre-LSCs from ER stress-induced apoptosis in vivo. To assess whether the adaptation to ER stress promotes pre-LSC clonal expansion, we transplanted wild-type or NrasGI2D CD45.2 whole bone marrow together with wild-type CD45.1 cells (at a 1:2 ratio) into lethally irradiated wild-type CD45.1 mice. Transplant recipients were then injected with three doses of LPS (2 mg kg−1 body mass) six weeks after transplantation and HSC chimerism was measured 2–4 weeks after LPS injection (Supplementary Fig. 4d). The PBS-injected recipients of NrasG12D cells exhibited higher donor HSC reconstitution than the PBS-injected recipients of wild-type cells (Fig. 2f), which is consistent with our previous report25. Injection with LPS induced a further increase of donor chimerism in NrasGI2D transplants (Fig. 2f), indicating that adaptation to LPS-induced ER stress provides NrasG12D pre-LSCs a selective advantage and promotes their clonal expansion.
N-RasG12D protects both quiescent and proliferative HSCs from ER stress-induced apoptosis.
Proliferation status is tightly linked to cell survival under stress27. To assess the response of relatively quiescent/dormant versus proliferative HSCs to ER stress, Col1α1-H2B-GFP; Rosa26-M2-rtTA mice were employed28, from which proliferative and quiescent/dormant HSCs can be separated by histone 2B (H2B)-green fluorescent protein (GFP) expression during 12–18 weeks of ‘off labelling’ chase after six weeks of doxycycline-mediated labelling (Supplementary Fig. 4e). Tunicamycin or thapsigargin treatment did not change the fraction of GFPhigh cells in H2B–GFP-labelled HSCs (Fig. 3a). As measured by annexin V staining, the quiescent (GFPhigh) HSCs exhibited a significant survival advantage compared with the proliferative (GFPlow) HSCs following treatment with either tunicamycin or thapsigargin (Fig. 3b,c). However, N-RasG12D significantly reduced ER stress-induced apoptosis in both quiescent (GFPhigh) and proliferative (GFPlow) HSCs (Fig. 3d,e).
Fig. 3 |. Hyperactive N-Ras-induced protection of pre-LSCs from ER stress is independent of HSC proliferation status but is blocked by MEK inhibition.
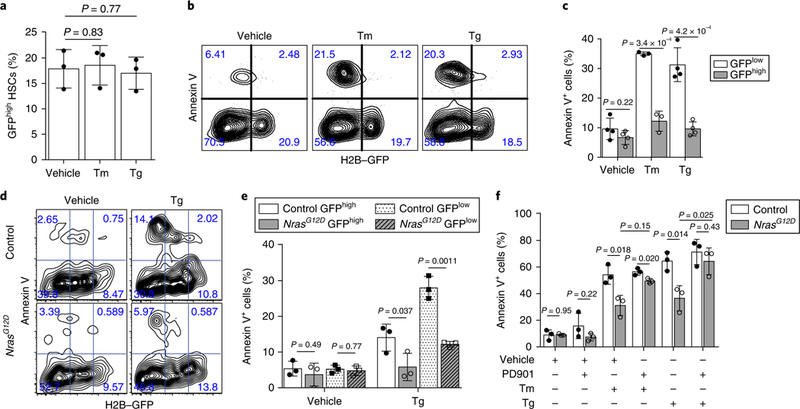
a, Percentage of GFPhigh HSCs after 18 h of treatment with 0.6 μl−1 Tm or 0.2 μM Tg (n = 3 biological replicates from 2 independent experiments). b, c, Representative FACS plots (b) and summary of the percentage of annexin V+ HSCs purified after 18 weeks off doxycycline and treated for 18 h with either 0.6μgml−1 Tm or 0.2 μM Tg (c; n = 4 biological replicates for vehicle- and Tg-treated, and n = 3 for Tm-treated HSCs from 2 independent experiments). The percentages of annexin V+ HSCs in each GFP fraction (GFPhigh or GFPlow) were normalized to the total percentage of HSCs within that particular GFP fraction, not the total HSCs. d,e, Representative FACS plots (d) and summary (e) of the annexin V staining of HSCs purified from Mx1-cre+ Nras+/+ (control) and NrasG12D mice 12 weeks off doxycycline, and then treated for 18 h with 0.2 μM Tg (e; n = 3 biological replicates from 2 independent experiments). f, Percentage of annexin V+ HSCs purified from Mx1-cre−; NrasG12D/+ (control) or NrasG12D mice and treated with 0.2 μM Tg together with 100nM PD901 for 18 h (n = 3 biological replicates from 3 independent experiments). Percentage of cells in each quadrant is indicated in FACS plots in b and d. Data represent the mean ± s.d. Two-sided Student’s t-tests were used for statistical analyses.
N-RasG12D-induced HSC survival under ER stress requires MEK-ERK-GSK3β but not STAT5 signalling.
Our previous studies demonstrated that N-RasG12D-induced HSC dysregulation depends on both MEK–ERK and JAK2–STAT5 signalling25. To test the involvement of MEK–ERK in the protective effect of N-RasG12D, we treated control and NrasG12D HSCs with the MEK inhibitor PD0325901 (PD901; 100 nM) together with tunicamycin or thapsigargin. PD901 blocked the survival advantage of NrasG12D pre-LSCs (Fig. 3f), therefore MEK is required for N-RasG12D-mediated protection. ERK has been reported to bind GSK3β and prime the deactivation of GSK3β by phosphorylation at Ser-9 (ref. 29). Inhibition of GSK3β promotes cell survival under stress30. Western blots showed that the Nras mutant CD48−LSK exhibited an increased level of Ser-9 phosphorylation of GSK3β (Supplementary Fig. 5a), suggesting GSK3β is inhibited in NrasG12D cells. Furthermore, inhibition of GSK3β with SB216763, which mimics GSK3β inhibition by MEK–ERK, promoted cell survival after thapsigargin treatment in wild-type HSCs but not NrasG12D HSCs (Supplementary Fig. 5b), suggesting that MEK–ERK–GSK3β mediates N-RasG12D-induced protection from ER stress.
To evaluate the role of JAK–STAT5 in N-RasG12D-mediated protection after ER stress, we mated Stat5a/bfl/+ mice31 with NrasG12D mice. Haploinsufficiency of Stat5 is not sufficient to block the protection of NrasGI2D HSCs from ER stress (Supplementary Fig. 5c). In addition, activation of STAT5 through haploinsufficiency of its negative regulator, suppressor of cytokine signalling 2 (Socs2)32, had no effect on HSC survival after treatment with tunicamycin or thapsigargin (Supplementary Fig. 5d,e). These results demonstrate that N-RasG12D-mediated protection from ER stress-induced apoptosis depends on MEK–ERK–GSK3β but not JAK–STAT5 signalling.
IRE1α-XBP1 branch of UPR is hyperactive in NrasG12D pre-LSCs after ER stress.
We next sought to examine the levels of UPR target genes in NrasGI2D HSCs treated with tunicamycin or thapsigargin (Supplementary Fig. 6a). NrasG12D pre-LSCs displayed similar or reduced levels of Grp78 and Chop, the main PERK pathway target, compared with controls. However, we observed much higher levels of the IRE1α-XBP1 target gene Erdj4 (Supplementary Fig. 6a) in NrasG12D pre-LSCs, suggesting N-RasG12D activates IRE1α signalling. Consistent with increased IRE1α activation, the levels of IRE1α-mediated degradation (RiDD) targets Blos1, Pdgfrb and Hgsnat were significantly reduced in NrasG12D pre-LSCs (Supplementary Fig. 6a). Furthermore, we detected increased levels of IRE1α phosphorylation and higher messenger RNA levels of Xbp1s but not total Xbp1 in NrasG12D HSCs compared with controls (Fig. 4a,b). Together, these data indicate that N-RasG12D hyperactivates IRE1α in HSCs.
Fig. 4 |. IRE1α-XBP1s branch of UPR signalling is hyperactivated in Nras mutant pre-LSCs after ER stress.
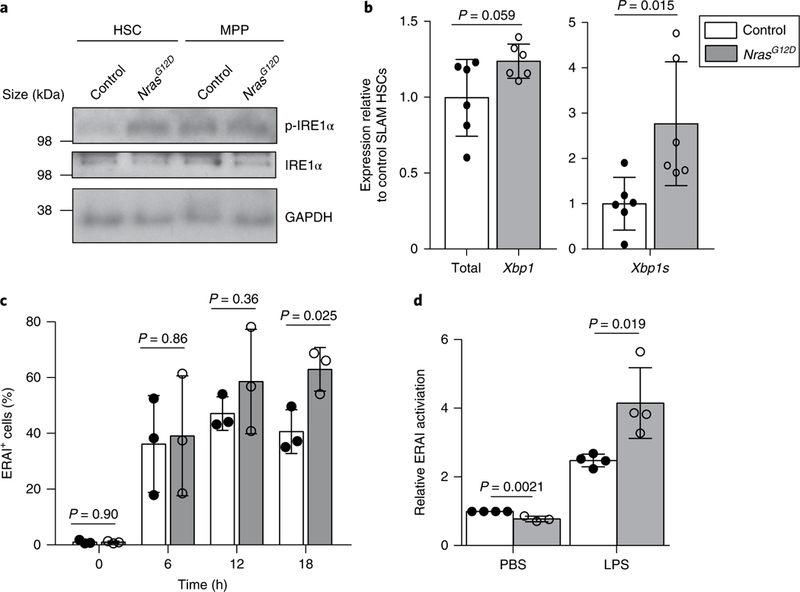
a, Representative western blot detecting the phosphorylation of IRE1α (p-IRE1α) at serine 724 in HSCs and MPPs that were purified from NrasG12D and control mice and stimulated with 10 ng ml−1 thrombopoietin (TPO) and 10 ng ml−1 stem cell factor (SCF) for 10min (n = 3 independent experiments). The original blot is shown in Supplementary Fig. 7. b, Total (spliced and unspliced) and spliced Xbp1 (Xbp1s) mRNA levels in HSCs purified from NrasG12D or control mice (n = 6 biological replicates from 3 independent experiments). c, Summary of the percentage of ERAI+ cells in HSCs purified from NrasG12D or control mice and treated with 0.2 μM Tg for 0–18 h (n = 3 biological replicates from 3 independent experiments). d, Summary of ERAI activation in HSCs purified from NrasG12D and control mice 24h after injection with either LPS or PBS (n = 4 biological replicates for the LPS-treated control, LPS-treated NrasG12Dand PBS-treated control groups, and n = 3 for the PBS-treated NrasG12Dgroup from 3 independent experiments). Data represent the mean ± s.d. Two-sided Student’s t-tests were used for statistical analyses. The legend in b also applies to c and d.
To confirm IRE1α activation in NrasGI2D cells, we crossed NrasG12D mice with ERAI mice and examined IRE1α activity after thapsigargin treatment. Both control and NrasGI2D HSCs responded to thapsigargin with significant increases in the ERAI signal 6 h after treatment (Fig. 4c). However, although the ERAI signal in control HSCs stabilized at 12 h, we observed continued increases in ERAI signal in Nras mutant HSCs, which was sustained at much higher levels at 18 h following thapsigargin treatment (Fig. 4c). We next determined activation of IRE1α-XBP1 in NrasG12D pre-LSCs in vivo following LPS (2 mg kg−1 body mass) treatment (Fig. 4d). LPS induced IRE1α activation in both control and Nras mutant HSCs, however, the ERAI signal was significantly higher in NrasGI2D HSCs (Fig. 4d). These results support the idea that N-RasG12D hyperactivates IRE1α-XBP1 in pre-LSCs in response to ER stress, both in vitro and in vivo. Notably, the N-RasG12D-mediated hyper-activation of IRE1α was probably restricted to HSCs and no significant increase in IRE1α activation was observed in NrasGI2D progenitors (Supplementary Fig. 6b).
IRE1α-XBP1 is required for N-RasG12D-induced resistance to ER stress and competitive advantage of pre-LSCs.
To interrogate the role of IRE1α in the N-RasG12D-mediated protection of pre-LSCs, we treated HSCs from control or NrasG12D mice that carry the ERAI reporter with thapsigargin and examined both levels of apoptosis and IRE1α activation (Fig. 5a). NrasG12D HSCs exhibited a higher percentage of ERAI+cells and a reduced percentage of annexin V+ cells compared with control HSCs (Fig. 5a), suggesting that N-RasG12D hyperactivates IRE1α signalling to protect HSCs from ER stress-induced apoptosis. In addition, small molecular RNase inhibitors specific to IRE1α, CD06 (compound 30 in ref. 33) or BI09 (ref. 34) blocked the increased survival of NrasG12D pre-LSCs after thapsigargin treatment (Fig. 5b and Supplementary Fig. 6c). N-RasG12D-induced HSC protection from ER stress-induced apoptosis, therefore, depends on IRE1α-XBP1 activation.
Fig. 5 |. IRE1α-XBP1s is required for N-RasG12D-induced resistance to ER stress and long-term reconstitution of pre-LSCs.
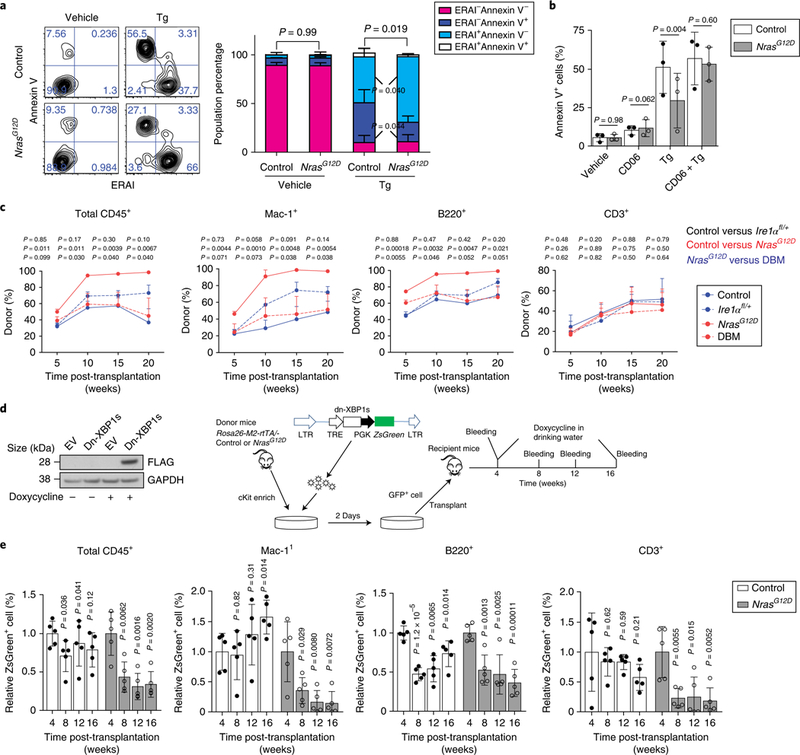
a, Representative FACS plots (left) and summary (right) of ERAI levels and annexin V staining of HSCs from NrasG12D and control mice 18 h after Tg treatment (n = 3 biological replicates from 3 independent experiments). Percentage of cells in each quadrant is indicated in FACS plots. b, Summary of the percentage of annexin V+ cells in HSCs that were purified from Mx1-cre−Nras+/+ (control) and NrasG12D mice and treated for 18h with 0.2 μM Tg together with 20 CD06 (n = 3 biological replicates from 3 independent experiments). c, Whole bone marrow cells (CD45.2; 0.5×106 cells) from Mx1-cre−; NrasG12D/+ (control), NrasG12D, Mx1-cre+; Ire1αfl/+ (Ire1αfl/+) and NrasG12D; Ire1αfl/+ double mutant (DBM) mice were transplanted into lethally irradiated recipients together with CD45.1 competitors (0.5×106 cells). The percentage of CD45.2+ cells in total CD45+, myeloid, B and T cells in peripheral blood was analysed for 20 weeks after transplantation (n = 5 biological replicates). d, Experimental scheme of the doxycycline-induced expression of a FLAG-tagged dn-XBP1s construct (right). The expression of dn-XBP1s is monitored by western blotting of the FLAG tag (left). The original blot is shown in Supplementary Fig. 7. LTR, long terminal repeat. e, Summary of the percentage of dn-XBP1s-transduced cells (ZsGreen+) in peripheral blood 4–16 weeks post-transplantation. Cells enriched for cKit from three donors were pooled and transduced with either control or dn-XBP1s cells and then transplanted into n = 5 recipients for each group. The expression levels were normalized to the average level of ZsGreen before induction with doxycycline. P values represent comparison to levels at 4 weeks. Data represent the mean ± s.d., except for c where data represent the mean ± s.e.m. Two-sided Student’s f-tests were used for statistical analyses.
To determine whether IRE1α inhibition affects the self-renewal of NrasG12D pre-LSCs, we generated the NrasG12D; Ireafl/+ double mutant mice. Notably, the loss of one Ire1α allele had minimal effects on steady-state haematopoiesis but blocked the N-RasG12D-mediated expansion of LSK and myeloid cells (Supplementary Fig. 6d–j). We then transplanted 5 × 105 whole bone marrow cells from wild-type, NrasG12D, Mx1-Cre+; Ire1αfl/+ or double mutant CD45.2 donors into lethally irradiated wild-type CD45.l recipients with 5 × 105 CD45.1 cells. As previously described25, NrasG12D cells gave significantly higher levels of long-term multi-lineage reconstitution than controls after transplantation (Fig. 5c). Loss of one Ire1α allele in the NrasG12D background completely blocked the increased reconstitution by NrasG12D cells, but had minimal effect on control HSCs (Fig. 5c). IRE1α activation is, therefore, required for N-RasG12D-induced HSC reconstitution.
To avoid confounding transplantation-related effects on ER stress signalling, we determined the result of IRE1α-XBP1 inhibition on HSCs that have established engraftment in transplant recipients. To do this, we transduced NrasG12D and control cKit+ cells with a doxycycline-inducible FLAG-tagged dominant negative XBP1s mutant (dn-XBP1s)35 and transplanted cells into lethally irradiated recipients (Fig. 5d). Four weeks after transplantation, recipients were started on doxycycline water to activate the expression of the dn-XBP1s mutant (verified by FLAG expression in Fig. 5d). The dn-XBP1s mutant significantly impaired the long-term multi-lineage repopulation potential of NrasG12D HSCs and we observed a significant decline in the percentage of dn-XBP1s-transduced NrasG12D cells (Fig. 5e). Dn-XBP1s expression, on the other hand, had no effect on the reconstitution of wild-type HSCs. NrasG12D pre-LSCs are therefore more dependent on IRE1α–XBP1 to sustain their self-renewal than control HSCs.
Our data showed that the N-RasG12D-mediated survival of HSCs under ER stress depends on MEK–ERK–GSK3β signalling (Fig. 3f and Supplementary Fig. 5a,b) and requires the activation of IRE1α (Fig. 5). To determine whether activation of IRE1α depends on MEK–ERK–GSK3β, we treated HSCs from ERAI mice with PD901 or the GSK3β inhibitor SB216763. Inhibition of MEK significantly reduced the level of ERAI signal and inhibition of GSK3β significantly increased the ERAI signal (Supplementary Fig. 5f,g), suggesting that MEK–ERK-induced GSK3β inhibition leads to IRE1α activation. Thus, we propose that N-RasG12D activates MEK–ERK, which phosphorylates and inhibits GSK3β, and leads to IRE1α activation to protect pre-LSCs from ER stress-induced apoptosis (Supplementary Fig. 5h).
Overexpression of DNAJB8 enhances HSC reconstitution.
The transcription factor XBP1s translocates to the nucleus following activation to regulate transcription of its target genes, which are important in protein folding and degradation. To identify potential downstream targets of IRE1α-XBP1s that mediate the protection of HSCs from ER stress, we examined the levels of genes previously proposed to be XBP1s targets in Ire1α knockout HSCs. We found that the level of Xbp1s, but not Grp78 or Erdj4, was significantly reduced in Ire1α knockout HSCs (Fig. 6a). In addition, Dnajb8, a molecular chaperone and potential XBP1s target, was significantly reduced (Fig. 6b). Our previously published gene expression profiles25 revealed an increased level of Dnajb8 in NrasG12D HSCs, a finding we confirmed with qRT–PCR (Fig. 6c). Consistent with an increased chaperone refolding activity, NrasG12D HSCs exhibited much lower protein aggregates after LPS treatment (Fig. 2d). Interestingly, Dnajb8 was enriched in HSCs and primitive progenitors (Fig. 6d). To determine the role of DNAJB8 in HSCs, we transduced cKit+ cells from wild-type bone marrow with a lentiviral construct that overexpresses DNAJB8 and transplanted the transduced HSCs into lethally irradiated recipients. Overexpression of DNAJB8 significantly enhanced the level of donor reconstitution in transplant recipients (Fig. 6e), suggesting that DNAJB8 promotes HSC self-renewal. Together, these results suggest that IRE1α-XBP1s activates DNAJB8 to protect NrasG12D pre-LSCs from ER stress-induced apoptosis and promote the self-renewal of pre-LSCs.
Fig. 6 |. The IRE1α-XBP1 target DNAJB8 is overexpressed in Nras mutant pre-LSCs and DNAJB8 overexpression promotes HSC reconstitution.
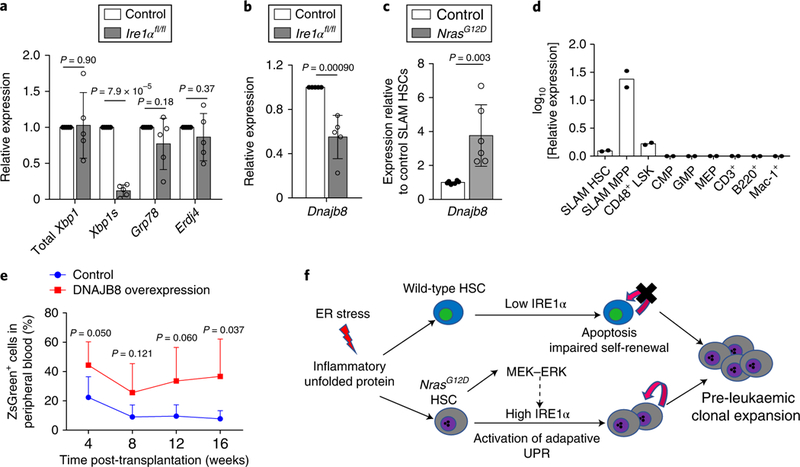
a, Total Xbp1, Xbp1s, Grp78 and Erdj4 mRNA expression in HSCs purified from Mx1-cre+; Ire1αfl/fl and Mx1-cre−; Ire1αfl/fl (control) mice (n = 5 independent experiments). b, Dnajb8 mRNA expression in HSCs purified from Mx1-cre+; Ire1αfl/fl and Mx1-cre−; Ire1αfl/fl (control) mice (n = 5 biological replicates from 5 independent experiments). c, Dnajb8 mRNA expression in HSCs purified from Mx1-cre−; NrasG12D/+ (control) and NrasG12D mice (n = 6 biological replicates from 3 independent experiments). d, Dnajb8 mRNA expression in different haematopoietic populations from the bone marrow of wild-type mice. Bone marrow cells were pooled from three mice and FACS sorted for different haematopoietic populations. n = 2 pools of samples were analysed for the levels of Dnajb8 mRNA by qRT-PCR. e, Cells enriched for cKit were infected with a lentivirus expressing DNAJB8 (together with a ZsGreen reporter) and transplanted into lethally irradiated recipient mice with 0.1×106 radioprotector cells and the donor contribution (measured by percentage of ZsGreen+ cells) in peripheral blood was then measured by FACS. Cells were pooled from three donors and transplanted into n = 5 recipient mice for each group. f, Schematic model of hyperactive N-Ras and activated adaptive ER stress IRE1α signalling promoting pre-LSC expansion. Data represent the mean ± s.d. Two-sided Student’s t-tests were used for statistical analyses.
Discussion
Conceptually, the longevity of long-term HSCs exposes them to various stress conditions over the lifetime of an individual, many of which lead to adverse impacts on HSC functions. For instance, inflammation directly impairs HSC function by causing ‘hyperproliferative stress’ via the TLR416,17,36,37. Here, we report that LPS triggers the activation of ER stress in long-term HSCs and induces HSC apoptosis. In addition, we show that activation of the protective branch of ER stress signalling, IRE1α–XBP1, maintains HSC clonogenicity and reconstitution capacity after exposure to LPS. We therefore provide evidence that adaptive ER stress signalling plays a critical role in HSC regulation and fate determination. Our results contrast a previous study7 that reported that human HSCs show attenuated IRE1α response and preferentially activate the pro-apoptotic PERK–eIF2α branch of UPR to sensitize human HSCs to ER stress-induced apoptosis in vitro7. In our studies, we observed a significant decrease of IRE1α activity following prolonged in vitro culture of HSCs, which may explain differences between our data and the previous report7, and emphasizes the importance of studying ER stress response under physiological conditions. Furthermore, conflicting data exist for the role of PERK–eIF2α in response to ER stress. The PERK–eIF2α pathway has been shown to maintain the stemness and prevent differentiation of muscle stem cells, by inhibiting global protein translation38. In a recent report, PERK–eIF2α activation reset global protein synthesis to foster aggressive tumour development in a prostate cancer model39. Here, using serial ATF4 reporters as indicators of PERK–eIF2α–activated stress translation, we show that the activation of PERK–eIF2α was not associated with increased apoptosis in human AML cell lines. Hence, the function of PERK–eIF2α in HSCs warrants further investigation.
In addition to the results supporting IRE1α signalling as critical to preserving HSC self-renewal, we show that its dysregulation plays an important role in the leukaemic transformation of HSCs. For pre-LSCs to gain a clonal advantage, they must adapt to and survive a variety of stress conditions more readily than normal cells. As HSCs persist in a bone marrow niche that harbours many known triggers of ER stress, adapting to ER stress more readily could allow pre-LSCs to escape depletion, thereby meeting the increased protein synthesis demands of increased proliferative stress imparted by oncogenic mutations40. In a previous study, Runx1 insufficiency was shown to confer resistance of pre-LSCs to many stress conditions, including ER stress, by inducing quiescence of HSCs and reducing ribosomal biogenesis to decrease overall protein synthesis27. However, whether and how Runx1 interacts with the ER stress signalling machinery is unclear. In addition, many other oncogenic mutations induce HSC hyperproliferation rather than quiescence and presumably subject the mutant HSCs to increased ER stress. Here, we report that N-RasG12D activates the IRE1α–XBP1 pathway in pre-LSCs to gain resistance to ER stress-induced apoptosis and to sustain enhanced competitiveness and self-renewal. We thus describe a role of UPR signalling in the promotion of pre-LSC expansion: activation of the adaptive IRE1α–XBP1 branch promotes survival of pre-LSCs under ER stress, re-balances protein homeostasis and maintains the stem-cell pool to promote pre-leukaemic HSC expansion (Fig. 6f). Here, we uncovered a signalling axis of N-Ras-MEK–ERK–GSK3β that activates adaptive UPR signalling IRE1α–XBP1 to protect pre-LSCs from ER stress-induced apoptosis. MEK–ERK signalling has been shown to uncouple the activation of ER stress signalling pathways and promote cell survival in human cell lines41. Whether IRE1α–XBP1 plays any role in human leukemogenesis and/or can be targeted for therapeutic intervention requires further investigation.
HSCs treated with chemical or biologic chaperones demonstrate enhanced self-renewal function in vitro and in vivo5–7. Here, we show that a protein chaperone, DNAJB8, is one of the downstream targets of IRE1α–XBP1 that mediate the protective effect in HSCs. Overexpression of DNAJB8 led to significantly enhanced HSC self-renewal. We therefore hypothesize that activation of IRE1α–XBP1 in Nras mutant pre-LSCs leads to the increased expression of protein chaperones, including DNAJB8, which in turn promote protein folding and adaptation to ER stress. This could ultimately enhance the competitive advantage of pre-LSCs by re-balancing protein homeostasis under ER stress. Further investigation will focus on delineating the function of DNAJB8 in HSCs under both physiologic conditions and during leukaemic transformation.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, statements of data availability and associated accession codes are available at https://doi.org/10.1038/s41556-019-0285-6.
Methods
Mice.
All mice were housed in the Unit for Laboratory Animal Medicine at the University of Michigan. All procedures of mouse use and care were in compliance with relevant ethical regulations and policies set forth by the University of Michigan Institutional Animal Care and Use Committee (IACUC), and all animal procedures and protocols were reviewed and approved by the IACUC. The animal protocol was initially approved in 2011 and has been renewed every three years since then. Loxp-STOP-Loxp-NrasGI2D/+ (ref. 26), Tet2+/− (ref. 42), Stat5ab+/− (ref. 31), Socs2+/− (ref. 32), Col1α1−H2B–GFP; Rosa26-M2-rtTA (ref. 1), Mx1-Cre (Jackson Laboratory, 003556) and ERAI11 (Riken biological resource, RBRC01099) mice were maintained in a C57BL/6 background. The B6;129S4-Ern1<tm2.1Tiw> (ref. 14; Ire1α knockout; RBRC05515) strain was obtained from Riken biological resource and backcrossed to a C57BL/Ka-CD45.2:Thy-1.1 background for at least six generations. Recipients in reconstitution assays were adult C57BL/Ka-CD45.1:Thy-1.2 mice that were at least eight weeks old at the time of irradiation. PBS was used to reconstitute pIpC (Amersham), which was administered at 0.5 μg g−1 body mass d−1 by intraperitoneal injection. All of the mice were then analysed at the age of 8–14 weeks, a minimum of two weeks after pIpC treatment and paired with sex- and age-matched controls. Doxycycline (Research Products International Corporation) was added to the water at a concentration of 0.2% (m/v) along with 1% sucrose (Fisher). LPS (Sigma) was reconstituted in PBS and administered at 2 μg g−1 body mass by intraperitoneal injection.
All analyses were performed on samples from age- and sex-matched mice. We performed separate analyses for male and female mice and did not observe any differences between genders. Thus, results represent analyses using both male and female mice. Equal numbers of male and female mice were used for analyses as much as possible.
Flow cytometry and isolation of haematopoietic cells.
Bone marrow cells were flushed from the long bones (tibias and femurs) of the mice with FACS buffer (Hank’s buffered salt solution (HBSS) without calcium or magnesium, supplemented with 2% heat-inactivated calf serum). Cells were triturated and filtered through a nylon screen (60 mm; Sefar America) to obtain a single-cell suspension. Haematopoietic cells, including CD150+CD48−LSK SLAM HSCs and CD150−CD48−LSK+ SLAM MPPs, were isolated as previously described2,3. For the isolation of HSCs, whole bone marrow cells were incubated with: antibodies to the lineage markers B220 (6B2), CD2 (RM2.5), CD3 (KT31.1), CD5 (53–7.3), CD8 (53–6.7), Gr-1 (8C5), CD41 (MWReg30) and Ter119 (Ter-119); anti-CD150 antibody (TC15–12F12.2); anti-CD48 antibody (HM48–1); anti-cKit antibody (2B8) conjugated to allophycocyanin (APC) and anti-Sca1 antibody (D7; all antibodies were purchased from Biolegend). After washing, cells were incubated with anti-APC conjugated to paramagnetic microbeads (Miltenyi Biotec). The microbead bound (cKit+) cells were then enriched using MS columns (Miltenyi Biotec). Non-viable cells were excluded from sorts and analyses using the viability dye 4′,6-diamidino-2-phenylindole (DAPI; 1 μg ml−1; Sigma). Cells were analysed with a BD Fortessa or sorted with a BD Aria. The other haematopoietic cell populations were defined as the following: LSK; common myeloid progenitor, CD16/32−CD34+Lin−Sca1−cKit+; granulocyte-macrophage progenitor, CD16/32+CD34+Lin−Sca1−cKit+ and megakaryocyte-erythroid progenitor, CD16/32−CD34−Lin−Sca1−cKit+. Additional information on antibodies is provided in Supplementary Table 1.
Genotyping.
To genotype mouse tail DNA for the presence of the Loxp-STOP-Loxp-NrasGI2D allele, primers R1 (5′-GCT GGA TCG TCAAGG CGC TTT TCC-3′) and F2 (5-AGA CGC GGA GAC TTG GCG AGC-3) were used in addition to the primer SD5′ (5′-AGC TAG CCA CCA TGG CTT GAG TAA GTC TGC A-3′). To verify the presence of the Mx1-cre, Rosa26-M2-rtTA, Col1a1-H2B-GFP and ERAI transgenes, the following primers were used, respectively: 5′ -ATT GCT GTC ACT TGG TCG TGG C-3′ (cre-F1) and 5′-GAA AAT GCT TCT GTC CGT TTGC-3′ (cre-R1); 5′-AAA GTC GCT CTG AGT TGT TAT-3′,5′-GCG AAG AGT TTG TCC TCA ACC-3′ and 5′-GGA GCG GGA GAA ATG GAT ATG-3′; 5′-CTG AAG TTC ATC TGC ACC ACC-3′ and 5′-GAA GTT GTA CTC CAG CTT GTG C-3′; 5′-GAA CCA GGA GTT AAG ACA GC-3′ and 5′-GAA CAG CTC CTC GCC CTT GC-3′. The following primers were used to genotype mice for the Stat5ab, Ire1α, Tet2 and Socs2 deletions, respectively: 5′-GAA AGC AGC ATG AAA GGG TTG GAG-3′, 5′ - AGC AGC AAC CAG AGG ACT AC-3′ and 5′-AAG TTA TCT CGA GTT AGT CAG G-3′; 5′ -CCA GTG CTC TTG AAA AGA GG-3′, 5′ -CCG AGC CAT GAG AA A CAA GG-3′ and 5′ -CCC TGC CAG GAT GGT CAT GG-3′; 5′ -AGA GCC TCA AGC AAC CAA AA-3′ and 5′ -ACA TCC CTG AGA GCT CTT GC-3′; 5′ -TGG T AC AGA ACA CGC AGG GCG GAG GAGT-3′, 5′ -GGC TCC AGG CAC CGC AGG GTC AT-3′ and 5′ -GTC ACG TTG GTG TAG ATG GGC GC-3′. To genotype for the presence of FLT3-ITD, the following primers were used: 5′-TCT GGT TCC ATC CAT CTT CC-3′ and 5′-TGG CTA CCC GTG ATA TTG CT-3′.
Long-term competitive repopulation assay.
Adult recipient mice (CD45.1) were irradiated with an Orthovoltage X-ray source delivering approximately 300 rad min−1 in two equal doses of 550 rad, delivered at least 3 h apart. Cells were injected into the tail veins of anaesthetized recipients. Starting at four or five weeks after transplantation and continuing for at least 16 weeks, blood was obtained from the tail veins of recipient mice, subjected to ammonium-chloride-potassium red-cell lysis and stained with directly conjugated antibodies to CD45.2 (104), CD45.1 (A20), B220 (6B2), Mac-1 (M1/70), CD3 (KT31.1) and Gr-1 (8C5) to monitor engraftment. Additional information on antibodies is provided in Supplementary Table 1.
Western blotting.
The same number of cells (10,000 CD48−LSK cells, and 5,000 SLAM HSCs and SLAM MPPs) from each population to be analysed were sorted into HBSS with 2% FCS. The cells were then washed and incubated with 10 ng ml−1 TPO and 10 ng ml−1 SCF at 37 °C for 10 min. The cells were then washed with PBS and precipitated with 10% trichloroacetic acid (final concentration). The extracts were incubated on ice overnight and spun down for 10 min at 16,000g and 4 °C. The supernatant was removed, and the precipitated pellets were washed twice with ice-cold acetone and then air-dried. The proteins in the pellets were solubilized with solubilization buffer (9 M urea, 2% Triton X-100, 1% dithiothreitol) before LDS loading buffer (Invitrogen) was added. Proteins were separated on a Bis-Tris polyacrylamide gel (Invitrogen) and transferred to a PVDF membrane (Millipore). The antibodies used were: anti-GAPDH (sc-25778, SCBT), anti-p-IRE1α (S724; PA1–16927, Thermo Scientific Pierce), anti-pStat5 (9356 S, CST), anti- SOCS2 (2779 S, CST), anti-IRE1α (3294 S, CST) and anti-FLAG (F1804, Sigma). Additional information on antibodies is provided in Supplementary Table 1.
Protein aggregation assay.
The protein aggregation assay was performed by collecting 20 × 106 whole bone marrow cells and staining them with FACS antibodies against the cell surface markers for HSCs and other haematopoietic populations as described above. After washing, cells were resuspended and fixed with Cytofix/Cytoperm Buffer (BD, 5227892) on ice for 10 min. The cells were then permeabilized with BD Cytoperm Permeabilization Buffer Plus (BD, 5227892). After washing with 1×Perm/Wash Buffer (BD, 5227892), the cells were resuspended with 1×Perm/Wash Buffer containing ProteoStat detection reagent (1/5,000; Enzo, ENZ-51023-KP050) and stained for 30 min. After washing with FACS Buffer, the cells were resuspended in FACS Buffer containing DAPI (2.5 μg ml−1). Aggregation was detected with channel 582/15(488) by flow cytometry with a BD Fortessa.
Caspase 3/7 activity assay.
The caspase3/7 assay was performed according to the manufacturer’s instructions (Promega, G8090). Briefly, 200 HSCs were FACS sorted into SF-03 media (Iwai North America Inc.,1303) contained 100 ng ml−1 TPO or SCF (PeptoTec, 315–14 and 250–03). Cells were recovered overnight and treated with tunicamycin (Sigma, T7766) or thapsigargin (Sigma, T9033) before the caspase 3/7 activation was measured.
In vivo and in vitro annexin V staining of HSCs.
The Annexin V FITC Kit I (BD, B556547) was used for in vivo and in vitro staining of annexin V. For in vivo staining, 20 × 106 whole bone marrow cells were collected and stained with the cocktail of FACS antibodies as described above for different haematopoietic populations including HSCs. After washing with FACS buffer, cells were washed with 1×Annexin V buffer and resuspended in 200 μl 1×Annexin V buffer. Fluorescein isothiocyanate (FITC)- or APC-conjugated anti-annexin V antibody was added at a concentration of 1:40 along with 2.5 μg ml−1 DAPI. For in vitro staining, at least 1,000 HSCs or CD48−LSK cells were sorted into SF-03 media contained TPO or SCF at 100 ng ml−1. Cells were recovered overnight before treatment with tunicamycin or thapsigargin for different time periods. The cells were then stained with fluorescence conjugated anti-annexin V antibody (1:40) along with 2.5 μg ml−1 DAPI.
Colony-forming unit assay.
For the colony formation assays, 200 SLAM HSCs were directly sorted into MethoCult GF M3434 media (Stem Cell technologies, 3434) supplemented with 100 μl SF-03 media containing 100 ng ml−1 TPO added on top. The cells were then mixed thoroughly by vortexing and plated onto culture dishes to grow for seven days before the colonies were counted.
Xbp1-splicing– and qRT–PCR.
RNA exaction, reverse transcription and qRT-PCR were performed as previously described4. The primers used here were: Grp78-F, 5′-CAG ATC TTC TCC ACG GCT-3′ and Grp78-R, 5′ -TGT CAC TCG GAG AAT ACC AT-3′; Chop-F, 5′ -CCA GAA TAA CAG CCG GAA CCT GA-3′ and Chop-R, 5′ -TCC TGC AGA TCC TCA TAC CAG; Erdj4-F, 5′-GGA TGG TTC TAG TAG ACA AAG G-3′ and Erdj4-R, 5′ -CTT CGT TGA GTG ACA GTC CTG C-3′; Blos1-F, 5′-CAA GGA GCT GCA GGA GAA GA-3′ and Blos1-R, 5′ -GCC TGG TTG AAG T TC TCC AC-3′; Xbp1s-F, 5′-GAG TCC GCA GCA GGT G-3′ and Xbp1s-R, 5′ -GTG TCA GAG TCC ATG GGA-3′; Xbp1 total-F, 5′ -AAA CAG AGT AGC AGC GCA GAC-3 ‘ and Xbp1 total-R, 5′-CAG GAT CCA GCG TGT CCA t-3′; β-actin-F, 5′ -GGC TGT ATT CCC CTC CAT CG-3′ and (β-actin-R, 5′ -CCA GTT GGT AAC AAT GC C ATG T-3′; Dnajb8-F, 5′ -TCG GTG CTG GTC ATC CCT T-3’ and Dnajb8-R, 5’ -GGG AAC TCA CCA AAG CCC GAA-3′; Pdgfrb-F, 5′ -AAC CCC CTT ACA GCT GTC C T-3′ and Pdgfrb-R, 5′ -TAA TCC CGT CAG CAT CTT CC-3 ‘; Hgsnat-F, 5′ -TCT CCG CTT TCT CCA TTT TG CT-3′ and Hgsnat-R, 5′ -CGC ATACAC GTG GAA AGT CA-3′. The XBP1 splicing assay was performed as previously described19 using the following primers: Xbp1 splicing-L, 5′ -ACA CGC TTG GGA ATG GA AC-3′ and Xbp1 splicing-R, 5′ -CCA TGG GAA GAT GTT CTG GG-3′.
Plasmids.
All plasmid construction was performed with overlapping PCR and exonuclease III-mediated ligation-independent cloning (LIC). Doxycycline-inducible vectors (pI2–3) were constructed based on pHAGE–CMV–MCS–IRES–ZsGreen (pHAGE; a gift from Z. Huang′s lab) backbones using overlapping PCR and LIC to replace the CMV–IRES with the TRE–PGK promotor. The dn-Xbp1s-FLAG DNA fragment35 was synthesized by GeneArt Gene Synthesis (Thermo Fisher) and ligated into pI2–3 using LIC. The TRE of pI2–3 was then replaced by the EF1α promotor to generate pHAGE EF1α (PE). PE was then used as a backbone for the subcloning of the ATF4 reporter constructs (PE A4RP1.2–3.2), similarly to as described previously7. Briefly, the Atf4 untranslated region and part of the coding DNA sequence (NM_009716,318–714) was amplified by overlapping PCR with mouse complementary DNA to generate wild-type Atf4 and uORF1 or uORF2 mutant fragments; these fragments were then fused to the synthesized DsRed-Express2 fragment (GeneArt Gene Synthesis, Thermo Fisher) by overlapping PCR and ligated to the PE vector through LIC. For the overexpression of DNAJB8, the human DNAJB8 cDNA to the coding DNA (MHS6278–202808924, GE Healthcare Dharmacon) was amplified, a FLAG tag sequence was added by PCR and the amplicon was ligated to PE by LIC. All plasmids are available on request.
Statistics and reproducibility.
Multiple independent experiments were performed to verify the reproducibility of all experimental findings. All quantitative data represent the mean ± s.d., unless otherwise stated. Unless otherwise indicated, two-tailed Student’s t-tests were used to assess statistical significance for comparisons between two groups and ANOVA tests were performed for comparisons of more than two groups using Prism 7 (GraphPad software). No randomization or blinding was used in any of the experiments. No mice were excluded in any of the experiments. For measurements in which the variation among experiments tends to be low (for example, HSC frequency), we generally examined between three and six mice. For measurements in which the variation among experiments tends to be higher (for example, reconstitution assays), we examined larger numbers of mice (that is, 5–20 mice). The sample sizes, defined as the number of independent experiments, are described in the figure legends.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgements
This work was supported by the University of Michigan Protein Folding Disease Initiative. Q.L. was supported by the NIH/NHLBI (grant no. 1R01HL132392), American Cancer Society (grant no. 125080-RSG-13–253-01-LIB), V Foundation for Cancer Research, Gabrielle’s Angel Foundation and Leukemia Research Foundation. J.D.V. is supported by the NIH (grant no. R01CA190860). Thanks to K. Haigis, T. Jacks, H. Hock, L. Hennighausen and M. Miura for providing LoxP-STOP-LoxP-NrasG12D, Col1a1-H2B-GFP; Rosa26-M2−rtTA, Stat5abfl and ERAI mice. We thank Z. Huang for the cloning vector for the ATF4 reporter constructs. We thank R. Signer (UCSD) for the critical reading and insightful feedback of our manuscript.
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41556-019-0285-6.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Data availability
The gene expression data re-analysed here (DNAJB8 levels) are available from GEO under the accession code GSE45194. Source data have been provided in Supplementary Table 2. All other data supporting the findings of this study are available from the corresponding author on reasonable request
References
- 1.Hetz C The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Ron D & Walter P Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Signer RA, Magee JA, Salic A & Morrison SJ Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wey S, Luo B & Lee AS Acute inducible ablation of GRP78 reveals its role in hematopoietic stem cell survival, lymphogenesis and regulation of stress signaling. PLoS ONE 7, e39047 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miharada K, Sigurdsson V & Karlsson S Dppa5 improves hematopoietic stem cell activity by reducing endoplasmic reticulum stress. Cell Rep. 7, 1381–1392 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Sigurdsson V et al. Bile acids protect expanding hematopoietic stem cells from unfolded protein stress in fetal liver. Cell Stem Cell 18, 522–532 (2016). [DOI] [PubMed] [Google Scholar]
- 7.van Galen P et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 510, 268–272 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C & Morrison SJ SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Oslowski CM & Urano F in Methods in Enzymology Vol. 490 (ed. Conn PM) 71–92 (Academic Press, Cambridge, 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshida H, Matsui T, Yamamoto A, Okada T & Mori K XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Iwawaki T, Akai R, Kohno K & Miura M A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat. Med. 10, 98–102 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Osorio F et al. The unfolded-protein-response sensor IRE-1α regulates the function of CD8α+ dendritic cells. Nat. Immunol. 15, 248–257 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Ghosh R et al. Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 158, 534–548 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iwawaki T, Akai R, Yamanaka S & Kohno K Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl Acad. Sci. USA 106, 16657–16662 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raetz CR & Whitfield C Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 71, 635–700 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takizawa H et al. Pathogen-induced TLR4-TRIF innate immune signaling in hematopoietic stem cells promotes proliferation but reduces competitive fitness. Cell Stem Cell 21, 225–240.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Zhang H et al. Sepsis induces hematopoietic stem cell exhaustion and myelosuppression through distinct contributions of TRIF and MYD88. Stem Cell Rep. 6, 940–956 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li S et al. Lipopolysaccharide induces autophagic cell death through the PERK-dependent ranch of the unfolded protein response in human alveolar epithelial A549 cells. Cell. Physiol. Biochem. 36, 2403–2417 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Martinon F, Chen X, Lee AH & Glimcher LH TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 11, 411–418 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y & Brandizzi F IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 23, 547–555 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Notta F et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 469, 362–367 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Rossi DJ, Jamieson CH & Weissman IL Stems cells and the pathways to aging and cancer. Cell 132, 681–696 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Welch JS et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 150, 264–278 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pandolfi A, Barreyro L & Steidl U Concise review: preleukemic stem cells: molecular biology and clinical implications of the precursors to leukemia stem cells. Stem Cells Transl. Med. 2, 143–150 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Q et al. Oncogenic Nras has bimodal effects on stem cells that sustainably increase competitiveness. Nature 504, 143–147 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Q et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood 117, 2022–2032 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai X et al. Runx1 deficiency decreases ribosome biogenesis and confers stress resistance to hematopoietic stem and progenitor cells. Cell Stem Cell 17, 165–177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foudi A et al. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat. Biotechnol. 27, 84–90 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding Q et al. Erk associates with and primes GSK-3β for its inactivation resulting in upregulation of β-catenin. Mol. Cell 19, 159–170 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Song L, De Sarno P & Jope RS Central role of glycogen synthase kinase-3β in endoplasmic reticulum stress-induced caspase-3 activation. J. Biol. Chem. 277, 44701–44708 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Cui Y et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol. Cell. Biol. 24, 8037–8047 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Metcalf D et al. Gigantism in mice lacking suppressor of cytokine signalling-2. Nature 405, 1069–1073 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Ranatunga S et al. Synthesis of novel tricyclic chromenone-based inhibitors of IRE-1 RNase activity. J. Med. Chem. 57, 4289–4301 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang CH et al. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J. Clin. Invest. 124, 2585–2598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee AH, Iwakoshi NN & Glimcher LH XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Megias J et al. Direct Toll-like receptor-mediated stimulation of hematopoietic stem and progenitor cells occurs in vivo and promotes differentiation toward macrophages. Stem Cells 30, 1486–1495 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Esplin BL et al. Chronic exposure to a TLR ligand injures hematopoietic stem cells. J. Immunol. 186, 5367–5375 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zismanov V et al. Phosphorylation of eIF2α is a translational control mechanism regulating muscle stem cell quiescence and self-renewal. Cell Stem Cell 18, 79–90 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Nguyen HG et al. Development of a stress response therapy targeting aggressive prostate cancer. Sci. Trans. Med. 10, eaar2036 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma Y & Hendershot LM The role of the unfolded protein response in tumour development: friend or foe? Nat. Rev. Cancer 4, 966–977 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Tay KH et al. Sustained IRE1 and ATF6 signaling is important for survival of melanoma cells undergoing ER stress. Cell. Signal. 26, 287–294 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Moran-Crusio K et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 20, 11–24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.