

Abstract
Objective
To estimate the prevalence of TOR1A sequence variants associated with DYT1 dystonia.
Methods
We determined the frequency of the common trinucleotide deletion that causes DYT1 in the Genome Aggregation Database and the Penn Medicine Biobank, totaling exomes from over 135,000 individuals. We also evaluated the prevalence of other possible pathogenic variants in this gene and asked whether the D216H polymorphism is linked to a higher diagnostic rate for dystonia independent of the DYT1-causing mutation.
Results
The estimated range of prevalence of the most common pathogenic variant that causes DYT1 is ∼17.6–26.1 carriers per 100,000 individuals. Based on the different data sets used, we predict that there are between 54,366 and 80,891 mutation carriers in the United States, which, due to the reduced penetrance of this variant, would translate into 16,475–24,513 DYT1 patients.
Conclusions
Our data provide a prevalence estimate of the most common DYT1 mutation in the general population. This information is specifically important for those with interest in the development of precision therapeutics for dystonia.
DYT1, an autosomal dominant form of dystonia, almost universally caused by an in-frame GAG deletion in the TOR1A gene1 with reduced clinical penetrance, reported at about 33%.2,3 In addition to the GAG deletion, a few private sequence variants in TOR1A have been linked to the appearance of dystonia,4,5 and biallelic mutations in this gene were recently identified in patients with a severe congenital syndrome.6 Recent studies have estimated the prevalence of dystonia in different populations.7,8 However, most focus on adult-onset isolated dystonia. With the advent of precision medicine, it is important to ascertain the prevalence of genetically defined forms. This epidemiologic information is important not only to extrapolate the burden of the disease but also to inform those with potential interest on therapeutic development about the target population.
Here, we designed a genetic ascertainment study that quantifies the prevalence of the GAG deletion and other rare pathogenic variants in TOR1A in the general population and interrogated the electronic health records of TOR1A variant carriers to estimate the prevalence of DYT1 dystonia.
Methods
We analyzed exome sequencing data from the Genome Aggregation Database (gnomAD) and Penn Medicine Biobank (PMBB). gnomAD is a coalition of investigators that have generated harmonized exome sequencing data freely available to the scientific community (from 125,748 unrelated individuals of various population genetic studies).9 The PMBB, approved by the University of Pennsylvania Institutional Review Board, consists of over 60,000 patients from clinical practice sites of the University of Pennsylvania Health System who have given appropriate consent regarding access to all available electronic health records (EHRs). Participants are recruited independent of diagnosis and at multiple locations, from clinical visits to those undergoing blood draws or radiologic studies for any reason, among others. This study included a subset of 11,451 individuals who have undergone whole-exome sequencing, for which we obtained whole-exome sequences as generated via DNA extracted from stored buffy coats by the Regeneron Genetics Center (Tarrytown, NY) and mapped these sequences to GRCh37 as previously described.10 Furthermore, International Classification of Diseases (ICD) Ninth Revision and Tenth Revision (ICD-10) disease diagnosis codes and procedural billing codes were extracted from the patients' EHR.
We first evaluated the frequency of the sequence variant responsible for almost all cases of DYT1 dystonia (exon5:c.907_909del:p.303_303del).1 Next, we queried the PMBB data set for other reported private pathogenic variants4,5 and to identify other novel predicted deleterious missense variants in TOR1A, evaluating their frequency in gnomAD. For prediction of deleterious missense variants, we used the Rare Exonic Variant Ensemble Learner (REVEL)11 selecting the top 20% REVEL scores to be considered deleterious. For all participants from the PMBB harboring known pathogenic or predicted deleterious variants, their EHR was reviewed for the presence of a diagnosis of dystonia, description of a phenotype suspected to be dystonia, or a family history of dystonia or similar neurologic syndrome.
Finally, we evaluated the PMBB data set for the frequency of the exon4:c.G646C:p.D216H single nucleotide polymorphism (SNP), known to influence the penetrance of the common DYT1 mutation3 but also proposed to be a risk factor for isolated dystonia independent of the GAG deletion.12 We identified carriers of the minor allele in heterozygous and homozygous state. As manual evaluation of the 11,451 medical charts is not feasible, we queried the diagnostic codes from the EMR of all PMBB participants with exome data for ICD-9 codes 333.6, 333.7, and 333.8 to correlate the p.D216H polymorphism with dystonia.
Data availability
Anonymized data not published within the article will be shared by request from any qualified investigator.
Results
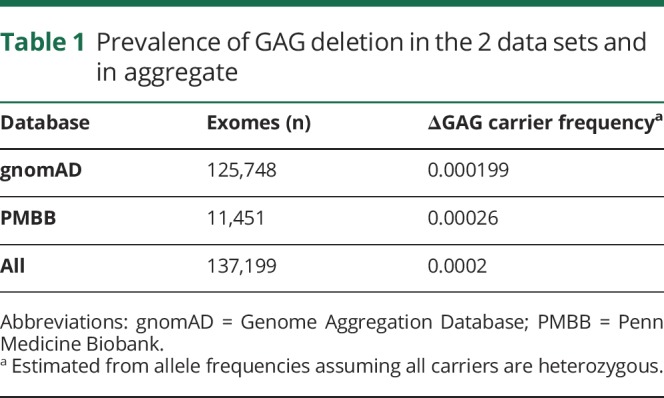
We used the minor allele frequencies reported for the GAG deletion in gnomAD, all in a heterozygous state, to calculate the carrier frequency (table 1). To confirm this prevalence in an independent US-based data set, we evaluated carrier frequency in the PMBB. Overall, the data were consistent between both data sets, with a frequency in the entire data set of 20 carriers per 100,000. Although there is no clinical information available for gnomAD participants, we reviewed the EHR for the 3 participants carrying the GAG deletion in the PMBB (54- and 85-year-old men and 88-year-old woman). None had any evidence of a neurologic disorder affecting motor function, with no mention of neurologic disease or disability in their family history (which was recorded). However, they had not been evaluated by a movement disorders neurologist.
Table 1.
Prevalence of GAG deletion in the 2 data sets and in aggregate
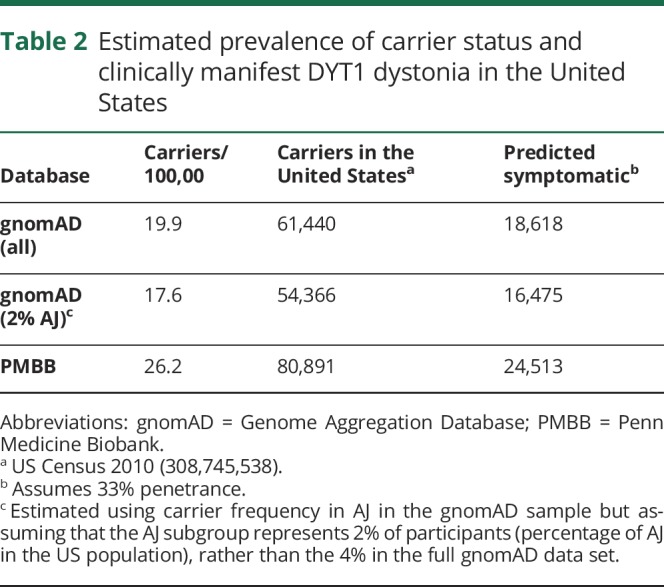
Next, we aimed to estimate the prevalence of this variant in the United States based on these data (table 2). However, this cannot be simply extrapolated from the full gnomAD data set. The GAG deletion is overrepresented in Ashkenazi Jewish (AJ),2 a subgroup that represents 4% of gnomAD participants but only 2% of the US population.13 This subgroup had a carrier frequency of 0.003. For that reason, we also estimated the expected prevalence of the GAG deletion based on both the PMBB data set (US based) and in gnomAD including an AJ subgroup adjusted to represent only 2% of the sample. As shown in table 2, this yields a range of prevalence of the GAG deletion in the United States of 17.6–26.2 carriers/100,000. Table 2 also shows the number of predicted carriers and symptomatic DYT1 patients, assuming a 33.3% penetrance. We did not collect information to identify an AJ subgroup in the PMBB data set, but 75% of that sample is of European descent.
Table 2.
Estimated prevalence of carrier status and clinically manifest DYT1 dystonia in the United States
We also queried the PMBB data set for other previously reported likely pathogenic variants in TOR1A4,5,14 and identified 3 participants, with 5 additional participants with predicted deleterious variants (table 3). EHR review disclosed that none of them had dystonia or any other motor or neurologic disorder. Similarly, there was no mention of family history of dystonia or other neurologic disorder in their records. None of the patients had been evaluated by a neurologist in our institution.
Table 3.
Prevalence of previously reported pathogenic variants and new predicted deleterious variants in TOR1A in PMBB, with carrier frequencies in gnomAD

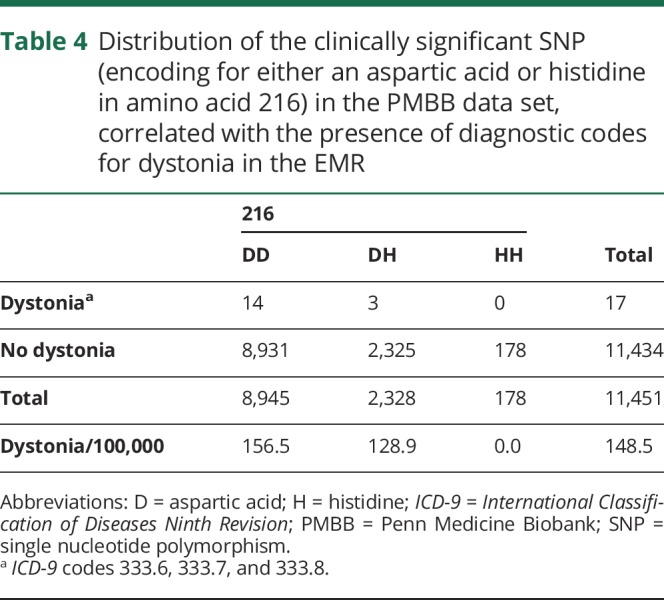
Finally, we evaluated for the frequency of the D216H polymorphism among the PMBB exome data set. In addition to influencing DYT1 penetrance, it has been proposed as a risk factor for isolated dystonia.12 The frequency of the SNP encoding for aspartic acid (88.3%) and histidine (11.7%) was as previously described,3 with the number of participants per genotype and frequency of diagnostic codes for dystonia in their EHR shown in table 4.
Table 4.
Distribution of the clinically significant SNP (encoding for either an aspartic acid or histidine in amino acid 216) in the PMBB data set, correlated with the presence of diagnostic codes for dystonia in the EMR
Discussion
In this work, we estimate the frequency of the most common DYT1-causing mutation in the general population using exome data sets representing over 135,000 individuals, making it a robust genetic ascertainment study. The only previous attempt to quantify the prevalence of the GAG deletion in the general population was genotyping of the GAG deletion in samples from a neonatal screen in South-Eastern France, identifying 1 mutation among 12,000 births.15 Our study includes 2 additional independent data sets. All yielded consistent findings. These data have very important implications for therapeutic development in DYT1, allowing to quantitatively estimate the target population. In addition, we evaluated the frequency of other rare potentially pathogenic and predicted deleterious variants in TOR1A, linking it to clinical information in PMBB participants. This additional piece of information was not used for our prevalence estimates, as the pathogenicity of some of these variants is still debated. However, it will help clarify their role in dystonia as their frequency and clinical correlates are evaluated in additional data sets.
Dystonia is clinically and etiologically very heterogeneous,16 and DYT1 represents a very small fraction of patients with dystonia. A well-designed study recently estimated the prevalence of adult-onset isolated idiopathic or hereditary dystonia in Finland at 405 per million,7 whereas a meta-analysis estimated the prevalence of isolated dystonia at 164 per 1,000,0008. However, genetic forms such as DYT1 represent a minority of those cases. The prevalence of the GAG deletion in TOR1A among patients of all ages presenting with idiopathic dystonia has been addressed following a forward genetics approach, with reported rates of 5% (France),17 3.4% (Japan),18 2.3% (Germany),19 1.5% (Taiwan),20 or 7% (Poland).21 If, based on these reports, we consider these dystonia prevalence data (405 per million) and the approximate frequency of the GAG deletion among those patients (5%), there would be approximately 6,252 patients with DYT1 in the United States. Another approach to evaluate DYT1 prevalence is to use epidemiologic data including only early-onset idiopathic dystonia, the most common phenotypical presentation of DYT1. The prevalence of early-onset idiopathic dystonia in the United States has been reported at 1 in 30,000,22 with about 70% of those predicted to carry the DYT1 mutation.23 Using these rates, the estimate would amount to 7,204 DYT1 patients in the United States. Both clinical ascertainment-based estimates are significantly lower than the range of 16,475 to 24,513 DYT1 patients yielded by this genetic ascertainment approach.
There are different potential reasons for this discrepancy. First, penetrance could be lower than the estimated 30%. The families used for penetrance studies were recruited through clinical ascertainment of affected family members followed by genetic screening of unaffected relatives. However, it is possible that many families carry the GAG deletion with no expression of dystonia. A second reason for the discrepancy between clinical and genetic ascertainment approaches is underdiagnosis (or misdiagnosis) of DYT1 of dystonia.
Finally, our data do not support an association of the D216H polymorphism with isolated dystonia. However, these data should be taken with caution because of the pitfalls of using diagnostic codes from EHR for genotype-phenotype correlations, especially in a challenging and heterogeneous syndrome such as dystonia.
In this work, we report the frequency of the most common DYT1 mutation in the general population using large genetic data sets, with additional data on rare pathogenic and a clinically significant polymorphism. Together with clinically ascertained information, we provide a solid prevalence estimate for DYT1. This information establishes the size of the target population for genetically driven precision therapies for DYT1.
Acknowledgment
The authors thank the Exome Aggregation Consortium, the Genome Aggregation Database, and the groups that provided exome variant data to these resources. A full list of contributing groups can be found at gnomad.broadinstitute.org/about. The data collection in this work was partially supported by the Regeneron Genetics Center. PGA is supported by the Neurogenetics Translational Center of Excellence, Department of Neurology, the University of Pennsylvania. SMD is supported by the US Department of Veterans Affairs (IK2-CX001780). This publication does not represent the views of the Department of Veterans Affairs or the US government.
Glossary
- EHR
electronic health record
- gnomAD
Genome Aggregation Database
- ICD
International Classification of Diseases
- PMBB
Penn Medicine Biobank
- REVEL
Rare Exonic Variant Ensemble Learner
- SNP
single nucleotide polymorphism
Appendix. Authors
Study funding
P. Gonzalez-Alegre is supported by the Neurogenetics Translational Center of Excellence, Department of Neurology, the University of Pennsylvania. S. Damrauer is supported by the US Department of Veterans Affairs (IK2-CX001780).
Disclosure
Disclosures available: Neurology.org/NG.
References
- 1.Ozelius LJ, Hewett JW, Page CE, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet 1997;17:40–48. [DOI] [PubMed] [Google Scholar]
- 2.Bressman SB, Sabatti C, Raymond D, et al. The DYT1 phenotype and guidelines for diagnostic testing. Neurology 2000;54:1746–1752. [DOI] [PubMed] [Google Scholar]
- 3.Risch NJ, Bressman SB, Senthil G, Ozelius LJ. Intragenic Cis and Trans modification of genetic susceptibility in DYT1 torsion dystonia. Am J Hum Genet 2007;80:1188–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hettich J, Ryan SD, de Souza ON, et al. Biochemical and cellular analysis of human variants of the DYT1 dystonia protein. TorsinA/TOR1A. Hum Mutat 2014;35:1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vulinovic F, Lohmann K, Rakovic A, et al. Unraveling cellular phenotypes of novel TorsinA/TOR1A mutations. Hum Mutat 2014;35:1114–1122. [DOI] [PubMed] [Google Scholar]
- 6.Reichert SC, Gonzalez-Alegre P, Scharer GH. Biallelic TOR1A variants in an infant with severe arthrogryposis. Neurol Genet 2017;3:e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ortiz R, Scheperjans F, Mertsalmi T, Pekkonen E. The prevalence of adult-onset isolated dystonia in Finland 2007-2016. PLoS One 2018;13:e0207729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steeves TD, Day L, Dykeman J, Jette N, Pringsheim T. The prevalence of primary dystonia: a systematic review and meta-analysis. Mov Disord 2012;27:1789–1796. [DOI] [PubMed] [Google Scholar]
- 9.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dewey FE, Murray MF, Overton JD, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016;354:aaf6814. [DOI] [PubMed] [Google Scholar]
- 11.Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an Ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 2016;99:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamm C, Fischer H, Garavaglia B, et al. Susceptibility to DYT1 dystonia in European patients is modified by the D216H polymorphism. Neurology 2008;70:2261–2262. [DOI] [PubMed] [Google Scholar]
- 13.Ostrer H, Skorecki K. The population genetics of the Jewish people. Hum Genet 2013;132:119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhagat SL, Qiu S, Caffall ZF, et al. Mouse model of rare TOR1A variant found in sporadic focal dystonia impairs domains affected in DYT1 dystonia patients and animal models. Neurobiol Dis 2016;93:137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frederic M, Lucarz E, Monino C, et al. First determination of the incidence of the unique TOR1A gene mutation, c.907delGAG, in a Mediterranean population. Mov Disord 2007;22:884–888. [DOI] [PubMed] [Google Scholar]
- 16.Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord 2013;28:863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brassat D, Camuzat A, Vidailhet M, et al. Frequency of the DYT1 mutation in primary torsion dystonia without family history. Arch Neurol 2000;57:333–335. [DOI] [PubMed] [Google Scholar]
- 18.Matsumoto S, Nishimura M, Kaji R, et al. DYT1 mutation in Japanese patients with primary torsion dystonia. Neuroreport 2001;12:793–795. [DOI] [PubMed] [Google Scholar]
- 19.Grundmann K, Laubis-Herrmann U, Bauer I, et al. Frequency and phenotypic variability of the GAG deletion of the DYT1 gene in an unselected group of patients with dystonia. Arch Neurol 2003;60:1266–1270. [DOI] [PubMed] [Google Scholar]
- 20.Lin YW, Chang HC, Chou YH, et al. DYT1 mutation in a cohort of Taiwanese primary dystonias. Parkinsonism Relat Disord 2006;12:15–19. [DOI] [PubMed] [Google Scholar]
- 21.Szczaluba K, Jurek M, Milewski M, et al. Clinical characteristics of carriers of a GAG deletion in the DYT1 gene amongst Polish patients with primary dystonia. Eur J Neurol 2007;14:659–662. [DOI] [PubMed] [Google Scholar]
- 22.Kamm C. Early onset torsion dystonia (Oppenheim's dystonia). Orphanet J Rare Dis 2006;1:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein C, Friedman J, Bressman S, et al. Genetic testing for early-onset torsion dystonia (DYT1): introduction of a simple screening method, experiences from testing of a large patient cohort, and ethical aspects. Genet Test 1999;3:323–328. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published within the article will be shared by request from any qualified investigator.