

Abstract
Objective
To measure the impact of JHU-083, a novel prodrug of the glutamine antagonist 6-diazo-5-oxo-l-norleucine, on immune cell proliferation and activation, along with physical and cognitive impairments associated with the experimental autoimmune encephalomyelitis (EAE) mouse model of MS.
Methods
Splenic-derived T cells and bone marrow–derived dendritic cells (DCs) were cultured, activated, and treated daily with vehicle or JHU-083. Proliferation and activation were measured via flow cytometry and IncuCyte live cell analysis. C57BL/6 mice were immunized for EAE. Vehicle or JHU-083 was administered orally every other day either from the time of immunization in the prevention paradigm or from the time of disease onset in the treatment paradigm. Disease scores and body weight were monitored. In the treatment paradigm, cognition was evaluated using the Barnes maze test.
Results
JHU-083 selectively inhibits T-cell proliferation and decreases T-cell activation, with no effect on DCs. In vivo, orally administered JHU-083 significantly decreases EAE severity in both prevention and treatment paradigms and reverses EAE-induced cognitive impairment.
Conclusions
JHU-083, a well-tolerated, brain penetrable glutamine antagonist, is a promising novel treatment for both the physical and cognitive deficits of MS.
MS is an immune-mediated disease that causes CNS damage in the form of demyelination and neurodegeneration. In addition to physical disability, learning and memory impairments occur in approximately half of all patients with MS.1,2 The exact cause of MS is unknown, but dysregulated glutamate signaling and aberrant T-cell activation are consistently implicated as contributors to both physical and cognitive disease pathogenesis.3–6 A number of trials have been conducted to identify either a disease-modifying therapy or a novel pharmacotherapy to treat MS-related cognitive impairment. To date, however, there are no approved treatments specifically targeting cognitive impairment in MS, as larger clinical trials often fail to replicate the initially reported positive findings,7,8 adverse effects are reported,9 and positive findings are either nonexistent10,11 or moderate at best.12,13
The enzyme glutaminase (GLS) is located throughout the body, including in neurons and glia in the CNS, and deamidates glutamine into glutamate. The glutamine analog 6-diazo-5-oxo-l-norleucine (DON) blocks glutamine-using reactions, including GLS, thereby inhibiting the formation of glutamate. Because rapidly proliferating cells require glutamine as a fuel source and excess glutamate can cause excitotoxic damage in the CNS, DON has dual therapeutic potential for treating diseases where excess cell proliferation and glutamate are pathogenic, such as MS. To our knowledge, only 1 study has examined the effects of DON in MS using a mouse model of the disease.14 The authors found that DON halted microglial activation and glutamate production, in turn conferring neuronal protection. The effects of DON on immune function, however, were not measured.
Although promising, DON is not clinically translatable. Its development for the treatment of cancer based on numerous positive preclinical and clinical studies15–21 was halted because of gastrointestinal (GI) toxicities, as the GI system is highly glutamine using. With regard to MS and other diseases of the CNS, DON is also not suitable for clinical translation because of its limited brain penetration. To overcome DON's peripheral toxicities and to enhance its delivery to the CNS, our laboratory created a series of lipophilic DON prodrugs. Prodrugs were designed to circulate intact as inert in plasma, but permeate and be cleaved to release DON once inside the brain. In numerous species, including swine and primates, prodrugs improved DON exposure in the CNS vs plasma (area under the curve [AUC]brain/AUCplasma) by 8- to 10-fold.22,23
In this report, we characterize the antiproliferative effects of the DON prodrug JHU-083 (ethyl 2-(2-amino-4-methylpentanamido)-DON) in vitro and evaluate chronic JHU-083 treatment in the experimental autoimmune encephalomyelitis (EAE) mouse model of MS using both physical and cognitive outcomes.
Methods
Animals
Male and female 7-week-old C57BL/6J mice (Jackson Laboratory) were housed in the Miller Research Building Johns Hopkins animal facility. All protocols were approved by the Johns Hopkins Institutional Animal Care and Use Committee.
T-cell isolation and proliferation
Spleens were removed from 3 naive mice, and single-cell suspensions were generated by passing cells through a 70-μM cell strainer (BD Biosciences). T cells were isolated from splenocytes using a CD4+ T-Cell Isolation Kit (Miltenyi Biotec) following the manufacturer's protocol and cultured in Roswell Park Memorial Institute media (RPMI) 1640 (Invitrogen) supplemented with 10% vol/vol fetal bovine serum (Invitrogen), 100 μg/mL penicillin and streptomycin (Quality Biological), 0.5 μm 2-mercaptoethanol (Invitrogen), 10 mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) buffer (Quality Biological), 1 mm sodium pyruvate (Sigma-Aldrich), minimal essential medium non-essential amino acid solution (Sigma-Aldrich), and glutamax (Thermo Fisher).
To measure proliferation via flow cytometry, 96-well flat-bottom plates were coated with 200 μL 1μg/mL α-CD3 (BD Biosciences) in phosphate buffered saline and incubated at 4°C overnight. The following day, plates were washed 3×, and 150,000 CD4+ T cells were plated with 20 μM eFlur450 cell proliferation dye (Thermo Fisher), 2 μg/mL α-CD28 (BD Biosciences), and ±JHU-083 (1 μm, 3 μM, 10 μM, 30 μM, or 100 μM, in triplicate) in 100 μM complete RPMI (cRPMI). Every 24 hours, cells were treated with 20 μM media ± JHU-083. After 72 hours in culture, cells were stained with fluorochrome-conjugated antibodies for CD69, CD25, CD62L, CD44 (Biolegend), and live/dead (L/D) aqua (Thermo Fisher) and quantified (MACSQuant Analyzer X, Miltenyi Biotec).
To measure proliferation and viability, splenic-derived CD4+ T cells were seeded, activated, and stained with IncuCyte Cytotox Green Reagent or NucLight Rapid Red Reagent (Sartorius, Essen BioScience). Cells were treated with vehicle or JHU-083 (1 μm, 10 μM, or 100 μM), media was changed every 24 hours, and cells were imaged by the Incucyte Live Cell Analysis System (Sartorius, Essen BioScience) every 2 hours. Negative control vehicle wells were included, in addition to separate 10% dimethyl sulfoxide (DMSO) wells to stimulate cell death. Five representative images were captured per well per hour. Data are reported as red object count to reflect the number of cells, green object count to reflect the number of dead cells, and red object count/green object count (red/green) to reflect cell number/dead and control for variability in the number of plated cells.
Dendritic cell isolation and activation
Bone marrow–derived dendritic cells (DCs) were generated as previously described24 with minor modifications. Briefly, femurs were removed from C57BL/6J mice, cut, and marrow flushed through the shaft with phosphate buffered saline. Cells were centrifuged, counted, and 2 million cells were plated in 10 mL plates in complete RPMI (cRPMI) media consisting of RPMI-1640 with GlutaMAX (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum, penicillin-streptomycin (Gibco), 50 μM 2-mercaptoethanol (Sigma-Aldrich), and 20 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF). Following 14 days in culture, 5 × 105 cells were seeded in a 24-well flat-bottom plate containing 1 mL cRPMI, 100 ng/mL lipopolysaccharide (LPS), and 100 ug/mL GM-CSF (Peprotech). DCs were treated with vehicle or JHU-083. Media were changed every 24 hours. Following 72 hours in culture, cells were stained with fluorochrome-conjugated antibodies against CD11c, CD86, and CD40 (BioLegend), and flow cytometry was conducted (MACSQuant Analyzer X, Miltenyi Biotec).
EAE immunizations and scoring
Female mice were immunized for EAE as previously described25 with minor modifications. Briefly, mice were administered murine myelin oligodendrocyte glycoprotein 35–55 (Johns Hopkins Peptide Synthesis Core Facility, Baltimore) in incomplete Freund adjuvant (Sigma-Aldrich) supplemented with heat-killed Mycobacterium tuberculosis (Sigma-Aldrich) via 2 subcutaneous flank injections. On days 0 and 2, mice received an intraperitoneal injection of 250 ng pertussis toxin (List Biological Laboratories). EAE disease scores were blindly assigned as previously described26 based on the following scale with 0.5 increments for intermediate scores: 0 = normal, 1 = limp tail, 2 = wobbly gait, 3 = dragging hind flank, 4 = hind limb paralysis, and 5 = quadriplegia. Experiments were conducted in duplicate.
JHU-083 treatment administration
Mice were administered vehicle or JHU-083 (1.83 mg/kg, corresponding to 1 mg/kg equivalent DON) via oral gavage every other day from the time of immunization (prevention paradigm) or from the time of EAE disease score ≥1 (treatment paradigm). Vehicle was made up of 5% ethanol/95% 50 mM HEPES buffer. Each week, fresh JHU-083 stock solutions were made by dissolving JHU-083 in 100% ethanol and stored at −20. On dosing days, stock solution was added to 50 mM HEPES buffer, and mice were immediately dosed.
Cognition studies
The Barnes maze cognitive test was administered to EAE mice from the treatment paradigm cohort as previously described.25 Vehicle or JHU-083 administration continued in the treatment cohort mice until disease scores normalized (p > 0.9), and the Barnes maze was conducted at 75–78 days postimmunization. Mice were trained to find the target platform for 2 trials per day over 3 consecutive days, and then, memory was tested on the fourth day. The researcher conducting cognition studies was blinded to the treatment groups.
Statistical analyses
Statistical analyses were completed using GraphPad Prism 6.0. Comparisons between groups for the in vitro T-cell proliferation and activation flow cytometry studies and DC activation studies were made with ordinary 1-way analysis of variance (ANOVA) with Sidak multiple comparison test. Repeated-measures 2-way ANOVA with Sidak multiple comparison post hoc test measured JHU-083 treatment effects in the in vitro T-cell proliferation Incucyte studies and body weight in the in vivo JHU-083 EAE prevention and treatment studies. Mann-Whitney t tests of rank-summed EAE scores were used to detect significance over time in EAE disease scores in the in vivo JHU-083 EAE prevention and treatment studies. Repeated-measures 2-way ANOVA with Sidak multiple comparison post hoc test were used to detect significance in EAE disease scores on individual days. Cognition studies were analyzed using an unpaired 2-tailed t test. p values <0.05 were considered statistically significant. Sample size was determined by power analyses with at least 80% power at the 0.05 level. Statistical analyses assistance and consultation were provided by the Johns Hopkins Institute for Clinical and Translational Research Biostatistics Program.
Data availability
Data sets generated in the present study are available from the corresponding authors on reasonable request.
Results
JHU-083 inhibits T-cell proliferation and activation, with no effect on DCs
In vitro studies were designed to evaluate whether JHU-083 had an effect on T-cell activation. Flow cytometry analyses revealed that JHU-083 significantly inhibited T-cell proliferation at the lowest tested concentration (1 μM) JHU-083 (p < 0.001, figure 1, A and B). A dose response effect was observed, with higher doses (10–100 μM) almost completely preventing cell proliferation as indicated by >96% of cells remaining undivided after 72 hours (p < 0.001). Following CD3/CD28 treatment, the majority of lymphocytes exhibit CD44, CD25, and CD69 surface markers indicating activation.27 Daily treatment of cells with 1 μM JHU-083 reduced levels of activation markers by approximately 60–70% (p < 0.001), with higher doses showing equivalent efficacy (figure 1, C–F).
Figure 1. Glutamine antagonist JHU083 inhibits T-cell proliferation in vitro.

T-cell proliferation and activation were measured by flow cytometry after 72 hours in culture. (A and B) JHU-083 administered to α-CD3/α-CD28-stimulated T cells every 24 hours significantly inhibited cell proliferation. JHU-083 administered to T cells every 24 hours significantly decreased cell activation as measured by CD44hi (C and D), and CD25+CD69+ (E and F). All doses in all conditions significantly different from control at p < 0.001 by ordinary 1-way analysis of variance with Sidak multiple comparison test. Data are mean ± standard error of the mean from 3 mice (M1, M2, and M3) in duplicate (M2 and M3) or triplicate (M1).
Lymphocyte proliferation was also measured by live cell analysis with imaging occurring every 2 hours (figure 2A), to both confirm flow cytometry results and also to verify that the observed antiproliferative effects of JHU-083 were not due to cytotoxic effects of the drug. Data are reported as red object count to reflect the number of cells, green object count to reflect the number of dead cells, and red object count/green object count (red/green) to reflect cell number/dead and control for variability in the number of plated cells.
Figure 2. The glutamine antagonist JHU 083 is not cytotoxic.

T-cell proliferation and viability were measured via IncuCyte live cell analysis. (A) Cells treated every 24 hours with JHU-083 were stained with dyes to mark cell number (red) and cell death (green). Cell counts were obtained from 5 areas per well every 2 hours for 72 hours. (B) Red counts marking cell number indicate that all doses of JHU-083 significantly inhibited proliferation and decreased cell number vs vehicle (repeated-measures 2-way analysis of variance [ANOVA] with Sidak multiple comparison post hoc test p < 0.05). (C) Green counts marking cell death indicate that DMSO was immediately cytotoxic, but JHU-083 was not. (D) Red counts normalized to green counts to control for any variances in plating indicate that doses of JHU-083 as low as 1 μM were effective at significantly inhibiting cell proliferation (repeated-measures 2-way ANOVA with Sidak multiple comparison post hoc test p < 0.001). Data are mean ± standard error of the mean, n = 2–3 per time point.
Significant inhibition of cell proliferation due to 1, 10, and 100 μM JHU-083, as measured by total number of red cells, was observed starting at 26, 22, and 32 hours, respectively, and continued through the end of the experiment (p < 0.05, figure 2B). Differences in timing for each dose were likely due to minor variations in plating and starting cell number, but all doses had approximate equivalent efficacy in halting proliferation. Green counts, corresponding to the number of dead cells, showed an immediate induction of cell death in DMSO wells with no change in cell number from baseline through 72 hours (figure 2C). The number of dead cells was lower in all JHU-083-treated groups vs vehicle control wells starting at 54 hours in culture (p < 0.05). This observation was likely due to the fact that cell proliferation was inhibited with the JHU-083 treatment, so there were a fewer number of cells to die. Normalizing the number of cells to the number of dead cells using red/green object count revealed significant reductions in red counts due to 1 μM and higher doses of JHU-083 starting at the first 2-hour time point (p < 0.05, figure 2D). Significant reductions vs control continued at all doses of JHU-083 from the 4-hour time point until the end of the experiment (p < 0.001). From the first time point, red/green object count remained at 1 in DMSO wells, indicating that nearly all cells were dead because of cytotoxic DMSO exposure. Red/green object count was significantly higher at all doses of JHU-083 vs DMSO (p < 0.001), indicating that JHU-083 halted proliferation but was not cytotoxic.
To determine whether DC activation and proliferation were similarly attenuated, JHU-083 (1–100 μM) was applied to DCs stimulated with GM-CSF. Of interest, the populations of CD11c+ CD40+ CD86+ DCs remained unaffected at all concentrations tested (figure 3). No LPS and no LPS+3 μM JHU-083 controls revealed that JHU-083 had no effect of DC activation and proliferation under unstimulated conditions.
Figure 3. Glutamine antagonist JHU083 does not affect dendritic cell (DC) activation.
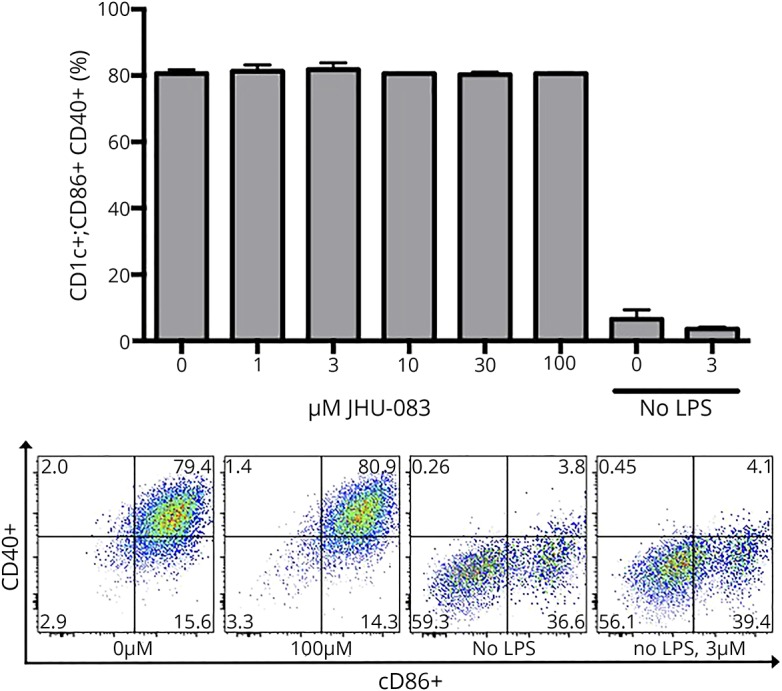
Bone marrow–derived DCs were activated with LPS and treated with JHU-083 daily for 72 hours. No dose of JHU-083 affected DC activation as measured by % CD86+CD40+ of the CD11c+ population using ordinary 1-way analysis of variance with Sidak multiple comparison test. Data are mean ± standard error of the mean.
JHU-083 decreases EAE severity in both prevention and treatment paradigms
Given its selective and robust effect on T-cell proliferation, and the fact that JHU-083 inhibits GLS without causing overt GI toxicities,28 we next tested whether JHU-083 could affect EAE development or progression in mice. EAE mice treated with 1.83 mg/kg oral JHU-083 every other day from the time of immunization exhibited decreased incidence, with only 2/7 mice displaying EAE scores >0, and significantly decreased severity of physical disabilities (p < 0.05, figure 4A). Furthermore, the JHU-083-treated mice did not experience the hallmark reduction in body weight associated with the acute stage of EAE (repeated-measures 2-way ANOVA treatment effect p < 0.01, figure 4B).
Figure 4. Oral JHU-083 administration prevents experimental autoimmune encephalomyelitis (EAE) disease and body weight loss when administered from the time of immunization or from the time of disease onset.

JHU-083 dosed at 1 mg/kg equivalent q.a.d from day 0 (A) prevents or delays the development of physical signs of EAE and (B) attenuates body weight loss associated with peak EAE symptoms. JHU-083 dosed at 1 mg/kg equivalent q.a.d from the day of disease onset when EAE score ≥1 (C) significantly improves disease severity as measured by the EAE disease score and (D) has no effect on body weight as compared to vehicle-treated EAE mice when administered 1 mg/kg equiv q.a.d. Mann-Whitney t tests of rank-summed EAE disease scores over time p < 0.05 (prevention) and p < 0.01 (treatment). Repeated-measures 2-way analysis of variance with Sidak multiple comparison significantly different from EAE + vehicle at *p < 0.05, **p < 0.01, ****p < 0.0001. Data are mean ± standard error of the mean, n = 7–8.
Given the robust effect on preventing EAE symptoms and the fact that JHU-083 delivers μM DON into nervous tissues,28 we followed with a treatment protocol. Mice treated with 1.83 mg/kg oral JHU-083 every other day beginning at the time of disease onset (EAE score ≥ 1) also had significantly reduced average severity of physical signs of EAE (p < 0.01, figure 4C). No impact on body weight was observed because of JHU-083 treatment (figure 4D).
JHU-083 attenuates EAE cognitive deficits
Given the ability of JHU-083 to deliver DON into the CNS and DON's positive treatment effects on cognition in other models of neuroinflammation,23 we next measured JHU-083 treatment effects on cognition in EAE mice. Every other day, JHU-083 treatment beginning from the time of disease onset continued for over 2 months, and by days 75–78 postimmunization, disease scores normalized between groups. Cognition was then tested using the Barnes maze. Latency to find the target hole on day 1 of training was compared with latency to find the target hole on the final day of testing, and mice receiving JHU-083 demonstrated significantly superior long-term memory vs EAE + vehicle controls (p < 0.05, figure 5).
Figure 5. Glutamine antagonist JHU-083 improves cognition in experimental autoimmune encephalomyelitis (EAE) mice.

(A) EAE mice administered JHU-083 demonstrated superior long-term memory with significantly increased primary latency deltas in the Barnes maze (day 1 trial 1 to day 4 trial 1). (B) Representative Barnes maze paths showing improved path efficiency in 6-diazo-5-oxo-l-norleucine prodrug-treated mice. Significantly different from EAE + vehicle at *p < 0.05 using an unpaired 2-tailed t test, n = 7–8.
Discussion
Prodrug development is a common strategy to alter the pharmacokinetic profile and/or toxicity profile of an otherwise promising small molecule therapeutic. In fact, prodrugs make up over 12% of the approved small molecule new chemical entities approved by the US Food and Drug Administration in the past 10 years.29 Using this tactic, our laboratory recently developed of a series of dual-moiety DON prodrugs that preferentially deliver DON to the CNS.22,23 By masking both the primary amine and carboxylate of DON with promoieties, which are stable in plasma, liver, and GI tissues, the prodrug circulates in the peripheral plasma intact and inert, but is transported and preferentially cleaved in the CNS. Compared with equimolar DON, this prodrug strategy led to an 8- to 10-fold improvement in the delivery of active DON to the CNS in both primates22 and swine,23 allowing for dose reductions and improved therapeutic index.
Although much of the focus of therapeutic application for glutamine antagonism is in oncology,21 there is equal applicability for diseases where glutamine-addicted lymphocytes are pathogenic and dysregulated glutamate signaling contributes to disease pathogenesis, such as MS. Rapidly dividing and activated inflammatory T cells are highly dependent on glutamine as a source of energy production. In fact, activated T cells significantly increase expression of glutamine transporters and import 5- to 10-fold more glutamine as compared to resting T cells,30,31 and intracellular levels of glutamine in activated peripheral blood mononuclear cells from patients with MS are 74% higher than healthy controls.32 To this end, levels of GLS are elevated in immune cells found in MS plaques, particularly at the center of active lesions,33 and levels of the excitotoxic GLS product glutamate are elevated in MS and associated with decreased neuroaxonal integrity as measured by N-acetylaspartate (NAA).4 Because decreased NAA levels are associated with learning and memory impairments in MS,6,34 it is also conceivable that aberrant GLS activity directly contributes to the development of cognitive dysfunction in MS.
The present study highlights the therapeutic potential of our newly synthesized DON prodrug, ethyl 2-(2-amino-4-methylpentanamido)-DON, also termed JHU-083,28 as a novel treatment for the autoimmune disease MS. Inhibition of glutamine-using reactions halts the proliferation of rapidly dividing cells that are dependent on glutamine and also curtails the synthesis of the excitotoxic GLS product glutamate. These dual actions of JHU-083 are ideal for targeting the pathogenic lymphocyte proliferation and glutamate excitotoxicity found in MS. A previously published EAE study showed a delay in the development of physical symptoms of disease when 1.6 mg/kg DON was dosed every other day from the day of immunization.14 The authors found that DON inhibits glutamate production and halts microglial activation, in turn conferring neuronal protection, but did not measure the impact of the drug on lymphocyte activation. Here, we measure the effects of JHU-083 on the immune system, showing that the DON prodrug inhibits T-cell proliferation and has positive therapeutic effects in the EAE model of MS. Taken together, these results support the hypothesis that DON and JHU-083 are protective in vivo through multiple mechanisms of action.
Here, we report that JHU-083 significantly inhibits T-cell proliferation in vitro at concentrations lower than previously reported from DON14,35 and that EAE symptoms and severity were improved because of JHU-083 treatment in both prevention and treatment dosing paradigms. Prevention data establish that JHU-083 works in the periphery, but the treatment paradigm data suggest that the drug might also be halting proliferation of pathogenic immune cells that have already migrated into in the CNS. A previous study from our laboratory showed that JHU-083 is CNS penetrant, with a brain to plasma ratio of 0.55, and reverses inflammation in the CNS caused by depression.28 Therefore, to functionally measure the impact of JHU-083 in the CNS, cognitive function was measured in EAE mice. Cognitive impairment is a well-characterized occurrence in both MS and EAE25,36 that is measurable in EAE using the hippocampal-dependent Barnes maze test. Because this land maze requires mobility, EAE mice were dosed with vehicle or JHU-083 until disease scores normalized and physical health was equal between groups. The significant improvement in cognition observed here provides additional evidence that JHU-083 has central in addition to peripheral action. We hypothesize that both a decrease in levels of pathogenic immune cells that contribute to cognitive impairment in MS5 and a decrease in levels of excitotoxic glutamate that causes tissue damage and cognitive impairment in MS4,6,34 could contribute to the observed positive behavioral effects.
As well as direct T-cell responses, we also evaluated the effects of JHU-083 on DC activation. DCs play a critical role in T-cell immune activation in both the periphery and in the CNS. Activation of CD11c+ DCs, also termed myeloid DCs (mDCs), was unaffected at concentrations up to 100 μM. This lack of effect is in line with findings from a recent study that showed no effect of DON on human mDC activation. Activation of mDCs causes a metabolic shift from oxidative phosphorylation to glycolysis,37 an energy generating process that is dependent on glucose, not glutamine. Therefore, it is likely that mDCs use glucose as a primary fuel source during activation, thus remaining unaffected by glutamine antagonism. Although no effect was observed on DC activation due to DON prodrug treatment in the present studies, previous studies have reported that activation of another type of DC, plasmacytoid DCs (pDCs), which causes an increase in intracellular glutamine levels that is reversible by DON treatment.38 The expression of genes related to oxidative phosphorylation is increased in activated pDCs but decreased in activated mDCs,38 supporting the idea that the metabolic demands of mDCs and pDCs differ and possibly explaining why pDCs but not mDCs are affected by glutamine antagonism.
In addition to MS, the DON prodrug JHU-083 has translational promise for other diseases wherein glutamine-addicted T cells and/or excess glutamate signaling are pathogenic. In fact, JHU-083 has demonstrated beneficial results in models of depression,28 a disease in which neuroinflammation is considered a critical component, and cerebral malaria.39 A study measuring the effect of DON on the development of alphavirus encephalomyelitis showed decreased leukocyte infiltration into the CNS and protection from paralysis and death following DON administration,35 and a study in a mouse model of HIV showed that DON prevents cognitive decline.23 Furthermore, deficiency or inhibition of GLS improves outcomes in a variety of preclinical models of autoimmune diseases, including inflammatory bowel disease, interleukin-17-dependent graft-versus-host disease,40 and rheumatoid arthritis.41
There are limitations to the present study. First, the positive treatment effects observed from JHU-083 in EAE are likely due, in large part, to decreased activation and proliferation of proinflammatory CD4+ T cells and decreased glutamate excitotoxicity, as both activated immune cells and increased glutamate neurotransmission contribute to physical and cognitive impairments in EAE.42 It is possible, however, that cell populations not studied in vitro here, such as pDCs, contributed to the attenuation of EAE symptoms. Future studies will focus on these outcomes, along with ex vivo analyses of immune cell populations and glutamate levels to provide additional mechanistic insight. A focus will be placed on CD8+ T cells in these future studies, as we expect JHU-083 to also halt proliferation of cytotoxic T cells based on previous findings using DON.43 We are also interested in exploring the effects of JHU-083 on oligodendrocyte protection, as previous studies have demonstrated that regulation of glutamate signaling via N-methyl D-aspartate antagonism not only improves EAE44 but also confers protection to oligodendrocytes and oligodendrocyte precursor cells against inflammatory and excitotoxic insults.45 Future studies will also include passive transfer EAE to examine the impact of JHU-083 on solely the efferent phase of disease, as we expect that GLS inhibition would independently inhibit both T-cell activation and proliferation. Second, it remains to be determined whether GLS inhibition and inhibition of glutamate production from glutamine are beneficial to treat cognitive impairment in MS. There is a body of research that underscores the importance of proper glutamate signaling for normal cognitive function, the cognitive enhancing effects of glutamatergic neurotransmission, and the cognitive impairing effects of disruptions in glutamate signaling (see reference 46 for review). In line with this, recent imaging studies have reported higher brain glutamate levels associated with better cognitive function in patients with MS,47,48 but a host of literature also exists showing increased glutamate in MS brains vs healthy controls49 and confirming the damaging effects of excess glutamate in MS (see reference 50 for review). Further investigation is required, but it is likely that these seemingly discrepant findings are due to the fact that high glutamate levels lead to neurodegeneration, and therefore, reductions in brain glutamate levels coincide with neuronal degeneration,51 which in turn coincides with cognitive impairment in MS. This interpretation is in part supported by work from Baranzini et al showing that patients with MS with a higher number of mutations in genes related to glutamate biology had higher levels of brain glutamate and experienced significantly more profound brain volume loss over the course of a year vs patients with MS with fewer glutamate-related mutations.52
The present study highlights the therapeutic potential of the well-tolerated, brain penetrable glutamine antagonist JHU-083, as a novel treatment for both the physical and cognitive deficits of MS. Inhibition of glutamine-using reactions including GLS halts proliferation of rapidly proliferating cells that are dependent on glutamine and curtails synthesis of the excitotoxic GLS product glutamate. These complementary actions of JHU-083 provide a path forward for targeting pathogenic proinflammatory glutamine-dependent T-cell activation and dysregulated glutamate signaling that is characteristic of MS.
Acknowledgment
The authors acknowledge support for the statistical analysis from the National Center for Research Resources and the National Center for Advancing Translational Sciences (NCATS) of the NIH through Grant Number 1UL1TR001079.
Glossary
- ANOVA
analysis of variance
- cRPMI
complete RPMI
- DC
dendritic cell
- DON
6-diazo-5-oxo-l-norleucine
- EAE
experimental autoimmune encephalomyelitis
- GI
gastrointestinal
- GLS
glutaminase
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- NAA
N-acetylaspartate
Author contributions
K.R. Hollinger, B.S. Slusher, and P.A. Calabresi conceived the concept, contributed to experimental designs, performed experiments, conducted data analyses, and wrote the manuscript. M.D. Smith assisted with experimental design and completion, data analyses, and manuscript revisions. L.A. Kirby assisted with experiments and data interpretation. E. Prchalova synthesized JHU-083. R. Rais designed the JHU083 formulations, and J. Alt conducted their stability analyses.
Study funding
This work was supported by P30MH075673, the Bloomberg Kimmel Institute for Cancer Immunotherapy, and R37NS041435 grants.
Disclosure
K.R. Hollinger, M.D. Smith, L.A. Kirby, and E. Prchalova report no disclosures. J. Alt and R. Rais report entitlement of royalty distributions related to technology used in the research described in this publication under a license agreement between Dracen Pharmaceuticals, Inc. and the Johns Hopkins University. Dr. Rais is a cofounder and holds equity in Dracen Pharmaceuticals, Inc. P.A. Calabresi reports consulting honoraria from Disarm Therapeutics and Biogen and grant support from Annexon and Biogen. B.S. Slusher reports entitlement of royalty distributions related to technology used in the research described in this publication under a license agreement between Dracen Pharmaceuticals, Inc. and the Johns Hopkins University. Dr. Slusher is a cofounder and holds equity in Dracen Pharmaceuticals, Inc. Go to Neurology.org/NN for full disclosures.
References
- 1.DiGiuseppe G, Blair M, Morrow SA. Short report: prevalence of cognitive impairment in newly diagnosed relapsing-remitting multiple sclerosis. Int J MS Care 2018;20:153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rao SM, Leo GJ, Bernardin L, Unverzagt F. Cognitive dysfunction in multiple sclerosis. I. Frequency, patterns, and prediction. Neurology 1991;41:685–691. [DOI] [PubMed] [Google Scholar]
- 3.Wu GF, Alvarez E. The immunopathophysiology of multiple sclerosis. Neurol Clin 2011;29:257–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azevedo CJ, Kornak J, Chu P, et al. . In vivo evidence of glutamate toxicity in multiple sclerosis. Ann Neurol 2014;76:269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger T. Immunological processes related to cognitive impairment in MS. Acta Neurol Scand 2016;134(suppl 200):34–38. [DOI] [PubMed] [Google Scholar]
- 6.Pan JW, Krupp LB, Elkins LE, Coyle PK. Cognitive dysfunction lateralizes with NAA in multiple sclerosis. Appl Neuropsychol 2001;8:155–160. [DOI] [PubMed] [Google Scholar]
- 7.Krupp LB, Christodoulou C, Melville P, Scherl WF, MacAllister WS, Elkins LE. Donepezil improved memory in multiple sclerosis in a randomized clinical trial. Neurology 2004;63:1579–1585. [DOI] [PubMed] [Google Scholar]
- 8.Krupp LB, Christodoulou C, Melville P, et al. . Multicenter randomized clinical trial of donepezil for memory impairment in multiple sclerosis. Neurology 2011;76:1500–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Villoslada P, Arrondo G, Sepulcre J, Alegre M, Artieda J. Memantine induces reversible neurologic impairment in patients with MS. Neurology 2009;72:1630–1633. [DOI] [PubMed] [Google Scholar]
- 10.Waubant E, Maghzi AH, Revirajan N, et al. . A randomized controlled phase II trial of riluzole in early multiple sclerosis. Ann Clin Transl Neurol 2014;1:340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lovera JF, Frohman E, Brown TR, et al. . Memantine for cognitive impairment in multiple sclerosis: a randomized placebo-controlled trial. Mult Scler 2010;16:715–723. [DOI] [PubMed] [Google Scholar]
- 12.Patti F, Amato MP, Bastianello S, et al. . Effects of immunomodulatory treatment with subcutaneous interferon beta-1a on cognitive decline in mildly disabled patients with relapsing-remitting multiple sclerosis. Mult Scler 2010;16:68–77. [DOI] [PubMed] [Google Scholar]
- 13.Maurer M, Ortler S, Baier M, et al. . Randomised multicentre trial on safety and efficacy of rivastigmine in cognitively impaired multiple sclerosis patients. Mult Scler 2013;19:631–638. [DOI] [PubMed] [Google Scholar]
- 14.Shijie J, Takeuchi H, Yawata I, et al. . Blockade of glutamate release from microglia attenuates experimental autoimmune encephalomyelitis in mice. Tohoku J Exp Med 2009;217:87–92. [DOI] [PubMed] [Google Scholar]
- 15.Eagan RT, Frytak S, Nichols WC, Creagan ET, Ingle JN. Phase II study on DON in patients with previously treated advanced lung cancer. Cancer Treat Rep 1982;66:1665–1666. [PubMed] [Google Scholar]
- 16.Earhart RH, Koeller JM, Davis HL. Phase I trial of 6-diazo-5-oxo-L-norleucine (DON) administered by 5-day courses. Cancer Treat Rep 1982;66:1215–1217. [PubMed] [Google Scholar]
- 17.Lynch G, Kemeny N, Casper E. Phase II evaluation of DON (6-diazo-5-oxo-L-norleucine) in patients with advanced colorectal carcinoma. Am J Clin Oncol 1982;5:541–543. [PubMed] [Google Scholar]
- 18.Magill GB, Myers WP, Reilly HC, et al. . Pharmacological and initial therapeutic observations on 6-diazo-5-oxo-1-norleucine (DON) in human neoplastic disease. Cancer 1957;10:1138–1150. [DOI] [PubMed] [Google Scholar]
- 19.Rahman A, Smith FP, Luc PT, Woolley PV. Phase I study and clinical pharmacology of 6-diazo-5-oxo-L-norleucine (DON). Invest New Drugs 1985;3:369–374. [DOI] [PubMed] [Google Scholar]
- 20.Sklaroff RB, Casper ES, Magill GB, Young CW. Phase I study of 6-diazo-5-oxo-L-norleucine (DON). Cancer Treat Rep 1980;64:1247–1251. [PubMed] [Google Scholar]
- 21.Lemberg KM, Vornov JJ, Rais R, Slusher BS. We're not “DON” yet: optimal dosing and prodrug delivery of 6-Diazo-5-oxo-L-norleucine. Mol Cancer Ther 2018;17:1824–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rais R, Jancarik A, Tenora L, et al. . Discovery of 6-Diazo-5-oxo-l-norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: a potential treatment for glioblastoma. J Med Chem 2016;59:8621–8633. [DOI] [PubMed] [Google Scholar]
- 23.Nedelcovych MT, Tenora L, Kim BH, et al. . N-(Pivaloyloxy)alkoxy-carbonyl prodrugs of the glutamine antagonist 6-Diazo-5-oxo-l-norleucine (DON) as a potential treatment for HIV associated neurocognitive disorders. J Med Chem 2017;60:7186–7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kornberg MD, Smith MD, Shirazi HA, Calabresi PA, Snyder SH, Kim PM. Bryostatin-1 alleviates experimental multiple sclerosis. Proc Natl Acad Sci U S A 2018;115:2186–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rahn KA, Watkins CC, Alt J, et al. . Inhibition of glutamate carboxypeptidase II (GCPII) activity as a treatment for cognitive impairment in multiple sclerosis. Proc Natl Acad Sci U S A 2012;109:20101–20106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hollinger KR, Alt J, Riehm AM, Slusher BS, Kaplin AI. Dose-dependent inhibition of GCPII to prevent and treat cognitive impairment in the EAE model of multiple sclerosis. Brain Res 2016;1635:105–112. [DOI] [PubMed] [Google Scholar]
- 27.Rabenstein H, Behrendt AC, Ellwart JW, et al. . Differential kinetics of antigen dependency of CD4+ and CD8+ T cells. J Immunol 2014;192:3507–3517. [DOI] [PubMed] [Google Scholar]
- 28.Zhu X, Nedelcovych MT, Thomas AG, et al. . JHU-083 selectively blocks glutaminase activity in brain CD11b(+) cells and prevents depression-associated behaviors induced by chronic social defeat stress. Neuropsychopharmacology 2019;44:683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rautio J, Meanwell NA, Di L, Hageman MJ. The expanding role of prodrugs in contemporary drug design and development. Nat Rev Drug Discov 2018;17:559–587. [DOI] [PubMed] [Google Scholar]
- 30.Carr EL, Kelman A, Wu GS, et al. . Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol 2010;185:1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brand K. Glutamine and glucose metabolism during thymocyte proliferation: pathways of glutamine and glutamate metabolism. Biochem J 1985;228:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Negrotto L, Correale J. Amino acid catabolism in multiple sclerosis affects immune homeostasis. J Immunol 2017;198:1900–1909. [DOI] [PubMed] [Google Scholar]
- 33.Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol 2001;50:169–180. [DOI] [PubMed] [Google Scholar]
- 34.Wood ET, Ronen I, Techawiboonwong A, et al. . Investigating axonal damage in multiple sclerosis by diffusion tensor spectroscopy. J Neurosci 2012;32:6665–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manivannan S, Baxter VK, Schultz KL, Slusher BS, Griffin DE. Protective effects of glutamine antagonist 6-Diazo-5-Oxo-l-Norleucine in mice with alphavirus encephalomyelitis. J Virol 2016;90:9251–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ziehn MO, Avedisian AA, Tiwari-Woodruff S, Voskuhl RR. Hippocampal CA1 atrophy and synaptic loss during experimental autoimmune encephalomyelitis. EAE Lab Invest 2010;90:774–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelly B, O'Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res 2015;25:771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basit F, Mathan T, Sancho D, de Vries IJM. Human dendritic cell subsets undergo distinct metabolic reprogramming for immune response. Front Immunol 2018;9:2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riggle BA, Sinharay S, Schreiber-Stainthorp W, et al. . MRI demonstrates glutamine antagonist-mediated reversal of cerebral malaria pathology in mice. Proc Natl Acad Sci U S A 2018;115:E12024–E12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson MO, Wolf MM, Madden MZ, et al. . Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell 2018;175:1780–1795.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takahashi S, Saegusa J, Sendo S, et al. . Glutaminase 1 plays a key role in the cell growth of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res Ther 2017;19:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mandolesi G, Grasselli G, Musumeci G, Centonze D. Cognitive deficits in experimental autoimmune encephalomyelitis: neuroinflammation and synaptic degeneration. Neurol Sci 2010;31:S255–S259. [DOI] [PubMed] [Google Scholar]
- 43.Lee CF, Lo YC, Cheng CH, et al. . Preventing allograft rejection by targeting immune metabolism. Cell Rep 2015;13:760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chechneva OV, Mayrhofer F, Daugherty DJ, Pleasure DE, Hong JS, Deng W. Low dose dextromethorphan attenuates moderate experimental autoimmune encephalomyelitis by inhibiting NOX2 and reducing peripheral immune cells infiltration in the spinal cord. Neurobiol Dis 2011;44:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lisak RP, Nedelkoska L, Benjamins JA. Effects of dextromethorphan on glial cell function: proliferation, maturation, and protection from cytotoxic molecules. Glia 2014;62:751–762. [DOI] [PubMed] [Google Scholar]
- 46.Dauvermann MR, Lee G, Dawson N. Glutamatergic regulation of cognition and functional brain connectivity: insights from pharmacological, genetic and translational schizophrenia research. Br J Pharmacol 2017;174:3136–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muhlert N, Atzori M, De Vita E, et al. . Memory in multiple sclerosis is linked to glutamate concentration in grey matter regions. J Neurol Neurosurg Psychiatry 2014;85:833–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O'Grady KP, Dula AN, Lyttle BD, et al. . Glutamate-sensitive imaging and evaluation of cognitive impairment in multiple sclerosis. Mult Scler Epub 2018 Sep 19. [DOI] [PMC free article] [PubMed]
- 49.Srinivasan R, Sailasuta N, Hurd R, Nelson S, Pelletier D. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain 2005;128:1016–1025. [DOI] [PubMed] [Google Scholar]
- 50.Macrez R, Stys PK, Vivien D, Lipton SA, Docagne F. Mechanisms of glutamate toxicity in multiple sclerosis: biomarker and therapeutic opportunities. Lancet Neurol 2016;15:1089–1102. [DOI] [PubMed] [Google Scholar]
- 51.Kaiser LG, Schuff N, Cashdollar N, Weiner MW. Age-related glutamate and glutamine concentration changes in normal human brain: 1H MR spectroscopy study at 4 T. Neurobiol Aging 2005;26:665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baranzini SE, Srinivasan R, Khankhanian P, et al. . Genetic variation influences glutamate concentrations in brains of patients with multiple sclerosis. Brain 2010;133:2603–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sets generated in the present study are available from the corresponding authors on reasonable request.