

Abstract
Objectives
To describe the neurologic spectrum and treatment outcomes for neurochondrin-IgG positive cases identified serologically in the Mayo Clinic Neuroimmunology Laboratory.
Methods
Archived serum and CSF specimens previously scored positive for IgGs that stained mouse hippocampal tissue in a nonuniform synaptic pattern by immunofluorescence assay (89 among 616,025 screened, 1993–2019) were reevaluated. Antibody characterization experiments revealed specificity for neurochondrin, confirmed by recombinant protein assays.
Results
IgG in serum (9) or CSF (4) from 8 patients yielded identical neuron-restricted CNS patterns, most pronounced in hippocampus (stratum lucidum in particular), cerebellum (Purkinje cells and molecular layer), and amygdala. All were neurochondrin-IgG positive. Five were women; median symptom onset age was 43 years (range, 30–69). Of 7 with clinical data, 6 presented with rapidly progressive cerebellar ataxia, brainstem signs, or both; 1 had isolated unexplained psychosis 1 year prior. Five of 6 had cerebellar signs, 4 with additional brainstem symptoms or signs (eye movement abnormalities, 3; dysphagia, 2; nausea and vomiting, 1). One patient with brainstem signs (vocal cord paralysis and VII nerve palsy) had accompanying myelopathy (longitudinally extensive abnormality on MRI; aquaporin-4-IgG and myelin oligodendrocyte glycoprotein-IgG negative). The 7th patient had small fiber neuropathy only. Just 1 of 7 had contemporaneous cancer (uterine). Six patients with ataxia or brainstem signs received immunotherapy, but just 1 remained ambulatory. At last follow-up, 5 had MRI evidence of severe cerebellar atrophy.
Conclusion
In our series, neurochondrin autoimmunity was usually accompanied by a nonparaneoplastic rapidly progressive rhombencephalitis with poor neurologic outcomes. Other phenotypes and occasional paraneoplastic causes may occur.
Autoimmune causes of movement disorders are increasingly recognized, particularly for ataxia. Identification of specific autoantibody biomarkers aids the neurologic diagnosis, expedites the search for occult cancer, and may also indicate most appropriate therapy and predict outcome. For example, Purkinje cell cytoplasmic antibody type 1 (also known as anti-Yo) is generally accompanied by subacute cerebellar ataxia, breast or gynecologic adenocarcinoma, and poor response to immunotherapy.1,2 In contrast, cerebellar ataxia caused by metabotropic glutamate receptor 1 (mGluR1) autoimmunity is occasionally accompanied by lymphoma, and the ataxia may respond robustly to immunotherapy.3
Herein, we describe 8 cases identified with an IgG autoantibody specific for a neuronal cytosolic protein, neurochondrin, in the course of characterizing unclassified neural-specific autoantibodies.4,5 Neurochondrin autoimmunity was previously described in 3 adult patients with cerebellar degeneration and in a child with chorea.6,7 All but 1 of the 7 patients with clinical information available in our report had predominance of rhombencephalitis (cerebellar and brainstem localization). Myelopathy and acute psychosis (accompanying ataxia) and neuropathy (without other features) were each encountered in single patients.
Methods
Standard protocol approvals, registrations, and patient consents
The Mayo Clinic Institutional Review Board approved human specimen acquisition and retrospective review of patients' histories (institutional review board #08–006647).
Study population
Between January 1, 1993 and June 7, 2018, the Mayo Clinic Neuroimmunology Laboratory tested by tissue-based indirect immunofluorescence assay (IFA), 616,025 serum and CSF specimens submitted for service testing for autoimmune encephalitic disorders or a suspected paraneoplastic neurologic disorder. IgG in 89 of those specimens (25 CSF; 64 serum) yielded a synaptic-type hippocampal staining pattern of nonuniform focal intensity. All 89 archived specimens were retested by IFA and classified in detail according to their respective staining patterns. Eight patients with an identical staining pattern are the subject of this report; clinical information was available in 7, 5 by review of Mayo Clinic records and 2 by contact with external physicians.
Laboratory methods
Indirect IFA, dual staining by confocal microscopy, Western blots, immunoprecipitation assay, and sequencing by mass spectrometry and neurochondrin-specific cell-based assays were undertaken, as previously described (appendix e-1, links.lww.com/NXI/A143).8
Results
Neural autoantigen characterization
Immunohistochemical distribution
An identical CNS-restricted pattern of immunoreactivity (synaptic and cytoplasmic regions) was detected in 12 specimens from 8 patients (8 serum; 4 CSF; figure 1, A–F). IgG bound robustly to several structures: hippocampus (particularly the stratum lucidum, continuous from CA2 and CA3 regions to the dentate gyrus granular cell layer), cerebellum (Purkinje neuronal cytoplasm and diffusely in molecular layer), and amygdala. Staining was less intense in the striatum, thalamus, and cerebral cortex. Myenteric nerves had faint staining, and parenchyma of kidney and gut were nonreactive (not shown). Median endpoint dilutions were 1:30,720 in serum (range, 1:3,840–1:61,440) and 1:64 in CSF (range, 1:2–1:256).
Figure 1. The distribution of neurochondrin immunoreactivity in mouse brain tissue revealed by binding of patient IgG.

(A–F) Patient serum IgG yields a synaptic-type immunofluorescence pattern in (A) hippocampus (Hi) CA3 layer and pyramidal cells, (B) cerebellar cortex, molecular layer (ML) more than granular layer (GL), and perikarya of Purkinje neurons (arrow), (C) basal ganglia (BG), (D) thalamus (Th), and (E) amygdalar neurons. (F) IgG purified from patient serum by adsorption to (and elution from) 75 kDa Western blot band recapitulates this staining pattern (hippocampus shown). (G) Coomassie staining of mouse cerebral protein extract immunoprecipitated with serum from patients 1, 4, and 6; purified IgG (from patient 1 and 4) and control serums (N1 and N2). Patient serum, but not healthy control serum, immunoprecipitated a protein of approximately 75 kDa. Scale bar = 100 μm. IgG = immunoglobulin G; kDa = kilodalton.
Autoantigen identification
Application of patient sera, but not healthy control sera, to immunoblots of mouse cerebral lysates, revealed a common immunoreactive band of approximate molecular weight 75 kDa (figure e-1, links.lww.com/NXI/A142). A candidate antigen was identified by protein G magnetic bead capture from mouse cerebral lysate using sera from patients 1, 4, and 6 (figure 1G). Mass spectrometry analysis of the electrophoretically separated proteins predicted the 75 kDa protein to be neurochondrin.
Dual IFA staining on mouse brain sections demonstrated colocalization of patient IgGs with a commercial neurochondrin polyclonal IgG, figure 2, A–D. Sera from patients 1–6, but not 3 healthy control sera, bound to recombinant full-length neurochondrin protein (Abcam [product number ab161548, glutathione S-transferases--tagged at N terminus], figure 2E). All patient specimens and 2 of 196 healthy control sera were positive by neurochondrin-specific cell-based assay; IgGs bound to neurochondrin-expressing cells, but not mock transfected cells (figure 2, F–G). Unlike the patient specimens, none of the 196 healthy control sera produced neural IgG staining by tissue-based IFA.
Figure 2. Confocal microscopy and recombinant protein assays (cell-based and Western blot) confirm the antigen is neurochondrin.
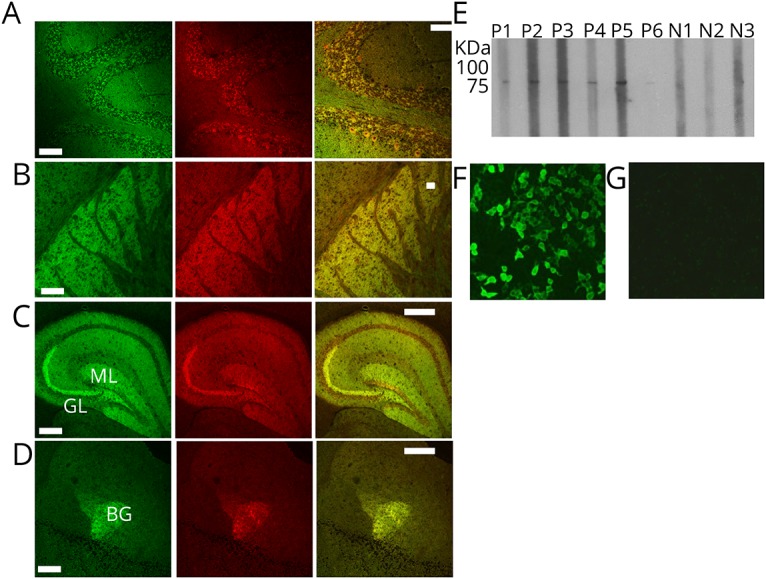
(A–D) Patient IgG colocalizes with neurochondrin immunoreactivity in mouse cerebellum, basal ganglia, hippocampus, and the amygdala. Left column: patient IgG is green. Middle column: rabbit antineurochondrin IgG is red. Right column: merged images. (E) In a Western blot utilizing recombinant neurochondrin full-length protein (with 26 kDa GST-tag at N-terminus; total construct molecular weight, 101 kDa), sera from patients 1–6, but not from 3 healthy control subjects, are reactive. (F–G) Patient IgG binding to HEK-293 cells transfected with cDNA encoding neurochondrin (F) and not to nontransfected control cells (G). Scale bar = 100 μm. HEK = human embryonic kidney; cDNA = complementary DNA; IgG = immunoglobulin G.
Summary of demographic and clinical findings
Six of 8 patients were identified retrospectively from the Mayo Clinic Neuroimmunology Laboratory archives (1997–2018) and 2 patients were identified prospectively after the study was underway. Five were female. The median symptom onset age was 43 years (range, 30–69).
Neurological manifestations
Clinical information was available for 7 of 8 patients (shown in table 1). Five of 7 were evaluated neurologically at the Mayo Clinic. The median follow-up period was 18 months (range, 2–60). Six of 7 patients presented with a rapidly progressive rhombencephalitis (cerebellar/brainstem) over the course of a few weeks. The remaining patient had symptoms of small fiber neuropathy and dysautonomia (pain, paresthesia, and constipation) without CNS findings.
Table 1.
Clinical information available for 7 neurochondrin-IgG positive patientsa


Among the 6 patients with CNS disorders, 5 had cerebellar signs including gait difficulties, dysarthria, and prominent dysmetria. One of those 5 patients had an isolated episode of unexplained psychosis 1 year prior to the onset of ataxia. She developed an unusual self-resolving paranoid state without a prior history of drug abuse or psychiatric illness. Brainstem symptoms or signs, present in 4 of those 5 patients, included eye movement abnormalities in all, dysphagia in 2, and nausea and vomiting in 1. The 6th patient, without cerebellar signs, had brainstem signs, vocal cord paralysis, and unilateral VIIth cranial nerve palsy accompanying myelopathy (longitudinally extensive T2 signal abnormality on MRI, though aquaporin-4-IgG and myelin oligodendrocyte glycoprotein-IgG serum tests were negative). The myelopathy was preceded by diffuse migratory itch 2 weeks prior to the onset of weakness.
Oncologic and autoimmune accompaniments
Of the 7 patients with records available, 1 had poorly differentiated uterine carcinoma contemporaneous with neurologic symptom onset. Five others had cancer-negative whole-body imaging (PET-CT in 4 and CT alone in 1). Two patients had coexisting autoimmune disease (Sjögren syndrome, 1; thyroiditis, 1).
CSF studies
CSF findings were abnormal in all 5 patients for whom data were available. All had signs of inflammation (increases in protein [all; median value, 80.5 mg/dL; range, 49–172], leukocyte count [3; all lymphocyte predominant, median value, 18/μL; range, 14–185]; IgG index and synthesis rate [3], or CSF-exclusive oligoclonal bands [3], table 1).
Imaging
Brain MRI images from early in the disease course were available for review in all patients with cerebellar or brainstem symptoms and signs, with the exception of patient 8 (figure 3). These early images were normal in 2 patients, demonstrated T2 abnormalities in 3 (cerebellum [2], brainstem [2], and frontal lobe [1]), and rhombencephalon-restricted postgadolinium abnormalities in 1, figure 3, A-E, and H. The patient with rhombencephalomyelitis, and cerebellar leptomeningeal enhancement, also had nonenhancing T2 abnormalities of the central thoracic cord, figure 3, F and G. Brain MRI acquired late in the disease course in 5 patients revealed severe cerebellar atrophy in all, figure 3, I–M. Patients 6 and 8, with ataxic neurologic phenotypes only, had bilateral hippocampal T2 signal abnormalities (image available for patient 6, figure 3, N). Thermoregulatory sweat test in the patient with small fiber neuropathy demonstrated hypohidrosis of the distal lower extremities.
Figure 3. MRI images of patients seropositive for neurochondrin-IgG acquired early (A–H) and late (I–M) in the disease course.

(A) Patient 6. T2 FLAIR axial of brain demonstrates T2 signal abnormality in gray-white matter junction of the left frontal lobe (arrow). (B) Patient 6. T2 FLAIR axial of brain demonstrates left cerebellar T2 hyperintensity (arrow). (C–D) Patient 3. FLAIR axial of brain demonstrates T2 hyperintensities, pancerebellar, pons, and in middle cerebellar peduncles. (E) Patient 1. T1 postgadolinium, sagittal of brain demonstrates cerebellar leptomeningeal enhancement. (F–G) Patient 1. T2 axial and sagittal thoracic of spine demonstrate intramedullary (F) and longitudinal hyperintensities (G). (H) Patient 1. T2 FLAIR axial of brain demonstrates dorsal pons hyperintensity (arrow). (I–M) Patients 1, 2, 3, 5, and 6. T1 sagittal of brain demonstrates cerebellar atrophy. (N) Patient 6. T2 FLAIR coronal of brain also had bilateral hippocampal T2 abnormalities at follow-up. FLAIR = fluid attenuated inversion recovery.
Treatment and outcomes
Five of 6 patients with an ataxic or brainstem syndrome were treated with immunotherapy within 1 month of symptom onset (corticosteroids, 5; IV immune globulin [IVIg], 3; plasma exchange, 1). Despite early intervention, only 1 patient (patient 6) remained ambulatory; she had received corticosteroids. Three patients became wheelchair-bound, and 1 died from complications of neurologic progression. One patient who commenced treatment 1 year after symptom onset remained wheelchair-bound. It was the opinion of the prescribing physicians that some immune therapies (rituximab, azathioprine, IVIg) slowed the rate of disease progression (3) or mildly improved symptoms (patient 6 who received both IV and oral corticosteroids, and azathioprine, table 1).
Discussion
We report 8 patients in whom we identified neurochondrin IgG incidentally in the course of characterizing an unclassified neural-specific IgG autoantibody. Specificity was confirmed by affinity purification of the antigen, mass spectrometry, confocal microscopy and protein-specific assays (Western blot and transfected cell-based).
Neurochondrin is a leucine-rich protein expressed in brain, bone (osteoblasts and osteoclasts), and cartilage.5,9 It enhances cell-surface localization of certain membrane-bound proteins, such as metabotropic glutamate receptors, but is itself a cytoplasmic antigen.5 While reliable as a biomarker of neurologic autoimmunity, neurochondrin-IgG is not pathogenic, but likely a proxy for a cytotoxic T cell-mediated pathogenesis. Consistent with our clinical findings, neurochondrin is highly expressed in cerebellar Purkinje cells, the brainstem, the lateral parts of the central amygdalar nuclei, the hippocampal pyramidal cells, and also the autonomic and peripheral nervous systems.4 Relevant to the psychosis, myelopathy, and neuropathy phenotypes, respectively, neurochondrin knockout mice have a schizophrenia-like behavioral phenotype, neurochondrin is required for the correct localization of a survival motor neuron protein, and is expressed in rat peripheral nerve.5,10,11 In bone, neurochondrin inhibits osteoclast- and macrophage-induced hydroxyapatite bone resorption.12 None of our patients was known to have had metabolic bone disease.
We confirm the principal clinical accompaniments originally described, namely, severe cerebellar ataxia and other features of rhombencephalitis, and usually without evidence of systemic cancer. Other phenotypes include chorea7 and small fiber neuropathy (reported herein). Two of our patients with rhombencephalitis had additional neuropsychiatric findings (psychosis or myelopathy). Neuroradiological findings were diverse, though were cerebellum and brainstem predominant. The diversity of cerebellar and extracerebellar T2 abnormalities, not typical for other autoimmune ataxias, could serve as a clue to neurochondrin CNS autoimmunity.
Neurochondrin IgG is one of several reported biomarkers of autoimmune ataxias with (usually) nonparaneoplastic, idiopathic autoimmune cause.13–15 These are listed in order of reported commonality in table 2.
Table 2.
Autoimmune (usually nonparaneoplastic) cerebellar/brainstem ataxias

The almost universal lack of neurologic improvements from immunotherapies in neurochondrin autoimmunity is consistent with other nonantibody-mediated, intracellular antigen-defined autoimmune ataxias, for some of which CD8+ cytotoxic T-cell infiltrative immunopathology has been demonstrated.6,16,17 We identified 1 patient who remained ambulatory after corticosteroid therapy and did not progress after treatment with azathioprine. It is possible that future patients treated early with high-dose steroids and T cell-directed therapy (cyclophosphamide) might have better outcomes. The most robust responses to immune therapies have been reported among autoimmune ataxia patients harboring antibodies reactive with cell surface antigens (table 2).16
For patients presenting with unexplained ataxia or brainstem symptoms and signs of subacute onset, comprehensive neural IgG screening with tissue-based immunohistochemistry permits simultaneous evaluation for a growing list of neural-specific IgGs, including neurochondrin-IgG.
Glossary
- IFA
immunofluorescence assay
- IVIg
IV immune globulin
Appendix. Authors
Study funding
This study was funded by the Mayo Clinic Center for Individualized Medicine and Euroimmun.
Disclosure
S. Shelly reports no disclosures. T.J. Kryzer has a financial interest in the following intellectual property: “Marker for Neuromyelitis Optica.” A patent has been issued for this technology, and it has been licensed to commercial entities. They have received cumulative royalties of greater than the federal threshold for significant financial interest from the licensing of these technologies, but receive no royalties from the sale of these tests by Mayo Medical Laboratories. L. Komorowski is employed by Euroimmun AG. R. Miske is employed by Euroimmun AG. M.D. Anderson reports no disclosures. E. Flanagan is a site principal investigator in a randomized placebo-controlled clinical trial of Inebilizumab (A CD19 inhibitor) in neuromyelitis optica spectrum disorders funded by MedImmune/Viela Bio and has served on its advisory board. S.R. Hinson reports no disclosures; V.A. Lennon has a patent application pending for septin-5 IgG as a biomarker of autoimmune neurologic disease. She has a financial interest in the following intellectual property: “Marker for Neuromyelitis Optica.” A patent has been issued for this technology, and it has been licensed to commercial entities. They have received cumulative royalties of greater than the federal threshold for significant financial interest from the licensing of these technologies, but receive no royalties from the sale of these tests by Mayo Medical Laboratories. S.J. Pittock has a patent application pending for septin-5 IgG as a biomarker of autoimmune neurologic disease, and holds patents that relate to functional AQP4/NMO-IgG assays and NMO-IgG as a cancer marker; consulted for Alexion and Medimmune; and received research support from Grifols, Medimmune, and Alexion. All compensation for consulting activities is paid directly to Mayo Clinic. A. McKeon has received research funding from Alexion, Grifols, and Medimmune, and has patents pending for the following IgGs as biomarkers of autoimmune neurologic disorders (septin-5, Kelch-like protein 11, GFAP, PDE10A, and MAP1B). Go to Neurology.org/NN for full disclosures.
References
- 1.Peterson K, Rosenblum MK, Kotanides H, Posner JB. Paraneoplastic cerebellar degeneration. I. A clinical analysis of 55 anti-Yo antibody-positive patients. Neurology 1992;42:1931–1937. [DOI] [PubMed] [Google Scholar]
- 2.McKeon A, Tracy JA, Pittock SJ, Parisi JE, Klein CJ, Lennon VA. Purkinje cell cytoplasmic autoantibody type 1 accompaniments: the cerebellum and beyond. Arch Neurol 2011;68:1282–1289. [DOI] [PubMed] [Google Scholar]
- 3.Lopez-Chiriboga AS, Komorowski L, Kumpfel T, et al. Metabotropic glutamate receptor type 1 autoimmunity: clinical features and treatment outcomes. Neurology 2016;86:1009–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Istvanffy R, Vogt Weisenhorn DM, Floss T, Wurst W. Expression of neurochondrin in the developing and adult mouse brain. Dev Genes Evol 2004;214:206–209. [DOI] [PubMed] [Google Scholar]
- 5.Wang H, Westin L, Nong Y, et al. Norbin is an endogenous regulator of metabotropic glutamate receptor 5 signaling. Science 2009;326:1554–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miske R, Gross CC, Scharf M, et al. Neurochondrin is a neuronal target antigen in autoimmune cerebellar degeneration. Neurol Neuroimmunol Neuroinflamm 2017;4:e307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rommel FR, Miske R, Stocker W, Arneth B, Neubauer BA, Hahn A. Chorea minor associated with anti-neurochondrin autoantibodies. Neuropediatrics 2017;48:482–483. [DOI] [PubMed] [Google Scholar]
- 8.Honorat JA, Lopez-Chiriboga AS, Kryzer TJ, et al. Autoimmune septin-5 cerebellar ataxia. Neurol Neuroimmunol Neuroinflamm 2018;5:e474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dateki M, Horii T, Kasuya Y, et al. Neurochondrin negatively regulates CaMKII phosphorylation, and nervous system-specific gene disruption results in epileptic seizure. J Biol Chem 2005;280:20503–20508. [DOI] [PubMed] [Google Scholar]
- 10.Thompson LW, Morrison KD, Shirran SL, et al. Neurochondrin interacts with the SMN protein suggesting a novel mechanism for spinal muscular atrophy pathology. J Cell Sci 2018;131:jcs211482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shinozaki K, Kume H, Kuzume H, Obata K, Maruyama K. Norbin, a neurite-outgrowth-related protein, is a cytosolic protein localized in the somatodendritic region of neurons and distributed prominently in dendritic outgrowth in Purkinje cells. Brain Res Mol Brain Res 1999;71:364–368. [DOI] [PubMed] [Google Scholar]
- 12.Ishiduka Y, Mochizuki R, Yanai K, et al. Induction of hydroxyapatite resorptive activity in bone marrow cell populations resistant to bafilomycin A1 by a factor with restricted expression to bone and brain, neurochondrin. Biochim Biophys Acta 1999;1450:92–98. [DOI] [PubMed] [Google Scholar]
- 13.Jarius S, Wildemann B. “Medusa head ataxia”: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 3: anti-Yo/CDR2, anti-Nb/AP3B2, PCA-2, anti-Tr/DNER, other antibodies, diagnostic pitfalls, summary and outlook. J Neuroinflamm 2015;12:168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarius S, Wildemann B. “Medusa head ataxia”: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 2: anti-PKC-gamma, anti-GluR-delta2, anti-Ca/ARHGAP26 and anti-VGCC. J Neuroinflamm 2015;12:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jarius S, Wildemann B. “Medusa-head ataxia”: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 1: anti-mGluR1, anti-Homer-3, anti-Sj/ITPR1 and anti-CARP VIII. J Neuroinflamm 2015;12:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones AL, Flanagan EP, Pittock SJ, et al. Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults. JAMA Neurol 2015;72:1304–1312. [DOI] [PubMed] [Google Scholar]
- 17.Albert ML, Darnell JC, Bender A, Francisco LM, Bhardwaj N, Darnell RB. Tumor-specific killer cells in paraneoplastic cerebellar degeneration. Nat Med 1998;4:1321–1324. [DOI] [PubMed] [Google Scholar]
- 18.Saiz A, Arpa J, Sagasta A, et al. Autoantibodies to glutamic acid decarboxylase in three patients with cerebellar ataxia, late-onset insulin-dependent diabetes mellitus, and polyendocrine autoimmunity. Neurology 1997;49:1026–1030. [DOI] [PubMed] [Google Scholar]
- 19.Pittock SJ, Yoshikawa H, Ahlskog JE, et al. Glutamic acid decarboxylase autoimmunity with brainstem, extrapyramidal, and spinal cord dysfunction. Mayo Clin Proc 2006;81:1207–1214. [DOI] [PubMed] [Google Scholar]
- 20.Sillevis Smitt P, Kinoshita A, De Leeuw B, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 2000;342:21–27. [DOI] [PubMed] [Google Scholar]
- 21.Boronat A, Gelfand JM, Gresa-Arribas N, et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol 2013;73:120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tobin WO, Lennon VA, Komorowski L, et al. DPPX potassium channel antibody: frequency, clinical accompaniments, and outcomes in 20 patients. Neurology 2014;83:1797–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sugiyama N, Hamano S, Mochizuki M, Tanaka M, Takahashi Y. A case of chronic cerebellitis with anti-glutamate receptor delta 2 antibody [in Japanese]. No To Hattatsu 2004;36:60–63. [PubMed] [Google Scholar]
- 24.Berridge G, Menassa DA, Moloney T, et al. Glutamate receptor delta2 serum antibodies in pediatric opsoclonus myoclonus ataxia syndrome. Neurology 2018;91:e714–e723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darnell RB, Furneaux HM, Posner JB. Antiserum from a patient with cerebellar degeneration identifies a novel protein in Purkinje cells, cortical neurons, and neuroectodermal tumors. J Neurosci 1991;11:1224–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honorat JA, Lopez-Chiriboga, Kryzer TJ, et al. Autoimmune gait disturbance accompanying adaptor protein-3B2-IgG. Neurology 2019;93:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jarius S, Scharf M, Begemann N, et al. Antibodies to the inositol 1,4,5-trisphosphate receptor type 1 (ITPR1) in cerebellar ataxia. J Neuroinflamm 2014;11:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alfugham N, Gadoth A, Lennon VA, et al. ITPR1 autoimmunity: frequency, neurologic phenotype, and cancer association. Neurol Neuroimmunol Neuroinflamm 2018;5:e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jarius S, Wandinger KP, Horn S, Heuer H, Wildemann B. A new Purkinje cell antibody (anti-Ca) associated with subacute cerebellar ataxia: immunological characterization. J Neuroinflamm 2010;7:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabater L, Gaig C, Gelpi E, et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol 2014;13:575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaig C, Graus F, Compta Y, et al. Clinical manifestations of the anti-IgLON5 disease. Neurology 2017;88:1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zuliani L, Sabater L, Saiz A, Baiges JJ, Giometto B, Graus F. Homer 3 autoimmunity in subacute idiopathic cerebellar ataxia. Neurology 2007;68:239–240. [DOI] [PubMed] [Google Scholar]