

Abstract
Objective
To assess the association of the degree of severity of motor impairment to that of cognitive impairment in a large cohort of patients with amyotrophic lateral sclerosis (ALS).
Methods
This is a population-based cross-sectional study on patients with ALS incident in Piemonte, Italy, between 2007 and 2015. Cognitive status was classified according to the revised ALS–FTD Consensus Criteria. The King system and the Milano Torino Staging system (MiToS) were used for defining the severity of motor impairment.
Results
Of the 797 patients included in the study, 163 (20.5%) had ALS–frontotemporal dementia (FTD), 38 (4.8%) cognitive and behavioral impairment (ALScbi), 132 (16.6%) cognitive impairment (ALSci), 63 (7.9%) behavioral impairment (ALSbi), 16 (2.0%) nonexecutive impairment, and 385 (48.2%) were cognitively normal. According to King staging, the frequency of cases with ALS-FTD progressively increased from 16.5% in stage 1–44.4% in stage 4; conversely, the frequency of ALSci, ALSbi, and ALScbi increased from King stage 1 to King stage 3 and decreased thereafter. A similar pattern was observed with the MiToS staging. ALS-FTD was more frequent in patients with bulbar involvement at time of cognitive testing. Patients with C9ORF72 expansion (n = 61) showed more severe cognitive impairment with increasing King and MiToS stages.
Conclusion
Our findings suggest that ALS motor and cognitive components may worsen in parallel, and that cognitive impairment becomes more pronounced when bulbar function is involved. Our data support the hypothesis that ALS pathology disseminates in a regional ordered sequence, through a cortico-efferent spreading model.
Amyotrophic lateral sclerosis (ALS) can no longer be considered a disease limited to the motor system but rather a multisystem disorder that involves cognitive domains in at least half of the cases.1 Longitudinal studies point to a relative stability of cognitive function in patients who are not impaired at first examination, and a progression of the impairment in patients already compromised,2–7 although such studies are complicated by the progressive loss of speech and motor function in the hands that hinders the accuracy of neuropsychological testing and causes a high patient attrition rate, further complicating our ability to evaluate the pattern of cognitive impairment in patients with ALS over time.2,3 A recent study performed on a cross-sectional clinical-based cohort of patients evaluated using the Edinburgh Cognitive and Behavioural ALS Screen (ECAS) has shown that cognitive and behavioural deficits are more frequent and severe with advanced disease.8
The aim of our study was to assess the relationship between patients' cognitive impairment, classified according to the recently published revised ALS-FTD Consensus Criteria,9 and patients' motor impairment, classified according to the King ALS staging system and Milano Torino Staging system (MiToS).10,11 To do this, we used a population-based series of patients with ALS, representing one of the largest and most complete cohorts evaluated to date.
Methods
Patients
The study population consisted of patients identified through the Piemonte and Valle d’Aosta Register for ALS (PARALS) incident in the 2007–2015 period. The PARALS is a prospective epidemiologic register established in 1995, whose characteristics have been published previously.12 Patients with history of neurologic disorders affecting cognition (major stroke, severe head injuries, mental retardation), alcohol and drug dependence, severe mental illness, and use of high-dose psychoactive medications were tested but their data were excluded from the analysis. Incident patients who were not native Italian speakers were assessed using an unstructured interview and therefore were excluded from the analysis. Patients whose cognitive testing was performed >12 months after diagnosis were excluded.
Neuropsychological evaluation
Patients with ALS underwent a battery of neuropsychological tests encompassing executive function, memory, visuospatial function, social cognition, and language, selected according to the Diagnostic Criteria for the Behavioural Variant of Frontotemporal Dementia13 and the ALS-FTD Consensus Criteria.9 All patients underwent the following neuropsychological batteries: Mini-Mental State Examination, Wisconsin Card Sorting Test, Trail-Making Test A and B, Digit Span Forward and Backward, Letter and Category fluency test, Boston Naming Test, Rey Auditory Verbal Learning Test, Babcock Story Recall Test, Rey-Osterrieth Complex Figure Test, Raven's Coloured Progressive Matrices, and Frontal Assessment Battery. The ECAS was added to the battery in 2014 when it became available. Raw data of each neuropsychological test were adjusted for age and years of education according to the Italian normative.
Neurobehavioral dysfunction was determined both by direct observation by the neuropsychologist and by patient history,13 and with the Frontal Systems Behavior Scale using the family form evaluated by a close relative/caregiver (scores: normal ≤59, borderline 60–64, pathologic ≥65).14 If a participant had scores reflecting an abnormality of frontal systems in both the premorbid and the postillness evaluations, pathology was considered only if there was an increase of ≥10 points in the T score between the 2 forms.15 Anxiety and depression were assessed using the Hospital Anxiety and Depression Scale (HADS); the item “I feel slowed down” was discussed with the patients in order to have them not refer to physical disability.16
The battery was administered following the same sequence in order to avoid the possible differential interference of the answers of one test over the others. The administration of the battery required about 1.5 hours,17 and was performed in the morning. If the participant felt too tired, a further session was scheduled to complete the battery within 2 weeks of the first session. Oxygen saturation at the time of the neuropsychological testing was measured with a pulse oximeter, and none of the patients displayed evidence of hypoxemia (oxygen saturation <92 mm Hg) based on this evaluation.
Cognitive categorization
Patients' cognitive status was classified according to the revised ALS-FTD Consensus Criteria.9 According to these criteria, patients may be classified into 4 main categories: (1) patients with ALS with a frontotemporal dementia (FTD) syndrome (ALS-FTD); (2) patients with ALS with behavioral impairment (ALSbi); (3) patients with ALS with cognitive impairment (ALSci); and (4) patients with ALS with combined cognitive and behavioral impairment (ALScbi), which includes patients who fulfill criteria for both ALSci and ALSbi. In addition, we designated patients with isolated nonexecutive impairment in the domains of memory and visuospatial function as ALSnex.9,15,18 Patients who did not fit into these categories were classified as cognitively normal (ALS-CN).
All patients were classified in a blind fashion by 2 experts in ALS neuropsychology. When there was disagreement, the case was discussed until a final diagnosis was agreed. The concordance rate was over 90% for all diagnoses.17
ALS staging
King staging is based on the spreading of motor symptoms in 3 different body regions (bulbar, upper limbs, and lower limbs), and on the use of noninvasive ventilation (NIV) and enteral nutrition. The 5 stages of the King staging system are as follows: 1: 1 region involved; 2: 2 regions involved; 3: 3 regions involved; 4A: patient needs gastrostomy; 4B: patient needs NIV.10 The stage can be derived from direct observation of the patients and also from the Amyotrophic Lateral Sclerosis Functional Rating Scale– Revised (ALSFRS-R) scale.19
In contrast to the region-based King system, the MiToS is aimed at determining the main milestones of patients' disability, based on the loss of 4 principal functions (communication, swallowing, ambulation, and breathing).11 Each of these represents a domain of the staging. The score, which ranges from 0 to 4, is given by the sum of the number of lost domains, with 0 representing no domain lost, and 4 the loss of all 4 domains. MiToS can be directly calculated from the ALSFRS-R scale.
ALSFRS-R decline
Disease severity was assessed with ALSFRS-R decline, calculated as the mean monthly number of points lost from onset to time of neuropsychological assessment:
 |
Genetics
Genetic assessment was performed in 749 cases (94.3%). All the coding exons and 50 bp of the flanking intron-exon boundaries of SOD1, of exon 6 of TARDBP, and of exons 14 and 15 of FUS were PCR amplified, sequenced using the Big-Dye Terminator v3.1 sequencing kit (Applied Biosystems Inc., Foster City, CA), and run on an ABIPrism 3130 genetic analyzer. These exons were selected as the vast majority of known pathogenic variants are known to lie within these mutational hotspots. A repeat-primed PCR assay was used to screen for the presence of the GGGGCC hexanucleotide expansion in the first intron of C9ORF72.20
Statistical methods
Comparisons between means were made with Student t test or analysis of variance. Comparisons between categorical variables were made with χ2 test. A p level <0.05 was considered significant. Statistical analyses were carried out using the SPSS 25.0 statistical package (SPSS, Chicago, IL).
Standard protocol approvals, registrations, and patient consents
The study was approved by the Ethical Committees of the 2 ALS centers involved in the study. All patients provided written informed consent before enrollment. The databases were anonymized according to the Italian law for the protection of privacy.
Data availability
Data will be available upon request by interested researchers.
Results
Out of the 1,311 patients with ALS incident in Piemonte and Valle d’Aosta in the 2007–2015 period, 797 (60.8%) have been included in the study. A flow chart of the sequence of participant selection is reported in figure 1. Nonincluded patients were older and more clinically impaired than those who underwent the examination (table 1). The mean number of education years of the enrolled patients is similar to that of the age- and sex-matched Piemonte and Valle d’Aosta population at the 2011 census (8.6 [SD 4.0] years vs 8.9 [SD 4.1] years) (http://www.sistemapiemonte.it/cms/pa/demografia‐e‐statistica). The median time from diagnosis to neuropsychological testing was 51 days (interquartile range 22–131).
Figure 1. Flowchart reporting enrollment.
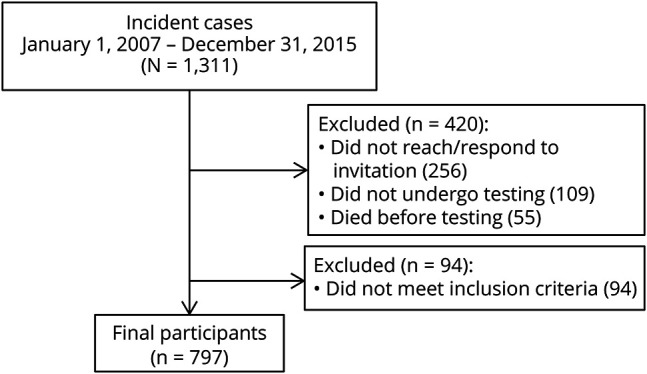
Table 1.
Demographic and clinical characteristics of included and nonincluded patients

Of the 797 patients included in the analysis, 163 (20.5%) were diagnosed with ALS-FTD, 38 (4.8%) ALScbi, 132 (16.6%) ALSci, 63 (7.9%) ALSbi, and 385 (48.2%) were cognitively normal (ALS-CN). Isolated nonexecutive impairment (ALSnex) was detected in 16 cases (2%), while in 6 patients, nonexecutive impairment was associated with executive impairment (ALSci).
The demographic and clinical characteristics of patients at the various stages according to the King and the MiToS classification systems are reported in tables 2 and 3. Mean age at onset and at time of neuropsychological testing was higher and mean number of years of education was lower in patients with more advanced disease. Interestingly, the mean time of testing since disease onset was similar in all stages. Moreover, frequency of bulbar symptoms at time of testing was higher as patients' stage reflected more advanced disease. Of the 797 patients, at the time of cognitive testing, 11 (1.4%) had severe depression (HADS score 15–21), 57 (7.2%) moderate depression (HADS score 11–14), 108 (13.6%) mild depression (HADS score 8–10), and 619 (77.7%) no depression. There was no correlation between the level of depression and Strong classification.
Table 2.
Demographic and clinical variables according to King stage

Table 3.
Demographic and clinical variables according to Milano Torino Staging system (MiToS) stage

Cognitive classification and King stages
According to King staging, the frequency of cases with ALS-FTD was progressively higher going from stage 1 (16.5% of cases) to stage 4 (44.4% of cases). The frequency of ALSci, ALSbi, and ALScbi increased from King stage 1 to King stage 3 and was lower in stage 4 (figure 2A). Conversely, the frequencies of ALSnex did not modify across the stages (King stage 1, 1.7%; King stage 2, 3.2%; King stage 3, 0.7%; King stage 4, 2.8%).
Figure 2. Cognitive classification and King stage or Milano Torino Staging system (MiToS) stage.
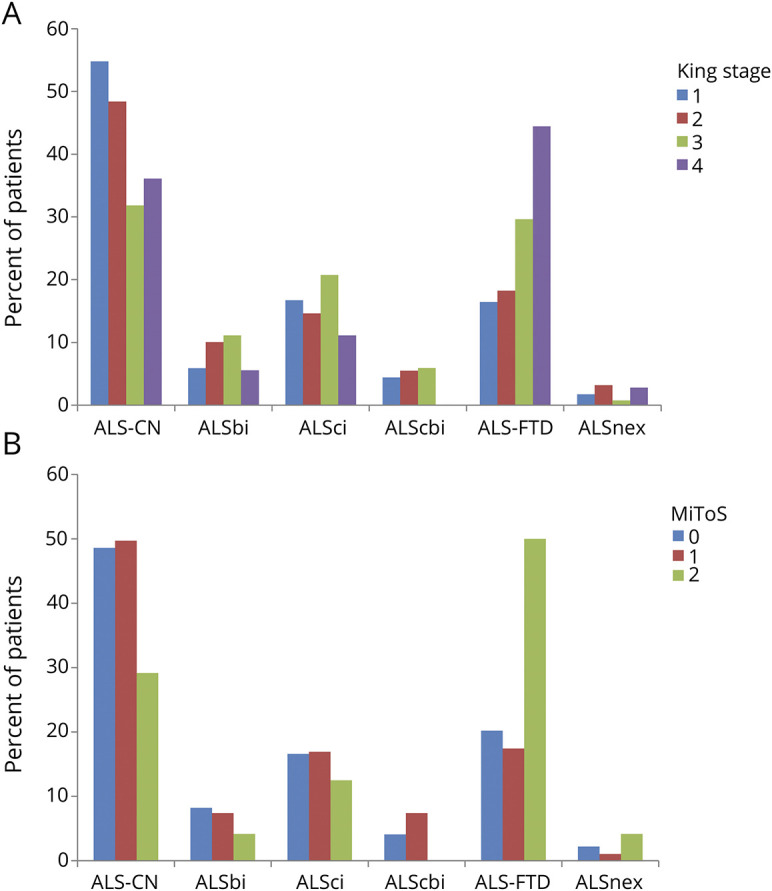
(A) Cognitive classification and King stage. Number of patients: King stage 1, 407; King stage 2, 219; King stage 3, 135; King stage 4, 36. (B) Cognitive classification and MiToS stage. Number of patients: MiToS stage 1, 584; MiToS stage 2, 189; MiToS stage 3, 24. ALS-CN = cognitively normal with amyotrophic lateral sclerosis; ALS-FTD = amyotrophic lateral sclerosis with a frontotemporal dementia syndrome; ALSbi = amyotrophic lateral sclerosis with behavioral impairment; ALScbi = amyotrophic lateral sclerosis with combined cognitive and behavioral impairment; ALSci = amyotrophic lateral sclerosis with cognitive impairment; ALSnex = amyotrophic lateral sclerosis with isolated nonexecutive impairment.
Cognitive classification and MiToS stages
A similar pattern was observed when the MiToS staging classification was applied. The frequency of ALS-FTD was progressively higher going from stage 0 (20.2% of cases) to stage 2 (50.0% of cases). The frequency of intermediate cognitive impairment (ALScbi, ALSci, and ALSbi) increased from stage 0 to stage 1 and decreased thereafter (figure 2B). No patients were tested in MiToS stages 3 and 4. The frequencies of ALSnex did not modify across the stages (MiToS stage 0, 2.2%; MiToS stage 1, 1.1%; MiToS stage 2, 4.2%).
Overall, cognitive impairment was more frequent in patients with more advanced disease based on both the King and MiToS staging systems: over 60% of patients manifested mild or severe cognitive impairment in more severe stages (King stage 4, 63.9%; MiToS stage 2, 70.8%), compared to 45.2% (King state 1) and 51.4% (MiToS stage 0) in the early stages.
Cognitive impairment and bulbar involvement
ALS-FTD was more frequent in patients with bulbar onset (figure 3, A–D) and in those with bulbar involvement at time of cognitive testing (figure 4, A–D). This pattern was observed with both classification systems, accounting for 70% of patients both in King stage 4 and in MiToS stage 2. The corresponding percentage of ALS-CN patients decreased from about 40% in King stages 1 and 2 and MiToS stage 0 to approximately 20% in King stage 4 and MiToS stage 2. This difference was even more marked in patients with ALS who did not show bulbar symptoms at time of cognitive evaluation: in fact, about 70% of this type of patient had preserved cognition at all King and MiToS stages.
Figure 3. Cognitive classification and amyotrophic lateral sclerosis (ALS) staging according to the site of onset.

(A) King staging, bulbar onset. (B) King staging, spinal onset. (C) Milano Torino Staging system (MiToS) staging, bulbar onset. (D) MiToS staging, spinal onset. ALS-CN = cognitively normal with amyotrophic lateral sclerosis; ALS-FTD = amyotrophic lateral sclerosis with a frontotemporal dementia syndrome; ALSbi = amyotrophic lateral sclerosis with behavioral impairment; ALScbi = amyotrophic lateral sclerosis with combined cognitive and behavioral impairment; ALSci = amyotrophic lateral sclerosis with cognitive impairment; ALSnex = amyotrophic lateral sclerosis with isolated nonexecutive impairment.
Figure 4. Cognitive classification and amyotrophic lateral sclerosis (ALS) staging according to the presence of bulbar symptoms at time of cognitive testing.

(A) King staging, bulbar sign present. (B) King staging, no bulbar signs. There are no patients in King stage 3 in the group of participants without bulbar signs at time of cognitive testing, in accordance with the method of classification. (C) Milano Torino Staging system (MiToS) staging, bulbar sign present. (B) MiToS staging, no bulbar signs. ALS-CN = cognitively normal with amyotrophic lateral sclerosis; ALS-FTD = amyotrophic lateral sclerosis with a frontotemporal dementia syndrome; ALSbi = amyotrophic lateral sclerosis with behavioral impairment; ALScbi = amyotrophic lateral sclerosis with combined cognitive and behavioral impairment; ALSci = amyotrophic lateral sclerosis with cognitive impairment; ALSnex = amyotrophic lateral sclerosis with isolated nonexecutive impairment.
Cognitive impairment, sex, and stage
Women were more likely to be cognitively impaired than men at each stage of the King and MiToS classification system (data not shown). However, this difference disappeared when patients were subdivided according to their site of onset, indicating that the observed differences in cognitive status across sexes were mostly due to the higher frequency of bulbar onset among women. In our cohort, 43.6% of women and 25.1% of men presented with bulbar-onset disease, and 60.8% of women and 43.6% of men had bulbar signs at time of cognitive evaluation.
C9ORF72 expansion and stage
Genetic testing has been performed in 751 cases (94.2% of study population). Of these, 61 (8.1%) carried a pathologic C9ORF72 expansion. These patients showed more severe cognitive impairment with increasing King and MiToS stages (data not shown). The number of patients with other mutations (SOD1, 15; TARDBP, 13; FUS, 3) was too low to allow for meaningful analyses.
Discussion
In our large population-based study, we found that cognitive impairment was more frequent among patients in the advanced stages of ALS based on 2 classification systems compared to earlier stages of the disease. Indeed, nearly two-thirds of cases had some evidence of cognitive impairment in more advanced stages. In contrast to previous publications,2–7 our data point to a correlation between the severity of motor impairment and the severity of cognitive deficits, and suggest that cognitive function may not remain stable in patients with ALS. Importantly, our data indicate that the presence of bulbar signs is strongly associated with more severe cognitive impairment at all stages of disease. Furthermore, cognitive impairment was more severe in patients at worse stage of their disease both in patients with and without the C9ORF72 expansion (known to be associated with FTD).
Our study adds novel findings to 2 previous cross-sectional studies assessing the correlation between ALS stages and cognition. A small clinical-based cohort study, which excluded patients with FTD and classified patients according to the 2009 ALS-FTD Consensus Criteria,21 reported that patients' cognition was more impaired with worsening of King stage.22 A larger multicenter cross-sectional study, which compared the domains of ECAS and King staging but did not classify patients according to the revised ALS-FTD Consensus Criteria, found that ALS-specific cognitive deficits and behavioral impairment were more frequent in advanced disease stages.8
Previously published longitudinal studies on ALS cognition purported that patients with normal cognition at first examination remained stable over time.2–7 However, these studies suffered severe attrition rates over the course of their follow-up (only 50% of patients were retested at 6 months and fewer than 25% at 1 year). Cognitive impairment is known to be associated with a more rapid decline of motor function in ALS.2,14,18,23 These studies failed to detect progression in cognitive impairment due to the selective loss to follow-up among the very patients most likely to manifest cognitive decline. Our population-based study allowed for a more accurate assessment of the relationship of these important clinical measures, but its cross-sectional design does not allow us to clarify this important issue.
The mean delay between symptoms onset and the diagnostic interview was similar in patients at all King and MiToS stages, suggesting that the cognitive categorization was related to the rate of motor decline. From a pathologic perspective, our findings logically support the notion that patients manifesting more advanced stages of disease at the time of diagnosis have experienced a faster spread of lesions involving both the motor cortex and the prefrontal and temporal cognitive cortices compared to patients at lower stages.24–26 Moreover, our data suggest that the rate of lesions spreading is different within subgroups of patients, but that, within each subgroup, this spread rate is the same within motor and cognitive cortices.
In general, the emergence of bulbar symptoms in patients with ALS is associated with a more severe impairment of cognition.8,15,27,28 We postulate that this association is related to connections between the prefrontal cognitive cortex and the cortical areas controlling facial and speech muscles, which could favor the dissemination of TDP-43 lesions from motor to cognitive area, or vice versa.24,25,29 Supporting this hypothesis is the observation emerging from follow-up studies that patients with FTD who subsequently develop ALS typically present with bulbar weaknesss.30,31
The ALS research community makes a distinction between the milder forms of cognitive impairment (ALSbi, ALSci, and ALScbi) and florid FTD dementia, maintaining that they represent separate processes. Supporting this notion is that milder forms of cognitive impairment are rarely seen among patients with FTD. Our data do not support this arbitrary distinction and instead strongly point to the milder forms being closely related to FTD. Notably, the occurrence of intermediate cognitive impairment was less frequent among our patients in more advanced stages of disease, while florid FTD was more frequent. The presence of motor impairment brings patients with ALS to the attention of neurologists, who then have the opportunity to diagnose milder forms of cognitive impairment among this patient population. In contrast, these more subtle forms of cognitive impairment are frequently overlooked in patients with FTD as their symptoms are not typically severe enough to bring them to medical attention. Our observations are consistent with the notion that intermediate forms of cognitive impairment should be considered the equivalent of mild cognitive impairment in Alzheimer disease.32
The demographics of patients in each King and MiToS stage are significantly different in a predictable template. First, mean age at onset was significantly higher from less severe to more severe King and MiToS stages. This is expected since more severe stages include patients with more rapidly declining motor function, who are likely to be older. Likewise, the increased frequency of cognitive impairment observed among patients in King stage 4 and MiToS stage 2 parallels the findings of epidemiologic studies, which have shown that FTD incidence increases with age.33,34 Second, the mean number of years of education significantly decreases with the increase in motor and cognitive severity. A lower level of education compared to cognitively normal controls has been reported in patients with Alzheimer disease35 and in patients with FTD.36 Education is thought to create a reserve capacity that allows some people to better endure brain damage, an effect that could also apply to the cognitive performance of ALS.
As expected in our series, the presence of a C9ORF72 expansion was related to more severe cognitive impairment at each stage of both staging systems.37 The cognitive picture of C9ORF72 patients is also characterized by the lower frequency of ALScbi and ALSbi, perhaps as patients are more likely to have progressed to florid FTD, although the relatively reduced number of patients with C9ORF72 expansion in this series does not allow us to draw definitive conclusions.
We found that non-ALS-related cognitive domains had almost the same frequency at each King and MiToS stage, supporting the notion that nonexecutive impairment is rare in isolation among patients with ALS. Our data challenge the introduction of this category in the classification.8,9 However, it has been reported that memory deficits in ALS are distinctly different from those observed in amnestic mild cognitive impairment (aMCI) and can be explained only to some extent in the context of comorbid executive dysfunction, indicating qualitative differences in temporal lobe dysfunction between ALS and aMCI.38 Therefore further research in this area of cognition is warranted.
King and MiToS staging systems clearly reflect the degree of motor impairment experienced by patients. However, these rating systems do not incorporate the severity of cognitive impairment, which can now be considered a central component of the ALS syndrome. Such cognitive impairment has a profound negative effect on patients' survival,15,18 ability to adapt to life-prolonging interventions,39 quality of life,40 and decision-making capacity.41,42 It also negatively affects caregivers' burden and quality of life.43,44 Based on these observations and our own data, we maintain that there is an urgent need to integrate cognitive measures into ALS clinical rating scales.
The cross-sectional design of our study may have limited our findings. Nevertheless, patients were tested early after diagnosis and the cognitive impairment at that time point likely reflects the rapidity of lesion spreading within nonmotor cortical areas of the brain. A second limitation of our study is that 40% of incident patients were not enrolled, primarily because they were not reached or did not respond to the invitation or because they were too disabled to be tested. Nonenrolled patients were older, had a lower education level than those who were included in the study, and had more severe disease. Alternative methods of cognitive testing in severely disabled patients, such as eye-tracker-controlled cognitive batteries and brain–computer interfaces, potentially represent important improvements in our ability to assess cognitive status among the ALS population.45,46 Third, the assumptions related to more severe stages (King 4 and MiToS 2) should be considered with caution due to the small number of patients tested in these stages.
This study has several strengths. First, it is population-based and includes incident patients tested for cognition shortly after the diagnosis. Second, it comprises the largest population of patients to be tested to date. Third, it is based on a large battery of tests evaluating the main domains of cognitive impairment, including those classified as non-ALS-specific. Fourth, it compares the King and MiToS staging to the revised ALS-FTD Consensus Criteria,9 allowing us to assess the association of the degree of severity of motor impairment to that of cognitive impairment. Fifth, it includes the genetics of ∼95% of patients.
Our findings have important implications in clarifying the dynamics of ALS progression, suggesting that its motor and cognitive components may worsen in parallel, and that cognitive worsening is more pronounced in patients with bulbar involvement. Our data support the hypothesis that ALS pathology disseminates in a regional ordered sequence, through a cortico-efferent spreading model.21,26 Moreover, our data show that intermediate cognitive categories may represent a transitional condition between normal cognition and FTD. In that regard, ALS represents a unique opportunity to study the initial clinical and pathologic stages of FTD. Further research on functional connectivity between motor and cognitive areas of the brain will be important to delineate the dynamics of the spreading of pathology in ALS.
Glossary
- ALS
amyotrophic lateral sclerosis
- ALS-CN
cognitively normal with amyotrophic lateral sclerosis
- ALSbi
amyotrophic lateral sclerosis with behavioral impairment
- ALScbi
amyotrophic lateral sclerosis with combined cognitive and behavioral impairment
- ALSci
amyotrophic lateral sclerosis with cognitive impairment
- ALSFRS-R
Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised
- ALSnex
amyotrophic lateral sclerosis with isolated nonexecutive impairment in the domains of memory and visuospatial function
- aMCI
amnestic mild cognitive impairment
- ECAS
Edinburgh Cognitive and Behavioural ALS Screen
- FTD
frontotemporal dementia
- HADS
Hospital Anxiety and Depression Scale
- MiToS
Milano Torino Staging system
- NIV
noninvasive ventilation
- PARALS
Piemonte and Valle d’Aosta Register for ALS
Appendix. Authors

Study funding
This work was supported in part by the Italian Ministry of Health (Ministero della Salute, Ricerca Sanitaria Finalizzata, grant RF-2016-02362405), the European Commission's Health Seventh Framework Programme (FP7/2007–2013 under grant agreement 259867), and the Joint Programme–Neurodegenerative Disease Research (Strength, ALS-Care and Brain-Mend projects), granted by Italian Ministry of Education, University and Research. This study was performed under the Department of Excellence grant of the Italian Ministry of Education, University and Research to the “Rita Levi Montalcini” Department of Neuroscience, University of Torino, Italy. The sponsor organizations had no role in data collections or analysis and did not participate in writing or approving the manuscript.
Disclosure
A. Chiò serves on scientific advisory boards for Mitsubishi Tanabe, Roche, and Cytokinetics, and has received a research grant from Italfarmaco. C. Moglia, A. Canosa, U. Manera, R. Vasta, M. Brunetti, M. Barberis, L. Corrado, S. D'Alfonso, E. Bersano, M. Sarnelli, V. Solara, J. Zucchetti, L. Peotta, B. Iazzolino, L. Mazzini, and G. Mora report no disclosures relevant to the manuscript. A. Calvo has received a research grant from Cytokinetics. Go to Neurology.org/N for full disclosures.
References
- 1.van Es MA, Hardiman O, Chio A, et al. Amyotrophic lateral sclerosis. Lancet 2017;390:2084–2098. [DOI] [PubMed] [Google Scholar]
- 2.Elamin M, Bede P, Byrne S, et al. Cognitive changes predict functional decline in ALS: a population-based longitudinal study. Neurology 2013;80:1590–1597. [DOI] [PubMed] [Google Scholar]
- 3.Poletti B, Solca F, Carelli L, et al. Cognitive-behavioral longitudinal assessment in ALS: the Italian Edinburgh cognitive and behavioral ALS screen (ECAS). Amyotroph Lateral Scler Frontotemporal Degener 2018;19:387–395. [DOI] [PubMed] [Google Scholar]
- 4.Kilani M, Micallef J, Soubrouillard C, et al. A longitudinal study of the evolution of cognitive function and affective state in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Mot Neuron Disord 2004;5:46–54. [DOI] [PubMed] [Google Scholar]
- 5.Schreiber H, Gaigalat T, Wiedemuth-Catrinescu U, et al. Cognitive function in bulbar- and spinal-onset amyotrophic lateral sclerosis: a longitudinal study in 52 patients. J Neurol 2005;252:772–781. [DOI] [PubMed] [Google Scholar]
- 6.Kasper E, Zydatiss K, Schuster C, et al. No change in executive performance in ALS patients: a longitudinal neuropsychological study. Neurodegener Dis 2016;16:184–191. [DOI] [PubMed] [Google Scholar]
- 7.Bock M, Duong YN, Kim A, Allen I, Murphy J, Lomen-Hoerth C. Progression and effect of cognitive-behavioral changes in patients with amyotrophic lateral sclerosis. Neurol Clin Pract 2017; 7:488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crockford C, Newton J, Lonergan K, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology 2018;91:e1370-e1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strong MJ, Abrahams S, Goldstein LH, et al. Amyotrophic lateral sclerosis: frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener 2017;18:153–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roche JC, Rojas-Garcia R, Scott KM, et al. A proposed staging system for amyotrophic lateral sclerosis. Brain 2012;135:847–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiò A, Hammond ER, Mora G, Bonito V, Filippini G. Development and evaluation of a clinical staging system for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2015;86:38–44. [DOI] [PubMed] [Google Scholar]
- 12.Chiò A, Mora G, Moglia C, et al. Secular trends of amyotrophic lateral sclerosis: the Piemonte and Valle d'Aosta register. JAMA Neurol 2017;74:1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grace J, Malloy P. Frontal Systems Behavior Scale (FrSBe): Professional Manual. Lutz, FL: Psychological Assessment Resources; 2001. [Google Scholar]
- 15.Montuschi A, Iazzolino B, Calvo A, et al. Cognitive correlates in amyotrophic lateral sclerosis: a population-based study in Italy. J Neurol Neurosurg Psychiatry 2015;86:168–173. [DOI] [PubMed] [Google Scholar]
- 16.Gibbons CJ, Mills RJ, Thornton EW, et al. Rasch analysis of the Hospital Anxiety and Depression Scale (HADS) for use in motor neurone disease. Health Qual Life Outcomes 2011;9:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iazzolino B, Pain D, Peotta L, et al. Validation of the revised classification of cognitive and behavioural impairment in ALS. J Neurol Neurosurg Psychiatry 2019;90:734–739. [DOI] [PubMed] [Google Scholar]
- 18.Phukan J, Elamin M, Bede P, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry 2012;83:102–108. [DOI] [PubMed] [Google Scholar]
- 19.Balendra R, Jones A, Jivraj N, et al. Estimating clinical stage of amyotrophic lateral sclerosis from the ALS Functional Rating Scale. Amyotroph Lateral Scler Frontotemporal Degener 2014;15:279–284. [DOI] [PubMed] [Google Scholar]
- 20.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-Linked ALS-FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strong MJ, Grace GM, Freedman M, et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2009;10:131–146. [DOI] [PubMed] [Google Scholar]
- 22.Trojsi F, Santangelo G, Caiazzo G, et al. Neuropsychological assessment in different King clinical stages of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2016;17:228–235. [DOI] [PubMed] [Google Scholar]
- 23.Oh SI, Park A, Kim HJ, et al. Spectrum of cognitive impairment in Korean ALS patients without known genetic mutations. PLoS One 2014;9:e87163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brettschneider J, Del Tredici K, Toledo JB, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol 2013;74:20–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt R, de Reus MA, Scholtens LH, van den Berg LH, van den Heuvel MP. Simulating disease propagation across white matter connectome reveals anatomical substrate for neuropathology staging in amyotrophic lateral sclerosis. Neuroimage 2016;124:762–769. [DOI] [PubMed] [Google Scholar]
- 26.Schulthess I, Gorges M, Müller HP, et al. Functional connectivity changes resemble patterns of pTDP-43 pathology in amyotrophic lateral sclerosis. Sci Rep 2016;6:38391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abrahams S, Goldstein LH, Al-Chalabi A, et al. Relation between cognitive dysfunction and pseudobulbar palsy in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 1997;62:464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmieri A, Mento G, Calvo V, et al. Female gender doubles executive dysfunction risk in ALS: a case-control study in 165 patients. J Neurol Neurosurg Psychiatry 2015;86:574–579. [DOI] [PubMed] [Google Scholar]
- 29.Brettschneider J, Del Tredici K, Irwin DJ, et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol 2014;127:423–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 2002;59:1077–1079. [DOI] [PubMed] [Google Scholar]
- 31.Van Langenhove T, Piguet O, Burrell JR, et al. Predicting development of amyotrophic lateral sclerosis in frontotemporal dementia. J Alzheimers Dis 2017;58:163–170. [DOI] [PubMed] [Google Scholar]
- 32.Kelley BJ, Petersen RC. Alzheimer's disease and mild cognitive impairment. Neurol Clin 2007;25:577–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knopman DS, Petersen RC, Edland SD, Cha RH, Rocca WA. The incidence of frontotemporal lobar degeneration in Rochester, Minnesota, 1990 through 1994. Neurology 2004;62:506–508. [DOI] [PubMed] [Google Scholar]
- 34.Garre-Olmo J, Genís Batlle D, del Mar Fernández M, et al. Incidence and subtypes of early-onset dementia in a geographically defined general population. Neurology 2010;75:1249–1255. [DOI] [PubMed] [Google Scholar]
- 35.Stern Y. Cognitive reserve in ageing and Alzheimer's disease. Lancet Neurol 2012;11:1006–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Placek K, Massimo L, Olm C, et al. Cognitive reserve in frontotemporal degeneration: neuroanatomic and neuropsychological evidence. Neurology 2016;87:1813–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irwin DJ, McMillan CT, Brettschneider J, et al. Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2013;84:163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Machts J, Bittner V, Kasper E, et al. Memory deficits in amyotrophic lateral sclerosis are not exclusively caused by executive dysfunction: a comparative neuropsychological study of amnestic mild cognitive impairment. BMC Neurosci 2014;15:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiò A, Ilardi A, Cammarosano S, Moglia C, Montuschi A, Calvo A. Neurobehavioral dysfunction in ALS has a negative effect on outcome and use of PEG and NIV. Neurology 2012;78:1085–1089. [DOI] [PubMed] [Google Scholar]
- 40.Goldstein LH, Atkins L, Leigh PN. Correlates of quality of life in people with motor neuron disease (MND). Amyotroph Lateral Scler Other Mot Neuron Disord 2002;3:123–129. [DOI] [PubMed] [Google Scholar]
- 41.Manes F, Torralva T, Ibáñez A, Roca M, Bekinschtein T, Gleichgerrcht E. Decision-making in frontotemporal dementia: clinical, theoretical and legal implications. Dement Geriatr Cogn Disord 2011;32:11–17. [DOI] [PubMed] [Google Scholar]
- 42.Bertoux M, Cova F, Pessiglione M, Hsu M, Dubois B, Bourgeois-Gironde S. Behavioral variant frontotemporal dementia patients do not succumb to the Allais paradox. Front Neurosci 2014;8:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiò A, Vignola A, Mastro E, et al. Neurobehavioral symptoms in ALS are negatively related to caregivers' burden and quality of life. Eur J Neurol 2010;17:1298–1303. [DOI] [PubMed] [Google Scholar]
- 44.Lillo P, Mioshi E, Hodges JR. Caregiver burden in amyotrophic lateral sclerosis is more dependent on patients' behavioral changes than physical disability: a comparative study. BMC Neurol 2012;12:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carelli L, Solca F, Faini A, et al. Brain-computer interface for clinical purposes: cognitive assessment and rehabilitation. Biomed Res Int 2017;2017:1695290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poletti B, Carelli L, Solca F, et al. An eye-tracker controlled cognitive battery: overcoming verbal-motor limitations in ALS. J Neurol 2017;264:1136–1145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be available upon request by interested researchers.