

Abstract
Objective: Provide information for pharmacists on idiopathic pulmonary fibrosis (IPF) and its treatment. Study Selection and Data Extraction: All articles with data from randomized controlled trials of nintedanib or pirfenidone were reviewed. Data Synthesis: IPF is a progressive and ultimately fatal interstitial lung disease characterized by decline in lung function and worsening dyspnea. It is uncommon and mainly occurs in individuals aged >60 years, particularly men with a history of smoking. Nintedanib and pirfenidone were approved in the United States for the treatment of IPF in 2014 and received conditional recommendations in the 2015 American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Association treatment guidelines. These drugs slow the progression of IPF by reducing the rate of decline in lung function. Their adverse event profile is characterized mainly by gastrointestinal events, which can be managed through dose adjustment and symptom management. Management of IPF should also include smoking cessation, vaccinations, and supportive care such as patient education, pulmonary rehabilitation, and the use of supplemental oxygen as well as optimizing the management of comorbidities. Relevance to Patient Care and Clinical Practice: This review provides clinical pharmacists with information on the course of IPF, what can be expected of current treatments, and how to help patients manage their drug therapy. Conclusions: IPF is a progressive disease, but treatments are available that can slow the progression of the disease. Clinical pharmacists can play an important role in the care of patients with IPF through patient education, monitoring medication compliance and safety, ensuring drugs for comorbidities are optimized, and preventive strategies such as immunizations.
Keywords: interstitial lung disease, disease management, drug trials, patient education, drug information
Idiopathic pulmonary fibrosis (IPF) is a progressive and ultimately fatal fibrosing interstitial lung disease (ILD) of unknown cause.1 IPF is one of the most common of the ILDs, a group of diseases that also includes hypersensitivity pneumonitis and “rheumatoid lung.” Although the course of IPF is variable, progression of the disease is characterized by deterioration in lung function, dyspnea, exercise capacity, and health-related quality of life.2-4 Hospitalizations are common events in patients with IPF, and in-hospital mortality rates are high.5 This review will provide an overview of best practice in the management of IPF, including an understanding of the disease and of the pharmacology and clinical studies of approved drugs, to assist pharmacists in the provision of care for individuals with this devastating disease.
Pathophysiology of IPF
Pulmonary fibrosis occurs when normal lung tissue is replaced with scar tissue, leading to impairment of gas exchange and a reduction in oxygenation of the blood (Figure 16). The histopathological hallmark of IPF is a temporally and spatially heterogeneous appearance with focal areas of fibrosis and honeycomb change alternating with areas of less affected or normal parenchyma.7 Fibroblastic foci—disordered collections of type II alveolar epithelial cells in association with fibroblasts—are considered a key pathological lesion in IPF.8
Figure 1.

The healthy lung (A) and lung damage in IPF (B).a
aSource: National Heart, Lung, and Blood Institute; National Institutes of Health; US Department of Health and Human Services.6
Diagnosis of IPF
IPF should be considered in middle-aged/elderly individuals with unexplained chronic exertional dyspnea. It commonly presents with cough and bibasilar inspiratory crackles (sometimes referred to as “Velcro rales” because of their sound) and less often with finger clubbing (spoon-shaped nails).1 Spirometry may be normal or show restriction9 (in contrast to obstruction such as in chronic obstructive pulmonary disease [COPD]). Diagnosis of IPF requires exclusion of known causes of ILD, including systemic autoimmune diseases and environmental exposures, and the presence of a usual interstitial pneumonia (UIP) pattern on high-resolution computed tomography (HRCT; Figure 2) or lung biopsy. Guidelines for the diagnosis of IPF published by American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Association (ATS/ERS/JRS/ALAT) in September 2018 provided new criteria for UIP patterns observed on HRCT.1 A UIP pattern on HRCT is characterized by features of honeycombing, with or without peripheral traction bronchiectasis or bronchiolectasis, and subpleural and basal predominance. A probable UIP pattern on HRCT consists of reticulation with peripheral traction bronchiectasis or bronchiolectasis, subpleural and basal predominance, and possibly mild ground glass opacities. In the appropriate clinical context, the presence of a typical or probable UIP pattern on HRCT is sufficient for a diagnosis of IPF to be made.1 In considering whether a surgical lung biopsy should be performed in patients with other patterns on HRCT, it is important to consider whether the benefits of securing a more confident diagnosis outweigh the risks of conducting the procedure in that patient.1,10 A review of medical records from more than 32 000 patients with ILDs in the United States found that in-hospital mortality was 1.7% following elective surgical lung biopsies and 16% following nonelective procedures.11 The risks of a surgical lung biopsy are greater in patients who are older or have more severe physiological impairment or comorbidities.10,11,12
Figure 2.
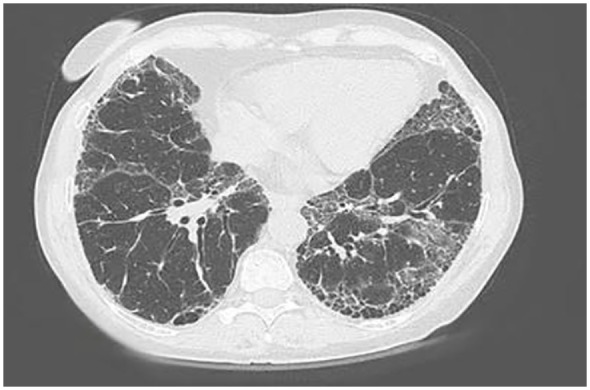
High-resolution computed tomography scan of individual with idiopathic pulmonary fibrosis.a Key features include a diffuse process affecting both lung fields with subpleural reticulation (fine lines heading into the lung from the periphery) and honeycomb cysts. The disease is located predominantly at the bases of the lung. There is a paucity of ground glass opacities (haziness that does not obscure the underlying lung architecture).
aSource: Pulmonary Fibrosis Foundation.
Epidemiology
IPF is more common in men than in women and typically presents in the sixth or seventh decade.13-15 Similar to COPD, about 70% of patients with IPF have a history of cigarette smoking.13-15 The reported incidence and prevalence of IPF vary widely depending on ascertainment and reporting methods. Based on a claims-based algorithm, the incidence of IPF in the United States has recently been estimated at 5.6 per 100 000 person-years.16
IPF rarely occurs in isolation: almost all patients with IPF have at least 1 comorbidity, and many have several.17-20 Common comorbidities include pulmonary hypertension, COPD, emphysema, gastroesophageal reflux disease (GERD), diabetes mellitus, and ischemic heart disease.17-20
Clinical Course
IPF has a variable clinical course. Some patients progress rapidly to death, whereas others experience a more gradual decline in lung function. A subset of patients suffer acute respiratory deteriorations, accompanied by new alveolar abnormalities, which are often of unknown cause. These are known as acute exacerbations and are associated with very high morbidity and mortality.21 Overall, the prognosis for patients with IPF is poor; data collected in the United States prior to the availability of drugs that slow the progression of IPF showed that the typical survival interval after diagnosis was 3 to 5 years.22,23
Treatment of IPF
Pharmacological Therapy
Until 2014, treatment of IPF focused on immunosuppressant therapy (despite a lack of evidence to support its efficacy or safety in this patient population), supplemental oxygen, and supportive care. In 2014, the results of the PANTHER-IPF trial showed that triple therapy with prednisone, azathioprine, and N-acetylcysteine, which at the time was widely regarded as being the standard of care for IPF, was in fact associated with an increased risk of hospitalizations and death.24 Chronic use of immunosuppressants is no longer recommended in patients with IPF, but steroids may be used in cases of acute respiratory deterioration.25 Results from the N-acetylcysteine monotherapy and placebo groups of the PANTHER-IPF trial showed that N-acetylcysteine had no effect on reducing lung function decline26; however, an exploratory analysis suggested that it may have an effect in patients with a particular genotype,27 and additional research on this agent is underway.
Drug development for IPF has focused on compounds believed to target fundamental pathways of fibrosis. After decades of failed clinical trials,28 2 drugs, nintedanib and pirfenidone, were shown in large randomized placebo-controlled trials to slow decline in lung function, with an acceptable safety and tolerability profile, and were approved by the US Food and Drug Administration in 2014.29,30 The latest ATS/ERS/JRS/ALAT clinical practice guidelines for the treatment of IPF, issued in July 2015, gave conditional recommendations for the use of nintedanib and pirfenidone, indicating that they would be an appropriate choice for a majority of patients, while recognizing that different choices will be appropriate for individual patients, and that the values and preferences of the patient should be considered when making decisions regarding their care.31 A conditional recommendation was also given for the use of anti-acid medications in patients with IPF and asymptomatic GERD, based on very low-quality evidence (no randomized controlled trials),31 but the risk-benefit of anti-acid medications in patients with IPF remains unknown, and there is some evidence that anti-acid medication use is associated with an increased risk of infection.32-34 The guidelines contained strong or conditional recommendations against several pharmacotherapies that had been shown to be ineffective as therapies for IPF, including drugs approved for use in other indications such as bosentan, ambrisentan, sildenafil, and warfarin.31
Nintedanib and pirfenidone are believed to inhibit fundamental pathways of fibrosis. Nintedanib is an intracellular inhibitor of tyrosine kinases, including the fibroblast growth factor receptor, platelet-derived growth factor receptor, and vascular endothelial growth factor (VEGF) receptor, as well as the nonreceptor members of the Src family.35,36 Nintedanib interferes with fibroblast proliferation, migration, and differentiation and the secretion of extracellular matrix components in the lung and has demonstrated antifibrotic and anti-inflammatory properties in animal models of lung fibrosis.35 The mechanism of action of pirfenidone in IPF is not fully understood. However, at high doses, pirfenidone has demonstrated antifibrotic, anti-inflammatory, and antioxidant properties in in vitro and in vivo models of lung fibrosis and been shown to inhibit fibroblast proliferation and differentiation and the synthesis of collagen.37
Data from the 2 INPULSIS trials of nintedanib38 and the ASCEND trial of pirfenidone39 in patients with IPF and mild or moderate impairment in lung function (forced vital capacity [FVC] > 50% predicted) at baseline showed that these drugs reduced the rate of decline in FVC over 1 year by approximately 50%. Key inclusion and exclusion criteria in these trials are summarized in Table 1.38-40 Both nintedanib and pirfenidone reduced FVC decline consistently across subgroups defined based on a variety of baseline characteristics, including age, gender, race, and FVC% predicted.33,34,41-50 Importantly, a post hoc analysis of pooled data from the 2 INPULSIS trials showed that patients with IPF and preserved lung volume (FVC > 90% predicted) at baseline had the same rate of FVC decline over 1 year and received the same benefit from nintedanib as patients with more impaired lung volume.45 In addition, a pooled analysis of data from 3 phase III trials of pirfenidone (ASCEND and the CAPACITY studies) showed that patients whose lung function had declined showed a benefit of continued treatment.51
Table 1.
Summary of Key Inclusion and Exclusion Criteria in the INPULSIS Trials of Nintedanib, the CAPACITY Trials of Pirfenidone, and the ASCEND Trial of Pirfenidone.38-40
| INPULSIS Trials of Nintedanib 150 mg bid | CAPACITY Trials of Pirfenidone 399 mg tid and Pirfenidone 801 mg tid | ASCEND Trial of Pirfenidone 801 mg tid | |
|---|---|---|---|
| Inclusion criteria | |||
| Age | ≥40 Years | 40-80 Years | 40-80 Years |
| Time since IPF diagnosis | <5 Years | <4 Years | 6-48 Months |
| FVC | ≥50% Predicted | ≥50% Predicteda | 50%-90% Predicted |
| FEV1/FVC ratio | ≥0.7 | ≥0.7 | ≥0.8 |
| DLco | 30%-79% Predicted | ≥35% Predicteda | 30%-90% Predicted |
| Radiological features | Honeycombing and/or a combination of traction bronchiectasis and reticulation in the absence of atypical features of UIP on HRCT | Definite UIP on HRCT, or probable UIP on HRCT plus definite or probable UIP on surgical lung biopsy | HRCT findings of definite UIP or possible UIP confirmed by surgical lung biopsy. Extent of fibrotic changes (honeycombing, reticular changes) greater than extent of emphysema on HRCT scan |
| 6MWT distance | — | ≥150 m | ≥150 m |
| Exclusion criteria | |||
| Lung transplant | Likely to receive lung transplant during the study | Likely to receive lung transplant during the study | Likely to receive lung transplant during the study |
| Excluded medications | Prednisone >15 mg/d, N-acetylcysteine; cyclophosphamide, cyclosporine A, pirfenidone, azathioprine, or any investigational drug | Use of any drug for the treatment of IPF except for short courses of azathioprine, cyclophosphamide, corticosteroids, or acetylcysteine | Use of any investigational, cytotoxic, immunosuppressive, cytokine-modulating, or receptor antagonist medications; daily use of fluvoxamine or sildenafil |
Abbreviations: 6MWT, 6-minute walk test; DLco, diffusion capacity of the lung for carbon monoxide; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; HRCT, high-resolution computed tomography; IPF, idiopathic pulmonary fibrosis; UIP, usual interstitial pneumonia.
Patients were also required to have FVC and/or DLco ≤90% predicted.
One potentially valuable benefit of pharmacological therapy for IPF is to decrease the risk of acute worsenings in respiratory function. Nintedanib had a statistically significant effect on reducing the rate of investigator-reported acute exacerbations in the phase II TOMORROW trial52 and in INPULSIS-2, but not in INPULSIS-1.38 Based on pooled data from the INPULSIS trials, the incidence rate of investigator-reported acute exacerbations was 5.3 versus 8.2 per 100 patient-years in patients treated with nintedanib versus placebo (risk ratio = 0.65; 95% CI = 0.40, 1.04; P = 0.07).53 In a pooled analysis of ASCEND and the 2 CAPACITY studies, patients treated with pirfenidone had a lower risk of respiratory-related hospitalization over 1 year compared with those on placebo (hazard ratio = 0.52; 95% CI = 0.36, 0.77; P = 0.001).54 There is no evidence to suggest that nintedanib or pirfenidone should be stopped during an acute exacerbation or during hospitalization. Pooled clinical trial data also suggest that nintedanib and pirfenidone improve life expectancy in patients with IPF.55,56
Meta-analyses evaluating pharmacotherapies across multiple studies suggest that nintedanib and pirfenidone have similar effects on reducing FVC decline.57-59 It is important that clinicians explain to patients that therapies for IPF slow but do not halt progression of the disease. Regardless of which therapy is chosen, early treatment of IPF is important to preserve lung function because lung function that is lost to fibrosis cannot be recovered with current therapies. It is also important to note that in clinical trials, neither nintedanib nor pirfenidone was associated with a significant improvement in clinical symptoms38,39; therefore, patients should be made aware that they may not see a change in their cough or shortness of breath as a consequence of treatment.
Given that IPF continues to progress in patients treated with nintedanib or pirfenidone, there is great interest among clinicians in whether a combination of these drugs could be used to provide greater efficacy. Trials of combinations of nintedanib and pirfenidone suggest that there is no pharmacokinetic interaction between these drugs60 and that combination therapy has a safety profile consistent with the adverse event profiles of the individual drugs.61,62 There was some evidence that nintedanib with add-on pirfenidone was associated with a reduced rate of FVC decline compared with nintedanib alone, but this finding should be interpreted with caution given the small number of patients included in the analysis (n = 104) and the short duration of treatment (12 weeks).61 Further data are needed on the risk-benefit profile of combination therapy with nintedanib and pirfenidone.
Nintedanib and pirfenidone have predominantly been investigated in patients with mild or moderate impairment in lung function, and more data are needed on the efficacy and safety of these drugs in patients with advanced disease. Data from the open-label INPULSIS-ON study of nintedanib and RECAP study of pirfenidone suggest that the efficacy and safety of these treatments are similar in patients with severe lung function impairment (FVC ≤50% predicted in INPULSIS-ON and FVC <50% and/or DLco <35% predicted in RECAP) as in those with less advanced disease.63,64 In the INSTAGE trial, which enrolled patients with IPF and DLco ≤35% predicted, the decline in FVC over 24 weeks in patients treated with nintedanib alone was similar to that observed in patients with less advanced disease in the INPULSIS trials.65 Treatment with nintedanib plus sildenafil did not provide a significant benefit on health-related quality of life compared with nintedanib alone but was associated with a reduction in FVC decline.65 The efficacy and safety of pirfenidone plus sildenafil in patients with DLco ≤40% predicted are under investigation.66
Management of Side Effects of IPF Therapy
The most common side effects associated with therapies for IPF are gastrointestinal events.67,68 Pooled data from the TOMORROW and INPULSIS trials showed that the most common adverse event associated with nintedanib 150 mg twice a day was diarrhea, which was reported in 61.5% of patients treated with nintedanib versus 17.9% treated with placebo and led to treatment discontinuation in 5.3% of patients treated with nintedanib.55 In most patients, the gastrointestinal adverse events associated with nintedanib can be managed through dose reduction (to 100 mg twice a day), treatment interruption, and measures to manage symptoms (eg, use of loperamide).29 The most common adverse event associated with pirfenidone in the CAPACITY and ASCEND trials was nausea (reported in 35.5% of patients treated with pirfenidone 2403 mg/d, the recommended dose for treatment of IPF, vs 15.1% of patients in the placebo group).43 Gastrointestinal toxicity associated with pirfenidone can be managed through dose reductions or treatment interruptions.30 Taking pirfenidone after meals may also be helpful.68 Photosensitivity and rash are also reported with pirfenidone, mostly within the first 6 months.68 In the CAPACITY and ASCEND trials, rash was reported in 29.2% of patients treated with pirfenidone versus 9.0% of patients treated with placebo.43 Patients should be advised to minimize sun exposure by wearing sunscreen and protective clothing.30
Nintedanib and pirfenidone may cause elevations in liver enzymes (alanine aminotransferase and aspartate aminotransferase) and bilirubin.29,30 Liver function tests should be conducted on treatment initiation and at regular intervals during treatment. Dose modification or treatment discontinuation may be required. In most cases, these reverse the hepatic enzyme elevations, but cases of drug-induced liver injury have been observed.29,69,70 Importantly, data from clinical trials have shown that the dose adjustments used to manage adverse effects did not have a significant impact on the efficacy of nintedanib or pirfenidone in reducing FVC decline.55,71,72
Safety data collected in real-world clinical practice73-79 appear to be consistent with the adverse events observed in clinical trials and described in the product labels. In a chart review of 186 patients at a single US center, the proportions of patients who discontinued pirfenidone or nintedanib because of an adverse drug reaction were similar to those observed in clinical trials (20.9% and 26.3%, respectively), with gastrointestinal problems being the events most likely to lead to discontinuation.75
Additional Warnings and Precautions
Nintedanib is not recommended for use in patients with moderate or severe hepatic impairment (Child-Pugh B or C).29 A lower dose (100 mg twice a day) is recommended in patients with mild hepatic impairment (Child-Pugh A).29 Pirfenidone is not recommended in patients with severe hepatic impairment and should be used with caution in patients with mild or moderate hepatic impairment.30
Because of its inhibition of the VEGF receptor, nintedanib is associated with a risk of bleeding and should be used in patients with known bleeding risk only after careful consideration of the potential benefit and risk.29 In the INPULSIS trials, bleeding adverse events were reported in 10.3% and 7.8% of patients treated with nintedanib and placebo, respectively, with epistaxis and contusion being the most frequently reported bleeding events.71 Postmarketing safety data collected in the United States found an incidence of bleeding events similar to that observed in the INPULSIS trials (119 vs 118 per 1000 patient-years).77 Inhibition of VEGF has also been linked to an increased risk of gastrointestinal perforation.80 In light of this, nintedanib should be used with caution in patients with recent abdominal surgery or diverticular disease or those who are receiving corticosteroids or nonsteroidal anti-inflammatory drugs and should be discontinued if perforation develops.29
Arterial thromboembolic events have been reported infrequently in patients treated with nintedanib.29,71 Nintedanib should be used with caution in patients at higher cardiovascular risk, including those with known coronary artery disease.29 In the INPULSIS trials, similar proportions of patients treated with nintedanib and placebo had cardiac disorder adverse events (10.0% and 10.6%, respectively) or serious cardiac disorder adverse events (5.0% and 5.4%, respectively), but events reported as a myocardial infarction or acute myocardial infarction were reported in a higher proportion of patients treated with nintedanib than placebo (1.6% vs 0.5%).71 Postmarketing safety data collected in the United States showed an incidence of myocardial infarction lower than that reported in nintedanib-treated patients in the INPULSIS trials (10 vs 17 per 1000 patient-years).77
Pharmacokinetics and Drug-Drug Interactions
The pharmacokinetics of nintedanib have been studied in healthy volunteers, patients with IPF and patients with lung cancer.29 After oral administration, peak blood levels occur within 2 to 4 hours, exhibiting an absolute bioavailability of 5%, largely related to transport mechanisms and extensive first-pass metabolism.81 Bioavailability increases by 20% when given with food. The volume of distribution is more than 1000 L, suggesting significant tissue distribution.81 Nintedanib is 98% bound to albumin. The predominant metabolism is via hydrolytic cleavage by esterases resulting in a carboxylate, which is glucuronidated by UGT enzymes. Whereas little biotransformation of nintedanib occurs via CYP pathways, it is a substrate for P-glycoprotein (P-gp). More than 90% of the dose is excreted via the biliary/fecal route.81 Mild and moderate hepatic impairment significantly affect drug clearance, whereas mild/moderate renal dysfunction has little effect. Tobacco smoking increases clearance by 20%.82 Nintedanib exhibits a half-life of ~10 hours, and steady state is achieved after 1 week of regular dosing.81,83 Coadministration of P-gp/CYP3A4 inhibitors (eg, ketoconazole, erythromycin) increases nintedanib exposure,84 and patients should be monitored closely for tolerability of nintedanib.29 Coadministration of P-gp/CYP3A4 inducers (eg, rifampicin, St John’s wort) decrease nintedanib exposure84 and should be avoided.29
The absolute oral bioavailability of pirfenidone has not been determined in humans. Peak blood levels are achieved within 4 hours, and food decreases absorption by ~15%.85 The half-life of pirfenidone is short—3 hours—thus, the 3 times daily administration. Plasma protein binding is modest (58%), with a volume of distribution of 60 to 70 L. In vitro profiling studies suggest that pirfenidone is primarily metabolized in the liver by CYP1A2 and multiple other CYPs (CYP2C9, 2C19, 2D6, 2E1), resulting in multiple metabolites, of which 5-carboxy-pirfenidone is present in plasma in significant quantities. In vitro data suggest that these metabolites are not pharmacologically active. Kidney disease increases levels of pirfenidone and 5-carboxy-pirfenidone. Moderate (eg, ciprofloxacin) and strong (eg, fluvoxamine) inhibitors of CYP1A2 increase pirfenidone exposure and may alter its adverse event profile.30 Use of strong CYP1A2 inhibitors should be avoided; if this is not possible, the dose of pirfenidone should be reduced to 267 mg 3 times a day.30 If use of ciprofloxacin 750 mg twice a day cannot be avoided, the dose of pirfenidone should be reduced to 534 mg three times a day.30 Coadministration of CYP1A2 inducers may decrease pirfenidone exposure and should be avoided.30
Overall Care of Patients With IPF
Putting a patient on nintedanib or pirfenidone may slow worsening of their symptoms but will not make their symptoms get better. Management of symptoms—particularly dyspnea, cough, and fatigue—is important for preserving quality of life in patients with IPF but is extremely challenging given the lack of evidence-based therapies.86,87 Other causes of dyspnea such as concurrent COPD or heart disease should be addressed. Smoking cessation and immunizations for common respiratory diseases such as pneumonia and influenza should be implemented where needed. The 2011 ATS/ERS/JRS/ALAT guidelines strongly recommended the use of supplemental oxygen in patients with IPF and significant resting hypoxemia.25 These guidelines also recommend pulmonary rehabilitation as part of the management of IPF,25 given its potential to improve exercise capacity, dyspnea, and quality of life.88 Lung transplant is an option for a minority of patients with IPF, and patients should be evaluated for transplant at an early stage to maximize their chances of being eligible.25,89
Cost of IPF and Its Treatment
Based on a recent systematic review, the estimated annual cost of health care resource use in patients with IPF in the United States was approximately $20 000 per patient, which was 3 times higher than the national average health care resource use.90 Acute exacerbations and associated hospitalizations are the key cost drivers. The mean cost of an IPF-related hospitalization has been estimated at $16 812.91 Nintedanib and pirfenidone are expensive drugs, typically approaching $10 000/month retail cost in the United States (http://www.goodrx.com). However, a recent cost-effectiveness analysis suggested that use of nintedanib may be associated with a reduction in medical costs, driven by fewer acute exacerbations.92 An analysis conducted in the United Kingdom (a single-payer system) suggested that nintedanib and pirfenidone are comparable in terms of costs and benefits on health-related quality of life.59
Relevance to Patient Care and Clinical Practice
Familiarity with this uncommon but impactful disease will assist the pharmacist in caring for patients with IPF. Medication compliance, addressing drug interactions with IPF therapies, smoking cessation, immunizations, and optimizing regimens for comorbidities such as COPD and GERD are key. In some settings, the pharmacist may be involved in formulary restrictions for IPF therapies; thus, understanding the disease and terminology to distinguish IPF from other ILDs will assist them in applying evidence-based guidelines to the drug approval process. Medication compliance is paramount for IPF because it is one of the most common lung diseases for which lung transplant is undertaken, and patients being screened for eligibility for lung transplant are excluded if significantly noncompliant with medications.
Nintedanib and pirfenidone are usually initiated on an outpatient basis through specialty pharmacies or other specialty distributors. If patients have commercial insurance, they may be eligible for the manufacturers’ copay programs. If the patient has government insurance, they may be eligible for copay assistance through independent foundations. Patients who are uninsured or underinsured may be eligible for assistance through the Boehringer Ingelheim Cares Foundation Patient Assistance Program93 or the Genentech Access to Care Foundation.94
Prior to initiating nintedanib or pirfenidone, pharmacists should highlight any potential drug-drug interactions or clinical issues (eg, renal or hepatic impairment) that may necessitate dose adjustment and counsel the patient regarding side-effects that may occur and how these should be managed. Utilizing dose reduction, or treatment interruption followed by dose reduction and uptitration, can help patients tolerate these drugs.
In the hospital setting, pharmacists can help manage drug therapy for IPF, including monitoring side-effects of courses of systemic corticosteroids given to treat an acute exacerbation. Because drug regimens are often complex in these patients, detailed medication reconciliation can be of significant value. Pharmacists may also be asked to assist with “patient taking own medications” procedures for the IPF therapies, which may not be on hospital formularies. There are no standard recommendations on whether to continue IPF therapies during hospitalization.
Education and emotional support are important components of the care of patients with IPF.95 Clinical pharmacists can assist patients with accurate information on their disease and its management and direct them to other reliable resources and care centers such as those provided by the Pulmonary Fibrosis Foundation (http://www.pulmonaryfibrosis.org). The American Thoracic Society provides a useful website on pulmonary rehabilitation, including a list of accredited programs (http://www.livebetter.org). In discussions with patients, pharmacists should be mindful that patients will likely have read material about IPF on the internet that may be inaccurate or outdated96 and may be highly fearful about their future. The timing and nature of palliative care should be individualized to the patient’s needs.97 Pharmacists involved in hospice care may encounter patients in whom profound dyspnea, high oxygen requirements, and the use of narcotics are common.
Summary
IPF is a progressive and ultimately fatal lung disease that usually affects middle-aged/elderly patients with several comorbidities. Nintedanib and pirfenidone are approved treatments for IPF, which slow the rate of decline in lung function. In addition to pharmacological therapy, management of IPF should include smoking cessation, vaccinations, and supportive care such as patient education, pulmonary rehabilitation, and the use of supplemental oxygen when required as well as optimizing the management of comorbidities. Familiarity with this uncommon disease and its management will assist the pharmacist in caring for patients with IPF.
Acknowledgments
The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). The authors received no direct compensation related to the development of this article. Writing support was provided by Julie Fleming, BSc, and Wendy Morris, MSc, of Fleishman-Hillard Fishburn, London, UK, which was contracted and funded by Boehringer Ingelheim Pharmaceuticals, Inc. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
Footnotes
Declaration of Conflicting Interests: The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Pleasants reports grants and personal fees from Boehringer Ingelheim, grants and personal fees from GlaxoSmithKline, and personal fees from AstraZeneca, Sunovion, and Teva. Dr Tighe reports grants and personal fees from Boehringer Ingelheim.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Roy Pleasants  https://orcid.org/0000-0002-9020-2487
https://orcid.org/0000-0002-9020-2487
References
- 1. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:e44-e68. doi: 10.1164/rccm.201807-1255ST [DOI] [PubMed] [Google Scholar]
- 2. Doubková M, Švancara J, Svoboda M, et al. EMPIRE Registry, Czech part: impact of demographics, pulmonary function and HRCT on survival and clinical course in idiopathic pulmonary fibrosis. Clin Respir J. 2018;12:1526-1535. doi: 10.1111/crj.12700 [DOI] [PubMed] [Google Scholar]
- 3. Glaspole IN, Chapman SA, Cooper WA, et al. Health-related quality of life in idiopathic pulmonary fibrosis: data from the Australian IPF Registry. Respirology. 2017;22:950-956. doi: 10.1111/resp.12989 [DOI] [PubMed] [Google Scholar]
- 4. Jo HE, Glaspole I, Moodley Y, et al. Disease progression in idiopathic pulmonary fibrosis with mild physiological impairment: analysis from the Australian IPF registry. BMC Pulm Med. 2018;18:19. doi: 10.1186/s12890-018-0575-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown AW, Fischer CP, Shlobin OA, et al. Outcomes after hospitalization in idiopathic pulmonary fibrosis: a cohort study. Chest. 2015;147:173-179. doi: 10.1378/chest.13-2424 [DOI] [PubMed] [Google Scholar]
- 6. National Heart, Lung, and Blood Institute. Idiopathic pulmonary fibrosis. https://www.nhlbi.nih.gov/health-topics/idiopathic-pulmonary-fibrosis. Accessed June 18, 2018.
- 7. Fernandez IE, Eickelberg O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet. 2012;380:680-688. doi: 10.1016/S0140-6736(12)61144-1 [DOI] [PubMed] [Google Scholar]
- 8. Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Ann Rev Pathol. 2014;9:157-179. doi: 10.1146/annurev-pathol-012513-104706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Martinez FJ, Flaherty K. Pulmonary function testing in idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3:315-321. doi: 10.1513/pats.200602-022TK [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018;6:138-153. doi: 10.1016/S2213-2600(17)30433-2 [DOI] [PubMed] [Google Scholar]
- 11. Hutchinson JP, Fogarty AW, McKeever TM, Hubbard RB. In-hospital mortality after surgical lung biopsy for interstitial lung disease in the United States. 2000 to 2011. Am J Respir Crit Care Med. 2016;193:1161-1167. doi: 10.1164/rccm.201508-1632OC [DOI] [PubMed] [Google Scholar]
- 12. Hutchinson JP, McKeever TM, Fogarty AW, Navaratnam V, Hubbard RB. Surgical lung biopsy for the diagnosis of interstitial lung disease in England: 1997-2008. Eur Respir J. 2016;48: 1453-1461. doi: 10.1183/13993003.00378-2016 [DOI] [PubMed] [Google Scholar]
- 13. Behr J, Kreuter M, Hoeper MM, et al. Management of patients with idiopathic pulmonary fibrosis in clinical practice: the INSIGHTS-IPF registry. Eur Respir J. 2015;46:186-196. doi: 10.1183/09031936.00217614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferrara G, Carlson L, Palm A, Einarsson J, Olivesten C, Sköld M. Idiopathic pulmonary fibrosis in Sweden: report from the first year of activity of the Swedish IPF-Registry. Eur Clin Respir J. 2016;3:31090. doi: 10.3402/ecrj.v3.31090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jo HE, Glaspole I, Grainge C, et al. Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian Idiopathic Pulmonary Fibrosis Registry. Eur Respir J. 2017;49:1601592. doi: 10.1183/13993003.01592-2016 [DOI] [PubMed] [Google Scholar]
- 16. Ley B, Urbania T, Husson G, et al. Code-based diagnostic algorithms for idiopathic pulmonary fibrosis: case validation and improvement. Ann Am Thorac Soc. 2017;14:880-887. doi: 10.1513/AnnalsATS.201610-764OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kreuter M, Ehlers-Tenenbaum S, Palmowski K, et al. Impact of comorbidities on mortality in patients with idiopathic pulmonary fibrosis. PLoS One. 2016;11:e0151425. doi: 10.1371/journal.pone.0151425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cottin V. The impact of emphysema in pulmonary fibrosis. Eur Respir Rev. 2013;22:153-157. doi: 10.1183/09059180.00000813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med. 2017;5:72-84. doi: 10.1016/S2213-2600(16)30222-3 [DOI] [PubMed] [Google Scholar]
- 20. Jo HE, Randhawa S, Corte TJ, Moodley Y. Idiopathic pulmonary fibrosis and the elderly: diagnosis and management considerations. Drugs Aging. 2016;33:321-334. doi: 10.1007/s40266-016-0366-1 [DOI] [PubMed] [Google Scholar]
- 21. Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis: an international working group report. Am J Respir Crit Care Med. 2016;194:265-275. doi: 10.1164/rccm.201604-0801CI [DOI] [PubMed] [Google Scholar]
- 22. Fernández Pérez ER, Daniels CE, Schroeder DR, et al. Incidence, prevalence, and clinical course of idiopathic pulmonary fibrosis: a population-based study. Chest. 2010;137:129-137. doi: 10.1378/chest.09-1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raghu G, Chen SY, Yeh WS, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001-11. Lancet Respir Med. 2014;2:566-572. doi: 10.1016/S2213-2600(14)70101-8 [DOI] [PubMed] [Google Scholar]
- 24. Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968-1977. doi: 10.1056/NEJMoa1113354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raghu G, Collard HR, Egan JJ, et al. ; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788-824. doi: 10.1164/rccm.2009-040GL [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Idiopathic Pulmonary Fibrosis Clinical Research Network; Martinez FJ, de Andrade JA, Anstrom KJ, King TE Jr, Raghu G. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2093-2101. doi: 10.1056/NEJMoa1401739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oldham JM, Ma SF, Martinez FJ, et al. TOLLIP, MUC5B, and the response to N-acetylcysteine among individuals with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2015;192:1475-1482. doi: 10.1164/rccm.201505-1010OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Raghu G. Idiopathic pulmonary fibrosis: lessons from clinical trials over the past 25 years. Eur Respir J. 2017;50:1701209. doi: 10.1183/13993003.01209-2017 [DOI] [PubMed] [Google Scholar]
- 29. OFEV® [package insert]. Ridgefield, CT: Boehringer Ingelheim Pharmaceuticals, Inc; 2018. http://bidocs.boehringer-ingelheim.com/BIWebAccess/ViewServlet.ser?docBase=renetnt&folderPath=/Prescribing+Information/PIs/Ofev/ofev.pdf. Accessed June 16, 2018. [Google Scholar]
- 30. ESBRIET® [package insert]. South San Francisco, CA: Genentech USA, Inc; 2017. https://www.gene.com/download/pdf/esbriet_prescribing.pdf. Accessed June 16, 2018. [Google Scholar]
- 31. Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192:e3-e19. doi: 10.1164/rccm.201506-1063ST [DOI] [PubMed] [Google Scholar]
- 32. Johannson KA, Strâmbu I, Ravaglia C, et al. Antacid therapy in idiopathic pulmonary fibrosis: more questions than answers? Lancet Respir Med. 2017;5:591-598. doi: 10.1016/S2213-2600(17)30219-9 [DOI] [PubMed] [Google Scholar]
- 33. Kreuter M, Spagnolo P, Wuyts W, et al. Antacid therapy and disease progression in patients with idiopathic pulmonary fibrosis who received pirfenidone. Respiration. 2017;93:415-423. doi: 10.1159/000468546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Costabel U, Behr J, Crestani B, et al. Anti-acid therapy in idiopathic pulmonary fibrosis: insights from the INPULSIS® trials. Respir Res. 2018;19:167. doi: 10.1186/s12931-018-0866-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wollin L, Wex E, Pautsch A, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1434-1445. doi: 10.1183/09031936.00174914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hilberg F, Tontsch-Grunt U, Baum A, et al. Triple angiokinase inhibitor nintedanib directly inhibits tumor cell growth and induces tumor shrinkage via blocking oncogenic receptor tyrosine kinases. J Pharmacol Exp Ther. 2018;364:494-503. doi: 10.1124/jpet.117.244129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ahluwalia N, Shea BS, Tager AM. New therapeutic targets in idiopathic pulmonary fibrosis: aiming to rein in runaway wound-healing responses. Am J Respir Crit Care Med. 2014;190:867-878. doi: 10.1164/rccm.201403-0509PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071-2082. doi: 10.1056/NEJMoa1402584 [DOI] [PubMed] [Google Scholar]
- 39. Noble PW, Albera C, Bradford WZ, et al. ; CAPACITY Study Group. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377:1760-1769. doi: 10.1016/S0140-6736(11)60405-4 [DOI] [PubMed] [Google Scholar]
- 40. King TE, Jr, Bradford WZ, Castro-Bernardini S, et al. ; ASCEND Study Group. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083-2092. doi: 10.1056/NEJMoa1402582 [DOI] [PubMed] [Google Scholar]
- 41. Albera C, Costabel U, Fagan EA, et al. Efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis with more preserved lung function. Eur Respir J. 2016;48:843-851. doi: 10.1183/13993003.01966-2015 [DOI] [PubMed] [Google Scholar]
- 42. Costabel U, Inoue Y, Richeldi L, et al. Efficacy of nintedanib in idiopathic pulmonary fibrosis across prespecified subgroups in INPULSIS. Am J Respir Crit Care Med. 2016;193:178-185. doi: 10.1164/rccm.201503-0562OC [DOI] [PubMed] [Google Scholar]
- 43. Noble PW, Albera C, Bradford WZ, et al. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. Eur Respir J. 2016;47:243-253. doi: 10.1183/13993003.00026-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taniguchi H, Xu Z, Azuma A, et al. Subgroup analysis of Asian patients in the INPULSIS® trials of nintedanib in idiopathic pulmonary fibrosis. Respirology. 2016;21:1425-1430. doi: 10.1111/resp.12852 [DOI] [PubMed] [Google Scholar]
- 45. Kolb M, Richeldi L, Behr J, et al. Nintedanib in patients with idiopathic pulmonary fibrosis and preserved lung volume. Thorax. 2017;72:340-346. doi: 10.1136/thoraxjnl-2016-208710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Raghu G, Wells AU, Nicholson AG, et al. Effect of nintedanib in subgroups of idiopathic pulmonary fibrosis by diagnostic criteria. Am J Respir Crit Care Med. 2017;195:78-85. doi: 10.1164/rccm.201602-0402OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Flaherty KR, Kolb M, Vancheri C, Tang W, Conoscenti CS, Richeldi L. Stability or improvement in forced vital capacity with nintedanib in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2018;52:1702593. doi: 10.1183/13993003.02593-2017 [DOI] [PubMed] [Google Scholar]
- 48. Brown KK, Flaherty KR, Cottin V, et al. Lung function outcomes in the INPULSIS® trials of nintedanib in idiopathic pulmonary fibrosis. Respir Med. 2019;146:42-48. doi: 10.1016/j.rmed.2018.11.012 [DOI] [PubMed] [Google Scholar]
- 49. Cottin V, Azuma A, Raghu G, et al. Therapeutic effects of nintedanib are not influenced by emphysema in the INPULSIS trials. Eur Respir J. 2019;53:1801655. doi: 10.1183/13993003.01655-2018 [DOI] [PubMed] [Google Scholar]
- 50. Xu Z, Li H, Wen F, et al. Subgroup analysis for Chinese patients included in the INPULSIS® trials of nintedanib in idiopathic pulmonary fibrosis. Adv Ther. 2019;36:621-631. doi: 10.1007/s12325-019-0887-1 [DOI] [PubMed] [Google Scholar]
- 51. Nathan SD, Albera C, Bradford WZ, et al. Effect of continued treatment with pirfenidone following clinically meaningful declines in forced vital capacity: analysis of data from three phase 3 trials in patients with idiopathic pulmonary fibrosis. Thorax. 2016;71:429-435. doi: 10.1136/thoraxjnl-2015-207011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079-1087. doi: 10.1056/NEJMoa1103690 [DOI] [PubMed] [Google Scholar]
- 53. Collard HR, Richeldi L, Kim DS, et al. Acute exacerbations in the INPULSIS trials of nintedanib in idiopathic pulmonary fibrosis. Eur Respir J. 2017;49:1601339. doi: 10.1183/13993003.01339-2016 [DOI] [PubMed] [Google Scholar]
- 54. Ley B, Swigris J, Day BM, et al. Pirfenidone reduces respiratory-related hospitalizations in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2017;196:756-761. doi: 10.1164/rccm.201701-0091OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Richeldi L, Cottin V, du Bois RM, et al. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS® trials. Respir Med. 2016;113:74-79. doi: 10.1016/j.rmed.2016.02.001 [DOI] [PubMed] [Google Scholar]
- 56. Nathan SD, Albera C, Bradford WZ, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med. 2017;5:33-41. doi: 10.1016/S2213-2600(16)30326-5 [DOI] [PubMed] [Google Scholar]
- 57. Canestaro WJ, Forrester SH, Raghu G, Ho L, Devine BE. Drug treatment of idiopathic pulmonary fibrosis: systematic review and network meta-analysis. Chest. 2016;149:756-766. doi: 10.1016/j.chest.2015.11.013 [DOI] [PubMed] [Google Scholar]
- 58. Fleetwood K, McCool R, Glanville J, et al. Systematic review and network meta-analysis of idiopathic pulmonary fibrosis treatments. J Manag Care Spec Pharm. 2017;23(3-b suppl):S5-S16. doi: 10.18553/jmcp.2017.23.3-b.s5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rinciog C, Watkins M, Chang S, et al. A cost-effectiveness analysis of nintedanib in idiopathic pulmonary fibrosis in the UK. Pharmacoeconomics. 2017;35:479-491. doi: 10.1007/s40273-016-0480-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Richeldi L, Fletcher S, Adamali H, et al. No relevant pharmacokinetic drug-drug interaction between nintedanib and pirfenidone. Eur Respir J. 2019;53:1801060. doi: 10.1183/13993003.01060-2018. [DOI] [PubMed] [Google Scholar]
- 61. Vancheri C, Kreuter M, Richeldi L, et al. ; INJOURNEY Trial Investigators. Nintedanib with add-on pirfenidone in idiopathic pulmonary fibrosis: results of the INJOURNEY trial. Am J Respir Crit Care Med. 2018;197:356-363. doi: 10.1164/rccm.201706-1301OC [DOI] [PubMed] [Google Scholar]
- 62. Flaherty KR, Fell CD, Huggins JT, et al. Safety of nintedanib added to pirfenidone treatment for idiopathic pulmonary fibrosis. Eur Respir J. 2018;52:1800230. doi: 10.1183/13993003.00230-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wuyts WA, Kolb M, Stowasser S, Stansen W, Huggins JT, Raghu G. First data on efficacy and safety of nintedanib in patients with idiopathic pulmonary fibrosis and forced vital capacity of ≤50 % of predicted value. Lung. 2016;194:739-743. doi: 10.1007/s00408-016-9912-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Costabel U, Albera C, Glassberg MK, et al. Effect of pirfenidone in patients with more advanced idiopathic pulmonary fibrosis. Respir Res. 2019;20:55. doi: 10.1186/s12931-019-1021-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kolb M, Raghu G, Wells AU, et al. Nintedanib plus sildenafil in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2018;379:1722-1731. doi: 10.1056/NEJMoa1811737 [DOI] [PubMed] [Google Scholar]
- 66. Behr J, Nathan SD, Harari S, et al. Sildenafil added to pirfenidone in patients with advanced idiopathic pulmonary fibrosis and risk of pulmonary hypertension: a PHASE IIb, randomised, double-blind, placebo-controlled study—rationale and study design. Respir Med. 2018;138:13-20. doi: 10.1016/j.rmed.2018.03.019 [DOI] [PubMed] [Google Scholar]
- 67. Cottin V. The safety and tolerability of nintedanib in the treatment of idiopathic pulmonary fibrosis. Expert Opin Drug Saf. 2017;16:857-865. doi: 10.1080/14740338.2017.1338268 [DOI] [PubMed] [Google Scholar]
- 68. Lancaster LH, de Andrade JA, Zibrak JD, et al. Pirfenidone safety and adverse event management in idiopathic pulmonary fibrosis. Eur Respir Rev. 2017;26:170057. doi: 10.1183/16000617.0057-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Verma N, Taneja S, Dhiman RK, Chawla Y. Pirfenidone related acute liver failure. J Clin Exp Hepatol. 2017;7 (suppl 2): S13. [Google Scholar]
- 70. Verma N, Kumar P, Mitra S, et al. Drug idiosyncrasy due to pirfenidone presenting as acute liver failure: case report and mini-review of the literature. Hepatol Commun. 2017;2:142-147. doi: 10.1002/hep4.1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Corte T, Bonella F, Crestani B, et al. Safety, tolerability and appropriate use of nintedanib in idiopathic pulmonary fibrosis. Respir Res. 2015;16:116. doi: 10.1186/s12931-015-0276-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Nathan SD, Lancaster LH, Albera C, et al. Dose modification and dose intensity during treatment with pirfenidone: analysis of pooled data from three multinational phase III trials. BMJ Open Respir Res. 2018;5:e000323. doi: 10.1136/bmjresp-2018-000323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Toellner H, Hughes G, Beswick W, et al. Early clinical experiences with nintedanib in three UK tertiary interstitial lung disease centres. Clin Transl Med. 2017;6:41. doi: 10.1186/s40169-017-0172-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cottin V, Koschel D, Guenther A, et al. Long-term safety of pirfenidone in a real-world setting: final results from the prospective, observational PASSPORT registry. Eur Respir J. 2017;50:PA2806. [Google Scholar]
- 75. Galli JA, Pandya A, Vega-Olivo M, Dass C, Zhao H, Criner GJ. Pirfenidone and nintedanib for pulmonary fibrosis in clinical practice: tolerability and adverse drug reactions. Respirology. 2017;22:1171-1178. doi: 10.1111/resp.13024 [DOI] [PubMed] [Google Scholar]
- 76. Hughes G, Toellner H, Morris H, Leonard C, Chaudhuri N. Real world experiences: pirfenidone and nintedanib are effective and well tolerated treatments for idiopathic pulmonary fibrosis. J Clin Med. 2016;5:E78. doi: 10.3390/jcm5090078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Noth I, Oelberg D, Kaul M, Conoscenti CS, Raghu G. Safety and tolerability of nintedanib in patients with IPF in the USA. Eur Respir J. 2018;52:1702106. doi: 10.1183/13993003.02106-2017 [DOI] [PubMed] [Google Scholar]
- 78. Tzouvelekis A, Karampitsakos T, Ntolios P, et al. Longitudinal “real-world” outcomes of pirfenidone in idiopathic pulmonary fibrosis in Greece. Front Med (Lausanne). 2017;4:213. doi: 10.3389/fmed.2017.00213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tzouvelekis A, Karampitsakos T, Kontou M, et al. Safety and efficacy of nintedanib in idiopathic pulmonary fibrosis: a real-life observational study in Greece. Pulm Pharmacol Ther. 2018;49:61-66. doi: 10.1016/j.pupt.2018.01.006 [DOI] [PubMed] [Google Scholar]
- 80. Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol. 2009;6:465-477. doi: 10.1038/nrclinonc.2009.94 [DOI] [PubMed] [Google Scholar]
- 81. Dallinger C, Trommeshauser D, Marzin K, Liesener A, Kaiser R, Stopfer P. Pharmacokinetic properties of nintedanib in healthy volunteers and patients with advanced cancer. J Clin Pharmacol. 2016;56:1387-1394. doi: 10.1002/jcph.752 [DOI] [PubMed] [Google Scholar]
- 82. Schmid U, Liesenfeld KH, Fleury A, Dallinger C, Freiwald M. Population pharmacokinetics of nintedanib, an inhibitor of tyrosine kinases, in patients with non-small cell lung cancer or idiopathic pulmonary fibrosis. Cancer Chemother Pharmacol. 2018;81:89-101. doi: 10.1007/s00280-017-3452-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Stopfer P, Rathgen K, Bischoff D, et al. Pharmacokinetics and metabolism of BIBF 1120 after oral dosing to healthy male volunteers. Xenobiotica. 2011;41:297-311. doi: 10.3109/00498254.2010.545452 [DOI] [PubMed] [Google Scholar]
- 84. Luedtke D, Marzin K, Jungnik A, von Wangenheim U, Dallinger C. Effects of ketoconazole and rifampicin on the pharmacokinetics of nintedanib in healthy subjects. Eur J Drug Metab Pharmacokinet. 2018;43:533-541. doi: 10.1007/s13318-018-0467-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rubino CM, Bhavnani SM, Ambrose PG, Forrest A, Loutit JS. Effect of food and antacids on the pharmacokinetics of pirfenidone in older healthy adults. Pulm Pharmacol Ther. 2009;22:279-285. doi: 10.1016/j.pupt.2009.03.003 [DOI] [PubMed] [Google Scholar]
- 86. Ferrara G, Luppi F, Birring SS, et al. Best supportive care for idiopathic pulmonary fibrosis: current gaps and future directions. Eur Respir Rev. 2018;27:170076. doi: 10.1183/16000617.0076-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. van Manen MJ, Geelhoed JJ, Tak NC, Wijsenbeek MS. Optimizing quality of life in patients with idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2017;11:157-169. doi: 10.1177/1753465816686743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vainshelboim B, Fox BD, Oliveira J, Kramer MR. Exercise training in idiopathic pulmonary fibrosis. Expert Rev Respir Med. 2016;10:69-77. doi: 10.1586/17476348.2016.1121104 [DOI] [PubMed] [Google Scholar]
- 89. Weill D, Benden C, Corris PA, et al. A consensus document for the selection of lung transplant candidates: 2014—an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2015;34:1-15. doi: 10.1016/j.healun.2014.06.014 [DOI] [PubMed] [Google Scholar]
- 90. Diamantopoulos A, Wright E, Vlahopoulou K, Cornic L, Schoof N, Maher TM. The burden of illness of idiopathic pulmonary fibrosis: a comprehensive evidence review. Pharmacoeconomics. 2018;36:779-807. doi: 10.1007/s40273-018-0631-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yu YF, Wu N, Chuang CC, et al. Patterns and economic burden of hospitalizations and exacerbations among patients diagnosed with idiopathic pulmonary fibrosis. J Manag Care Spec Pharm. 2016;22:414-423. doi: 10.18553/jmcp.2016.22.4.414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Yu Y, Park DH, Corman S. Financial impact of treating idiopathic pulmonary fibrosis patients with nintedanib [abstract]. J Manag Care Spec Pharm. 2018;24:S76 [Google Scholar]
- 93. Boehringer Ingelheim Pharmaceuticals, Inc. Save on your OFEV capsules prescription. https://www.ofev.com/prescription-savings. Accessed June 21, 2019.
- 94. Genentech USA, Inc. Esbriet access solutions. https://www.genentech-access.com/patient/brands/esbriet.html. Accessed June 21, 2019.
- 95. Holland AE, Fiore JF Jr, Goh N, et al. Be honest and help me prepare for the future: what people with interstitial lung disease want from education in pulmonary rehabilitation. Chron Respir Dis. 2015;12:93-101. doi: 10.1177/1479972315571925 [DOI] [PubMed] [Google Scholar]
- 96. Fisher JH, O’Connor D, Flexman AM, Shapera S, Ryerson CJ. Accuracy and reliability of internet resources for information on idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;194:218-225. doi: 10.1164/rccm.201512-2393OC [DOI] [PubMed] [Google Scholar]
- 97. Lanken PN, Terry PB, Delisser HM, et al. ; ATS End-of-Life Care Task Force. An official American Thoracic Society clinical policy statement: palliative care for patients with respiratory diseases and critical illnesses. Am J Respir Crit Care Med. 2008;177:912-927. doi: 10.1164/rccm.200605-587ST [DOI] [PubMed] [Google Scholar]