

Abstract
Aim
To infer the geological and climatic factors that have shaped the genetic diversity and structure of a willow species (Salix humboldtiana) in three basins of Central Mexico.
Location
Central Mexico.
Methods
We collected samples from 11 populations across two hydrological basins (Balsas and Lerma) and one population from another basin (Ameca) within the Mexican Central Plateau (MCP). Individuals were analyzed using sequences of two chloroplast DNA (cpDNA) regions and eight nuclear simple sequence repeats (nSSR). Population genetic diversity and structure were determined from these data. To evaluate whether genetic structure was associated with ecological niche differentiation, we determined whether there is niche equivalence, overlap, or divergence between the Balsas and Lerma basins. Also, we evaluated the relative contributions of geographic distribution and climatic variation on population genetic structuring through redundancy analysis (RDA) and partial RDA.
Results
Both cpDNA and nSSRs data indicated the presence of three highly differentiated genetic groups, mostly geographically congruent with the three main hydrological basins. According to nSSRs, the three genetic groups can be further subdivided into eight subgroups corresponding to different rivers within the main basins. The niche equivalency test showed that the niches of the species in the Balsas and Lerma basins are significantly nonequivalent. The RDA indicated a significant association of genetic variation among populations with climate variables (particularly those related to the precipitation regime), while controlling for geographic distribution.
Main conclusions
The genetic structure of S. humboldtiana is strongly associated with the historical and current geological configuration of the basins and the rivers within basins. The observed hierarchical genetic differentiation can be due to gene flow limitation resulting from physical barriers to the dispersal of S. humboldtiana, but also to some degree of isolation by environment, as suggested by the significant association between genetic variation among populations and precipitation regime.
Keywords: ecological niche modeling, genetic diversity, genetic structure, niche divergence, riparian ecosystem
1. INTRODUCTION
The genetic structure of populations and the establishment of new evolutionary lineages are strongly mediated by processes associated with the exchange of genetic information within and among populations (Hamrick & Schnabel, 1985). Understanding how the environment affects the processes of diversification and genetic differentiation has been considered a fundamental aim in evolutionary biology (Kozak & Wiens, 2006; Pease et al., 2009; Wiens & Graham, 2005). In recent years, the use of ecological niche modeling (ENM) in combination with genetic data has allowed biologists to infer how the environment influences the differentiation among species and genealogical lineages (Alvarado‐Serrano & Knowles, 2014; Costion et al., 2015; Kalvik, Stout, Doonan, & Parkinson, 2012; Kozakn & Wiens, 2006). This kind of analysis has caused a debate on how the niches of closely related species evolve. For example, when two early divergent lineages that have passed to allopatric divergence (i.e., sister lineages) retain similar ecological traits, we refer to it as niche conservatism. On the other hand, environmental differences across a species' distribution may cause morphological and genetic differentiation on populations and also niche divergence among them, even without the presence of a physical barrier (Costion et al., 2015; Makowsky, Marshall, McVay, Chippindale, & Rissler, 2010; Peterson & Holt, 2003; Raxworthy, Ingram, Rabibisoa, & Pearson, 2007; Wiens & Harrison, 2004; Wiens & Graham, 2005). In widely distributed species, it may be difficult to distinguish if a physical barrier (niche conservatism) or adaptation to a new environment (niche divergence), or a combination of both, is responsible for the divergence. However, these two scenarios can be distinguished by an analysis of niche similarity combined with the assessment of the geographic distribution of genetic diversity. Previous analysis of divergence within widely distributed tree species with populations living in different environments has shown that climate can influence genetic and morphological variation (Riordan et al., 2016; Sork et al., 2010), what could lead to differentiation through local adaptation or simply because of variation in flowering phenology (Cavender‐Bares & Pahlich, 2009; Huang et al., 2015; Sork et al., 2016).
Central Mexico has a very complex geological and climatic history. One of the most prominent physiographical features within this area is the Mexican Central Plateau (MCP), a massive uplift that has been modified by the intense volcanism present since the Miocene, product of the activity of the Transmexican Volcanic Belt (TMVB; Tamayo & West, 1964). During the periods of volcanic activity, the lake basins and rivers that run along the MCP (mainly the river Lerma and tributaries of the Balsas and the Ameca) have suffered multiple events of fragmentation and connection (Ferrari, Orozco‐Esquivel, Manea, & Manea, 2012; Tamayo & West, 1964). Also, the climatic changes during the Pleistocene modified the extension and water levels of the lakes, during alternating humid and dry periods of the glacial cycles, producing events of connection and vicariance for the populations of associated species (see Figure 1). For example, there is paleolimnological evidence that a connection between the Ameca and Lerma rivers was possible through different lakes like Chapala and San Marcos. During some periods of the Pliocene and Pleistocene, these lakes were wider than today making the exchange of different species possible (Barbour, 1973; Tamayo & West, 1964). Other basins that were interconnected with the Lerma were the upper parts of rivers Tuxpan, Cupatizio, and Ario de Rosales, that now are part of the Balsas Basin (Álvarez, 1972; Tamayo & West, 1964). All these changes have greatly influenced the evolution of different aquatic taxa (Domínguez‐Domínguez, Alda, León, García‐Garitagoitia, & Doadrio, 2008; Domínguez‐Domínguez, Doadrio, & Pérez‐Ponce de León, 2006). Several phylogeographic studies have shown that many species of fish, principally of the family Goodeidae, and their parasites, have experienced multiple events of speciation product of these processes of connection and fragmentation between rivers (Corona‐Santiago, 2013; Domínguez‐Domínguez, Pérez‐Rodríguez, Escalera‐Vázquez, & Doadrio, 2009; Huidrobo, Morrone, Villalobos, & Álvarez, 2006; Mejia‐Madrid, Vázquez‐Domínguez, & Pérez‐Ponce De León, 2007; Pérez‐Rodríguez, Domínguez‐Domínguez, León, & Doadrio, 2009). Nevertheless, there is a lack of phylogeographic studies about how these changes have affected other species associated with rivers, like trees that form riparian forests.
Figure 1.
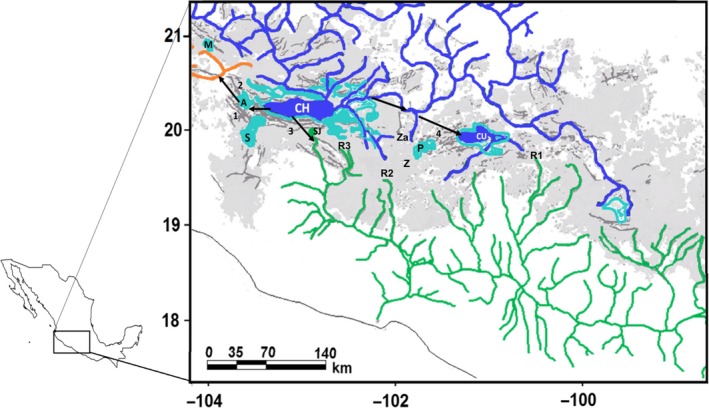
Main basins in the Mexican Central Plateau (MCP) and geological events. The Transmexican Volcanic Belt (TMBV) is represented in light gray. Miocene‐Pliocene paleolakes extension is represented in light blue and in bold blue their current size. Arrows show possible historical connections between basins. Lakes: S, Sayula; M, Magdalena; A, Zacoalco‐Ameca; SJ, Sn. Juanico; P, Pátzcuaro; CU, Cuitzeo; CH, Chapala; Z, Zirahuén; Za, Zacapu. Rivers: R1, Tuxpan; R2, Ario de Rosales; R3, Cupatitzio. Faults and trenches are represented by darker gray lines and numbers. 1, San Marco; 2, Ameca; 3, Chapala‐Tula; 4, Corredor Volcánico Tarasco. Color lines represent current water courses of the basins (green = Balsas; orange = Ameca; blue = Lerma) (modified from Domínguez‐Domínguez et al., 2008)
Riparian forests are defined as the arboreal vegetation that grows near rivers, lakes, and floodplains. These ecosystems are characterized by a linear distribution of populations along the river, so they are also susceptible to changes in the distribution of watercourses (Mitsui, Isagi, & Setoguchi, 2010). However, contrary to fish fauna, whose dispersal patterns are confined to river courses, riparian plants usually can disperse their seeds and pollen through wind and water, enabling gene flow between populations of different rivers. Among riparian trees, willows (genus Salix) outstand as one of the most important ecological components of these ecosystems in boreal and temperate areas of the Northern hemisphere (Newsholme, 1992). Willows are usually dioecious trees or shrubs that discharge little plumed seeds primarily dispersed by wind and secondarily by water (Gage & Cooper, 2005; Karrenberg, Edwards, & Kollmann, 2002; Seiwa et al., 2008). These attributes make them an attractive model for studying the evolution and dynamics of freshwater ecosystems.
Salix humboldtiana is a diploid (2n = 38) riparian pioneer tree that can reach a height of up to 25 m. As the majority of willows, S. humboldtiana is an insect‐pollinated dioecious species that reproduces sexually by seed, but also shows frequent vegetative propagation through the establishment of branch fragments. It is the single Salix species distributed from the south of the United States to the Patagonia in Argentina in tropical and subtropical climates. In Mexico, it is commonly found from sea level up to 2,300 m, forming patches of riparian forest along river courses. Salix humboldtiana is commonly found in the Lerma and Balsas river systems within the MCP. Therefore, this species represents a good model to infer how topography and climate have affected the patterns of structure and gene flow of riparian forests in this region. The first goal of the present study was to determine the genetic structure of S. humboldtiana in two basins within the MCP using both nuclear and chloroplast DNA markers. Then, by combining genetic data, ecological niche modeling, and spatial analyses, we aimed at elucidating the consequences of historical changes in the distribution of the rivers. The specific questions addressed were as follows: (a) How is the genetic structure of S. humboldtiana related to the past or the current geological configuration of these two basins within the MCP? (b) Are climatic conditions in the two basins different and how is this related to the genetic structure within this species?
2. MATERIALS AND METHODS
Fully detailed methodology, sampling, molecular protocols, and species records are available in Appendix S1.
2.1. Sampling and laboratory procedures
Leaf tissue was collected from a total of 133 individuals representing 12 sampling sites located in the basins of the rivers Balsas and Lerma and one additional sampling site from the Ameca river basin (Table S1 in Appendix S1). At each site, a minimum distance of 15 m was kept between successive samples. Leaves were dried in silica gel and stored in paper bags. Genomic DNA was extracted from dried tissue using the cetyl‐trimethyl ammonium bromide protocol (CTAB) described by Doyle and Doyle (1987).
A total of 17 nuclear simple sequence repeats (nSSRs) markers previously designed for Salix burjatica (Barker, Pahlich, Trybush, Edwards, & Karp, 2003) and Salix arbutifolia (Hoshikawa, Kikuchi, Nagamitsu, & Tomaru, 2009) were assayed for amplification and polymorphism. Also, we tested 10 universal chloroplast simple sequence repeats (cpSSRs) designed by Weising and Gardner (1999). Parameters of the thermal cycling programs and primers selected to be used in S. humboldtiana are specified in Appendix S1 (PCR protocols). Amplification products were analyzed at the Roy J. Carver Biotechnology Center in the Illinois University using an ABI Prism 3730 xl capillary sequencer, with the Liz 500® (Applied Biosystems) size standard. Resulting electropherograms were analyzed in peak scanner 1.0 (Perkin Elmer Applied Biosystems) for allele sizing. Only eight nSSRs and one cpSSRs were polymorphic for this species and were used for the whole sample (Table S2 in Appendix S1).
2.2. Phylogenetic analysis
To assess historical relationships among S. humboldtiana populations in the three basins, we sequenced 444 bp from the psbA‐trnH and the 5′‐trnS intergenic spacer regions of the cpDNA from four individuals from each basin (Ameca, Balsas, and Lerma) chosen to represent the main river systems within them (detailed amplification and sequence analysis are specified in Appendix S1). No further samples were included in this analysis since initial data revealed very limited polymorphism and only among‐basin variation.
We performed a phylogenetic analysis using the 12 psbA‐trnH (GenBank accession numbers MH919415–MH919426) and 5′‐trnS (GenBank accession numbers MH924999–MH925010) sequences of S. humboldtiana individuals. A Salix babylonica sequence (GenBank accession number EU750539.1) was used as outgroup. Alignment of the sequences was performed manually with BioEdit 7.0.5 (Hall, 1999), and then, the psbA‐trnH and 5′‐trnS matrices were concatenated with Mesquite 3.31 (Madison & Madison, 2002). Both markers displayed only length variation, and therefore, indels were considered as a fifth character state. Levels of genetic variation for each basin, that is, the number of segregating sites (S), the number of haplotypes (h), and haplotype diversity (Hd), were estimated in dnasp5.10.01 (Librado & Rozas, 2009). A maximum‐likelihood (ML) method was used to construct the phylogenetic tree in MEGA 7.0.18 (Kumar, Stecher, & Tamura, 2016) with 10,000 bootstrap replicates.
2.3. Genetic diversity and structure
Since Salix is well known to reproduce clonally by cuttings, a distance matrix was used to identify individuals with identical multilocus nSSRs genotypes using the program GenoDive v 2.0b23 (Meirmains & Van Tienderen, 2004). We detected 16 clonal individuals in a total of 133 samples. Individuals with identical genotypes were always found within the same river, and particularly in two sites, Queréndaro and Jiquilpan (see Appendix S1 and Table S1). To avoid biases in the genetic diversity and differentiation estimates, only a single individual for each clonal multilocus genotype was retained for further analysis resulting in a total of 117 unique genets.
The nSSRs data were analyzed with the program FreeNa v1 (Chapuis & Estoup, 2007) to estimate the frequency of null alleles. The expectation maximization algorithm (Demster, Laird, & Rubin, 1977) was used, and FreeNa was run with 1,000 bootstraps. This program was also used to estimate differentiation (F ST) values among populations with the method of Weir (1996) implementing the ENA correction to account for null alleles (Chapuis & Estoup, 2007). Hardy–Weinberg (HW) equilibrium for each loci and linkage disequilibrium for all pairs of loci were examined in GENEPOP v 4.07 using an exact test based on MCMC with 1,000 iterations and 200 batches. The false discovery rate (FDR) was used to eliminate significance by chance in multiple comparisons (Benjamini & Hochber, 1995). This method was preferred over Bonferroni corrections because it has been proven that it is more effective in rejecting type‐one errors (Verhoeven, Simonsen, & McIntyre, 2005). Expected (H E) and observed (H O) heterozygosity and the inbreeding coefficient (F IS) in each locality were estimated using the software ARLEQUIN v 3.5.1.2 (Excoffier & Lischer, 2010) with 10,000 permutations.
The STRUCTURE v. 2.3 (Pritchard, Stephens, & Donnelly, 2000) program was run with the nSSRs data set, to assign populations and individuals into differentiated genetic groups. To run the program, the following setup was used: K varied from 1 to 12 with 50 iterations for each K, with a burn‐in of 105 followed by 106 Markov chain Monte Carlo (MCMC) iterations. To determine the most probable K value from the analysis, the highest value of ΔK was calculated implementing the Evanno method (Evanno, Regnaut, & Goudet, 2005) in the online version of Structure Harvester v 0.6.9 (Earl & vonHoldt, 2012). Additionally, the spatial Bayesian clustering algorithm implemented in GENELAND v 3.1.4. (Guillot, Leblois, Coulon, & Frantz, 2009; Guilliot, Mortier, & Estoup, 2005) was used to delimit the genetic groups while simultaneously considering the geographical location of the samples. First, we performed 15 independent runs with a number of K min = 1 to K max = 12 to estimate the number of genetic groups. Then, we fixed K value at 8, the maximum number of possible genetic groups with a probability of each individual assigned to a genetic group higher than .6. We ran 50 additional independent runs and chose the 10 runs with the highest mean logarithms of posterior probability. Runs were performed with 1 × 106 MCMC iterations and a thinning value of 1,000. The STRUCTURE and GENELAND runs were summarized using CLUMPP (Jakobsson & Rosenberg, 2007), and the results were plotted in DISTRUCT (Rosenberg, 2004).
Analyses of molecular variance (AMOVA) using both the infinite alleles and the stepwise mutation models (IAM and SMM, respectively) were performed with ARLEQUIN v 3.5.1.2 (Excoffier & Lischer, 2010) with 10,000 permutations. These analyses were based on different population groupings to understand the partitioning of genetic diversity at different levels (among and within populations, between the Balsas and Lerma basins, and according to the group assignment obtained with the programs STRUCTURE and GENELAND). Pairwise values of F ST and R ST among populations were also obtained as well as their significance levels.
2.4. Ecological niche modeling
To assess differentiation in the ecological niche between S. humboldtiana populations in the Balsas and Lerma basins, an ecological niche modeling (ENM) approach was used. We collected environmental data from 65 S. humboldtiana spatially unique presence records from our own sampling and from botanical collection records (www.conabio.gob.mx/remib/doctos/remib_esp.html). To avoid biases resulting from unequal sampling across the area, we only considered records that were separated at least 10 km from each other. Final analyses included 24 records from the Balsas Basin and 33 from the Lerma Basin. To reduce the overprediction of area suitability as well as to perform a better model validation (Barve et al., 2011), we first defined an area of accessibility (sensu Soberón, 2007), considering the consensus hydrographic basins of Mexico (Priego‐Santander, Moreno Casasola, Palacio Prieto, López Portillo, & Geissert Kientz, 2003) to select those polygons assigned to the Balsas and Lerma basins.
For the ENM analysis, the 19 bioclimatic variables from the World Clim database were used (http://www.worldclim.org/bioclim; Hijmans, Cameron, Parra, Jones, & Jarvis, 2005), as well as one topographic variable (slope) derived from the elevation grid from the Hydro1k database (https://webgis.wr.usgs.gov/; U.S. Geological Survey, 2001), with a resolution of 30” (≈1 km2). To avoid over‐parameterization of the models, we performed Pearson's correlation tests among all pairs of variables. From each pair of highly correlated variables (r ≥ .8), we chose the more temporally inclusive one (e.g., preferring annual mean temperature over mean temperature of the warmest quarter). These analyses were performed with XLSTAT Pro 7.5 (www.xlstat.com/en/products-solutions.html). As a result, 10 climatic variables (Table 3B) and slope were selected. Slope has been regarded as an important factor for seed and juvenile establishment in many willow species (Azami et al., 2013).
Table 3.
(A) Results of redundancy analyses (RDA) for association between genetic variation, geography, and climate for Salix humboldtiana populations. Two partial analysis are shown pRDA1 (effect of climatic variables while controlling for geographic effects) and pRDA2 (effect of geographic distribution while controlling for climatic variation). The proportion of variance explained by both geographic and climatic effects is also indicated (climatic/geographic). Proportion constrained corresponds to the partitioned variance relative to the constrained variance of the full RDA model. (B) Loadings of climate variables into the first two axes of pRDA1 and their significance
| Microsatellite genetic variation | Partitioned variance | Proportion | p |
|---|---|---|---|
| (A) | |||
| Full model: geography and climate (constrained variance) | 0.08024 | 1 | .001 |
| pRDA1 | 0.01646 | 0.2052 | .001 |
| pRDA2 | 0.003281 | 0.04089 | .002 |
| Climatic/geographic | 0.060499 | 0.24609 | NA |
| Variable | df | pRDA1 | ||
|---|---|---|---|---|
| Axis 1 | Axis 2 | F | ||
| (B) | ||||
| Bio 2 | 1 | 0.21 | 0.33 | 5.4*** |
| Bio 3 | 1 | −0.14 | −0.31 | 2.3 |
| Bio 4 | 1 | 0.20 | 0.22 | 3.2 |
| Bio 7 | 1 | 0.22 | 0.36 | 1.02 |
| Bio 12 | 1 | 0.12 | −0.23 | 14.8*** |
| Bio 13 | 1 | 0.42 | −0.15 | 13.5*** |
| Bio 15 | 1 | 0.42 | 0.12 | 2.4 |
| Bio 16 | 1 | 0.31 | −0.17 | 0.72 |
| Bio 18 | 1 | −0.07 | −0.21 | 7*** |
| Bio 19 | 1 | −0.05 | −0.17 | 0.31 |
| Slope | 1 | −0.02 | −0.25 | 5.1*** |
p = .0001; **.001; *.01.
The maximum entropy algorithm implemented in MAXENT 3.3.3 (Phillips, Anderson, & Schapire, 2006) was used for ENM. Analyses were performed with a convergence threshold of 1 × 10–5 with 500 iterations. Because MAXENT produces maps in a logistic probability format (from 0 to 1), we converted each model into binary (presence–absence or 1–0) format using as a threshold the 10‐percentile training presence, that is, the probability assigned to the 10th percentile of training occurrence records (Pearson, 2007). We used 75% of occurrence records from each basin for model training and 25% for model testing. To evaluate overall classification accuracy in the model performance, MAXENT estimates the area under the curve (AUC) from the receiver operating characteristic (ROC) plot (Phillips et al., 2006). We used an alternative and external transformation of this AUC into partial‐ROC graphics, as suggested based on AUC drawbacks (Lobo, Jiménez‐Valverde, & Real, 2008; Peterson, Papeş, & Soberón, 2008). The statistical significance of partial‐ROC graphics, and thus the AUC, was assessed using null distributions of expectations created via 1,000 bootstrap replicates of 50% of the total available points and calculating the probability that the mean AUC ratio was ≤1.
To evaluate whether the ecological niches of populations in the Lerma and Balsas basins are equivalent, we reciprocally projected each of the two models into the other's geographical space; that is, the model of the Balsas Basin was projected into the Lerma area and vice versa (Appendix S2 and Figure S1). Then, we estimated the overlap index between the two niche models (OI). This index represents the fraction of the niche model of one basin predicted by the other (Martínez‐Gordillo, Rojas‐Soto, & Espinosa de los Monteros, 2010). Second, to determine whether the ecological niches from both basins were significantly different and whether their niche spaces were interchangeable, we used a modification of the method proposed by Warren, Glor, Turelli, and Funk (2008). The niche similarity and equivalency tests are directly performed from environmental space with no need to build an ENM. These statistics allow the identification of environmental differences between population groups (Broenimann et al., 2011; Petipierre et al., 2012). The first test estimates the empirical values of overlap between ecological niches (environmental spaces), generating values of niche overlap (D) and niche similarity (I) with ranges from 0 (no overlap) to 1 (identical niche models). The second test, the niche equivalency (or identity), determines whether the niches of, in this case, the populations in the two basins are more different than expected by chance even if they are drawn from the same underlying distribution. The latter was done based on D and I values randomly generated by 100 pseudo replicates and compared against empirical values with a one‐tailed t test (Warren, Glor, & Turelli, 2010; Warren et al., 2008). Finally, we evaluated the background similarity, an analysis that incorporates environmental heterogeneity to test whether the observed niche models with partially or entirely nonoverlapping distributions are any more different from one another than expected by chance (null hypothesis), or as a result of differences in habitat suitability or selection (Glor & Warren, 2010; Warren et al., 2008). The modified method estimates the use of the environment with a density of occurrence layer in environmental space, which is the result of a smoothed kernel density function of observed occurrences (Broenimann et al., 2011).
We used the Ecospat package in R to estimate the density of occurrences and to compare the environmental spaces of populations in the Balsas and Lerma basins. A density Gaussian kernel was constructed from the given set of occurrences in environmental space in both basins following Broenniman et al. (2011). The Kernel function was used with a band width equal to 0.9 times the minimum of the standard deviation and the interquartile range of the data divided by 1.34 times the sample size to the negative one‐fifth power (Silverman, 1986). We set the resolution of environmental space to a graticule of 100 × 100 pixels, so that each environmental pixel would be defined by approximately a 60‐mm interval in precipitation and a 0.3°C interval in temperature (assuming these variables were completely related to orthogonal components). One hundred and 1,000 iterations were performed to test niche equivalency and niche similarity, respectively. Significance was evaluated by the position of the observed value in relation to the distribution of simulated values under the null models of each hypothesis.
Finally, to determine whether geographic distance, climatic variation, or both have an influence on the genetic structuring among populations, we carried out redundancy and partial redundancy analyses (RDA and pRDA, respectively) using the “vegan” library in R (Oksanen, 2015). The environmental variables chosen for this analysis were the 10 climatic variables and slope used in the ENM analyses (Table 3B). As genetic variables, we used the individual scores on the first two axis of a principal coordinates analysis (PCoA) performed in GenAlex v 6.5 (Peakall & Smouse, 2006, 2012) from the genotypes at the eight nSSR loci. The spatial coordinates from each individual were used as geographic variables. We ran three different models, a full model including both geographic and environmental explanatory variables, and two partial models, firstly using only environmental variables and controlling for geographic effects (pRDA1), and secondly including geographic variables and controlling for environmental effects (pRDA2).
3. RESULTS
3.1. Phylogenetic analysis
A total of 444 bp were aligned from chloroplast sequence data. Sequence variation corresponded only two indels that defined three different haplotypes. Total haplotype diversity was moderate (Hd = 0.691). Each of the three haplotypes corresponded to one of the basins, Ameca, Balsas, and Lerma (Figure 2).
Figure 2.
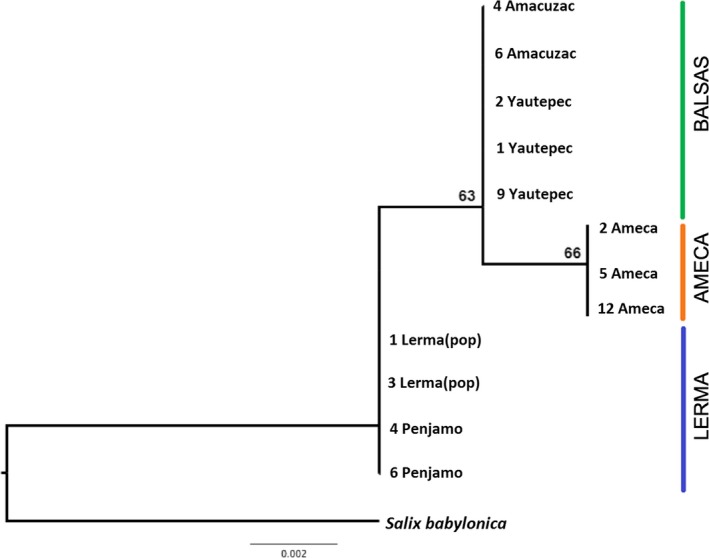
Phylogenetic reconstruction of S. humboldtiana haplotypes using cpDNA sequences of psbA‐trnH and 5′‐trnS. Colored lines refer to the three different basins (green = Balsas; orange = Ameca; blue = Lerma). Codes are composed by individual number and population of origin. Bootstrap values are shown only for Ameca and Balsas clades. Consensus tree based on 1,000 trees
3.2. Genetic diversity and structure
Eight nSSRs and one cpSSR were polymorphic for S. humboldtiana (Table S2 in Appendix S2). In the cpSSR, we found only three different alleles whose frequencies in each population are shown in Figure 3a. The H1 allele was found in all populations sampled in the Lerma Basin and in two populations (6 and 7) from the headwaters of the Balsas Basin. H2 was rare and only found in a few individuals from the Lerma and the Balsas basins, but from widely separated populations. Finally, H3 was found in most populations from the Balsas Basin and in the single population sampled in the Ameca Basin.
Figure 3.
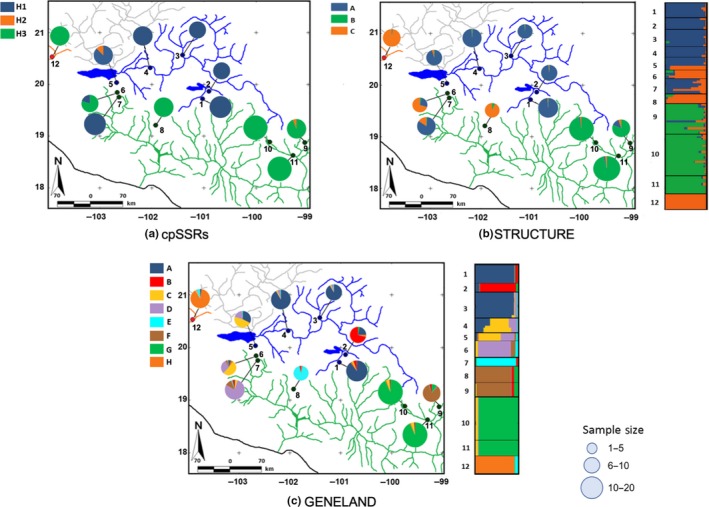
Geographic structure of genetic variation in one cpSSR and eight nSSR loci in 12 populations of S. humboldtiana. Colored lines indicate sampled basins in this study (green = Balsas; orange = Ameca; blue = Lerma). Gray lines represent the Santiago Basin (part of the Lerma‐Santiago system), but not included in this study. In each panel, colored pay charts represent the genetic composition of the populations, with size of the circle proportional to sample size after removing clones. (a) Frequency of three alleles (H1, H2, H3) at a cpSSRs locus. (b) STRUCTURE and (c) GENELAND proportion of assignment to each genetic group within each sampling locality based on eight nSSRS. Assignment is represented at both the population (pay chart) and individual (right bar plot) levels
For the eight nSSRs, we found 101 alleles, 28 of which are private alleles to one locality (Table 1). According to the FreeNA analysis, null alleles are present in two populations (Yautepec and La Parota; Table S3 in Appendix S2), but the ENA correction did not indicate an overall effect product of null alleles on the calculation of F ST (not using ENA = 0.269, using ENA = 0.263; Table S4 in Appendix S2). There were statistically significant deviations from Hardy–Weinberg equilibrium due to heterozygote deficiency in the populations Ameca (loci Cha 437 and Sb 233), Yautepec (Cha 475, Cha 528, Sb 201, Sb233), B. Amacuzac (Cha 475, 433, 437 and Sb 233), and La Parota (all loci except, Sb 201). Mean values of the expected (H E) and observed (H O) heterozygosity, inbreeding coefficient (F), number of alleles per locus (Na), number of effective alleles per locus (Ne), allelic richness (Ar), and number of private alleles (Pa) per population are listed in Table 1.
Table 1.
Genetic diversity in 12 populations of S. humboldtiana
| ID | Pop | Basin | N | Na | Ne | Ar | Pa | H E | Ho | F |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Charo | L | 9 | 5 | 3.5 | 4.09 | 3 | 0.78 | 0.79 | 0.048 |
| 2 | Queréndaro | L | 6 | 3.9 | 3.2 | 3.69 | 1 | 0.742 | 0.857 | −0.243 |
| 3 | Lerma | L | 9 | 5 | 3.8 | 4.34 | 1 | 0.768 | 0.653 | −0.057 |
| 4 | Penjamo | L | 6 | 4.9 | 3.5 | 4.47 | 4 | 0.703 | 0.667 | −0.082 |
| 5 | Jiquilpan | L | 8 | 4.9 | 3.6 | 4.28 | 2 | 0.723 | 0.719 | −0.411 |
| 6 | Sn. Juanico | B | 5 | 3.9 | 2.9 | 3.87 | 1 | 0.631 | 0.65 | −0.224 |
| 7 | Tocumbo | B | 9 | 5.1 | 3.6 | 4.30 | 2 | 0.734 | 0.667 | −0.001 |
| 8 | La Parota | B | 5 | 1.6 | 1.5 | 1.62 | 1 | 0.361 | 0.1 | 0.905 HD |
| 9 | Yautepec | B | 17 | 4.6 | 2.4 | 3.08 | 5 | 0.534 | 0.354 | 0.712 HD |
| 10 | B. Amacuzac | B | 19 | 3.5 | 2.0 | 2.77 | 4 | 0.502 | 0.323 | 0.092 HD |
| 11 | A. Amacuzac | B | 15 | 3.7 | 2.4 | 2.66 | 2 | 0.521 | 0.482 | −0.188 |
| 12 | Ameca | A | 9 | 3.5 | 2.3 | 2.99 | 2 | 0.543 | 0.407 | 0.629 HD |
| Total | 117 | 101 | 4.146 | 5.061 | 28 | 0.778 | 0.5 | 0.36 |
HD: Hardy–Weinberg disequilibrium (significant heterozygote deficit; p < .05).
Abbreviations: Ar: allelic richness (rarefacted); Basin: L: Lerma, B: Balsas, A: Ameca; F: inbreeding coefficient; H E: expected heterozygosity; H o: observed heterozygosity; ID: identification number of population; N: number of individuals analyzed; Na: mean number of alleles per locus; Ne: mean number of effective alleles per locus; Pa: number of private alleles; Pop: population name.
Populations with significant heterozygote deficit were evaluated with the INest v2.2 software (Chybicky & Burczyk, 2009) to separate actual inbreeding from the effect of null alleles on deviations from Hardy–Weinberg equilibrium. For populations, La Parota, B. Amacuzac, and Sn Matute inbreeding is the significant component of the model, while in the case of Yautepec, null alleles seem to have more weight (Table S5 in Appendix S2).
We identified K = 3 in STRUCTURE as the most probable number of clusters after calculating ΔK (Figure 3b). These clusters greatly corresponded to the Lerma Basin and upper part of west Balsas (blue cluster), upper part of east Balsas and southeastern Balsas (green cluster) and Ameca, and upper part of west Balsas (orange cluster). In the spatially explicit analyses performed in GENELAND, the most probable number of clusters was K = 8 (Figure 3c). These eight clusters presented a more detailed spatial grouping of populations. In both STRUCTURE and GENELAND analyses, individuals corresponding to the populations 6 (Tocumbo) and 7 (Sn. Juanico) that belong to the upper west part of the Balsas Basin revealed a similar genetic composition to individuals from location 5 (Jiquilpan) situated in the southwestern portion of the Lerma Basin (Figure 3).
The results of the AMOVAs for the different groupings are shown in Table 2. The analyses with the IAM (F ST) indicated that 13.65% of the variation is found among the Balsas and Lerma basins, while 18.41% is among populations within basins, and 67.94% is within populations. In turn, the grouping of individuals according to the three genetic groups identified by STRUCTURE indicated that 18.7% of the variation is among groups, 12.93% among populations, and 68.38% within populations. Finally, the grouping according to the eight genetic groups identified by GENELAND indicated that 21.38% of the variation is among the genetic groups, 7.37% among populations, and 71.25% within populations. The analysis with the SMM (R ST) showed similar trends, but the proportion of the genetic variation explained at the among‐groups level was considerably higher, indicating that mutations are contributing to the differentiation among population groups. Pairwise genetic differentiation values using R ST and F ST were statistically significant (p < .05) for almost all pairs of populations and ranged between 0.036 and 0.524 for F ST and between −0.01 and 0.846 for R ST (Table S6 in Appendix S2).
Table 2.
Results of analyses of molecular variance (AMOVA) for Salix humboldtiana in central Mexico based on eight nSSRs loci with different population groupings: among and within all populations and grouped according to basin (Balsas/Lerma) and according to the results of the Bayesian clustering algorithms GENELAND and STRUCTURE
| Grouping | df | R ST | F ST | ||||
|---|---|---|---|---|---|---|---|
| SS | VC | % | SS | VC | % | ||
| Basin | |||||||
| Among groups | 1 | 44,098.3 | 354.7 | 28.9*** | 66.8 | 0.5 | 13.6*** |
| Among populations within groups | 10 | 75,334.8 | 368.5 | 30.1*** | 139.3 | 0.6 | 18.4*** |
| Within populations | 222 | 111,525.5 | 502.4 | 41 | 505.1 | 2.3 | 67.9 |
| Total | 233 | 230,958.6 | 1,225.5 | 711.2 | 3.3 | ||
| Structure | |||||||
| Among groups | 2 | 85,404.2 | 531.2 | 44*** | 113.6 | 0.6 | 18.7*** |
| Among populations within groups | 9 | 34,029 | 173.8 | 14*** | 95.2 | 0.4 | 12.9*** |
| Within populations | 222 | 111,525.5 | 502.4 | 42 | 509.2 | 2.3 | 68.4 |
| Total | 233 | 230,958.6 | 1,207.4 | 718.0 | 3.4 | ||
| Geneland | |||||||
| Among groups | 7 | 115,947.5 | 564.5 | 50.9*** | 178.8 | 0.7 | 21.38*** |
| Among populations within groups | 4 | 4,552.5 | 32.4 | 2.9*** | 27.3 | 0.2 | 7.37*** |
| Within populations | 222 | 112,631.4 | 513 | 46.2 | 505.2 | 2.3 | 71.25 |
| Total | 233 | 233,232.3 | 1,109.9 | 711.2 | 3.2 | ||
| Populations | |||||||
| Among populations | 11 | 120,499.9 | 543.7 | 51.8*** | 208.8 | 0.87 | 27.47*** |
| Among individuals within populations | 222 | 112,631.4 | 512.9 | 48.3 | 509.194 | 2.32 | 72.52 |
| Total | 233 | 233,131.3 | 1,063 | 711.2 | 3.1 | ||
Analyses were performed with both the infinite alleles (F ST) and stepwise (R ST) mutation models.
Abbreviations: df, degrees of freedom; SS, sum of squares; VC, variance components.
p < .001.
3.3. Ecological niche modeling and niche differentiation
The performance of the two models (corresponding to the Balsas and Lerma basins) produced by MAXENT was significantly better than expected by chance alone according to the partial‐ROC bootstrap tests (Figure 4a), which showed significant ratio values of empirical AUC over null expectations (Balsas = 1.742, p < .001; Lerma = 1.714, p < .001). Regarding estimates of relative contributions of the environmental variables to the MAXENT models, results indicated that the topographic variable (slope) and the precipitation of the warmest quarter (Bio 18) were highly informative for both Balsas and Lerma models (slope = 45.3 and 14%, Bio 18 = 7.5 and 37%, respectively). Precipitation of the wettest quarter (Bio 16 = 28.9%) resulted highly informative for the Balsas model, while temperature seasonality was informative for the Lerma model (Bio 4 = 25%).
Figure 4.
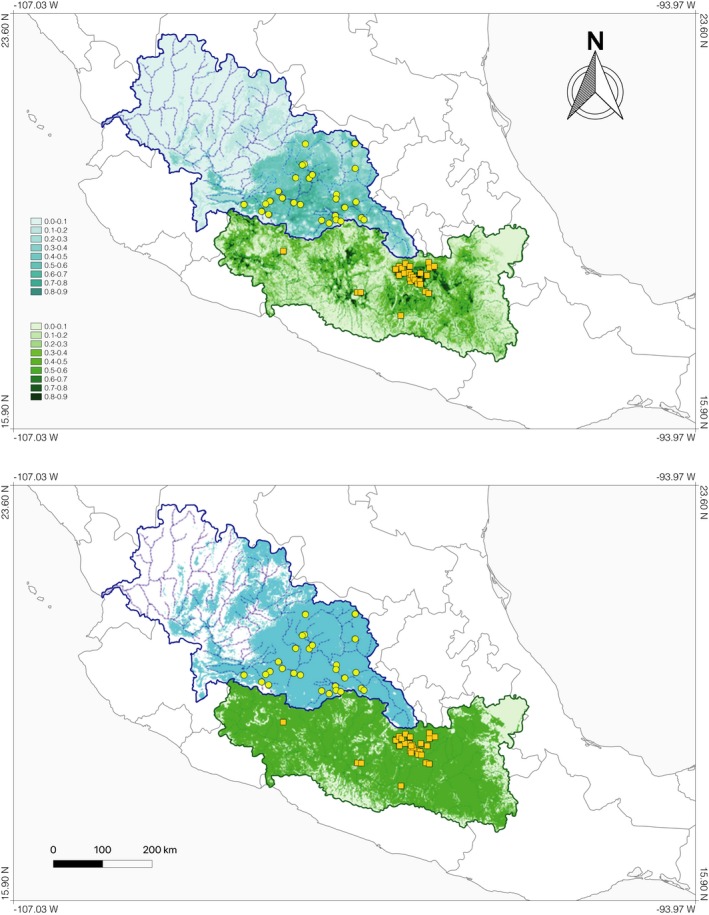
Ecological niche models (ENMs) obtained with MAXENT for Salix humboldtiana in basins Lerma = blue and Balsas = green. The map on the upper panel shows the logistic probability of environmental suitability. Darker areas represent higher probability of species occurrence. Circles and squares represent unique records of S. humboldtiana for the two basins obtained from REMIB data. ENMs were obtained using record data that were at least 10 km separated from each other (Balsas = 24) (Lerma = 32). Map on the lower panel shows the transformation of suitability probability into a binary (presence–absence) map using as a decision threshold the 10% of training presence
Our results indicated that the niche models for the two basins exhibited an overlap index of 54.37 for the Balsas to Lerma comparison and 45.62% for the Lerma to Balsas comparison (Appendix S2). Interpredictability (IP) was 90.62% considering the Balsas to Lerma comparison, while the totality of the Balsas records was predicted by the Lerma model (IP Lerma to Balsas = 100%).
Results of the principal components analysis showed that ~85% of the environmental variation is explained by the first two principal components. All 10 climatic variables (previously chosen) and the slope were negatively correlated to the first PCA axis. Precipitation seasonality (Bio 15) was correlated positively to the second PCA axis, and precipitation of the warmest quarter (Bio 18) was negatively correlated to the second axis (Figure 5a). The occurrence density surfaces in environmental space indicated a separation in the environmental spaces of the populations in the two basins in at least one direction along the first PCA axis (Figure 5b,c, shaded areas).
Figure 5.
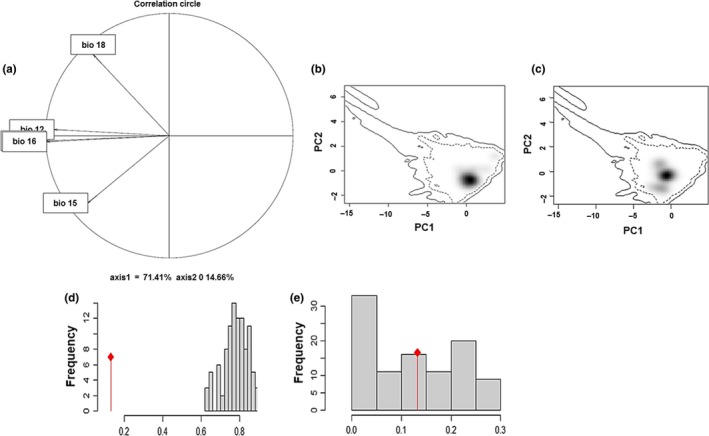
Differentiation of niche space between the Lerma and Balsas basins. (a) Correlation circle and the direction of the arrows indicate the correlation of each variable to each of the two principal components. (b) Occurrence density surfaces for the Lerma Basin and c) the Balsas Basin. The shaded area represents the density of environmental space. Niche equivalency (d) and niche similarity (e) test results. The similarity between two probability surfaces, measured by Hellinger's D, is shown. Values of D on the x‐axis and the corresponding histogram represent the expected distribution based on the randomization of the data under different null models. The first null model randomizes the identities of the records from the two basins (equivalency test), and the second one (similarity test) randomizes the distribution of the samples of each of the two basins within their accessibility area. The observed D value is shown by the red diamond
The niche equivalency test showed that the niches of the Balsas and Lerma basins are not identical, showing a low similarity value of D = 0.134 (p = .02; Figure 5d). Furthermore, the niche similarity test showed the environmental spaces of both basins were not more similar than expected under the null model (Figure 5b,c; D = 0.4, p = .455).
3.4. Redundancy analysis test climatic and genetic association
The full RDA model considering the effect of both geography and climate on genetic structuring among populations was significant (p = .001) and accounted for a considerable proportion of the variance (R 2 = 57.3%; Table 3A). The pRDA1 for the association between genetic variation and climate while controlling for geography was also significant (R 2 = 21%, p = .001). In this analysis, the most important variables were annual precipitation and precipitation of the wettest month, followed by precipitation of the warmest quarter, temperature mean diurnal range, and slope (Table 3B). The pRDA2 indicated that geography alone explained a much smaller proportion of genetic variation but was still significant (R 2 = 4%, p = .003). Finally, climate and geography together accounted for 25% of the variation (p = .001).
4. DISCUSSION
In the present study, we found a hierarchical pattern of genetic structure among populations of S. humboldtiana at regional and local scales in the MCP. Regional scale differentiation could be explained by the historical and current geological and hydrological configuration of the main basins, Balsas, Lerma, and Ameca. Local structure is probably related to population dynamics inside watersheds and rivers where populations are found. Finally, evidence of ecological differentiation may suggest the influence of scenopoetic variables, mainly topography and precipitation, on the genetic structure and differentiation of S. humboldtiana populations located in these basins.
4.1. Geological history and genetic differentiation
Both nuclear and chloroplast DNA data are congruent with the idea that S. humboldtiana populations in the study area have experienced several vicariant events (Figures 2 and 3). There have been multiple connections and disconnections between rivers within and between basins at different times that have been important for delimiting gene flow among populations of organisms associated with water courses (Domínguez‐Domínguez et al., 2008; Mejia‐Madrid, Domínguez‐Domínguez, & Pérez‐Ponce De León, 2005; Rosas‐Elguera et al., 2003; Smith & Miller, 1986). For example, Domínguez‐Domínguez et al. (2008) found that the divergence between two main lineages of Zoogoneticus quitzeoensis (Goodeidae) distributed in an east–west gradient in the MCP occurred ca. 3.3 Mya based on the molecular clock calibration and was associated with the disconnection of water bodies during a dry period and the formation of geological isolation barriers. But dispersal of these organisms is restrained to river courses, and these connections have never been examined in an organism that is dispersed by both wind and water. Our results indicate very restricted gene flow among S. humboldtiana populations situated in different basins. Phylogenetic analysis of the psbA‐trnH and 5′trnS sequences revealed only three haplotypes, each restricted to one of the three basins, with a closer relationship between the haplotypes in the Ameca and Balsas basins (Figure 2). We must take this result with caution because of the reduced number of individuals used (12 individuals of S. humboldtiana) and the scarce genetic variation found. A similar pattern was obtained, though, from nuclear and chloroplast SSRs from all individuals, supporting in general a clear genetic differentiation among basins, with only a few populations showing evidence of admixture among basins. Particularly, in the case of the cpSSR locus, populations Tocumbo (6) and Sn. Juanico (7) belonging to the Balsas Basin, showed the haplotype present in all populations of the Lerma Basin, and population Ameca (12, Ameca basin) was fixed for the same haplotype present in almost all populations in the Balsas Basin (Figure 3a). For nuclear markers, Bayesian assignment into three genetic groups also indicated the sharing of genetic variation involving populations 5, 6, 7, 8, and 12 (Figure 3b,c).
These patterns are probably related to an ancient connection between the Chapala‐San Marcos and Zacoalco‐Atotonilco lake systems (Figure 1; Smith & Miller, 1986). It has been proposed that many headwaters that currently form the Balsas Basin were originally part of the Lerma. This is the case for the Tuxpan, Cupatitzio, and Ario de Rosales rivers, which were captured by the Balsas in different geological events (Rosas‐Elguera et al., 2003; Tamayo & West, 1964). The connections between hydrological systems are common, particularly in headwaters, where erosion and volcanic activity have changed the water course from one basin to another (Burr & Page, 1986; Kozak, Wiens, & Pfennig, 2006). These phenomena have been widely documented in the MCP rivers (Corona‐Santiago, 2013; Doadrio & Domínguez, 2004; Domínguez‐Domínguez et al., 2006; Domínguez‐Domínguez, 2009; Mateos, Sanjur, & Vrijenhoek, 2002; Smith & Miller, 1986; Webb et al., 2004). It has been stated that Sn. Juanico Lake was part of the ancient Chapala Lake, but during the Plio‐Pleistocene, an intense volcanic activity isolated part of the Chapala Lake that today is recognized as a part of the Balsas Basin (Álvarez, 1963; Barbour, 1973).
4.2. Genetic diversity and within basin differentiation
Levels of genetic variability in S. humboldtiana are similar to those found in other Salix species using the same nSSRs markers (Baker et al., 2003; Kikuchi, Suzuki, & Sashimura, 2011). Nevertheless, four populations showed significant levels of heterozygote deficit. High values of inbreeding could be associated with a small effective population size resulting from a high proportion of clonal individuals or an unequal sex ratio (Ueno, Suyama, & Seiwa, 2007; Crawford & Balfour, 1990). But also, a high frequency of null alleles in certain loci could inflate inbreeding values. Salix humboldtiana is dioecious and therefore an obligate outcrossing species, and even though it can reproduce by asexual propagation, our results indicated a relatively low frequency of clones. After analyzing genotypes with the Bayesian algorithm implemented in INEsT, significant inbreeding was detected in three populations (Ameca, B. Amacuzac, and La Parota) and only Yautepec showed a high frequency of null alleles (Table S3 in Appendix S2). Therefore, it seems likely that inbreeding in this study system is resulting from low effective population sizes associated with the frequent extinction and founding of populations. Like other riparian tree groups, willows are pioneer species with short generation times (no more than 20 years; Karrenberg et al., 2002; Liotta, 2001). Short life span in riparian ecosystems is related to a metapopulation dynamics in which populations establish in streambeds that are eroded periodically by water, causing recurrent extinction and establishment of populations along rivers (Karrenberg et al., 2002; Liotta, 2001; Oliveira‐Filho, Vilela, Gavilanes, & Carvalho, 1994). Usually, new populations are founded by few individuals, which could lead to frequent local inbreeding and genetic drift. These patterns have also been found in other willows like S. daphnoides (Sochor, Vašut, Bártová, Majeský, & Mráček, 2013) and S. hukaona (Kikuchi et al., 2011). These dynamics are probably more accentuated in upstream populations where erosion is more intense than downstream (Imbert & Lefévre, 2003; Kikuchi et al., 2011; Stamati, Hollingsworth, & Russell, 2007). In our case, Ameca and La Parota are upstream populations, which is congruent with this explanation.
We also found lower but significant levels of genetic structure (F ST) associated with different rivers within basins. In other Salicaceae species, lack of genetic structure is commonly attributed to the absence of geographic barriers and long‐distance dispersal of seeds and pollen through wind (Perdereau, Kelleher, Douglas, & Hodkinson, 2014). In contrast to Populus, Salix is insect pollinated, and dispersal of seeds by wind is limited to about 200 m from the source plant (Gage & Cooper, 2005). Also, as stated before, the geographical area studied has an extremely complex topography, making long‐distance wind‐mediated gene flow difficult and limiting it even among spatially close populations. In these circumstances, hydrochory could be an effective way of secondary dispersion, but restricting gene flow to populations downstream in the same system (Kikuchi et al., 2011; Pollux, Luteijn, Groenendael, & Ouborg, 2009; Seiwa et al., 2008).
4.3. Ecological differentiation
Both cpDNA and nuclear markers indicated a strong differentiation and low gene flow among populations situated in different basins. As previously explained, this differentiation could be related to the historical and current topographical configuration of the basins. However, it is also possible that environmental differences are also contributing to limit gene flow. To explore this idea, we performed niche differentiation tests among basins and determined whether genetic differentiation is related to environmental distances more than to spatial distances.
Our results indicated that slope and precipitation of the warmest quarter (Bio 18) were highly informative for both Balsas and Lerma, while precipitation of the wettest quarter (Bio 16) was highly informative for the Balsas model and temperature seasonality (Bio 4) was informative for the Lerma model. Considering the differentiation of niches, the combination of a high proportion of overlap (54.37%–45.62%) and interpredictability (90.62%–100%) of potential distributions, and the results of the equivalency and similarity tests, in which Lerma and Balsas ecological spaces are significantly nonidentical and also significantly not similar, a complex biogeographical scenario can be proposed in which probably vicariance primarily drove the spatial isolation of S. humboldtiana, followed by the effect of ecological factors as a second force of differentiation among populations. It is plausible that slope and precipitation seasonality are important variables that could determine the establishment of S. humboldtiana in these basins of the MCP, while the differences between precipitation in the wettest period of the year and temperature seasonality could be acting as factors of differentiation between basins. The latter agrees with the ecological and environmental differences among basins: While warmer temperatures, contrasting elevations, and tropical dry forest vegetation characterize the Balsas Basin, the Lerma Basin shows more constant elevations, more temperate conditions, and mainly oak‐pine forest vegetation (Challenger, 1998).
The RDA analysis also showed an association between climate and genetic variation, with precipitation variables as the most important ones (annual precipitation, precipitation of the wettest month, and precipitation of the warmest quarter) followed by temperature mean diurnal range and slope. It has been shown that morphological and genetic variation is associated with climatic variables in other tree species like oaks (Riordan et al., 2016; Sork et al., 2016). In our case, the fact that genetic differentiation among populations is related to differences in precipitation regimes across localities seems reasonable given the importance of these variables for the establishment of willows and also probably for their reproductive phenology. As previously mentioned, slope is also determinant for the establishment of willows along river banks (Azami et al., 2013). These results are also in agreement with the ecological niche models and comparisons.
In conclusion, the complex geological and climatic history of the MCP, including the volcanic activity of the TMVB, has had a profound impact on gene flow patterns in S. humboldtiana and many other organisms. However, this is the first study in the MCP to examine patterns of genetic differentiation in a riparian tree that is dispersed by wind and water. We found that even for an organism like Salix, characterized in general by a high dispersal capacity, gene flow in this region is restricted between basins and that genetic structure is strongly mediated by river configuration, limiting genetic exchange even between close populations. These results contrast with other studies of Salix species in the world, which show efficient gene flow though wind‐dispersed seeds and scarce isolating barriers between populations (Perdereau et al., 2014).
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
Mariana S. Hernández‐Leal and Daniel Piñero designed the study and collected the data. Marco Suárez‐Atilano and Mariana S. Hernández‐Leal analyzed the data and wrote the manuscript with input from all authors. All authors discussed the results and commented on the manuscript. Antonio González‐Rodríguez and Daniel Piñero supervised the project.
Supporting information
ACKNOWLEDGMENTS
This work was supported by Grants from Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT IN203411) and Consejo Nacional de Ciencia y Tecnología (CONACYT 130773) to AGR and IEUNAM. M.S Hernández‐Leal is a doctoral student from Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM), and was supported by CONACYT scholarship No. 339623. We thank J.P. Jaramillo‐Correa and G. Pérez‐Ponce de León for their comments during the development of this work. Also, we thank A. Morales for her laboratory assistance and A. Mastretta‐Yañez, A. Ortiz‐Medrano, A. Romero‐Mariscal, A. Vazquez‐Lobo, C.L. Rodriguez‐Enriquez, R. Ortiz and S. Nicasio‐Arzeta, R. Salas‐Lizana, and L. Jardón‐Barbolla for field work.
Hernández‐Leal MS, Suárez‐Atilano M, Piñero D, González‐Rodríguez A. Regional patterns of genetic structure and environmental differentiation in willow populations (Salix humboldtiana Willd.) from Central Mexico. Ecol Evol. 2019;9:9564–9579. 10.1002/ece3.5475
DATA AVAILABILITY STATEMENT
DNA sequences: Genbank accessions MH919415–MH919426 (psbA‐trnH) and MH924999–MH925010 (5′‐trnH). nSSRs and cpSSRs Dryad repository identification https://doi.org/10.5061/dryad.vg11d5s/1
REFERENCES
- Alvarado‐Serrano, D. F. , & Knowles, L. L. (2014). Ecological niche models in phylogeographic studies: Applications, advances and precautions. Molecular Ecology Resources, 14(2), 233–248. 10.1111/1755-0998.12184 [DOI] [PubMed] [Google Scholar]
- Álvarez, J. (1963). Ictiología Michoacana III. Los peces de San Juanico y Tocumbo, Mich. Anales De La Escuela Nacional De Ciencias Biológicas, 12, 111–138. [Google Scholar]
- Álvarez, J. (1972). Ictiología Michoacana V. Origen y distribución de la ictiofauna dulceacuícola de Michoacán. Anales De La Escuela Nacional De Ciencias Biológicas, 19, 155–161. [Google Scholar]
- Azami, K. , Fukuyama, A. , Asaeda, T. , Takechi, Y. , Nakazawa, S. , & Tanida, K. (2013). Conditions of establishment for the Salix community at lower‐than‐normal water levels along a dam reservoir shoreline. Landscape and Ecological Engineering, 9(2), 227–238. 10.1007/s11355-012-0200-9 [DOI] [Google Scholar]
- Barbour, C. D. (1973). A biogeographical history of chirostoma (Pisces: Atherinidae): A species flock from the Mexican Plateau. Copeia, 1973(3), 533–556. 10.2307/1443118 [DOI] [Google Scholar]
- Barker, J. H. A. , Pahlich, A. , Trybush, S. , Edwards, K. J. , & Karp, A. (2003). Microsatellite markers for diverse Salix species. Molecular Ecology Notes, 3(1), 4–6. 10.1046/j.1471-8286.2003.00332.x [DOI] [Google Scholar]
- Barve, N. , Barve, V. , Jiménez‐Valverde, A. , Lira‐Noriega, A. , Maher, S. P. , Peterson, A. , … Villalobos, F. (2011). The crucial role of the accessible area in ecological niche modeling and species distribution modeling. Ecological Modelling, 222(11), 1810–1819. 10.1016/j.ecolmodel.2011.02.011 [DOI] [Google Scholar]
- Benjamini, Y. , & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society: Series B (Methodological), 57(1), 289–300. [Google Scholar]
- Broennimann, O. , Fitzpatrick, M. C. , Pearman, P. B. , Petitpierre, B. , Pellissier, L. , Yoccoz, N. G. , … Guisan, A. (2011). Measuring ecological niche overlap from occurrence and spatial environmental data. Global Ecology and Biogeography, 21(4), 481–497. 10.1111/j.1466-8238.2011.00698.x [DOI] [Google Scholar]
- Burr, B. M. , & Page, L. M. (1986). Zoogeography of fishes of the lower Ohio‐Upper Mississippi basin In Hocutt E. O., & Wiley C. H. (Eds.), Zoogeography of North American Fishes (pp. 287–324). Chichester, UK: John Wiley & Sons Ltd. [Google Scholar]
- Cavender‐Bares, J. , & Pahlich, A. (2009). Molecular, morphological and ecological niche differentiation of sympatric sister oak species Quercus virginiana and Q. geminata (Fagaceae). American Journal of Botany, 96(9), 1690–1702. [DOI] [PubMed] [Google Scholar]
- Challenger, A. (1998). Utilización y conservación de los ecosistemas terrestres de México: pasado, presente y futuro (847 pp.). México, D.F., México: Comisión Nacional para el Uso y Conocimiento de la Biodiversidad. [Google Scholar]
- Chapuis, M.‐P. , & Estoup, A. (2007). Microsatellite null alleles and estimation of population differentiation. Molecular Biology and Evolution, 24(3), 621–631. 10.1093/molbev/msl191 [DOI] [PubMed] [Google Scholar]
- Chybicki, I. J. , & Burczyk, J. (2009). Simultaneous estimation of null alleles and inbreeding coefficients. Journal of Heredity, 100(1), 106–113. 10.1093/jhered/esn088 [DOI] [PubMed] [Google Scholar]
- Corona‐Santiago, D. K. (2013). Filogeografía de tres species de Goodeidos endémicos del centro de México: Allotoca diazi, A. meeki y A. catarinae (Cyprinodontiformes: Goodeidae). Universidad Michoacana De San Nicolas Hidalgo. [Google Scholar]
- Costion, C. M. , Edwards, W. , Ford, A. J. , Metcalfe, D. J. , Cross, H. B. , Harrington, M. G. , … Crayn, D. M. (2015). Using phylogenetic diversity to identify ancient rain forest refugia and diversification zones in a biodiversity hotspot. Diversity and Distributions, 21(3), 279–289. 10.1111/ddi.12266 [DOI] [Google Scholar]
- Crawford, R. M. M. , & Balfour, J. (1990). Female‐biased sex ratios and differential growth in arctic Willows. Flora, 184(4), 291–302. 10.1016/S0367-2530(17)31625-0 [DOI] [Google Scholar]
- Dempster, A. P. , Laird, N. M. , & Rubin, D. B. (1977). Maximum likelihood from incomplete data via the EM algorithm. Journal of the Royal Statistical Society. Series B (Methodological), 39(1), 1–38. [Google Scholar]
- Doadrio, I. , & Domínguez, O. (2004). Phylogenetic relationships within the fish family Goodeidae based on cytochrome b sequence data. Molecular Phylogenetics and Evolution, 31(2), 416–430. 10.1016/j.ympev.2003.08.022 [DOI] [PubMed] [Google Scholar]
- Domínguez‐Domínguez, O. , Alda, F. , de León, G.‐P.‐P. , García‐Garitagoitia, J. L. , & Doadrio, I. (2008). Evolutionary history of the endangered fish Zoogoneticus quitzeoensis (Bean, 1898) (Cyprinodontiformes: Goodeidae) using a sequential approach to phylogeography based on mitochondrial and nuclear DNA data. BMC Evolutionary Biology, 8(1), 161 10.1186/1471-2148-8-161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez‐Domínguez, O. , Doadrio, I. , & Pérez‐Ponce de León, G. (2006). Historical biogeography of some river basins in central Mexico evidenced by their goodeine freshwater fishes: A preliminary hypothesis using secondary Brooks parsimony analysis. Journal of Biogeography, 33(8), 1437–1447. 10.1111/j.1365-2699.2006.01526.x [DOI] [Google Scholar]
- Domínguez‐Domínguez, O. , Pérez‐Rodríguez, R. , Escalera‐Vázquez, L. H. , & Doadrio, I. (2009). The genus Notropis rafinesque, 1817 (Actinopterygii, Cyprinidae) from Lerma River Basin in Central Mexico: Dos nuevas especies del género Notropis rafinesque, 1817 (Actinopterygii, Cyprinidae), de la cuenca del río Lerma, México Central. Hidrobiológica, 19(2), 159–172. [Google Scholar]
- Doyle, J. J. , & Doyle, J. L. (1987). A rapid DNA isolation procedure for samll quantities of fresh leaf tissue. Phytochemical Bulletin, 19, 11–15. [Google Scholar]
- Earl, D. A. , & vonHoldt, B. M. (2012). STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources, 4(2), 359–361. 10.1007/s12686-011-9548-7 [DOI] [Google Scholar]
- Evanno, G. , Regnaut, S. , & Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Molecular Ecology, 14 10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- Excoffier, L. , & Lischer, H. E. L. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3), 564–567. [DOI] [PubMed] [Google Scholar]
- Ferrari, L. , Orozco‐Esquivel, T. , Manea, V. , & Manea, M. (2012). The dynamic history of the Trans‐Mexican Volcanic Belt and the Mexico subduction zone. Tectonophysics, 522–523, 122–149. 10.1016/j.tecto.2011.09.018 [DOI] [Google Scholar]
- Gage, E. A. , & Cooper, D. J. (2005). Patterns of willow seed dispersal, seed entrapment, and seedling establishment in a heavily browsed montane riparian ecosystem. Canadian Journal of Botany, 83(6), 678–687. 10.1139/b05-042 [DOI] [Google Scholar]
- Glor, R. E. , & Warren, D. (2010). Testing ecological explanations for biogeographic boundaries. Evolution, 65(3), 673–683. 10.1111/j.1558-5646.2010.01177.x [DOI] [PubMed] [Google Scholar]
- Guillot, G. , Leblois, R. , Coulon, A. , & Frantz, A. C. (2009). Statistical methods in spatial genetics. Molecular Ecology, 18(23), 4734–4756. 10.1111/j.1365-294X.2009.04410.x [DOI] [PubMed] [Google Scholar]
- Guillot, G. , Mortier, F. , & Estoup, A. (2005). Geneland: A computer package for landscape genetics. Molecular Ecology Notes, 5(3), 712–715. 10.1111/j.1471-8286.2005.01031.x [DOI] [Google Scholar]
- Hall, T. A. (1999). BioEdit: A user‐friendly biologycal sequence alignment editor and analysis program for windows 95/98/NT. In Nucleic Acids Symposium Series (pp. 95–98). [Google Scholar]
- Hamrick, J. L. , & Schnabel, A. (1985). Understanding the genetic structure of plant populations: Some old problems and a new approach BT In Gregorius H.‐R. (Ed.), Population Genetics in Forestry (pp. 50–70). Berlin, Heidelberg, Germany: Springer. [Google Scholar]
- Hijmans, R. J. , Cameron, S. E. , Parra, J. L. , Jones, P. G. , & Jarvis, A. (2005). Very high resolution interpolated climate surfaces for global land areas. International Journal of Climatology, 25(15), 1965–1978. 10.1002/joc.1276 [DOI] [Google Scholar]
- Hoshikawa, T. , Kikuchi, S. , Nagamitsu, T. , & Tomaru, N. (2009). Eighteen microsatellite loci in Salix arbutifolia (Salicaceae) and cross‐species amplification in Salix and Populus species. Molecular Ecology Resources, 9(4), 1202–1205. [DOI] [PubMed] [Google Scholar]
- Huang, C. L. , Chang, C. T. , Huang, B. H. , Chung, J. D. , Chen, J. H. , Chiang, Y. C. , & Hwang, S. Y. (2015). Genetic relationships and ecological divergence in Salix species and populations in Taiwan. Tree Genetics & Genomes, 11, 39 10.1007/s11295-015-0862-1 [DOI] [Google Scholar]
- Huidobro, L. , Morrone, J. J. , Villalobos, J. L. , & Álvarez, F. (2006). Distributional patterns of freshwater taxa (fishes, crustaceans and plants) from the Mexican Transition Zone. Journal of Biogeography, 33(4), 731–741. 10.1111/j.1365-2699.2005.01400.x [DOI] [Google Scholar]
- Imbert, E. , & Lefèvre, F. (2003). Dispersal and gene flow of Populus nigra (Salicaceae) along a dynamic river system. Journal of Ecology, 91(3), 447–456. 10.1046/j.1365-2745.2003.00772.x [DOI] [Google Scholar]
- Jakobsson, M. , & Rosenberg, N. A. (2007). CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics, 23(14), 1801–1806. 10.1093/bioinformatics/btm233 [DOI] [PubMed] [Google Scholar]
- Kalkvik, H. M. , Stout, I. J. , Doonan, T. J. , & Parkinson, C. L. (2012). Investigating niche and lineage diversification in widely distributed taxa : Phylogeography and ecological niche modeling of the Peromyscus maniculatus species group. Ecography, 35(1), 54–64. [Google Scholar]
- Karrenberg, S. , Edwards, P. J. , & Kollmann, J. (2002). The life history of Salicaceae living in the active zone of floodplains. Freshwater Biology, 47(4), 733–748. 10.1046/j.1365-2427.2002.00894.x [DOI] [Google Scholar]
- Kikuchi, S. , Suzuki, W. , & Sashimura, N. (2011). Gene flow in an endangered willow Salix hukaoana (Salicaceae) in natural and fragmented riparian landscapes. Conservation Genetics, 12(1), 79–89. 10.1007/s10592-009-9992-z [DOI] [Google Scholar]
- Kozak, K. H. , Wiens, & J. J. (2006). Does niche conservatism promote speciation? A case study in north american salamanders. Evolution, 60(12), 2604–2621. 10.1111/j.0014-3820.2006.tb01893.x [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , & Tamura, K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33(7), 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado, P. , & Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25(11), 1451–1452. 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- Liotta, J. (2001). Rasgos biológicos de Salix humboldtiana willd. Y régimen de pulsos de inundación. Interciencia, 26(9), 397–403. [Google Scholar]
- Lobo, J. M. , Jiménez‐Valverde, A. , & Real, R. (2008). AUC: A misleading measure of the performance of predictive distribution models. Global Ecology and Biogeography, 17(2), 145–151. 10.1111/j.1466-8238.2007.00358.x [DOI] [Google Scholar]
- Maddison, W. P. , & Maddison, D. R. (2002). Mesquite: A modular system for evolutionary analysis. version 0.992, 2002. Retrieved from http://mesquiteproject.org [Google Scholar]
- Makowsky, R. , Marshall, J. C. , McVay, J. , Chippindale, P. T. , & Rissler, L. J. (2010). Phylogeographic analysis and environmental niche modeling of the plain‐bellied watersnake (Nerodia erythrogaster) reveals low levels of genetic and ecological differentiation. Molecular Phylogenetics and Evolution, 55(3), 985–995. 10.1016/j.ympev.2010.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐Gordillo, D. , Rojas‐Soto, O. , & Espinosa de los Monteros, A. (2010). Ecological niche modelling as an exploratory tool for identifying species limits: An example based on Mexican muroid rodents. Journal of Evolutionary Biology, 23(2), 259–270. 10.1111/j.1420-9101.2009.01897.x [DOI] [PubMed] [Google Scholar]
- Mateos, M. , Sanjur, O. I. , & Vrijenhoek, R. C. (2002). Historical biogeography of the livebearing fish genus Poeciliopsis (Poeciliidae: Cyprinodontiformes). Evolution, 56(5), 972–984. 10.1111/j.0014-3820.2002.tb01409.x [DOI] [PubMed] [Google Scholar]
- Meirmans, P. G. , & Van Tienderen, P. H. (2004). genotype and genodive: Two programs for the analysis of genetic diversity of asexual organisms. Molecular Ecology Notes, 4(4), 792–794. 10.1111/j.1471-8286.2004.00770.x [DOI] [Google Scholar]
- Mejía‐Madrid, H. H. , Domínguez‐Domínguez, O. , & Pérez‐Ponce De León, G. (2005). Adult endohelminth parasites of Goodeinae (Cyprinodontiformes: Goodeidae) from México with biogeographical considerations. Comparative Parasitology, 72(2), 200–211. 10.1654/4169 [DOI] [Google Scholar]
- Mejía‐Madrid, H. H. , Vázquez‐Domínguez, E. , & Pérez‐Ponce De León, G. (2007). Phylogeography and freshwater basins in central Mexico: Recent history as revealed by the fish parasite Rhabdochona lichtenfelsi (Nematoda). Journal of Biogeography, 34(5), 787–801. 10.1111/j.1365-2699.2006.01651.x [DOI] [Google Scholar]
- Mitsui, Y. , Isagi, Y. , & Setoguchi, H. (2010). Multiple spatial scale patterns of genetic diversity in riparian populations of Ainsliaea faurieana (Asteraceae) on Yakushima Island, Japan. American Journal of Botany, 97(1), 101–110. [DOI] [PubMed] [Google Scholar]
- Newsholme, C. (1992). Willows: The genus Salix. London, UK: BT Batsford Ltd. [Google Scholar]
- Oksanen, J. (2015). Vegan: an introduction to ordination.http://cran.rproject.org/web/packages/vegan/vignettes/introvegan.pdf [Google Scholar]
- Oliveira‐Filho, A. T. , Vilela, E. A. , Gavilanes, M. L. , & Carvalho, D. A. (1994). Effect of flooding regime and understorey bamboos on the physiognomy and tree species composition of a tropical semideciduous forest in Southeastern Brazil. Vegetatio, 113(2), 99–124. [Google Scholar]
- Peakall, R. , & Smouse, P. E. (2006). GenAlex 6: Genetic analysis in excel. Population genetic software for teaching and research. Molecular Ecology Notes, 6(1), 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall, R. , & Smouse, P. E. (2012). GenAlEx 6.5: Genetic analysis in excel. Population genetic software for teaching and research‐an update. Bioinformatics, 28, 2537–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, R. G. (2007). Species' distribution modeling for conservation educators and practitioners. Lessons in Conservation, 3, 54–89. [Google Scholar]
- Pease, K. M. , Freedman, A. H. , Pollinger, J. P. , Mccormack, J. E. , Buermann, W. , Rodzen, J. , … Wayne, R. K. (2009). Landscape genetics of California mule deer (Odocoileus hemionus): The roles of ecological and historical factors in generating differentiation. Molecular Ecology, 18(9), 1848–1862. [DOI] [PubMed] [Google Scholar]
- Perdereau, A. C. , Kelleher, C. T. , Douglas, G. C. , & Hodkinson, T. R. (2014). High levels of gene flow and genetic diversity in Irish populations of Salix caprea L. inferred from chloroplast and nuclear SSR markers. BMC Plant Biology, 14(1), 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Rodríguez, R. , Domínguez‐Domínguez, O. , de León, G. P. P. , & Doadrio, I. (2009). Phylogenetic relationships and biogeography of the genus Algansea Girard (Cypriniformes: Cyprinidae) of central Mexico inferred from molecular data. BMC Evolutionary Biology, 9(1), 223 10.1186/1471-2148-9-223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, A. T. , & Holt, R. D. (2003). Niche differentiation in Mexican birds: Using point occurrences to detect ecological innovation. Ecology Letters, 6(8), 774–782. 10.1046/j.1461-0248.2003.00502.x [DOI] [Google Scholar]
- Peterson, A. T. , Papeş, M. , & Soberón, J. (2008). Rethinking receiver operating characteristic analysis applications in ecological niche modeling. Ecological Modelling, 213(1), 63–72. 10.1016/j.ecolmodel.2007.11.008 [DOI] [Google Scholar]
- Petitpierre, B. , Kueffer, C. , Broennimann, O. , Randin, C. , Daehler, C. , & Guisan, A. (2012). Climatic niche shifts are rare among terrestrial plant invaders. Science, 335(6074), 1344–1348. [DOI] [PubMed] [Google Scholar]
- Phillips, S. J. , Anderson, R. P. , & Schapire, R. E. (2006). Maximum entropy modeling of species geographic distributions. Ecological Modelling, 190(3), 231–259. 10.1016/j.ecolmodel.2005.03.026 [DOI] [Google Scholar]
- Pollux, B. J. A. , Luteijn, A. , Van Groenendael, J. M. , & Ouborg, N. J. (2009). Gene flow and genetic structure of the aquatic macrophyte Sparganium emersum in a linear unidirectional river. Freshwater Biology, 54(1), 64–76. [Google Scholar]
- Priego Santander, Á. , Moreno Casasola, P. , Palacio Prieto, J. L. , López Portillo, J. , & Geissert Kientz, D. (2003). Relación entre la heterogeneidad del paisaje y la riqueza de especies de flora en cuencas costeras del estado de Veracruz, México. Investigaciones Geográficas. scielomx. [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raxworthy, C. J. , Ingram, C. M. , Rabibisoa, N. , & Pearson, R. G. (2007). Applications of ecological niche modeling for species delimitation: A review and empirical evaluation using day Geckos (Phelsuma) from Madagascar. Systematic Biology, 56(6), 907–923. 10.1080/10635150701775111 [DOI] [PubMed] [Google Scholar]
- Riordan, E. C. , Gugger, P. F. , Ortego, J. , Smith, C. , Gaddis, K. , Thompson, P. , & Sork, V. L. (2016). Association of genetic and phenotypic variability with geography and climate in three southern California oaks. American Journal of Botany, 103(1), 73–85. 10.3732/ajb.1500135 [DOI] [PubMed] [Google Scholar]
- Rosas‐Elguera, J. , Alva‐Valdivia, L. M. , Goguitchaichvili, A. , Urrutia‐Fucugauchi, J. , Ortega‐Rivera, M. A. , Prieto, J. C. S. , & Lee, J. K. W. (2003). Counterclockwise rotation of the Michoacan block: Implications for the tectonics of Western Mexico. International Geology Review, 45(9), 814–826. 10.2747/0020-6814.45.9.814 [DOI] [Google Scholar]
- Rosenberg, N. A. (2004). Distruct: A program for the graphical display of population structure. Molecular Ecology Notes, 4(1), 137–138. 10.1046/j.1471-8286.2003.00566.x [DOI] [Google Scholar]
- Seiwa, K. , Tozawa, M. , Ueno, N. , Kimura, M. , Yamasaki, M. , & Maruyama, K. (2008). Roles of cottony hairs in directed seed dispersal in riparian willows. Plant Ecology, 198(1), 27–35. 10.1007/s11258-007-9382-x [DOI] [Google Scholar]
- Silverman, B. W. (1986). Density estimation for statistics and data analysis. London, UK: Chapman and Hall. [Google Scholar]
- Smith, M. L. , & Miller, R. R. (1986). The evolution of the Rio Grande Basin as inferred from its fish fauna In Hocutt E. O., & Wiley C. H. (Eds.), The zoogeography of North American freshwater fishes (pp. 457–485). New York, NY: John Wiley & Sons Ltd. [Google Scholar]
- Soberón, J. (2007). Grinnellian and Eltonian niches and geographic distributions of species. Ecology Letters, 10(12), 1115–1123. 10.1111/j.1461-0248.2007.01107.x [DOI] [PubMed] [Google Scholar]
- Sochor, M. , Vašut, R. J. , Bártová, E. , Majeský, Ľ. , & Mráček, J. (2013). Can gene flow among populations counteract the habitat loss of extremely fragile biotopes? An example from the population genetic structure in Salix daphnoides . Tree Genetics & Genomes, 9(5), 1193–1205. 10.1007/s11295-013-0628-6 [DOI] [Google Scholar]
- Sork, V. , Squire, K. , Gugger, P. , Steele, E. , Levy, E. D. , & Eckert, A. J. (2016). Landscape genomic analysis of candidate genes for climate adaptation in a California endemic oak, Quercus lobata . American Journal of Botany, 103, 33–46. [DOI] [PubMed] [Google Scholar]
- Sork, V. L. , Davis, F. W. , Westfall, R. , Flint, A. , Ikegami, M. , Wang, H. , & Grivet, D. (2010). Gene movement and genetic association with regional climate gradients in California valley oak (Quercus lobata Née) in the face of climate change. Molecular Ecology, 19, 3806–3823. [DOI] [PubMed] [Google Scholar]
- Stamati, K. , Hollingsworth, P. M. , & Russell, J. (2007). Patterns of clonal diversity in three species of sub‐arctic willow (Salix lanata, Salix lapponum and Salix herbacea). Plant Systematics and Evolution, 269(1‐2), 75–88. [Google Scholar]
- Tamayo, J. L. , & West, R. C. (1964). The hydrography of Middle America In Wauchope R. C., & West R. (Eds.), Handbook of middle American Indians. (pp. 84–121). Austin, TX: University of Texas Press. [Google Scholar]
- Ueno, N. , Suyama, Y. , & Seiwa, K. (2007). What makes the sex ratio female‐biased in the dioecious tree Salix sachalinensis? Journal of Ecology, 95(5), 951–959. 10.1111/j.1365-2745.2007.01269.x [DOI] [Google Scholar]
- U.S. Geological Survey (2001). SRTM DEM digital elevation data-base. http://srtm.csi.cgiar.org/ (Accessed September 21, 2016) [Google Scholar]
- Verhoeven, K. J. F. , Simonsen, K. L. , & McIntyre, L. M. (2005). Implementing false discovery rate control: Increasing your power. Oikos, 108(3), 643–647. 10.1111/j.0030-1299.2005.13727.x [DOI] [Google Scholar]
- Warren, D. L. , Glor, R. E. , & Turelli, M. (2010). ENMTools: A toolbox for comparative studies of environmental niche models. Ecography, 33(3), 607–611. 10.1111/j.1600-0587.2009.06142.x [DOI] [Google Scholar]
- Warren, D. L. , Glor, R. E. , Turelli, M. , & Funk, D. (2008). Environmental niche equivalency versus conservatism: Quantitative approaches to niche evolution. Evolution, 62(11), 2868–2883. 10.1111/j.1558-5646.2008.00482.x [DOI] [PubMed] [Google Scholar]
- Webb, S. A. , Graves, J. A. , Macias‐Garcia, C. , Magurran, A. E. , Foighil, D. Ó. , & Ritchie, M. G. (2004). Molecular phylogeny of the livebearing Goodeidae (Cyprinodontiformes). Molecular Phylogenetics and Evolution, 30(3), 527–544. 10.1016/S1055-7903(03)00257-4 [DOI] [PubMed] [Google Scholar]
- Weir, B. S. (1996). Genetic Data Analysis II. Sunderland, MA: Sinauer & Associates. [Google Scholar]
- Weising, K. , & Gardner, R. C. (1999). A set of conserved PCR primers for the analysis of simple sequence repeat polymorphisms in chloroplast genomes of dicotyledonous angiosperms. Genome, 42(1), 9–19. 10.1139/g98-104 [DOI] [PubMed] [Google Scholar]
- Wiens, J. J. , & Graham, C. H. (2005). Niche conservatism: Integrating evolution, ecology, and conservation biology. Annual Review of Ecology, Evolution, and Systematics, 36(1), 519–539. 10.1146/annurev.ecolsys.36.102803.095431 [DOI] [Google Scholar]
- Wiens, J. J. , & Harrison, R. (2004). Speciation and ecology revisited: Phylogenetic niche conservatism and the origin of species. Evolution, 58(1), 193–197. 10.1111/j.0014-3820.2004.tb01586.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA sequences: Genbank accessions MH919415–MH919426 (psbA‐trnH) and MH924999–MH925010 (5′‐trnH). nSSRs and cpSSRs Dryad repository identification https://doi.org/10.5061/dryad.vg11d5s/1