

Supplemental Digital Content is available in the text
Abstract
Previous retroviral and knock-in approaches to model human t(11;19)+ acute mixed-lineage leukemia in mice resulted in myeloproliferation and acute myeloid leukemia not fully recapitulating the human disease. The authors established a doxycycline (DOX)-inducible transgenic mouse model “iMLL-ENL” in which induction in long-term hematopoietic stem cells, lymphoid primed multipotent progenitor cells, multipotent progenitors (MPP4) but not in more committed myeloid granulocyte-macrophage progenitors led to a fully reversible acute leukemia expressing myeloid and B-cell markers. iMLL-ENL leukemic cells generally expressed lower MLL-ENL mRNA than those obtained after retroviral transduction. Disease induction was associated with iMLL-ENL levels exceeding the endogenous Mll1 at mRNA and protein levels. In leukemic cells from t(11;19)+ leukemia patients, MLL-ENL mRNA also exceeded the endogenous MLL1 levels suggesting a critical threshold for transformation. Expression profiling of iMLL-ENL acute leukemia revealed gene signatures that segregated t(11;19)+ leukemia patients from those without an MLL translocation. Importantly, B220+iMLL-ENL leukemic cells showed a higher in vivo leukemia initiation potential than coexisting B220− cells. Collectively, characterization of a novel transgenic mouse model indicates that the cell-of-origin and the fusion gene expression levels are both critical determinants for MLL-ENL-driven acute leukemia.
Introduction
The t(11;19)(q23;p13.3) translocation leading to the expression of an MLL-ENL fusion protein is one of the most prevalent alterations affecting the mixed lineage leukemia 1 (MLL1) gene.1 The ENL fusion partner is a transcriptional elongation factor involved in controlling transcription in lymphoid and myeloid cells.2 Although the majority of the human MLL-ENL+ leukemia cases are diagnosed as B-cell acute lymphoblastic leukemia (ALL), several studies have shown that t(11;19) positive tumor cells simultaneously express lymphoid and myeloid markers characterizing the disease as mixed phenotype or mixed-lineage leukemia.1,3,4 These observations suggest that the disease initiates by malignant transformation of a hematopoietic stem (HSC) or early progenitor cell with lymphoid and myeloid potential rather than of a lineage-restricted progenitor cell.
Previous studies modeling the oncogenic activity of MLL-ENL by retroviral overexpression in mouse bone marrow (BM) cells revealed induction of aberrant self-renewal in vitro, shown by serial replating in methylcellulose (MC) and upon subsequent transplantation into lethally irradiated recipients, the development of acute myeloid leukemia (AML) but not of ALL or mixed-lineage leukemia.5,6 These myeloid leukemia phenotypes are considerably influenced by viral integration events potentially leading to the activation of cooperating oncogenes or to the loss of tumor suppressors. In addition, in vitro expansion of cells in growth factor-containing medium likely influences cellular lineage fate decisions, and thereby biases the resulting leukemic phenotype. Finally, the oncogenic expression levels driven by retroviral vectors like the murine stem cells virus are beyond those observed in primary patients’ cells.7,8
To overcome such limitations, several knock-in mouse lines have been established.9–14 Surprisingly, MLL-ENL expression in most of these models resulted in chronic myeloproliferative disorders rather than in lymphoblastic or mixed-lineage acute leukemia suggesting that the fusion oncogene may not have been expressed in the natural hematopoietic target cell at the appropriate level. One more recent study demonstrated that transgenic doxycycline (DOX) inducible MLL-ENL expression in granulocyte-macrophage progenitors (GMP) resulted in AML, whereas the HSC compartment appeared to be inherently protected from transformation.15
To address the impact of the cellular origin of MLL-ENL-driven leukemia, we established a DOX-regulated transgenic mouse model “iMLL-ENL,” in which activation of the oncogene in long-term hematopoietic stem cells (LT-HSC), multipotent progenitors cells, or lymphoid primed multipotent progenitor cells (LMPP) but not GMP cells resulted in fully penetrant transplantable and reversible mixed-lineage acute leukemia. Interestingly, iMLL-ENL expression surpassed endogenous Mll1 at mRNA and protein levels in tumor cells from diseased mice. Likewise, MLL-ENL mRNA levels exceeded those of MLL1 in 5 t(11;19)+ ALL patients suggesting that the cell-of-origin and oncogene expression levels are both critical determinants for the transforming capacity of MLL-ENL.
Materials and methods
Establishment of iMLL-ENL transgenic mice
The human MLL-ENL cDNA16 was cloned into p2Lox, and electroporated into A2Lox-Cre ES cells.17,18 The transgene was targeted in a region upstream of the Hprt locus. Standard procedures were used to generate the transgenic line that was backcrossed to C57BL/6 for over 10 generations. Double heterozygous female iMLL-ENL mice and cells originating from them were used throughout this study. All experiments were done in adherence to Swiss laws for animal welfare and approved by the Swiss Cantonal Veterinary Office of Basel-Stadt.
Liquid cultures and colony forming assays
Cells were grown in liquid cultures containing RPMI-1640, 10% fetal bovine serum, 1% Pen/Strep supplemented with 10 ng/mL of human interleukin-6 (hIL-6), 6 ng/mL of murine mIL-3, 10 ng/mL of murine mIL-7, 100 ng/mL of murine stem cell factor (mSCF), and 100 ng/mL of mouse Flt3-ligand (mFlt-3L) (PeproTech EC, London, UK) in the presence of 0.5 μg/mL DOX where indicated. Cells were kept in culture for up to 25 days, and the cell number was scored every 2nd day. Total BM and/or progenitors cells were isolated from transgenic mice and plated (104) in 2 mL MC culture (Methocult M3534 or M3434; StemCell Technologies, Vancouver, BC, Canada), containing IL-3, IL-6, mSCF. The medium was further supplemented with IL-7, and Flt-3L and with 0.5 μg/mL DOX where indicated. Colonies were scored microscopically after 5 to 7 days, then harvested and replated (104) for up to 5 rounds. Cellular differentiation was examined by Wright-Giemsa staining of cytospin preparations.
Flow cytometric analysis and sorting of hematopoietic stem and progenitor cells
BM cells were first stained with a lineage cocktail that contains antibodies specific for the following murine BM cell markers: Cd5, Cd11b, CD45R/B220, Ly-6G (Gr-1), and Ter119 (MAGM209, R&D Systems, Minneapolis, MN). Labeled cells were depleted by magnetic separation and the remaining cells were stained with a streptavidin Pacific Blue-conjugated (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA), a biotin-conjugated antimouse IL-7Rα (A7R34, eBioscience, San Diego, CA), a phycoerythrin (PE)-conjugated antimouse FcγRII/III (93, eBioscience) or CD150 (mShad150, eBioscience) or Flt-3L (eBioscience), an Alexa Fluor 647-conjugated antimouse Cd34 (RAM34, BD Biosciences, San Jose, CA), an allophycocyanin-conjugated antimouse c-Kit (2B8, BD Biosciences, Allschwil, Switzerland) and a phycoerythrin-Cy7 (PE-Cy7)-conjugated antimouse Sca-1 (BioLegend, San Diego, CA) monoclonal antibody, a fluorescein isothiocyanate (FITC)-conjugated Cd48 (HM48-1, eBioscience). GMP and common myeloid progenitors (CMPs) were fluorescence-activated cell sorted as IL-7Rα− Lin− Sca-1− c-Kit+ Cd34+ FcγRII/IIIhigh and IL-7Rα− Lin− Sca-1− c-Kit+ Cd34+ FcγRII/IIIlow, respectively; LT-HSC were sorted as Lin− Cd34− Sca-1− c-Kit+ Cd150+ Cd48−; LSK were sorted as Lin− Sca-1− c-Kit+; MPP4s as Lin− Sca-1+ c-Kit+ Flt-3L+ Cd150− Cd48+; and LMPPs as Lin− Sca1+ c-Kit+ Cd34− Flt-3L+. For BM cell fluorescence activated cell sorting (FACS) analysis, single-cell suspensions from leukemic mice with >95% infiltration were stained with the following monoclonal antibodies: PE-labeled Cd16/32 (FcγRII/III), PE-Cy7-conjugated c-Kit, an allophycocyanin labeled (APC) B220, an allophycocyanin-Cy7-conjugated (APC-Cy7) Gr-1, a hydroxycoumarin (BV510)-conjugated Mac1, and an FITC-labeled Cd34, or Cd19 (all from Bioscience, San Diego, CA).
RNA sequencing of in vivo mouse samples
RNA was isolated from 3 × 104 total BM cells (n = 3) or BM cells cultured ex vivo for 48 hours and sorted as B220+ (n = 3) and B220− (n = 3) from leukemic iMLL-ENL mice with over 95% blast infiltration. RNA was also obtained from sorted wt LSK (n = 3) and wt GMP cells (n = 3) using the PicoPure RNA Isolation Kit (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA). The RNA sequencing library was prepared using the NuGen Ovation v2 RNA-Seq kit and sequenced using an Illumina HiSeq 2000 machine. The RNA-sequencing data are available from GEO (GSE84988). For target validation by Q-polymerase chain reaction (PCR) assays we used standard protocols that together with the methodology for the statistical analysis of mouse and human datasets are described in the Supplemental Material section (Supplemental Digital Content 1).
Results
A novel reversible transgenic mouse model of MLL-ENL-induced leukemia
We generated “iMLL-ENL” transgenic mice in which the expression of the human MLL-ENL fusion cDNA16 is driven by the reverse-type tetracycline-controlled transactivator (rtTA) integrated in the Rosa26 gene locus (Fig. 1A).17,18 Induction of iMLL-ENL resulted in a rapid expansion of linage marker-depleted BM cells in liquid cultures (Fig. 1B). iMLL-ENL cells cultured in growth factor containing MC formed mostly round and dense colonies previously referred as type-I colonies (Fig. 1C).19,20 DOX removal resulted in rapid cellular differentiation mainly toward monocytes and macrophages but also toward cells expressing high levels of lymphoid markers, as shown by colony morphology (Fig. 1D), Wright-Giemsa staining (Fig. S1A, Supplemental Digital Content 2) and flow cytometry measuring the expression of FcγRII/III, Cd34, Mac-1, and B220 surface markers (Fig. S1B, Supplemental Digital Content 2). Exposure of BM cells to increasing doses of DOX (0–2 μg/mL) resulted in enhanced iMLL-ENL mRNA expression returning to background levels 24 hours after DOX removal (Fig. 1E). In the presence of factors supporting myeloid and lymphoid cell growth (mIL-3, hIL-6, mSCF, mFlt-3L, and mIL-7) expression of iMLL-ENL resulted in efficient serial replating activity for >4 rounds (Fig. 1F). Collectively, DOX-regulated transgenic iMLL-ENL expression reversibly induced aberrant self-renewal, blocked differentiation, and significantly expanded mouse BM-derived hematopoietic stem and progenitor cells (HSPC) ex vivo.
Figure 1.
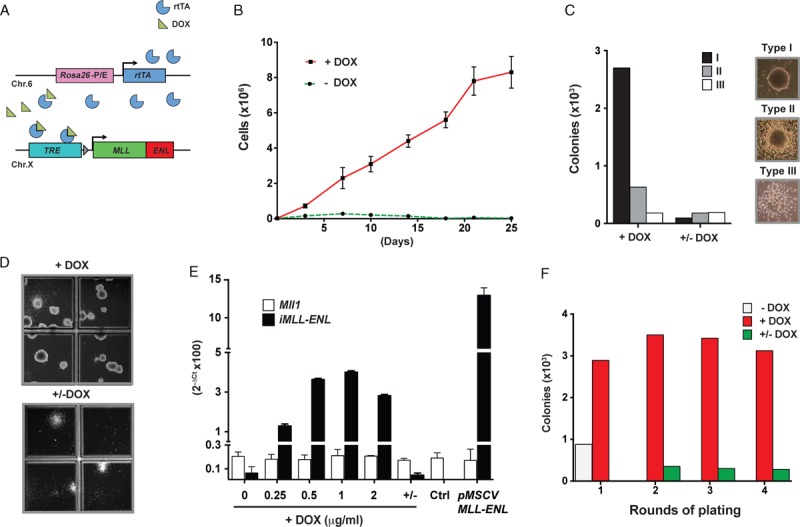
Ex vivo induction of iMLL-ENL provides aberrant self-renewal and blocks differentiation of HSPCs in a fully reversible manner. (A) MLL-ENL expression is controlled by a Tet-responsive element (TRE). The reverse tetracycline transactivator (rtTA) cassette was integrated into the Rosa26 locus and the MLL-ENL fusion gene was targeted into the Hprt locus. (B) Proliferation of iMLL-ENL BM cells grown in liquid cultures with growth factors (10 ng/mL of IL-3, IL-6, and IL-7, 100 ng/mL of SCF and Flt3L) in presence (red) of absence (green) of DOX (1 μg/mL) for up to 25 days. (C) Quantification of colony types formed in the 1st replate in the presence or absence of DOX. (D) Reversibility of transformation, loss of immature compact colony formation capacity, and differentiation of blasts into mature macrophage/monocyte like cells upon removal of DOX as shown by the representative MC cultures. (E) Expression of iMLL-ENL mRNA in ex vivo cultured BM cells exposed to increasing amounts of DOX compared with cells immortalized by retroviral MLL-ENL expression, as assessed by quantitative (TaqMan) RT-PCR analysis. (F) Number of colonies of serial replatings of iMLL-ENL BM cells in growth factor-containing MC with and without DOX, and upon DOX removal. BM = bone marrow, DOX = doxycycline, MC = methylcellulose.
To study the effects of iMLL-ENL expression in vivo, DOX (0.4 mg/mL) was administered to the drinking water of the mice. After a mean latency of 104.3 ± 16.9 days, all animals developed signs of disease (Fig. 2A). Importantly, DOX-exposed mice that carried only the iMLL-ENL or the rtTA allele, or iMLL-ENL mice maintained in the absence of DOX never developed any disease. Transplantation of 1 × 106 total BM cells from naive iMLL-ENL mice into lethally irradiated wild type (wt) mice receiving DOX also induced the disease in all recipients (Fig. 2A). Transfer of 1 × 105 leukemic cells, either originating from directly induced iMLL-ENL mice or originating from transplanted mice into sublethally irradiated DOX-pretreated secondary recipients also consistently developed the disease after a shorter latency (Fig. 2A). Upon disease development in 50% of a mouse cohort (Fig. 2B, arrow), discontinuation of DOX administration DOX resulted in the survival of all the remaining mice with the exception of 1 mouse with extensive organ infiltration (not shown). All symptomatic mice presented with elevated white blood cell counts (WBC), hepatosplenomegaly, lymphadenopathy, and excessive multiorgan infiltration by leukemic blasts (Fig. 2C and D, top right panel, and data not shown). Symptomatic mice that survived after DOX removal had normal WBC, blood smears, spleen, and liver weights measured after 60 to 120 days (Fig. 2C), with occasional focal infiltrations of cells with pyknotic appearing nuclei (Fig. 2D, bottom right panel).
Figure 2.

In vivo induction of iMLL-ENL results in a reversible acute leukemia expressing myeloid and lymphoid markers. (A) Kaplan-Meier survival plot of animals reconstituted with 106 naive BM from iMLL-ENL mice (median latency 72.0 ± 5.3 days, n = 10) or transplanted with 105 blasts from leukemic mice (median latency 15.4 ± 0.9 days, n = 5). Wild type recipients of naive BM cells were lethally irradiated (137Cs, 9Gy) and DOX-induced 2 weeks after reconstitution. Recipients of leukemic BM were sublethally irradiated (4.5Gy) and received DOX 2 days prior to bone marrow transplantation and onward. (B) DOX administration in the drinking water induced acute leukemia in iMLL-ENL (median latency of 104.3 ± 16.9 days, n = 10). Upon DOX removal (arrow) the remaining animals (n = 10) survived up to 300 days without developing the disease, except of 1 mouse with extensive multiorgan leukemic infiltration (cross). (C) White blood cell counts and spleen weight of diseased mice on DOX (+DOX) and after DOX removal (+/− DOX) measured at different time points. (D) Wright-Giemsa-stained blood smears and histopathology of the liver showing infiltration of leukemic blasts of different sizes. Upon DOX removal the blood smears were normal and only small residual infiltrations were found occasionally in the livers. (E) Top panel: representative flow cytometric analysis of leukemic blasts isolated from highly infiltrated BM grouped according to different size (“large,” “intermediate,” and “small”). Bottom panel: quantification of the representative FACS plot showing the percentage of “large,” “intermediate,” and “small” cells. The bars represent the percentage of mean of size measured from BM cells of 3 independently analyzed diseased iMLL-ENL mice. (F) Detailed flow cytometric analysis of BM cells from a representative diseased iMLL-ENL mouse (E), showing the percentage of cell populations expressing lymphoid (B220 and Cd19) versus myeloid markers (Mac-1 and Gr-1), grouped according to size. Cells were costained with antibodies for all the markers. Different populations were gated according to size (SSC-A vs FSC-A), singlet (FSC-A vs FSC-H), live (DAPI−), and expression of the respective markers. (G) Polymerase chain reaction analysis of the IgH D-J gene rearrangements using DNA isolated from the BM cells of a leukemic mouse (input) of single B220+ and double B220+/Cd19+ flow-sorted cells versus control DNA samples isolated from the ear, the spleen of a wild type (B6) mouse, and from a lymphoma of Eμ-c-myc transgenic mice (GL: 2.1 kb, DJH1: 1.4 kb, DJH2: 1.1 kb, DJH3: 0.7 kb, and DJH4: 0.2 kb). BM = bone marrow, DOX = doxycycline, FACS = fluorescence activated cell sorting, GL = germline.
Inspection of blood smears from diseased mice revealed the presence of large, intermediate-sized, and small leukemic blasts (Fig. 2D, top left panel). In symptomatic mice with highly (>95%) infiltrated BM, the majority of the tumor cells were of intermediate size (86.8 ± 7.4%), with minor fractions (1.7 ± 0.9% and 1.4 ± 0.3%) of cells of large and small size, respectively (Fig. 2E). Notably, 37% (±7.5, n = 3) of the small cells expressed the lymphoid marker B220, some also Cd19. A minority of the B220+ cells also expressed the myeloid markers Mac1 (8.1 ± 4.4%, n = 3) or Gr1 (4.4 ± 3.3%, n = 3) (Fig. S1C, Supplemental Digital Content 2). By contrast, the large- and intermediate-sized cells showed little expression of B220 or Cd19, but were mostly Mac1 or Mac-1/Gr1 positive (Figs. 2F and S1D, Supplemental Digital Content 2). PCR analysis showed that a fraction of B220+iMLL-ENL leukemic blasts underwent IgH D-J gene rearrangement.20 Whereas complete IgH D-J rearrangement was seen in normal controls, incomplete rearrangement, indicated by the presence of a germline (GL) band, was observed in B220+/Cd19+ cells from diseased iMLL-ENL mice (Fig. 2G). Collectively, iMLL-ENL is a novel robust inducible and reversible transgenic mouse model for acute leukemia expressing myeloid and lymphoid markers.
Expression signatures of iMLL-ENL mouse leukemia separate ALL patients carrying MLL rearrangements
To characterize the iMLL-ENL phenotype, we compared the gene expression programs of leukemic cells from diseased mice with FACS-sorted LSK from normal mice. Overall, we identified 1935 differentially expressed genes (absolute logFC > 2, false discovery rate [FDR] < 0.001) of which 780 genes showed increased and 1155 decreased levels in leukemic cells (Fig. 3A and B). Among the genes significantly upregulated in iMLL-ENL leukemic cells, we found many previously proposed MLL-fusion targets including Hoxa9, Hoxa10, Meis1, Eya1, Pbx3, S100a4, S100a8, and S100a9,19,20 as well as genes implicated in ALL and B-cell maintenance (Table S1, Supplemental Digital Content 3). We performed gene set enrichment analysis (GSEA) to compare the set of genes differentially expressed between leukemic iMLL-ENL and LSK cells with previously established expression signatures. We observed significant enrichments with signatures derived from mouse leukemia induced by overexpression of Hoxa9 and Meis1 (Fig. 3C).21 In addition, the signatures also showed similarities to those previously established from late hematopoietic progenitors obtained from normal mice (Fig. 3C). We validated MLL-fusion-dependent expression of Hoxa9 and Meis1 in blasts from iMLL-ENL and iMLL-AF9 mice cultured for 24 hours with or without DOX (Fig. 3D). Collectively, leukemic cells from diseased iMLL-ENL mice express many known MLL-fusion targets but also genes associated with ALL that were previously not described MLL-ENL+ mouse leukemia models.
Figure 3.

Cross-species transcriptome analysis of iMLL-ENL-derived leukemias revealed common genes that segregate ALL patients with from those without MLL-rearrangements. (A) Volcano plot showing fold change and statistical significance between wt LSK and iMLL-ENL-derived leukemic cells. The 1155 significantly lower expressed genes are plotted in blue and the 780 more highly expressed genes are plotted in red. Cutoffs used for differential expression: FDR < 0.001, absolute log fold change > 2 (same in panels B, C, E, F). (B) Heatmap showing relative expression (log2 level over mean) of 1935 genes differentially expressed between wt LSK and iMLL-ENL samples. Average expression is shown on the right as logCPM (green). (C) Gene set enrichment analysis (GSEA) results for 2 signatures of the C2 Molecular Signature Database (MsigDB) enriched in the iMLL-ENL/wt LSK signature. Left: “Immortalized by Hoxa9 and Meis1 Up” represents genes upregulated in mouse leukemia by overexpression of Hoxa9 and Meis1 (normalized enrichment score [NES]: 2.0880373; nominal P value: 0.0; FDR q value: 5.326164E−4; family-wise error rate [FWER] P value: 0.006). Right: “Hematopoiesis late progenitor” represents genes upregulated in BM-derived late hematopoietic progenitors (NES: 2.2291334; nominal P value: 0.0; FDR q value: 0.0; FWER P value: 0.0). (D) Hoxa9 and Meis1 mRNA expression measured by qRT-PCR in BM cells isolated from naive wt GMP and wt LSK progenitors and iMLL-ENL or iMLL-AF9 leukemic cell cultured in vitro for 24 hours with DOX or overnight without DOX. Relative expression levels were normalized to Gapdh mRNA levels expression and to expression in naive murine GMP. Results are the mean ± standard deviation of triplicates in the indicated AML groups (n = 3 per group). (E) Pair-wise correlation maps and hierarchical clustering of human patients with the t(11;19) (in purple) and germline MLL (in gray) genotypes. Correlations and clustering of patient samples were computed using expression values of human genes that are orthologous to mouse genes being part of the following 3 gene sets. Left panel: genes significantly differentially expressed between iMLL-ENL cells and wt LSKs. Middle panel: a control set of genes with the same expression distribution as the genes from the iMLL-ENL signature. Right panel: all genes expressed in iMLL-ENL and wt LSK samples. (F) Distribution of normalized expression levels (log2 CPM) in the 3 gene sets used in (E). ALL = acute lymphoblastic leukemia, AML = acute myeloid leukemia, DOX = doxycycline, GMP = granulocyte-macrophage progenitors, LSK = Lin− Sca-1+ c-Kit+.
We next asked whether the expression signature from iMLL-ENL mouse leukemia versus LSK would also be informative in human datasets. We tested whether the signatures would be able to distinguish ALL patients potentially carrying MLL gene rearrangements from a cohort of primary infant ALL expression profiles typified by translocations t(4;11) (n = 29), t(11;19) (n = 21), and t(9;11) (n = 8), or the absence of MLL translocations (n = 14).22 We found that the iMLL-ENL expression signatures fully segregated MLL-ENL+ patients, to some extent also MLL-AF4+ patients and less efficiently MLL-AF9+ patients from patients with no MLL rearrangements (Figs. 3E and S2A and B, Supplemental Digital Content 4). By contrast, expression signatures derived from populations of randomly selected genes with similar expression-level distributions or from all genes did not segregate patients according to their genetic aberrations (Figs. 3E and F and S2A and B, Supplemental Digital Content 4). Thus, this analysis shows that genes differentially expressed in iMLL-ENL versus wt LSK are enriched for genes differentially expressed in human MLL-rearranged leukemia. The resulting signature of differentially expressed genes did, however, not allow clustering of human ALLs according to their MLL-status (not shown). In summary, these findings support the notion that iMLL-ENL mice develop a disease characterized by a gene expression signature matching to signatures of t(11;19)+ acute leukemia in patients.
The cellular origin determines the leukemogenic activity of iMLL-ENL
Next, we determined the transformation potential of iMLL-ENL in different cell populations of the hematopoietic hierarchy. First, we compared in vitro growth of flow-sorted iMLL-ENL hematopoietic LSK and GMP in the presence of myeloid and lymphoid growth factors and DOX. We found that LSK cells expanded significantly faster than GMP (Fig. 4A). However, fusion gene expression levels did not significantly differ between LSK- and GMP-derived iMLL-ENL cells (Fig. 4B). To address the cellular origin in vivo, we transplanted LT-HSC and GMP from naive iMLL-ENL mice into lethally irradiated recipients pretreated with DOX. Hereby we observed that 500 LT-HSC were sufficient to induce the leukemic phenotype after a median latency of 169.6 ± 27.5 days whereas 5000 GMP never resulted in any disease within an observation period of >400 days (Fig. 4C). This difference might not solely be attributed to inherent differences in engraftment capacities of LT-HSC and GMP, as transplantation of the same number of GMP from iMLL-AF9 mice was sufficient to induce AML.18 We also transplanted equal numbers (1 × 104) of sorted MPP4, LMPP, and CMP progenitor cells and found that, although the disease could be induced by all of these populations, the latency was shorter upon transfer of LMPP (54.4 ± 11.7, n = 5) than of MPP4 (61.0 ± 13.7, n = 5). Notably, only 3 out of 5 mice transplanted with CMP developed the disease within the observation period of 120 days (Fig. 4D). The induced disease was similar to directly induced mice with extensive multiorgan infiltration by leukemic blasts of different sizes expressing myeloid and lymphoid markers (Fig. 4E). Notably, the amount of B220+ tumor cells was significantly higher in iMLL-ENL tumors originating from LT-HSC, LMPP, or MPP4 than from CMP (Fig. 4F). Collectively, our data suggest that iMLL-ENL preferentially immortalizes hematopoietic stem or early multipotent precursor cells rather than more committed myeloid progenitors.
Figure 4.

iMLL-ENL preferentially transforms hematopoietic stem cells and pluripotent rather than more committed myeloid progenitor cells. (A) Ex vivo expansion of naive iMLL-ENL LSK- and GMP-derived cells in liquid cultures containing hIL-6 (10 ng/mL), mIL-3 (6 ng/mL), mIL-7 (10 ng/mL), mSCF (100 ng/mL), and mFlt-3L (100 ng/mL) in the presence of DOX. (B) Expression of iMLL-ENL mRNA of sorted LSK and GMP cells cultured ex vivo in MC for 3 rounds with growth factors in the presence or absence of DOX. Relative mRNA expression levels were normalized to Gapdh expression and are expressed as 2−ΔCt. Results the mean values ± standard deviation of duplicates. (C) Average survival of lethally irradiated mice transplanted with the indicated numbers (1000, 500, 250, and 125) of naive iMLL-ENL LT-HSCs or GMPs (5000, 2500, and 1250) into wild type recipients on DOX. Mice receiving 500 to 1000 LT-HSCs developed acute leukemia (median latency 169.6 ± 27.3 days, n = 10) whereas mice receiving 125 to 250 LT-HSCs (n = 10) or GMPs (n = 15) did not develop the disease within 400 days. Mice transplanted with the indicated cells but kept off DOX served as controls (n = 10). (D) Average survival of lethally irradiated mice transplanted with 104 of transgenic MPP4s or LMPPs or CMPs on DOX. Mice receiving MPP4s and LMPPs developed acute leukemia with a median latency of 61.0 ± 13.7 days and 54.4 ± 11.7, respectively (n = 5 per group), whereas 3 out of 5 mice receiving CMPs (n = 5) develop the disease within 120 days. (E) Wright-Giemsa-stained blood smear showing presence of leukemic blasts of different size; and histopathology of the spleen showing infiltration. (F) Quantification of flow cytometry analysis of “small” cell fraction showing the percentage of B220+ in BM of leukemic mice transplanted with different hematopoietic progenitor populations. Notably LT-HSC-derived disease was associated with a significantly higher fraction of B220+ BM cells than CMP-derived leukemia (P = 0.002, n = 2, unpaired t test). BM = bone marrow, CMP = common myeloid progenitor, DOX = doxycycline, GMP = granulocyte-macrophage progenitors, hIL-6 = human interleukin-6, LMPP = lymphoid primed multipotent progenitor cells, LSK = Lin− Sca-1+ c-Kit+, LT-HSC = long-term hematopoietic stem cells, MC = methylcellulose, MPP4 = multipotent progenitors, mSCF = murine stem cell factor.
B220+iMLL-ENL leukemic blasts are enriched for leukemia initiating cells
The leukemic disease in iMLL-ENL mice was composed by different populations of cells that could be distinguished by the expression level of the B220 surface marker (Fig. 5A, left panel). As B220+ cells were previously proposed in another mouse model to contain leukemic stem cells,23 we compared the leukemia initiation potential by transplanting 1 × 105 B220+ or B220− leukemic blasts sorted from total mBM ex vivo cultures (48 hours) into sublethally irradiated recipients. Transplantation of B220+ leukemic blasts propagated the disease after a significantly shorter latency (Fig. 5A, right panel) than the B220− cells (15.6 ± 0.5 vs 22.5 ± 1.1 days, P ≤ 0.0028). Both B220+ and B220− leukemic cells isolated from terminally diseased mice expressed similar levels of iMLL-ENL mRNA and remained fully DOX-dependent (Fig. S3A, Supplemental Digital Content 5). Leukemic mice transplanted with B220+ cells showed more extensive cell infiltrations in the BM, spleen, liver, intestines, kidney, and lungs as compared to the recipients of B220− cells. All mice, however, ultimately succumb to the same disease as directly induced iMLL-ENL mice, characterized by leukemic blasts of different size expressing myeloid and lymphoid surface markers (Fig. 5B and not shown). Similar to primary induced iMLL-ENL mice, the tumor cells of diseased mice expressed myeloid and lymphoid surface markers. Interestingly, tumor cells of mice transplanted with B220+ cells again expressed higher B220 levels than those transplanted with B220− cells (Fig. S3B, Supplemental Digital Content 5 and data not shown). These findings suggest that B220+ cells have higher leukemia initiation potential than B220−iMLL-ENL cells.
Figure 5.

B220+iMLL-ENL leukemic blasts are enriched for leukemia initiating cells. (A) BM-derived leukemic blasts were cultured overnight in medium with hIL-6 (10 ng/mL), mIL-3 (6 ng/mL), mIL-7 (10 ng/mL), mSCF (100 ng/mL), and mFlt-3L (100 ng/mL) and DOX, then sorted (left panel) according to B220+ versus B220− expression and transferred into secondary recipients. Kaplan-Meier plot (right panel): the difference of the latency periods was assessed by a log-rank test (P < 0.0028). (B) Histopathology of spleen, lungs, and liver revealed massive infiltration of leukemic blasts in mice transplanted with 1 × 105iMLL-ENL B220− versus B220+ ex vivo cultured mBM cells with complete loss of normal organ architecture in the latter. (C) Pair-wise correlation maps and hierarchical clustering of human patients with the t(11;19) (in purple) and germline MLL (in gray) genotypes. Correlations and clustering of patient samples were computed using expression values of human genes that are orthologous to mouse genes being part of the following 3 gene sets. Left panel: genes significantly differentially expressed between iMLL-ENL B220+ and iMLL-ENL B220− samples. Right panel: a control set of genes with the same expression distribution as the genes from the iMLL-ENL B220+/B220− signature. (D) Distribution of normalized expression levels (log2 CPM) in the 2 gene sets used in (C). The distribution of normalized expression values of all genes expressed in both samples is given as a comparison. BM = bone marrow, DOX = doxycycline, hIL-6 = human interleukin-6, mSCF = murine stem cell factor.
To better characterize the biological differences between iMLL-ENL leukemia induced by B220+ or B220− cells, we compared their gene expression signatures. We found 335 differentially expressed genes of which 313 genes were expressed at significantly higher levels (absolute logFC > 1; FDR < 0.01) and only 22 genes at significantly lower levels in B220+ compared with B220− cells (Fig. S3C and Table S2, Supplemental Digital Contents 5 and 6 and respectively). Gene ontology (GO) analysis revealed terms including “regulation of cell motility,” “positive regulation of cellular component movement,” “positive regulation of locomotion,” “granulocyte migration,” and “regulation of cell activation” (P < 10−15) (Fig. S3D, Supplemental Digital Content 5). GSEA indicated significant overlaps between the genes more highly expressed in B220+ versus by B220− cells with genes up-regulated in solid cancers harboring activating mutations in the K-RAS oncogene or the STK33 serine-threonine kinase, previously implied in RAS-mediated transformation, and with genes upregulated in pathways involved in K-RAS and p53 signaling (Fig. S3E, Supplemental Digital Content 5).24,25
Finally, we investigated whether the B220+ versus B220−iMLL-ENL expression signature would enable identification of ALL patients carrying MLL gene rearrangements within a cohort of primary infant ALL expression profiles.22 The genes significantly differentially expressed in B220+ versus B220− enabled clustering of MLL-ENL+ patients (n = 21) from those without MLL1 gene rearrangements (n = 14) (Fig. 5C, left panel), contrary to a control gene set or to all genes (Fig. 5D). By contrast, the signature was less effective in separating MLL-AF4+ ALL patients (n = 29) and not effective in segregating MLL-AF9+ ALL patients (n = 9) from patients without MLL rearrangements (Fig. S3F and G, Supplemental Digital Content 5). Thus, the B220+iMLL-ENL signature is enriched for genes that are differentially expressed in MLL-ENL+ as compared to leukemia patients without any MLL-gene rearrangements.
MLL-ENL protein levels and leukemic transformation
To address whether the expression levels correlate with transformation activity we compared transgenic iMLL-ENL mRNA levels with the expression level of endogenous Mll1. In the absence of DOX or within 24 hours after DOX removal, iMLL-ENL mRNA levels were below Mll1 expression (Fig. 1E), a condition in which we observed rapid cell differentiation (Fig. S1A and B, Supplemental Digital Content 2). In leukemic iMLL-ENL cells from diseased mice, the estimated fusion mRNA expression level consistently exceeded Mll1 (Fig. 6A). Notably, expression of MLL-ENL in retrovirally transduced BM cells (rMLL-ENL) appeared 5- to 10-fold higher than iMLL-ENL mRNA expression in BM derived in vitro immortalized cell lines or in leukemic cells from diseased iMLL-ENL mice. In similar, we found that the relative fusion transcript levels exceeded those of endogenous MLL1 in leukemic blasts from 5 t(11;19)+ ALL patients (Fig. 6B). In addition, MLL-ENL protein expression clearly exceeded the expression of wild type MLL in the MLL-ENL+ KOPN8 cells, and about equal to wild type MLL in BM and spleen cells of diseased iMLL-ENL mice (Fig. 6C). These data suggest that MLL-ENL protein levels equal to or exceeding that of the nonrearranged MLL is necessary for leukemic transformation by the MLL-ENL fusion.
Figure 6.

Expression of iMLL-ENL levels determine cellular transformation in vitro and in vivo. (A) The expression of Mll1 and MLL-ENL mRNA was measured in naive and leukemic BM cells from transgenic mice and BM cells from symptomatic mice transplanted with retrovirally (pMSCV-MLL-ENL) transduced cells. Relative mRNA expression levels were normalized to Gapdh expression and are expressed as 2−ΔCt. Results are shown as mean values ± standard deviation of duplicates. (B) Expression of MLL1 and MLL-ENL mRNA in leukemic cells of human patients diagnosed with ALL and >80% infiltration. Relative mRNA expression levels were normalized to Gapdh expression and are expressed as 2−ΔCt. Results are shown as mean values ± standard deviation of duplicates. (C) Western blot analysis of total cell lysates of K562 (CML, wild type MLL), KOPN8 (ALL, t(11;19)+), BM of healthy mice, and BM and spleen cells of diseased iMLL-ENL mice. The blot was probed with an antibody recognizing the MLL-N-terminus. KOPN8 cells express an MLL-ENL fusion of calculated size of about 170 kDa,29 while the iMLL-ENL fusion results in a protein of 220 kDa.16 Bands were quantified according to intensity, and calculated as fold change normalized to wild type MLL. ALL = acute lymphoblastic leukemia, BM = bone marrow, CML = chronic myelogenous leukemia.
Discussion
We established an inducible transgenic mouse for MLL-ENL-driven acute mixed lymphomyeloid leukemia. Transplanting naive LT-HSCs, MPP4, or LMPP progenitors into DOX-treated recipients efficiently induced the disease phenotype. By contrast, transplantation of CMPs resulted in disease only in some animals and transfer of even more committed GMPs failed to induce the disease. These results contrast strongly to a comparable inducible transgenic mouse model of MLL-AF9-driven AML: here the leukemic phenotype was induced by transplanting iMLL-AF9 expressing LT-HSCs, short-term HSCs, CMPs, or GMPs, which supports a concept for an MLL fusion-specific transformation potential of particular cellular compartments.18
Intriguingly, a recent study claimed that HSCs are intrinsically protected against MLL-ENL-mediated transformation.15 This conclusion emerged from experiments with a different DOX inducible transgenic mouse strain in which expression of the identical human MLL-ENL fusion cDNA,16 integrated in a DOX responsive operon in the 3′ UTR of the murine Col1a1 gene, is controlled by the M2 reverse tetracycline transactivator (M2-rtTA), the latter constitutively expressed from the Rosa26 locus. In contrast to iMLL-ENL, transplantation of Col1a1-tetO-MLL/ENL LT-HSCs never induced a disease in DOX-treated recipients. Likewise, leukemia induction by transferring MPPs was very poor, whereas GMPs efficiently induced the disease. Interestingly, the expression levels of the MLL-ENL fusion transcripts from the Col1a1-tetO-MLL/ENL locus in HSCs and MPPs remained clearly below endogenous Mll1, while it exceeded Mll1 in GMPs. In contrast to our iMLL-ENL model, the induced disease was consistently pure AML.15
Although both models (iMLL-ENL and Col1a1-tetO-MLL/ENL) are limited in pinpointing the disease origin to specific cell types, our work suggests that expression of MLL-ENL mRNA exceeding Mll1 might be important to potently induce LT-HSCs or MPP cell-derived disease. Low iMLL-ENL mRNA expression (comparable or lower than endogenous Mll1) in unexposed naive iMLL-ENL mice (due to the inherent leakiness of the rtTA transcriptional transactivator) never resulted in disease during a normal lifespan, suggesting that a certain threshold level is necessary for the leukemogenic activity in vivo. Importantly, akin to the iMLL-ENL mouse model, we also found that MLL-ENL fusion transcripts were generally higher than endogenous MLL1 levels in 5 MLL-ENL+ leukemia patients. Importantly, we observed abundant amounts of the fusion protein in human t(11;19)+ KOPN8 cells but also in leukemic blasts from diseased iMLL-ENL mice. This finding extends previous work by others in other human MLL-ENL+ cell lines suggesting that fusion protein expression indeed exceeds MLL1 in leukemic blasts.26 While this observation warrants further validation in larger patients cohorts, it is intriguing to postulate that the relative expression levels of MLL fusions versus endogenous MLL1 might be a critical factor for the transforming activity in different cells of the hematopoietic hierarchy. Recent work indeed suggested that stabilization of the wild type MLL1 protein displaces MLL fusions from critical targets and impairs transformation, which suggests competition between wild type and rearranged MLL1.27
GSEA identified some similarities with gene expression profiles derived from previous studies on MLL fusions verifying that iMLL-ENL leukemic cells also express a common signature, similar to what we observed in the iMLL-AF9 model.18 Besides the known MLL fusion targets, gene expression profiling of leukemic cells from diseased iMLL-ENL mice identified multiple putative target genes never described in previous mouse models for this leukemia. This difference may result from the fact that iMLL-ENL model causes mixed-lineage leukemia composed of blasts expressing myeloid and lymphoid surface markers mostly likely originating from multipotent HSPCs.
Cross-species comparison of iMLL-ENL expression signatures with those from ALL patients was able to segregate patients with and without MLL1 gene rearrangements.22 Interestingly, clustering of ALL patients according to their MLL rearrangement status could only be achieved by signatures generated from the comparison of the gene expression of the leukemic cells with normal LSK but not with normal GMP. As activation of the fusion in stem cells but not in GMP initiated the disease, this observation supports furthermore that the cell of origin for the iMLL-ENL leukemia is within the stem cell and/or early hematopoietic progenitor rather than in more committed myeloid progenitor compartment (not shown).
Patients with t(11;19)+ leukemia often display immature immunoglobulin rearrangements and present with a pro-B cell lymphoblastic or mixed-lineage leukemia phenotype.3,4iMLL-ENL induced acute leukemia was characterized by the coexistence of blasts of various sizes that expressed myeloid and/or lymphoid markers or exclusively lymphoid markers in a small fraction of the cells. This intriguing mixed phenotype differs from previous mouse models expressing MLL-ENL by retroviral transduction, interchromosomal recombination or from conditional transgenes that resulted in MPN and/or AML. However, retrovirally MLL-ENL expressing mouse BM cells cultured under B-cell lineage favoring conditions resulted in the outgrowth of cells expressing B-cell markers Cd19 and B220. Transplanting immature Cd19−/B220+ cells resulted in a leukemic phenotype in which the blasts still maintained a monocytoid morphology suggesting that MLL-ENL transformed a lymphoid/myeloid precursor leading to a bi-phenotypic-like leukemia.28 By transplanting leukemic blasts from diseased iMLL-ENL mice we found that B220+ leukemic cells were more potent to propagate leukemia than B220− cells. This situation is somehow similar to mice that developed acute leukemia by transplanting BM retrovirally expressing the CALM-AF10 fusion gene with leukemia propagating cells expressing B220 with IgH D-J gene rearrangements.23
Acknowledgments
The authors thank R. Slany (Erlangen, Germany) for providing the pMSCV-MLL-ENL expression plasmid and the KOPN8 cell line; D. Labes, E. Traunecker, and P. Tspaogas for FACS assistance; U. Schneider and his team for animal husbandry; B. Kuchemann for his assistance in establishing the transgenic line; T. Roloff and his team for transcriptional profiling (FMI functional genomics); P. Matthias and Vincent Pionel (FMI, Basel) for providing material; and R.W. Stam (Rotterdam) and J. De Boer (London) for practical technical support.
Author contributions
Vaia Stavropoulou established the transgenic mouse line, designed and performed experiments, and wrote the manuscript.
Marwa Almosailleakh: designed and performed experiments and wrote the manuscript.
Hélène Royo: designed and performed bioinformatic data analysis.
Jean-François Spetz and Patrick Kopp: established the transgenic mouse line.
Sabine Juge and Laurent Brault: designed and performed experiments.
Michelina Iacovino and Michael Kyba: provided essential material.
Alexandar Tzankov: supervised histopathology analysis.
Michael B. Stadler: supervised bioinformatic data analysis.
Gianni Cazzaniga: provided patient-derived materials.
Antoine H.F.M. Peters: designed, supervised experiments and data analysis, and wrote the manuscript.
Juerg Schwaller: designed, performed, supervised experiments and data analysis, and wrote the manuscript.
Supplementary Material
Supplementary Material
{kind=link}
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Footnotes
Citation: Stavropoulou V, Almosailleakh M, Royo H, Spetz J-F, Juge S, Brault L, Kopp P, Iacovino M, Kyba M, Tzankov A, Stadler MB, Cazzaniga G, Peters AHFM, Schwaller J. A Novel Inducible Mouse Model of MLL-ENL-driven Mixed-lineage Acute Leukemia. HemaSphere, 2018;2:4. http://dx.doi.org/10.1097/HS9.0000000000000051
Funding/support: This work was supported by grants from the Swiss National Science Foundation (SNF-31003A_130661 and 31003A_149714/1), Swiss Cancer League (OCS-2357-02-2009, OCS-02778-02-2011, and KFS-3019-08-2012), and SystemsX.ch (Cell plasticity), the Novartis Research Foundation (14B058). HR was supported by grants of the Peter und Traudl Engelhorn foundation and the Marie-Curie program (FP7-PEOPLE-2013-IEF). MBS and AHFMP were supported by the Novartis Research Foundation. JS was supported by the Gertrude Von Meissner Foundation Basel.
Disclosure: The authors have indicated they have no potential conflicts of interest to disclose.
Marwa Almosailleakh and Hélène Royo have shared second authorship.
Antoine H.F.M. Peters and Juerg Schwaller have shared senior authorship.
References
- 1.Meyer C, Burmeister T, Groger D, et al. The MLL recombinome of acute leukemias in 2017. Leukemia 2018; 23:273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubnitz JE, Morrissey J, Savage PA, et al. ENL, the gene fused with HRX in t(11;19) leukemias, encodes a nuclear protein with transcriptional activation potential in lymphoid and myeloid cells. Blood 1994; 84:1747–1752. [PubMed] [Google Scholar]
- 3.Hayashi Y, Kobayashi Y, Hirai H, et al. Immunoglobulin heavy chain gene rearrangements and mixed lineage characteristics in acute leukemias with the 11;19 translocation. Cancer 1988; 61:712–720. [DOI] [PubMed] [Google Scholar]
- 4.Hudson MM, Raimondi SC, Behm FG, et al. Childhood acute leukemia with t(11;19) (q23;p13). Leukemia 1991; 5:1064–1068. [PubMed] [Google Scholar]
- 5.Lavau C, Szilvassy SJ, Slany R, et al. Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX-ENL. EMBO J 1997; 16:4226–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cozzio A, Passegue E, Ayton PM, et al. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev 2003; 17:3029–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ren R. Modeling the dosage effect of oncogenes in leukemogenesis. Curr Opin Hematol 2004; 11:25–34. [DOI] [PubMed] [Google Scholar]
- 8.Haviernik P, Bunting KD. Safety concerns related to hematopoietic stem cell gene transfer using retroviral vectors. Curr Gene Ther 2004; 4:263–276. [DOI] [PubMed] [Google Scholar]
- 9.Cano F, Drynan LF, Pannell R, et al. Leukaemia lineage specification caused by cell-specific Mll-Enl translocations. Oncogene 2008; 27:1945–1950. [DOI] [PubMed] [Google Scholar]
- 10.Chambers JS, Tanaka T, Brend T, et al. Sequential gene targeting to make chimeric tumor models with de novo chromosomal abnormalities. Cancer Res 2014; 74:1588–1597. [DOI] [PubMed] [Google Scholar]
- 11.Drynan LF, Pannell R, Forster A, et al. Mll fusions generated by Cre-loxP-mediated de novo translocations can induce lineage reassignment in tumorigenesis. EMBO J 2005; 24:3136–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forster A, Pannell R, Drynan LF, et al. Engineering de novo reciprocal chromosomal translocations associated with Mll to replicate primary events of human cancer. Cancer Cell 2003; 3:449–458. [DOI] [PubMed] [Google Scholar]
- 13.Ono R, Masuya M, Nakajima H, et al. Plzf drives MLL-fusion-mediated leukemogenesis specifically in long-term hematopoietic stem cells. Blood 2013; 122:1271–1283. [DOI] [PubMed] [Google Scholar]
- 14.Takacova S, Slany R, Bartkova J, et al. DNA damage response and inflammatory signaling limit the MLL-ENL-induced leukemogenesis in vivo. Cancer Cell 2012; 21:517–531. [DOI] [PubMed] [Google Scholar]
- 15.Ugale A, Norddahl GL, Wahlestedt M, et al. Hematopoietic stem cells are intrinsically protected against MLL-ENL-mediated transformation. Cell Rep 2014; 9:1246–1255. [DOI] [PubMed] [Google Scholar]
- 16.Slany RK, Lavau C, Cleary ML. The oncogenic capacity of HRX-ENL requires the transcriptional transactivation activity of ENL and the DNA binding motifs of HRX. Mol Cell Biol 1998; 18:122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iacovino M, Bosnakovski D, Fey H, et al. Inducible cassette exchange: a rapid and efficient system enabling conditional gene expression in embryonic stem and primary cells. Stem Cells 2011; 29:1580–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stavropoulou V, Kaspar S, Brault L, et al. MLL-AF9 expression in hematopoietic stem cells drives a highly invasive AML expressing EMT-related genes linked to poor outcome. Cancer Cell 2016; 30:43–58. [DOI] [PubMed] [Google Scholar]
- 19.Somervaille TC, Cleary ML. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006; 10:257–268. [DOI] [PubMed] [Google Scholar]
- 20.Krivtsov AV, Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006; 442:818–822. [DOI] [PubMed] [Google Scholar]
- 21.Wang QF, Wu G, Mi S, et al. MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood 2011; 117:6895–6905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stam RW, Schneider P, Hagelstein JA, et al. Gene expression profiling-based dissection of MLL translocated and MLL germline acute lymphoblastic leukemia in infants. Blood 2010; 115:2835–2844. [DOI] [PubMed] [Google Scholar]
- 23.Deshpande AJ, Cusan M, Rawat VP, et al. Acute myeloid leukemia is propagated by a leukemic stem cell with lymphoid characteristics in a mouse model of CALM/AF10-positive leukemia. Cancer Cell 2006; 10:363–374. [DOI] [PubMed] [Google Scholar]
- 24.Azoitei N, Hoffmann CM, Ellegast JM, et al. Targeting of KRAS mutant tumors by HSP90 inhibitors involves degradation of STK33. J Exp Med 2012; 209:697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scholl C, Frohling S, Dunn IF, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 2009; 137:821–834. [DOI] [PubMed] [Google Scholar]
- 26.Yokoyama A, Lin M, Naresh A, et al. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 2010; 17:198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang K, Volk AG, Haug JS, et al. Therapeutic targeting of MLL degradation pathways in MLL-rearranged leukemia. Cell 2017; 168:59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeisig BB, Garcia-Cuellar MP, Winkler TH, et al. The oncoprotein MLL-ENL disturbs hematopoietic lineage determination and transforms a biphenotypic lymphoid/myeloid cell. Oncogene 2003; 22:1629–1637. [DOI] [PubMed] [Google Scholar]
- 29.Wilkinson AC, Ballabio E, Geng H, et al. RUNX1 is a key target in t(4;11) leukemias that contributes to gene activation through an AF4-MLL complex interactions. Cell Rep 2013; 3:116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.