

Abstract
Inhibitors of human α-l-fucosidases, tissue α-l-fucosidase (tFuc), and plasma α-l-fucosidase reportedly play roles in multiple diseases, suggesting their therapeutic potential for gastric disease associated with Helicobacter pylori and fucosidosis. Terminal fucose linkages on glycoproteins and glycolipids are a natural substrate for both enzymes; however, there are currently no fluorogenic substrates allowing their cellular evaluation. Here, we described the development of novel three-color fluorogenic substrates for lysosome-localized tFuc that exhibited excellent specificity and sensitivity in three human cell lines. Additionally, we developed a cell-based high-throughput inhibitor screening system in a 96-well format and a cell-based inhibitory activity evaluation system in a 6-well format for tFuc inhibitors using this substrate, which allowed accurate quantification of the inhibition rate. Moreover, analysis of significant changes in gene expression resulting from 30% inhibition of tFuc in HeLa cells revealed potential roles in gastric disease.
Keywords: α-l-Fucosidase, fluorogenic substrate, inhibitor, high-throughput screening, DNA microarray
The FUCA1 and FUCA2 genes constitute the two α-l-fucosidase genes in the human genome and encode tissue α-l-fucosidase (tFuc) and plasma α-l-fucosidase (pFuc), respectively. tFuc plays an important role in hydrolyzing α-1,6-linked fucose linkages at the nonreducing end of N-acetylglucosamine (GlcNAc) on glycoproteins and glycolipids presented on lysosomes, with tFuc mutations associated with fucosidosis as an autosomal-recessive lysosomal-storage disease.1 By contrast, pFuc is a secreted protein that hydrolyzes α-1,6-linked fucose linkages of glycans in the extracellular region, with pFuc critical for Helicobacter pylori adhesion, growth, and pathogenicity related to the development of gastric cancer.2 A variety of physiological and pathological events are associated with fucose-containing glycoconjugates, with studies of fucosidase inhibition reporting their relevance to inflammation,3 antigenic determination,4 cystic fibrosis,5 and tumor progression.6 Such inhibitors could potentially be used to study tFuc and pFuc functions to promote the development of therapeutic agents.7 Although inhibitor studies have used commercially available α-l-fucosidase from bovine kidney for in vitro evaluation,8,9 few studies report inhibition of human α-l-fucosidase in vitro.10 To develop α-l-fucosidase inhibitors into lead compounds for drug development, it is necessary to evaluate their inhibitory activities against human-origin enzymes. Similarly, to provide an appropriate membrane permeability for inhibitors, suitable screening methods are required using human cultured cells. Importantly, there are currently no reports of purified human tFuc and pFuc exhibiting α-l-fucosidase activity, and although recombinant α-l-fucosidase derived from FUCA1 is commercially available, it is expensive and can only be obtained in small amounts. Therefore, in this study, our goal was to develop a method for screening and evaluating human tFuc and pFuc inhibitors using a cell-based high-throughput screening (HTS) system. As the first target inhibitor enzyme, we selected tFuc expressed in lysosomes from adherent human cultured cells. By contrast, the pFuc, which is secreted into the extracellular space and potentially shows a deficient activity in cells, was selected as the next target. In a previous report, pFuc was secreted only under coculture conditions wherein host human cells were infected with H. pylori.11
Previous inhibitor studies used commercially available nitrophenyl α-l-fucopyranoside derivatives as colorimetric tFuc substrates and 4-methylumbelliferyl α-l-fucopyranoside as a fluorogenic tFuc substrate.8−10 At the cellular level, only an α-l-fucopyranosyl anthracenecarboximide derivative in human embryonic kidney 293T cells and murine aneuploid fibrosarcoma cells has been reported as a substrate.12 Additionally, an enzyme inhibitor screening and enzyme activity-based cell imaging of adherent cultured cells generally require a fluorogenic substrate; however, use of 4-methylumbelliferone as a fluorophore will be ineffective at low pH, given the location of tFuc, an acidic organelle (pH ≈ 3.5).13 Therefore, we chose resorufin (excitation, emission: 571 and 585 nm, respectively), 2-methyl TokyoGreen (2MeTG; 491 and 510 nm, respectively), and 4-trifluoromethylumbelliferone (TFMU; 385 and 502 nm, respectively) as red, green, and blue fluorophores based on their distinct excitation and emission wavelengths, excellent fluorescence quantum yields, and low pH-dependent changes in fluorescence.
For use as tFuc-specific fluorogenic substrates, it is common to conjugate these fluorophores at the C1 position of α-l-fucopyranoside similar to the commercial substrates. However, we previously raised a concern about the poor binding affinity of such designed substrates for the active site of tFuc, which is an exo-acting glycan hydrolase.14,15 The active sites of hydrolases are bound by their respective substrates at the subsite −1, which recognizes a sugar residue at the nonreducing end of the substrate, and the subsite +1, which recognizes the penultimate sugar residue at the nonreducing end. Therefore, fluorophores representing these substrates might be incompatible with the subsite +1 due to steric hindrance, thereby requiring a different approach to design fluorogenic substrates for tFuc. In our previous study, we developed a quinone methide cleavage (QMC) substrate-design platform to investigate the activities of Golgi-localized β-galactosidase14 and β-allosidase15 as exo-acting glycan hydrolases. In the present study, we used this platform to design fluorogenic substrates for tFuc, and they exhibited improved fluorescence intensity at low pH and binding affinity to subsite +1 of the active site. We expected that the designed substrates (Figure 1) would effectively report tFuc activity based on the ability of the α-l-fucopyranoside residue and benzyl moieties to occupy the subsite −1 and subsite +1, respectively, while the attached fluorophore would remain located outside of the active site.
Figure 1.

Structure of QMC platform-based fluorogenic substrates for tFuc localized in the lysosome in adherent cultured cells.
The hydrolysis reaction catalyzed by tFuc will release the fluorophore via cleavage of the α-l-fucopyranoside linkage, thereby initiating a signal in the lysosome reporting tFuc location. To improve the cell-membrane permeability of the substrates (4–6), we incorporated acetyl modifications to obtain substrates 1 through 3, which returned distribution coefficient (clog D7.4) values of 4.54, 3.70, and 7.78, respectively, at physiological pH in cells, indicating good membrane permeability based on a previous study using Caco-2 cells.16 Furthermore, we previously reported that acetyl modification of substrate hydroxyl groups effectively improved membrane permeability without influencing the detection of glycosidase activity in living cells.14,15
To assess the affinity of the designed substrates for the tFuc active site, we performed molecular docking simulations using the crystallographic structure of Thermotoga maritima α-l-fucosidase (PDB ID: 2ZXD(17)) and three types of ligands: QMC platform-based substrates 4 through 6, α-1,6-fucose-linked GlcNAc as a natural substrate, and 2MeTG α-l-fucopyranoside (a model compound with 2MeTG instead of a commercial substrate). Due to the lack of a tFuc crystal structure, we used the T. maritima enzyme from the same glycoside hydrolase family as tFuc.17 The docking models of substrates 4 through 6 and a natural substrate (Figure 2 and Figure S4) demonstrated potential interactions between each ligand and the subsite −1 and subsite +1 in the active site. The 1-position carbon atom of the fucopyranoside moiety of substrate 6 and that of the natural substrate were located 3.8 and 3.6 Å from the carboxyl group of the Asp224 residue as a catalytic nucleophile amino acid residue, respectively, with both ligands predicted to occupy both subsite −1 and subsite +1 (Figure 2A,B). By comparing the ligand conformations of bound substrates 4 through 6 and the natural substrate, the configuration of the fucopyranoside moiety and the glycosidic bond hydrolyzed by the enzyme were found to show a very substantial overlap (Figure 2D left). By contrast, the fucopyranoside moiety of 2MeTG α-l-fucopyranoside did not sufficiently occupy the subsite −1 (Figure 2C), and the position of the glycosidic bond between the catalytic residue, Asp224, and the decomposition was inappropriate (Figure 2D right) due to the incompatibility of sizable molecular size fluorophores to subsite +1. The simulation results suggested the efficacy of the designed substrates to appropriately bind the target active site, thereby suggesting their efficacy as tFuc-specific fluorogenic substrates. Additional details concerning the computational analyses are presented in the Supporting Information.
Figure 2.
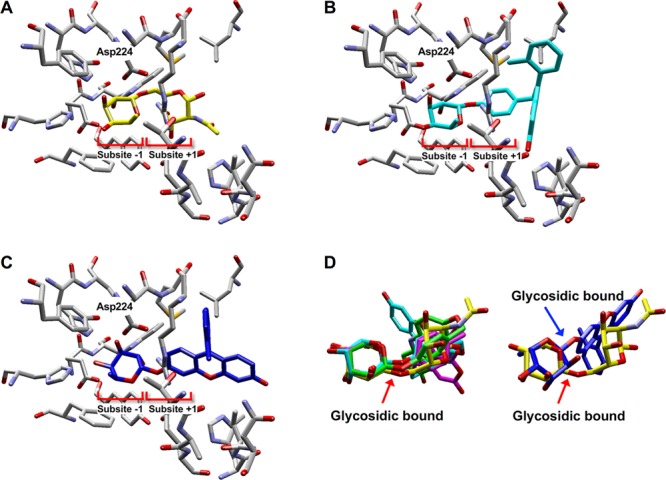
Evaluation of the suitability of the fluorogenic substrates for α-l-fucosidase by docking simulations. Docking models using (A) the natural substrate as a native ligand (yellow), (B) substrate 6 (light blue), and (C) 2MeTG α-l-fucopyranoside (blue). (D, left) Superposition of the natural substrate, substrate 4 (green), substrate 5 (purple), and substrate 6 in the active site. (D, right) Superposition of the natural substrate and 2MeTG α-l-fucopyranoside in the active site.
Substrates 1 through 6 were synthesized according to Figure 3. l-Fucose as the starting material was acetylated to obtain compound 7, followed by selective deacetylation at the C1 position to obtain compound 8. Compound 8 was converted to glycosyl imidate 9. Compound 10 was synthesized by Schmidt glycosylation18 of compound 9, and the tert-butyldimethylsily group of 10 was subsequently deprotected to obtain compound 11. Compound 11 and a fluorophore (resorufin, TFMU, or 2MeTG) were reacted under Mitsunobu reaction conditions14,15 to obtain acetylated fluorogenic substrates 1 through 3, after which substrates 4 through 6 were synthesized by deacetylation of substrates 1 through 3, respectively. Synthesis details, including 1H NMR, 13C NMR, MS, and elemental analysis (C, H, N, and F), are provided in the Supporting Information. Effectiveness of fluorogenic substrates requires the fluorescence to show a minimal emission state before hydrolysis. We confirmed this characteristic in substrates 1 through 6 in phosphate-buffered saline, ensuring their status in a quenched state before hydrolysis (Figures S1–S3).
Figure 3.

Synthesis of fluorogenic substrates 1 through 6.
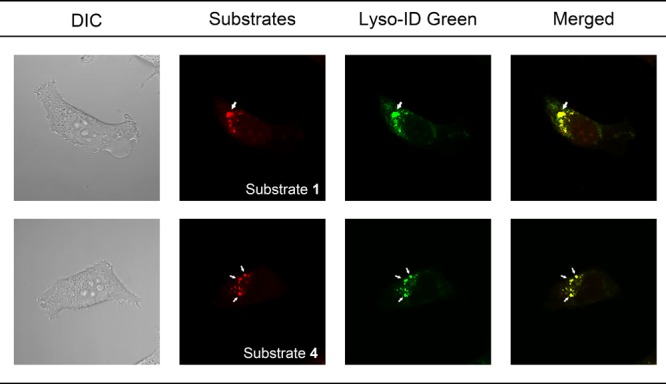
To detect tFuc activity in lysosomes in the adherent cultured cells, synthesized fluorogenic substrates 1 through 6 were evaluated in a human fibrosarcoma cell line (HT1080), a human cervical cancer cell line (HeLa), and a human neuroblastoma cell line (SK-N-SH) at final concentrations of 10 μM, respectively (Figure 4). To avoid the possibility of cross activity contamination of pFuc activity, the medium containing the pFuc used for cell culture was removed before performing the cell-based tFuc assay. We observed bright fluorescence signals from acetylated substrates 1 through 3, and substrates 4 through 6 revealed intercellular accumulation of each fluorophore at the nuclear periphery accompanied by a small-dot pattern typical of lysosome staining and not indicative of the endoplasmic reticulum, Golgi apparatus, or cytosol. Moreover, the fluorescence intensities of substrates 1 through 6 in each cell line were independent of fluorophore structure and acetyl modification. To distinguish subcellular localization of the observed tFuc activity, we evaluated costaining with substrates 1 and 4 (resorufin; red) and a commercially available lysosome-specific fluorescent dye (Lyso-ID Green; excitation, 473 nm; emission, 543 nm) in three cell lines, revealing that the resorufin fluorophores exhibited similar signals as those of Lyso-ID Green (Figure 5). These findings verified that substrates 1 and 4 successfully targeted lysosomes. However, they cannot be substrates for pFuc as secreted enzymes according to this assay method as pFuc is properly removed from the assay medium and the endogenous pFuc activity is deficient.
Figure 4.
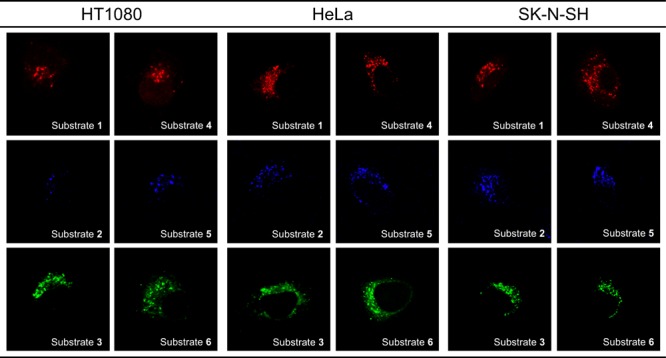
Fluorescence images of fluorogenic substrates 1 through 6 based on tFuc activity in three human cell lines.
Figure 5.
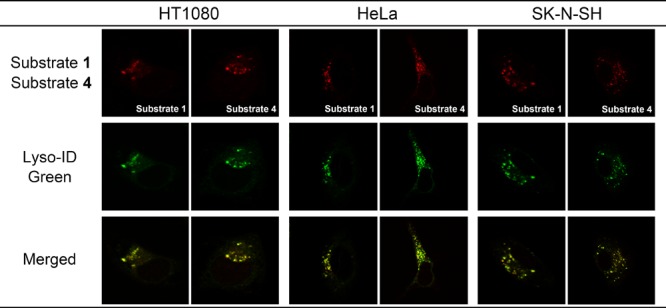
Fluorescence and merged images of tFuc substrates 1 and 4, as well as those of Lyso-ID Green as a lysosome-specific fluorescent dye, in three human cell lines.
Ezawa et al.19 reported that FUCA1 localizes to an organelle surrounding the nucleus in addition to lysosomes in H1299 cells. This potential discrepancy with our results prompted us to examine tFuc subcellular localization by treating the stained lysosomes with chloroquine, which results in their enlargement by inhibiting lysosomal function.20 After costaining HT1080 cells with substrates 1, 4, and Lyso-ID Green, we found that chloroquine treatment enlarged the fluorescence-stained lysosomes (Figure 6). Given these observations of fluorophore localization in lysosomes of human cells based on endogenous tFuc enzyme activity, the discrepancy with previous studies might be explained by the differences in detection objects. The previous study detected the FUCA1 gene-derived protein itself, while we detected the enzyme activity of tFuc derived from the FUCA1 gene. Details of cell-based imaging results are provided in the Supporting Information.
Figure 6.
Differential interference contrast microscopy, fluorescence, and merged images of chloroquine-treated HT1080 cells stained with tFuc substrates 1 and 4, as well as Lyso-ID Green as a lysosome-specific fluorescent dye. Arrows represent the enlarged lysosome following chloroquine treatment.
By ensuring that the recombinant human tFuc hydrolyzes these substrates, we provide a molecular-level confidence that these substrates are substrates for tFuc. To detect α-l-fucosidase activity from commercial recombinant human tFuc, synthesized fluorogenic substrates 4 through 6 were respectively evaluated in 50 mM sodium acetate buffer including 5 mM MgCl2 (pH 4.5) at 37 °C at final concentrations of 50 μM (Figure S5). As a result, tFuc reliably hydrolyzed these substrates regardless of the structure of the three fluorophores. Details of the in vitro assay results are provided in the Supporting Information.
Cell-based HTS represents a powerful method for identifying inhibitors from a compound library. We constructed the human-cell-based inhibitor HTS system to identify tFuc inhibitors using fluorogenic substrate 3 and HeLa cells on a 96-well plate. The 96-well format inhibitor HTS system is useful for primary inhibitor screening against a compound library that especially requires a screening speed and efficiency. The quality and suitability of the constructed system were evaluated using the Z′ factor.21,22 The Z′ factor for the system was calculated to be 0.78 using the fluorescence intensity from substrate 3 based on signal and background controls evaluated. Besides, the calculated interwell variability was CV < 6.8%, and the signal-to-background ratio obtained was >10. Together, these statistical parameters confirm the high quality and suitability of the system. Next, we constructed and evaluated a 6-well format inhibitor evaluation system using changes in fluorescence intensity and area of the hydrolyzing activity of substrate 3 by tFuc in cultured HeLa cells. These data were processed according to our previous study.23 This system made it possible to eliminate false positive compounds as final hit compounds of inhibitor screening to observe morphological changes of the cells by cytotoxicity.
To determine changes in gene expression resulting from tFuc inhibition, we performed assays using the tFuc inhibitor deoxyfuconojirimycin (DFJ; 250 μM) and HeLa cells in the 6-well format system, which resulted in 30% tFuc inhibition. The reason why the DFJ indicated a low inhibition rate despite its high concentration (250 μM) was that the DFJ had a low permeability to the cell membrane based on its high hydrophilic property.24 Details of these experiments are provided in the Supporting Information. Analysis of gene expression under conditions that precisely define the enzyme-inhibition rate is important for assessing the intracellular function of the inhibitor. We used DNA microarray analysis to evaluate changes in gene expression at 30% inhibition of tFuc activity in HeLa cells following treatment with 250 μM DFJ for 18 h, with resulting microarray data submitted to the GEO repository (accession number: GSM991004). This is the first report of changes in gene expression associated with tFuc inhibition, with comparisons of differentially expressed genes relative to a negative control and according to upregulated (fold change > 2.5) and downregulated (fold change > 0.45) genes and their associated pathways (Tables S1–S3). The most highly expressed gene, pepsinogen III/A (PEP3), exhibited a 6.51-fold change in expression relative to the control. A previous study reported PEP3 as a biomarker of atrophic chronic gastritis, H. pylori-related corpus-predominant or multifocal atrophy.25 Furthermore, pathways associated with the upregulated genes included those related to small-ligand G-protein-coupled receptors and Kit-receptor signaling, which are associated with gastric cancer26 and gastrointestinal stromal tumors,27 respectively. Additional details related to the microarray analysis are provided in the Supporting Information.
In summary, we developed three-color fluorogenic substrates, an inhibitor HTS system, and an inhibitor evaluation system to identify inhibitors of tFuc at the cellular level. These represent the first in a class of new substrates and systems that can be utilized to investigate inhibitor efficacy as therapeutic agents in human cells. Additionally, this system allowed gene-expression analysis based on tFuc inhibition, revealing significant changes in gene expression and the regulation of associated pathways related to gastric diseases. Our future work will focus on identifying tFuc inhibitors from a compound library to provide a basis for developing drugs for gastric diseases in the future.
Acknowledgments
We thank A. Sato of A-Rabbit-Science Japan Co., Ltd., for performing the elemental analyses. We thank K. Nagata (Toray New Frontiers Research Laboratories) for assisting with DNA microarray analysis. The illustrations of a 96-well plate, HeLa cells, and a stomach in graphical abstract were obtained from TogoTV (©2016 DBCLS TogoTV).
Glossary
ABBREVIATIONS
- 2MeTG
2-methyl TokyoGreen
- GlcNAc
N-acetylglucosamine
- TBS
tert-butyldimethylsilyl
- DFJ
deoxyfuconojirimycin
- HTS
high-throughput screening
- pFuc
plasma α-l-fucosidase
- QMC
quinone methide cleavage
- TFMU
4-trifluoromethylumbeliferone
- tFuc
tissue α-l-fucosidase
- PEP3
pepsinogen III/A
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00259.
Experimental procedures and spectral data for the test compounds (PDF)
Author Contributions
K.M., T.H., T.N., and W.H. designed the experiments. K.M. and W.H. performed in silico analyses. T.T. and K.M. synthesized the substrates. K.M performed all cell-based assays. T.T. and W.H. performed gene-expression analysis using a DNA microarray. W.H. and K.M. wrote and corrected the manuscript. All authors discussed the results and commented on the manuscript. All authors read and approved the final manuscript
This work was funded in part by a Sasagawa Scientific Research Grant from The Japan Science Society (grant No. 27–304), JSPS KAKENHI (grant Nos. 26460157 and 17K08375), and a Nihon University College of Bioresource Sciences Research Grant for 2012.
The authors declare no competing financial interest.
Supplementary Material
References
- Willems P. J.; Seo H. C.; Coucke P.; Tonlorenzi R.; O’Brien J. S. Spectrum of mutations in fucosidosis. Eur. J. Hum. Genet. 1999, 7, 60–67. 10.1038/sj.ejhg.5200272. [DOI] [PubMed] [Google Scholar]
- Liu T. W.; Ho C. W.; Huang H. H.; Chang S. M.; Popat S. D.; Wang Y. T.; Wu M. S.; Chen Y. J.; Lin C. H. Role for α-l-fucosidase in the control of Helicobacter pylori-infected gastric cancer cells. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 14581–14586. 10.1073/pnas.0903286106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperandio M. Selectins and glycosyltransferases in leukocyte rolling in vivo. FEBS J. 2006, 273, 4377–4389. 10.1111/j.1742-4658.2006.05437.x. [DOI] [PubMed] [Google Scholar]
- Arosio D.; Chiodo F.; Reina J. J.; Marelli M.; Penadés S.; van Kooyk Y.; Bernardi A. Effective targeting of DC-sign by α-fucosylamide-functionalized gold nanoparticles. Bioconjugate Chem. 2014, 25, 2244–2251. 10.1021/bc500467u. [DOI] [PubMed] [Google Scholar]
- Glick M. C.; Kothari V. A.; Liu A. H.; Stoykova L. I.; Scanlin T. F. Activity of fucosyltransferases and altered glycosylation in cystic fibrosis airway epithelial cells. Biochimie 2001, 83, 743–747. 10.1016/S0300-9084(01)01323-2. [DOI] [PubMed] [Google Scholar]
- Korekane H.; Hasegawa T.; Matsumoto A.; Kinoshita N.; Miyoshi E.; Taniguchi N. Development of an antibody-lectin enzyme immunoassay for fucosylated α-fetoprotein. Biochim. Biophys. Acta, Gen. Subj. 2012, 1820, 1405–1411. 10.1016/j.bbagen.2011.12.015. [DOI] [PubMed] [Google Scholar]
- Tu Z. J.; Lin Y. N.; Lin C. H. Development of fucosyltransferase and fucosidase inhibitors. Chem. Soc. Rev. 2013, 42, 4459–4475. 10.1039/c3cs60056d. [DOI] [PubMed] [Google Scholar]
- Prichard K.; Campkin D.; O’Brien N.; Kato A.; Fleet G. W.; Simone M. I. Biological activities of 3,4,5-trihydroxypiperidines and their N- and O-derivatives. Chem. Biol. Drug Des. 2018, 92, 1171–1197. 10.1111/cbdd.13182. [DOI] [PubMed] [Google Scholar]
- Bathula C.; Ghosh S.; Hati S.; Tripathy S.; Singh S.; Chakrabarti S.; Sen S. Bioisosteric modification of known fucosidase inhibitors to discover a novel inhibitor of α-l-fucosidase. RSC Adv. 2017, 7, 3563–3572. 10.1039/C6RA24939F. [DOI] [Google Scholar]
- Jiang J.; Kallemeijn W. W.; Wright D. W.; van den Nieuwendijk A. M.; Rohde V. C.; Folch E. C.; van den Elst H.; Florea B. I.; Scheij S.; Donker-Koopman W. E.; Verhoek M.; Li N.; Schürmann M.; Mink D.; Boot R. G.; Codée J. D. C.; van der Marel G. A.; Davies G. J.; Aerts J. M. F. G.; Overkleeft H. S. In vitro and in vivo comparative and competitive activity-based protein profiling of GH29 α-l-fucosidases. Chem. Sci. 2015, 6, 2782–2789. 10.1039/C4SC03739A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. W.; Ho C. W.; Huang H. H.; Chang S. M.; Popat S. D.; Wang Y. T.; Wu M. S.; Chen Y. J.; Lin C. H. Role for α-l-fucosidase in the control of Helicobacter pylori-infected gastric cancer cells. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 14581–14586. 10.1073/pnas.0903286106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X.; Peng J.; Zeng F.; Yu C.; Wu S. An anthracenecarboximide fluorescent probe for in vitro and in vivo ratiometric imaging of endogenous alpha-l-fucosidase for hepatocellular carcinoma diagnosis. Mater. Chem. Front. 2017, 1, 660–667. 10.1039/C6QM00112B. [DOI] [Google Scholar]
- Wu L.; Wang Y.; James T. D.; Jia N.; Huang C. A hemicyanine-based ratiometric fluorescence probe for mapping lysosomal pH during heat stroke in living cells. Chem. Commun. 2018, 54, 5518–5521. 10.1039/C8CC02330A. [DOI] [PubMed] [Google Scholar]
- Miura K.; Hakamata W.; Tanaka A.; Hirano T.; Nishio T. Discovery of human Golgi β-galactosidase with no identified glycosidase using a QMC substrate design platform for exo-glycosidase. Bioorg. Med. Chem. 2016, 24, 1369–1375. 10.1016/j.bmc.2016.02.010. [DOI] [PubMed] [Google Scholar]
- Hakamata W.; Miura K.; Tanaka A.; Hirano T.; Nishio T. Identification of a novel glycan-processing enzyme with exo-acting β-allosidase activity in the Golgi apparatus using a new platform for the synthesis of fluorescent substrates. Bioorg. Med. Chem. 2015, 23, 73–79. 10.1016/j.bmc.2014.11.023. [DOI] [PubMed] [Google Scholar]
- Waring M. J. Lipophilicity in drug discovery. Expert Opin. Drug Discovery 2010, 5, 235–248. 10.1517/17460441003605098. [DOI] [PubMed] [Google Scholar]
- Wu H. J.; Ho C. W.; Ko T. P.; Popat S. D.; Lin C. H.; Wang A. H. J. Structural basis of α-fucosidase inhibition by iminocyclitols with Ki values in the micro-to picomolar range. Angew. Chem., Int. Ed. 2010, 49, 377–340. 10.1002/anie.200905597. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Schmidt R. R. New principles for glycoside-bond formation. Angew. Chem., Int. Ed. 2009, 48, 1900–1934. 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]
- Ezawa I.; Sawai Y.; Kawase T.; Okabe A.; Tsutsumi S.; Ichikawa H.; Kobayashi Y.; Tashiro F.; Namiki H.; Kondo T.; Semba K.; Aburatani H.; Taya Y.; Nakagama H.; Ohki Remba K. Novel p53 target gene FUCA1 encodes a fucosidase and regulates growth and survival of cancer cells. Cancer Sci. 2016, 107, 734–745. 10.1111/cas.12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi T.; Kuwahara T.; Sakurai M.; Komori T.; Fujimoto T.; Ito G.; Yoshimura S. I.; Harada A.; Fukuda M.; Koike M.; Iwatsubo T. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E9115–E9124. 10.1073/pnas.1812196115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatokun A. A.; Liu J. O.; Dawson V. L.; Dawson T. M. Identification through high-throughput screening of 4′-methoxyflavone and 3′,4′-dimethoxyflavone as novel neuroprotective inhibitors of parthanatos. Br. J. Pharmacol. 2018, 169, 1263–1278. 10.1111/bph.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. H.; Chung T. D.; Oldenburg K. R. A simple statistical parameter for use in evaluation and validation of high-throughput screening assays. J. Biomol. Screening 1999, 4, 67–73. 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Koyama R.; Hakamata W.; Hirano T.; Nishio T. Identification of small-molecule inhibitors of human Golgi mannosidase via a drug repositioning screen. Chem. Pharm. Bull. 2018, 66, 678–681. 10.1248/cpb.c17-01009. [DOI] [PubMed] [Google Scholar]
- Onda M.; Hakamata W. Antiviral activity and mechanism of action of endoplasmic reticulum glucosidase inhibitors: a mini review. Trends Glycosci. Glycotechnol. 2018, 30, E139–E145. 10.4052/tigg.1753.1E. [DOI] [Google Scholar]
- Broutet N.; Plebani M.; Sakarovitch C.; Sipponen P.; Megraud F. Pepsinogen A, pepsinogen C, and gastrin as markers of atrophic chronic gastritis in European dyspeptics. Br. J. Cancer 2003, 88, 1239–1247. 10.1038/sj.bjc.6600877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H. Q.; Cai A. Z.; Wu X. S.; Cui J. X.; Shen W. S.; Bian S. B.; Wang N.; Li J. Y.; Lu C. R.; Song Z.; Wei B.; Chen L. Leucine-rich repeat-containing G-protein-coupled receptor 5 is associated with invasion, metastasis, and could be a potential therapeutic target in human gastric cancer. Br. J. Cancer 2014, 110, 2011–2020. 10.1038/bjc.2014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J.; Wu Y. L.; Chen B. J.; Zhang W.; Tanaka Y.; Sugiyama H. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int. J. Biol. Sci. 2013, 9, 435–202. 10.7150/ijbs.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.