

Abstract

The Bromodomain and Extra Terminal (BET) family of proteins recognize post-translational N-ε-acetylated lysine modifications, regulating transcription as “reader” proteins. Bromodomain inhibitors are interesting targets for the development of potential cancer, inflammation, and heart disease treatments. Several dual kinase-bromodomain inhibitors have been identified by screening kinase inhibitor libraries against BET proteins. Although potentially useful from a polypharmacology standpoint, multitarget binding complicates deciphering molecular mechanisms. This report describes a systematic approach to mitigating kinase activity in a dual kinase-bromodomain inhibitor based on a 1,2,3-triazole-pyrimidine core. By modifying the triazole substituent and altering the pyrimidine core, this structure–activity relationship study enhanced BET activity while reducing the p38α kinase activity >90,000-fold. A BRD4-D1 cocrystal structure indicates that the 1,2,3-triazole is acting as a N-ε-acetylated lysine mimic. A BRD4 sensitive cell line, MM.1S, was used to demonstrate activity in cells, which is further supported by reduced c-Myc expression.
Keywords: Bromodomains, BET, BRD4, p38α, kinases
Bromodomains act as “readers” for epigenetic modifications. Specifically, they bind N-ε-acetylated lysine residues on histones and transcription factors through protein–protein interactions to regulate cellular processes including the cell cycle, cell proliferation, and cellular differentiation.1 The 61 human bromodomains, which are contained in 46 proteins, have been subdivided into eight classes based on structural or sequence similarities.2 The Bromodomain and Extra Terminal (BET) proteins, which include BRD2, BRD3, BRD4, and BRDT, all contain a similar domain architecture, including two tandem bromodomains and an extra-terminal domain.
Inhibiting protein–protein interactions between BET bromodomains and N-ε-acetylated lysine residues is a potential method for treating BET related diseases, including cancer, inflammation, and heart disease.2−5 For example, BRD4 can recognize N-ε-acetylated K310 in the RelA subunit of NF-κB, which is important for the activation of NF-κB, following inflammatory signaling.6 Additionally, the bromodomains of BRD4 can recognize N-ε-acetylated lysine residues on histones and recruit transcription factors to superenhancer regions. Inhibition of BRD4 at superenhancer regions can reduce c-Myc expression, which could be a therapeutic strategy for treating cancer.7 Given the significant roles that BET bromodomains play in oncogene expression and inflammation, 19 clinical trials are underway to assess the therapeutic effects of BET inhibition.
Several dual kinase-bromodomain inhibitors were discovered by screening kinase inhibitor libraries against BRD4-D1 (Figure 1A).8−13 In 2014, both Ciceri et al. and Ember et al. identified several dual kinase-bromodomain inhibitors including BI-2536 (1), a PLK1 inhibitor with high affinity for BRD4-D1.8,11 The dihydropteridinone carbonyl and the methylamino group function as the N-ε-acetylated lysine mimic. CDK inhibitor Dinaciclib (2) functions as a BRDT inhibitor.12 The pyridine oxide serves as the N-ε-acetylated lysine mimic and is recognized by N109 on BRDT. Although modest in BRDT affinity, this molecule is of historical note, being the first dual kinase-bromodomain inhibitor reported. Other p38-BET inhibitors, including SB-202190 and SB-203580, were likewise identified. Since these reports, developing bromodomain inhibitors by kinase library screening has attracted more attention.8,11,13,14 Urick et al. screened a library of 229 small molecules by protein-observed fluorine NMR and subsequently developed a dual p38α-BET inhibitor named V (3).13,15 While compounds 1 and 2 are pan-BET inhibitors, compound 3 was shown to selectively inhibit the N-terminal BET bromodomains with highest affinity for BRD4. Although dual kinase-bromodomain inhibitors may produce synergistic effects in some cases,9,11,14,16 selective inhibition is ideal for understanding the physiological and pharmacological effect of BET inhibition as well as minimizing potential side effects.17
Figure 1.

Dual kinase-bromodomain inhibitors.
In our previous work, a trisubstituted 1,2,3-triazole-based dual kinase-bromodomain inhibitor was reported (Figure 1b, compound 4).15 The 1,2,3-triazole is known to engage BET targets as an N-ε-acetylated lysine mimic.18 This study describes the systematic removal of the p38α kinase activity displayed by compound 4. A selective BET inhibitor is reported (5), which is >15-fold more potent against BRD4-D1 and >90,000-fold less potent against p38α (>1,000,000-fold change in relative selectivity vis-à-vis compound 4). A BRD4-D1 cocrystal structure and cellular data are provided that further support the BET activity of the reported compounds.
This study began by studying how compounds like 4 bind kinases. Cocrystal structures of p38α and similarly structured hinge-binding kinase inhibitors provided hypotheses for a systematic structure–activity relationship (SAR) study aimed at mitigating p38α activity. Cocrystal structures with p38α indicate that one of the inhibitor’s pyrimidine nitrogen atoms serves as a hydrogen bond acceptor, and the exocyclic N–H is a hydrogen bond donor (Figure 2); both interactions are made to the M109 peptide backbone in p38α (Figure 2).19−21 If the exocyclic N–H was replaced by an ether linkage, there would no longer be a hydrogen bond donor, which would weaken binding to p38α.21 Hypothetically, binding to M109 could be further minimized by replacing the pyrimidine nitrogen with a C–H unit (pyridine), which would not be a suitable hydrogen bond acceptor.
Figure 2.

Hydrogen bonding interactions in p38α cocrystal structure and proposed analogs.
Given the significance of these two interactions, four inhibitors were synthesized, each with one of the four possible X/Y substitution combinations (Table 1). Switching from an exocyclic amine (4) to an ether (6) led to a ∼50-fold reduction in p38α binding. A ∼1000-fold reduction in kinase binding was observed when the analogous change was made to compound 1.17 When the pyrimidine was replaced with a pyridine (4 vs 7), the p38α affinity was reduced by a factor of ∼3,000. Thus, in this scaffold, removing the pyrimidine nitrogen appears to be more important to mitigating the p38α activity than installing an ether linkage. This was supported by examining a compound with both an ether linkage and a pyridine ring (8). This compound displayed significantly diminished p38α binding relative to the pyrimidine (6), but it was on a par with the analog with the corresponding exocyclic nitrogen (7).
Table 1. Systematically Minimizing Binding to M109 in p38α.
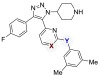
| compound | X | Y | p38α Kd (nM)a |
|---|---|---|---|
| 4 | N | NH | 2.2 ± 0.63 |
| 6 | N | O | 120 ± 51 |
| 7 | CH | NH | 6500 ± 6300 |
| 8 | CH | O | 5000 ± 6100 |
Kd values were determined by KINOMEscan. Data represents the mean and standard error from two independent trials.
In addition to hydrogen bonding to M109, the p38α binding affinity is likely enhanced by van der Waals interactions to the 4-fluorophenyl group in compounds 4 and 6–8. An analogous p38α inhibitor, SB220025, demonstrates this interaction with the 4-fluorophenyl ring residing in a hydrophobic pocket between L104 and T106.20−22 A reduction in p38α activity was observed when the 4-fluorophenyl group was replaced with an ethyl group.23 Minimizing this van der Waals interaction would likely further mitigate the p38α activity and produce a selective BET inhibitor if bromodomain binding could be maintained.
Inhibitors containing a variety of alkyl chains were synthesized to investigate this hypothesis (Scheme 1). The sequence began with a CuAAC reaction,24 which afforded triazole 11. The triazole could be directly coupled to an aryl halide through a metalation/Negishi cross-coupling reaction. The direct C–H arylation provided facile access to fully substituted triazoles in a regiocontrolled manner. The resulting product 13 could be deprotected to yield amine 14. Subsequent guanidinylation (15) and deprotection afforded inhibitors 19–22. The guanidine motif was added to increase solubility at the highest concentrations assayed. The key piperidyl amine (14) can be synthesized in three linear steps. This allows much quicker access to a wide variety of analogs relative to the 10-step route required for compound 3.15
Scheme 1. Synthesis of Inhibitors.

Compound 13d was synthesized through an alternate three-step route with an overall yield of 35%. See SI for details.
The activity of these compounds was determined by fluorescence anisotropy against BRD4-D1 (Table 2). The 4-fluorophenyl containing compound (6) was a weak BRD4-D1 ligand in this assay (>100 μM). The low solubility of compounds 6, 16, 17, and 18 was an issue in the fluorescence anisotropy assay. Herein, the guanidine group was added to increase the solubility. Most of the alkyl groups yielded little activity (16–21) with the exception of a methyl group. The methyl triazole (22) was more potent. This is similar to the results in a SAR study with compound 1 that demonstrated methyl was the optimal N-substituent in the N-ε-acetylated lysine mimic.17
Table 2. Significance of R Group to BRD4-D1 Binding Affinity.
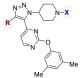
| compound | R | X | BRD4-D1 IC50 (μM)a |
|---|---|---|---|
| 6 | 4–F-C6H4 | H | >100b,c |
| 16 | c-hexyl | H | >100b |
| 17 | c-pentyl | H | >100b |
| 18 | t-Bu | H | >100b |
| 19 | i-Pr | C(NH)NH2 | >100b |
| 20 | n-Pr | C(NH)NH2 | >100b |
| 21 | c-Pr | C(NH)NH2 | 61 ± 8.1 |
| 22 | Me | C(NH)NH2 | 9.7 ± 1.8 |
IC50 values were determined by fluorescence anisotropy. Data represents the mean and standard deviation of three independent trials.
The IC50 value was determined to be >100 μM by fluorescence anisotropy.
The IC50 value was determined to be 27 μM by AlphaScreen by Reaction Biology.
Having identified that a methyl substituent significantly enhanced BRD4-D1 binding, attention was turned toward determining the pyrimidine ring’s significance with regard to BRD4-D1 binding. This seemed prudent given the findings on p38α activity in Table 1. Based on a cocrystal structure of BRD4-D1 and compound 3, the 3,5-dimethylphenyl ring appears to provide an edge-to-face π–π interaction with W81, and the pyrimidine ring interacts with the WPF shelf through a nonspecific van der Waals interaction.15 This analysis indicated that, unlike binding to p38α, neither nitrogen atom in the pyrimidine ring provided a key binding interaction. Replacing the pyrimidine ring (22) with a benzene ring (23) had minimal impact on BRD4-D1 binding (Table 3), which is consistent with this analysis. Comparing the pyrimidine to a pyridine ring provided mixed results. Improved binding was observed when the nitrogen atom was at the 1 position (5). Placing a nitrogen atom at the 2 position was tolerated (24), while placing the nitrogen atom at either the 3 (25) or 4 (26) position resulted in complete loss of affinity. By comparison to compound 22, the loss in affinity of compounds 25 and 26 may indicate an unfavorable interaction of the nitrogen lone pair. The enhanced binding observed with compound 5 was exciting in light of the diminished p38α binding observed with the 2,6-disubstituted pyridine (7–8, Table 1).
Table 3. Effect of Nitrogen Atom(s) on BRD4-D1 Affinity.
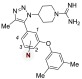
| compound | N atom location | BRD4-D1 IC50 (μM)a |
|---|---|---|
| 22 | 1,2 | 9.7 ± 1.8 |
| 23 | none | 12 ± 1.1 |
| 5 | 1 | 3.6 ± 0.05 |
| 24 | 2 | 97 ± 2.6 |
| 25 | 3 | >100 |
| 26 | 4 | >100 |
IC50 values were determined by fluorescence anisotropy. Data represents the mean and standard deviation of three independent trials.
The selectivity of compounds 5 and 27 was further characterized (Table 4). The BRD4-D1 Kd of these two compounds was determined by BROMOscan and was consistent with the fluorescence anisotropy assay.25,26 The methyl triazole demonstrated minimal selectivity for the two domains in BRD4, which is consistent with other PAN-BET inhibitors. Minimal binding to SMARCA2 and CBP was observed (Table 4). SMARCA2 and CBP were selected as representative non-BET family bromodomains. Prior work with compound 3 (Figure 1) demonstrated modest binding to SMARCA2 and CBP.15 Most significantly, compound 5 did not demonstrate any detectable activity toward p38α (Kd > 200 μM). This indicates that this SAR study was able to systematically reduce p38α affinity by >90,000-fold (relative to compound 4, Kd = 2.2 nM) while maintaining BET selectivity and affinity.
Table 4. Demonstrating Selectivity for Compound 5 and 27.
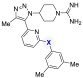
| protein target | X = O (5) | X = NH (27) |
|---|---|---|
| BRD4-D1 IC50 (μM)a | 3.6 ± 0.05 | 4.5 ± 0.35 |
| BRD4-D1 Kd (μM)b | 1.1 ± 0.63 | 1.2 ± 0.62 |
| BRD4-D2 Kd (μM)b | 0.94 ± 0.48 | 2.1 ± 1.1 |
| SMARCA2 Kd (μM)b | >100 | >100 |
| CBP (μM)b | >100 | 78 ± 48 |
| p38α Kd (μM)c | >200 | >25 |
IC50 values were determined by fluorescence anisotropy. Data represents the mean and standard deviation of three independent trials.
Kd values were determined by BROMOscan. Data represents the mean and standard error from two independent trials.
Kd values were determined by KINOMEscan. Data represents the mean and standard error from two independent trials.
To gain a structural understanding, a BRD4-D1 cocrystal structure was obtained with compound 27. This information could be compared to a cocrystal structure of compound 3 (PDB ID 6MH1).13 The central triazole nitrogen (N2) in compound 27 serves as the N-ε-acetylated lysine mimic by forming a direct hydrogen bond to N140 (Figure 3a). This was somewhat surprising because the terminal nitrogen (N3) has been calculated to be more basic than the central nitrogen (N2).27 One explanation for this binding mode is that the terminal nitrogen (N3) in compound 27 is forming a hydrogen bond to Y97 via a bridging structured water molecule in the cocrystal structure (Figure 3a). Similar interactions are observed for methyl isoxazoles and pyrazoles.26,28,29 In the cocrystal structure with compound 3, the 4-fluorophenyl group displaces three of four structured water molecules in this binding pocket relative to a DMSO cocrystal structure (PDB ID 4IOR), including the water bridging to Y97.15 This mode of interaction was used to support the BRD4-D1 selectivity for compound 3. In the absence of this structured water molecule, diminished binding affinity would be expected due to (i) the energy required to displace the bound water and (ii) the loss of the bridging hydrogen bond. This is consistent with the increase in potency observed for compound 22 relative to compound 6 (Table 2). Comparing BRD4-D1 cocrystal structures containing compound 27 vs 3 demonstrates that the pyrimidine ring and pyridine ring occupy similar binding conformations relative to the WPF shelf (Figure 3b). This supports the notion that while the pyrimidine nitrogen atom in compound 3 is essential for p38α binding (Table 1), replacing the nitrogen with a C–H unit only minimally affects the BRD4-D1 binding.
Figure 3.

BRD4-D1 compound 27 cocrystal structure and comparative analysis to dual p38α-BRD4 inhibitor 3. (A) Cocrystal structure of 27 with BRD4-D1. Key interactions include a hydrogen bond of N2 of 27 to the amine of N140 (3.1 Å) and a water-mediated hydrogen bond of N3 of 27 to the hydroxyl of Y97 (2.9 Å, 2.6 Å). (B) Overlay of 27 (green) with 3 (gray). (C) Structure of 27. (D) Structure of 3.
The inhibitors described above were assayed in MM.1S cells. Multiple myeloma cell strains from hematological cancers have a strong dependence on c-Myc.7 The survival of MM.1S cells are highly BRD4 dependent because of an IgH and MYC rearrangement.7 Reports have shown that BET inhibitors effectively decrease c-Myc transcription, which results in decreased cell viability.15,30 In MM.1S cells, guanidinylated compounds 22, 5, and 27 displayed weak activity (Table 5, EC50 > 50 μM). This weak activity may be due to decreased cell permeability. To test this hypothesis, several analogs with a free piperidine NH were assayed. Compounds 28, 29, and 30 demonstrated clear antiproliferative effects in MM.1S cells (EC50 = 6.6, 2.9, and 6.3 μM, respectively). The observed EC50 values loosely correlate to the BRD4-D1 in vitro fluorescence anisotropy assay IC50 values. Additionally, decreased levels of c-Myc were observed by Western blot after treatment with compounds 28, 29, and 30 for 6 h (see Supporting Information).
Table 5. Viability of MM.1S Cells Treated with Compounds.
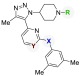
| compound | R | X | Y | BRD4-D1 IC50 (μM)a | MM.1S EC50 (μM)b |
|---|---|---|---|---|---|
| 22 | C(NH)NH2 | O | N | 9.7 ± 1.8 | >50 |
| 5 | C(NH)NH2 | O | CH | 3.6 ± 0.05, 1.7 ± 0.30c | >50 |
| 27 | C(NH)NH2 | NH | CH | 4.5 ± 0.35 | >50 |
| 28 | H | O | N | 11 ± 2.5 | 6.6 ± 1.9 |
| 29 | H | O | CH | 2.9 ± 0.07, 1.3 ± 0.25c | 2.9 ± 0.6 |
| 30 | H | NH | CH | 3.9 ± 0.35 | 6.3 ± 1.1 |
IC50 values were determined by fluorescence anisotropy. Data represents the mean and standard deviation of three independent trials.
Data reported are mean ± SEM of three biological replicates, with three technical replicates each. EC50 values were determined using the Nonlinear fit algorithm on GraphPad Prism.
IC50 values were determined by AlphaScreen by Reaction Biology.
In conclusion, we have successfully reversed the p38α/BRD4-D1 selectivity observed with compound 4. Originally, compound 4 demonstrated a >10,000-fold selectivity for p38α over BRD4-D1. By modifying the hinge binding motif and removing a hydrophobic 4-fluorophenyl group, compound 5 demonstrated a selectivity for BRD4-D1 and BRD4-D2 over p38α by >100-fold. This represents a >1,000,000 relative difference in binding. A cocrystal structure and cellular assays further support the binding mode of the triazole inhibitor and the BET activity. In the future, this molecular design approach may be applied to other previously reported dual p38-BET inhibitors including 3, SB-202190, and SB-203580.
Acknowledgments
We acknowledge Daniel A. Harki for his helpful discussions during this project. MM.1S cells were a gift from Dr. Brian Van Ness at the University of Minnesota. This work was supported by the NIGMS (Grant R35GM124718), the American Cancer Society (# 129819-IRG-16-189-58-IRG88), the University of Minnesota Masonic Cancer Center (Pre-R01 pilot grant), and the University of Minnesota. J.J. was supported by a National Institutes of Health Biotechnology training grant 5T32GM008347-23. A.D. was supported by the NIH chemistry–biology interface training grant at the University of Minnesota, 5T32GM008700-18.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00227.
Experimental procedures and data (PDF)
Author Contributions
† A.S.C. and H.C. contributed equally to this study. A.S.C. synthesized the molecules with H.C. and R.M.B. Biophysical experiments were performed by H.C. Cellular assays were performed by A.D. The cocrystal structure was solved by J.A.J. Research was directed by J.J.T. and W.C.K.P. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): The authors submitted a provisional patent on the composition of matter for the compounds disclose here.
Supplementary Material
References
- Filippakopoulos P.; Knapp S. The Bromodomain Interaction Module. FEBS Lett. 2012, 586, 2692–2704. 10.1016/j.febslet.2012.04.045. [DOI] [PubMed] [Google Scholar]
- Pervaiz M.; Mishra P.; Günther S. Bromodomain Drug Discovery - the Past, the Present, and the Future. Chem. Rec. 2018, 18, 1808–1817. 10.1002/tcr.201800074. [DOI] [PubMed] [Google Scholar]
- Gallinari P.; Di Marco S.; Jones P.; Pallaoro M.; Steinkühler C. HDACs, Histone Deacetylation and Gene Transcription: From Molecular Biology to Cancer Therapeutics. Cell Res. 2007, 17, 195–211. 10.1038/sj.cr.7310149. [DOI] [PubMed] [Google Scholar]
- Andrieu G.; Belkina A. C.; Denis G. V. Clinical Trials for BET Inhibitors Run Ahead of the Science. Drug Discovery Today: Technol. 2016, 19, 45–50. 10.1016/j.ddtec.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S.; Piya S.; Borthakur G. Bromodomain Inhibitors: What Does the Future Hold?. Clin. Adv. Hematol. Oncol. 2018, 16, 504–515. [PubMed] [Google Scholar]
- Huang B.; Yang X.-D.; Zhou M.-M.; Ozato K.; Chen L.-F. Brd4 Coactivates Transcriptional Activation of NF-κB via Specific Binding to Acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore J. E.; Issa G. C.; Lemieux M. E.; Rahl P. B.; Shi J.-W.; Jacobs H. M.; Kastritis E.; Gilpatrick T.; Paranal R. M.; Qi J.; Chesi M.; Schinzel A. C.; Mckeown M. R.; Heffernan T. P.; Vakoc C. R.; Bergsagel P. L.; Ghobrial I. M.; Richardson P. G.; Young R. A.; Hahn W. C.; Anderson K. C.; Kung A. L.; Bradner J. E.; Mitsiades C. S. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ember S. W. J.; Zhu J.-Y.; Olesen S. H.; Martin M. P.; Becker A.; Berndt N.; Georg G. I.; Schonbrunn E. Acetyl-Lysine Binding Site of Bromodomain-Containing Protein 4 (BRD4) Interacts with Diverse Kinase Inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. 10.1021/cb500072z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlino L.; Rastelli G. Dual Kinase-Bromodomain Inhibitors in Anticancer Drug Discovery: A Structural and Pharmacological Perspective. J. Med. Chem. 2016, 59, 9305–9320. 10.1021/acs.jmedchem.6b00438. [DOI] [PubMed] [Google Scholar]
- Boehm J. C.; Bower M. J.; Gallagher T. F.; Kassis S.; Johnson S. R.; Adams J. L. Phenoxypyrimidine Inhibitors of p38α Kinase Synthesis and Statistical Evaluation of the p38 Inhibitory Potencies of a Series of 1-(piperidin-4-yl)-4-(4-fluorophenyl)-5-(2-phenoxypyrimidine-4-yl) Imidazoles. Bioorg. Med. Chem. Lett. 2001, 11, 1123–1126. 10.1016/S0960-894X(01)00163-9. [DOI] [PubMed] [Google Scholar]
- Ciceri P.; Müller S.; O’Mahony A.; Fedorov O.; Filippakopoulos P.; Hunt J. P.; Lasater E. A.; Pallares G.; Picaud S.; Wells C.; Martin S.; Wodicka L. M.; Shah N. P.; Treiber D. K.; Knapp S. Dual Kinase-Bromodomain Inhibitors for Rationally Designed Polypharmacology. Nat. Chem. Biol. 2014, 10, 305–312. 10.1038/nchembio.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. P.; Olesen S. H.; Georg G. I.; Schönbrunn E. Cyclin-Dependent Kinase Inhibitor Dinaciclib Interacts with the Acetyl-Lysine Recognition Site of Bromodomains. ACS Chem. Biol. 2013, 8, 2360–2365. 10.1021/cb4003283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urick A. K.; Hawk L. M. L.; Cassel M. K.; Mishra N. K.; Liu S.; Adhikari N.; Zhang W.; dos Santos C. O.; Hall J. L.; Pomerantz W. C. K. Dual Screening of BPTF and Brd4 Using Protein-Observed Fluorine NMR Uncovers New Bromodomain Probe Molecules. ACS Chem. Biol. 2015, 10, 2246–2256. 10.1021/acschembio.5b00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ember S. W.; Lambert Q. T.; Berndt N.; Gunawan S.; Ayaz M.; Tauro M.; Zhu J.-Y.; Cranfill P. J.; Greninger P.; Lynch C. C.; Benes C. H.; Lawrence H. R.; Reuther G. W.; Lawrence N. J.; Schönbrunn E. Potent Dual BET Bromodomain-Kinase Inhibitors as Value-Added Multitargeted Chemical Probes and Cancer Therapeutics. Mol. Cancer Ther. 2017, 16, 1054–1067. 10.1158/1535-7163.MCT-16-0568-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaran A.; Talluri S. K.; Ayoub A. M.; Mishra N.; Cui H.; Widen J. C.; Berndt N.; Zhu J.-Y.; Carlson A. S.; Topczewski J. J.; Schönbrunn E.; Harki D. A.; Pomerantz W. C. K. Molecular Basis for the N-Terminal Bromodomain and Extra Terminal (BET) Family Selectivity of a Dual Kinase-Bromodomain Inhibitor. J. Med. Chem. 2018, 61, 9316–9334. 10.1021/acs.jmedchem.8b01248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts E.; Heidenreich D.; Tucker E.; Raab M.; Strebhardt K.; Chesler L.; Knapp S.; Bellenie B.; Hoelder S. Designing Dual Inhibitors of Anaplastic Lymphoma Kinase (ALK) and Bromodomain-4 (BRD4) by Tuning Kinase Selectivity. J. Med. Chem. 2019, 62, 2618–2637. 10.1021/acs.jmedchem.8b01947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Yap J. L.; Yoshioka M.; Lanning M. E.; Fountain R. N.; Raje M.; Scheenstra J. A.; Strovel J. W.; Fletcher S. BRD4 Structure-Activity Relationships of Dual PLK1 Kinase/BRD4 Bromodomain Inhibitor BI-2536. ACS Med. Chem. Lett. 2015, 6, 764–769. 10.1021/acsmedchemlett.5b00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S.; Feutrill J. T.; Garnham A.; Dawson M. A.; Glaser S.; Wilks A. F.; Hatfaludi T.; Walkley C. R.; Segal D.; Hall P.; Czabotar P.; Jousset H.; Huang D. C. S.; Tyler D.; Cuzzupe A.; Garnier J.-M.; Jarman K. E.; Xu Z.; Burns C. J.; Sharp P. P. Design, Synthesis, and Biological Activity of 1,2,3-Triazolobenzodiazepine BET Bromodomain Inhibitors. ACS Med. Chem. Lett. 2017, 8, 1298–1303. 10.1021/acsmedchemlett.7b00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabat M.; VanRens J. C.; Clark M. P.; Brugel T. A.; Maier J.; Bookland R. G.; Laufersweiler M. J.; Laughlin S. K.; Golebiowski A.; De B.; Hsieh L. C.; Walter R. L.; Mekel M. J.; Janusz M. J. The Development of Novel C-2, C-8, and N-9 Trisubstituted Purines as Inhibitors of TNF-α Production. Bioorg. Med. Chem. Lett. 2006, 16, 4360–4365. 10.1016/j.bmcl.2006.05.050. [DOI] [PubMed] [Google Scholar]
- Simard J. R.; Getlik M.; Grutter C.; Pawar V.; Wulfert S.; Rabiller M.; Rauh D. Development of a Fluorescent-Tagged Kinase Assay System for the Detection and Characterization of Allosteric Kinase Inhibitors. J. Am. Chem. Soc. 2009, 131, 13286–13296. 10.1021/ja902010p. [DOI] [PubMed] [Google Scholar]
- Townes J. A.; Golebiowski A.; Clark M. P.; Laufersweiler M. J.; Brugel T. A.; Sabat M.; Bookland R. G.; Laughlin S. K.; VanRens J. C.; De B.; Hsieh L. C.; Xu S. C.; Janusz M. J.; Walter R. L. The Development of New Bicyclic Pyrazole-Based Cytokine Synthesis Inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 4945–4948. 10.1016/j.bmcl.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Canagarajah B. J.; Boehm J. C.; Kassisa S.; Cobb M. H.; Young P. R.; Abdel-Meguid S.; Adams J. L.; Goldsmith E. J. Structural Basis of Inhibitor Selectivity in MAP Kinases. Structure 1998, 6, 1117–1128. 10.1016/S0969-2126(98)00113-0. [DOI] [PubMed] [Google Scholar]
- Gallagher T. F.; Seibel G. L.; Kassis S.; Laydon J. T.; Blumenthal M. J.; Lee J. C.; Lee D.; Boehm J. C.; Fier-Thompson S. M.; Abt J. W.; Soreson M. E.; Smietana J. M.; Hall R. F.; Garigipati R. S.; Bender P. E.; Erhard K. F.; Krog A. J.; Hofman G. A.; Sheldrake P. L.; McDonnell P. C.; Kumar S. K.; Young P. R.; Adams J. L. Regulation of Stress-Induced Cytokine Production by Pyridinylimidazoles; Inhibition of CSBP Kinase. Bioorg. Med. Chem. 1997, 5, 49–64. 10.1016/S0968-0896(96)00212-X. [DOI] [PubMed] [Google Scholar]
- Meldal M.; Tornøe C. W. Cu-Catalyzed Azide–Alkyne Cycloaddition Cu-Catalyzed Azide-Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015. 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- Wu Q.; Heidenreich D.; Ackloo S.; Krämer A.; Nakka K.; Lima-Fernandes E.; Deblois G.; Duan S.; Vellanki R. N.; Li F.; Vedadi M.; Dilworth J.; Lupien M.; Brennan P. E.; Arrowsmith C. H.; Muller S.; Fedorov O.; Filippakopoulos P.; Knapp S. A Chemical Toolbox for the Study of Bromodomains and Epigenetic Signaling. Nat. Commun. 2019, 10, 1915–1929. 10.1038/s41467-019-09672-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M.; Measures A. R.; Wilson B. G.; Cortopassi W. A.; Alexander R.; Höss M.; Hewings D. S.; Rooney T. P. C.; Paton R. S.; Conway S. J. Small Molecule Inhibitors of Bromodomain-Acetyl-Lysine Interactions. ACS Chem. Biol. 2015, 10, 22–39. 10.1021/cb500996u. [DOI] [PubMed] [Google Scholar]
- Dinér P.; Andersson T.; Kjellén J.; Elbing K.; Hohmann S.; Grøtli M. Short Cut to 1,2,3-Triazole-Based p38 MAP Kinase Inhibitors via [3 + 2]-Cycloaddition Chemistry. New J. Chem. 2009, 33, 1010–1016. 10.1039/B818909A. [DOI] [Google Scholar]
- Albrecht B. K.; Gehling V. S.; Hewitt M. C.; Vaswani R. G.; Côté A.; Leblanc Y.; Nasveschuk C. G.; Bellon S.; Bergeron L.; Campbell R.; Cantone N.; Cooper M. R.; Cummings R. T.; Jayaram H.; Joshi S.; Mertz J. A.; Neiss A.; Normant E.; O’Meara M.; Pardo E.; Poy F.; Sandy P.; Supko J.; Sims R. J.; Harmange J. C.; Taylor A. M.; Audia J. E. Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials. J. Med. Chem. 2016, 59, 1330–1339. 10.1021/acs.jmedchem.5b01882. [DOI] [PubMed] [Google Scholar]
- Gehling V. S.; Hewitt M. C.; Vaswani R. G.; Leblanc Y.; Côté A.; Nasveschuk C. G.; Taylor A. M.; Harmange J.-C.; Audia J. E.; Pardo E.; Joshi S.; Sandy P.; Mertz J. A.; Robert J.; Sims I.; Bergeron L.; Bryant B. M.; Bellon S.; Poy F.; Jayaram H.; Sankaranarayanan R.; Yellapantula S.; Srinivasamurthy N. B.; Birudukota S.; Albrecht B. K. Discovery, Design, and Optimization of Isoxazole Azepine BET Inhibitors. ACS Med. Chem. Lett. 2013, 4, 835–840. 10.1021/ml4001485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayoub A. M.; Hawk L. M. L.; Herzig R. J.; Jiang J.; Wisniewski A. J.; Gee C. T.; Zhao P.; Zhu J.-Y.; Berndt N.; Offei-Addo N. K.; Scott T. G.; Qi J.; Bradner J. E.; Ward T. R.; Schönbrunn E.; Georg G. I.; Pomerantz W. C. K. BET Bromodomain Inhibitors with One-Step Synthesis Discovered from Virtual Screen. J. Med. Chem. 2017, 60, 4805–4817. 10.1021/acs.jmedchem.6b01336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.