

Abstract
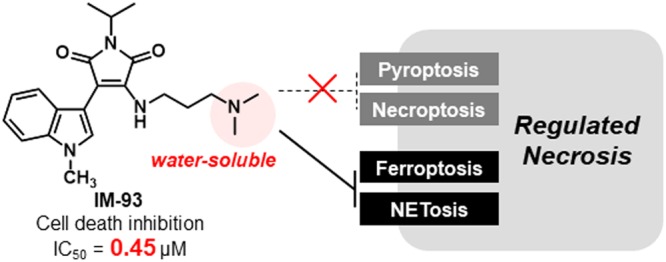
The indolylmaleimide (IM) derivative IM-17 shows inhibitory activity against oxidative-stress-induced necrotic cell death and cardioprotective activity in rat ischemia-reperfusion injury models. In order to develop a more potent derivative, we conducted a detailed structure–activity relationship study of IM derivatives and identified IM-93 as the most potent derivative with good water solubility. IM-93 inhibited ferroptosis and NETosis, but not necroptosis or pyroptosis. In contrast, ferrostatin-1 (Fer-1), a ferroptosis inhibitor, did not inhibit NETosis, although the accompanying lipid peroxidation was partially inhibited by Fer-1, as well as by IM-93. Thus, IM derivatives have a unique activity profile and appear to be promising candidates for in vivo application.
Keywords: Indolylmaleimide, necrosis, cell death, ferroptosis, NETosis
Cell death plays a critical role in maintaining the homeostasis of multicellular organisms. Classically, only apoptosis had been considered as representing programmed cell death for the removal of unwanted cells,1 and after the identification of caspase as a key player, the major signaling pathways of apoptosis were clarified.2,3 However, various types of nonapoptotic cell death were subsequently found to play important roles under both physiological and pathological conditions.4−7
Recently, necrosis/necrotic cell death, which had initially been considered as accidental and uncontrolled cell death, was redefined as regulated cell death.8−10 Morphologically, necrosis involves the rapid permeabilization of plasma/organelle membranes, leading to cellular swelling. Recent studies have identified several machineries leading to membrane permeabilization,11,12 and these provide the molecular basis of “regulated” necrosis. Membrane permeabilization induces the release from cells of cytosolic components known as damage-associated molecular patterns (DAMPs), which activate the inflammatory responses and cause severe damage to surrounding cells and tissues.12 Therefore, the control of necrosis is pathophysiologically important.
Regulated necrosis has been classified into several categories, mainly necroptosis,8,12 pyroptosis,12,13 mitochondrial permeability transition (MPT)-driven necrosis,11,14 parthanatos,14,15 ferroptosis,16 and NETosis.17,18 Specific inhibitors of each type of necrosis have been developed (Figure 1), and some of them show protective effects in various disease models,10 indicating the potential of necrosis inhibitors as therapeutic agents.
Figure 1.

Structures of typical necrosis inhibitors.
Recent studies indicate independent pathways for each type of regulated necrosis,9,10 even though the resulting damage due to membrane permeabilization is generally similar. Therefore, multiple inhibition by a common inhibitor of regulated necrosis might be a promising strategy to treat necrosis-related diseases. In this study, we developed dual inhibitors of ferroptosis, necrotic cell death induced by iron-dependent lipid peroxidation,16 and NETosis, necrotic cell death accompanying with neutrophil extracellular traps (NETs) formation in neutrophils.17,18
We have been developing indolylmaleimide (IM) derivatives as inhibitors of oxidative-stress-induced necrosis (Figure 2).19−23 A representative derivative, IM-54, inhibited necrotic cell death induced by H2O2 or tert-butyl hydroperoxide (TBHP), which was not inhibited by Nec-1, CsA, or DPQ (Figure 1).20,23 In contrast, IM-54 did not inhibit apoptosis or necroptosis induced by physiological death ligand or apoptosis induced by antitumor reagents.20,23 These results suggest that IM derivatives are selective inhibitors of some kinds of necrosis other than necroptosis, parthanatos, or MPT-driven necrosis. In addition to their unique cell death inhibition profile, IM-12 and IM-17 were found to show cardioprotective effects against ischemia-reperfusion injury in rat isolated heart model.23 Moreover, a water-soluble derivative, IM-17, inhibited sudden death by ischemia-reperfusion-induced arrhythmia in an in vivo rat model.23 Taking these results together, IM derivatives seem to be promising lead compounds for therapeutic agents to treat ischemic diseases, such as stroke, heart attack and so on. However, the cytoprotective activity of IM-17, as a representative IM derivative, was weak (Figure 2). Therefore, in this study, we conducted a detailed structure–activity relationship study of IM derivatives, aiming to identify a more potent, water-soluble derivative. We also examined the inhibitory profile toward various types of regulated necrosis.
Figure 2.

Structures and cell death inhibitory activities of IM derivatives.
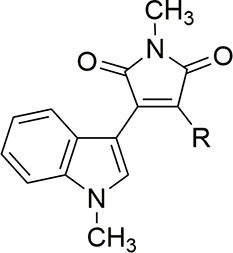
First, the alkylamino side chain of IM derivatives was varied (Scheme S1), and the inhibitory activity of the resulting derivatives against necrotic cell death induced by 100 μM H2O2 was determined by means of lactate dehydrogenase (LDH) assay (Table 1). This assay quantifies necrotic cells based on the leakage of cytosolic LDH, which reflects cellular membrane permeabilization, a typical hallmark of necrosis. IC50 values were determined as the concentration of IM derivatives causing 50% inhibition of the LDH leakage induced by 100 μM H2O2. Previous SAR studies20−22 suggested that electron delocalization from the alkylamino group to the maleimide and indole rings is important for the activity. We expected that addition of an alkyl chain to the nitrogen would increase the electron density of the nitrogen and thus the activity. However, disappointingly, the activity was not improved (IM-39 and IM-21) compared with mono- or nonsubstituted derivatives (IM-20 and IM-38). Introduction of the second alkyl group on the nitrogen might disrupt the coplanarity of the maleimide and indole rings. However, branching of the alkyl group is tolerated (IM-24, IM-42), and the cyclopentyl derivative IM-36 showed improved activity. The cyclopentyl group showed the best activity (IM-36) compared with a cyclopropyl (IM-30) or cyclohexyl (IM-35) group, indicating the importance of appropriate bulkiness and size of the alkyl chain.
Table 1. Cell Death Inhibitory Activities of IM Derivatives: Effects of Structures of the Alkylamino Group.

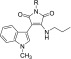
Next, from the viewpoint of flexibility for incorporating various substituents, we planned to introduce a phenyl ring. Since IM-60 having a cyclohexyl ring showed comparable activity to IM-35, we considered that a phenyl ring might be tolerated at this position. Indeed, replacement of the cyclohexyl group with a phenyl group did not decrease the activity (IM-57). However, further elongation of the alkyl chain decreased the activity (IM-76), indicating that there is a size restriction on the alkyl chain. Moreover, introduction of an additional group at the benzylic position of IM-57 seemed not to be beneficial (IM-75, IM-68). In the case of IM-75, both enantiomers were evaluated separately, but there was a little difference in activity between them ((S)-IM-75, (R)-IM-75). These results imply that a benzyl group has appropriate size and bulkiness for the activity. To examine the effects of electron density of the phenyl ring, we introduced methoxy (IM-62, IM-63, IM-64) as an electron-donating group or chlorine (IM-65, IM-66, IM-67) as an electron-withdrawing group, on the phenyl ring and tested the cell death inhibitory activity. Although the electronic effects of two groups are different, ortho-substitution decreased the activity in both cases (IM-62, IM-65). The presence of an ortho-substituent may change the preferred conformation of the side chain and thus affect the activity. In contrast, in the cases of para- and meta-substitution, the chloro group improved the activity and the methoxy group decreased it (IM-63, IM-64, IM-66, IM-67). Although the substitution effects were not strong, these results imply that the π electron density of the phenyl ring might contribute to the activity. A furan ring maintained moderate activity (IM-92). However, regardless of the substitution position, a pyridine ring decreased the activity (IM-84, IM-85, IM-86), indicating that polarity of the pyridyl nitrogen might lead to an unfavorable interaction. These results imply the existence of a hydrophobic pocket recognizing the aminoalkyl chain. Considering the importance of hydrophobic interaction for activity, we finally examined the replacement of the amino group of IM-17 with a dimethylamino group, which is expected to improve the hydrophobicity of the alkyl chain. In addition, compared with a pyridyl ring, the flexibility of the alkyl chain could allow a polar substituent to take a position away from hydrophobic region. Indeed, as expected, a dimethylamino group improved the activity (IM-31), and elongation of the alkyl chain further increased the activity (IM-87).
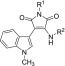
Next, we further examined the substituent effect at nitrogen of the maleimide ring. Starting from a known synthetic intermediate,24 we synthesized IM derivatives with various alkyl groups on the nitrogen of the maleimide ring (Scheme S2) and evaluated their cell death inhibitory activities (Table 2). We found that a moderately long alkyl chain improved the activity compared to the methyl group (IM-79 and IM-82). Moreover, derivatives with a branched isopropyl or cyclopentyl group were more active (IM-50, IM-53) than IM-12. However, bulkier branched groups were not favorable for the activity (IM-70, IM-77, IM-44), indicating that there is also an appropriate size and/or hydrophobicity of the substituent on the maleimide nitrogen. Considering its moderate hydrophobicity, an isopropyl group was selected as the best substituent at maleimide nitrogen (IM-50).
Table 2. Cell Death Inhibitory Activities of IM Derivatives: Effects of Substituents on the Maleimide Ring.

Based on the SAR of the maleimide ring and alkylamino side chain, we synthesized IM-93 having both isopropyl and dimethylaminopropyl groups (Scheme S3) and tested its cell death inhibitory activity (Table 3). IM-93 showed more potent activity than IM-87, i.e., it showed the greatest activity among IM derivatives having an amino group. Furthermore, IM-93 was easily transformed into its HCl salt, which showed 10 times higher water solubility than IM-17 (Scheme S4 and Table 3). Therefore, IM-93 has both higher cytoprotective activity and greater water solubility than IM-17, and it showed no significant inhibitory activities against a panel of 456 kinases (Table S1). Furthermore, IM-93 was stable in the presence of H2O2 or TBHP (Figures S1 and S2), indicating that IM-93 did not directly react with these oxidants. These results suggest that IM-93 would be a promising lead compound for developing therapeutic agents.
Table 3. Cell Death Inhibitory Activity and Water Solubility of IM Derivatives.

Previously reported23 IC50 values.
HCl salt was used.
With IM-93 in hand, we next examined its cytoprotective activity against well-characterized types of regulated necrosis. Necroptosis is a form of necrotic cell death induced by physiological death ligand, such as TNF-α or Fas ligand.8 In the previous study, IM-54 was found to have no effect on necroptosis induced by Fas ligand in combination with Z-VAD and cycloheximide (CHX). Similarly, IM-93 did not affect necroptosis (Figure 3A). We next examined the effect on pyroptosis (Figure 3B,C). Pyroptosis is caspase-1-dependent necrotic cell death of macrophages, induced by bacterial infection.12,13 It is thought to activate inflammatory response and to play an important role in immune protection against infectious diseases. To induce pyroptosis, human THP-1 cells were treated with two different bacteria, Staphylococcus aureus and Pseudomonas aeruginosa (Figure 3B,C), in the presence or absence of test compounds. CA-074Me, a cathepsin B inhibitor that was reported to inhibit pyroptosis,25 was used as a positive control. IM-93 did not inhibit pyroptosis induced by these bacteria at all, whereas CA-074Me strongly inhibited pyroptosis (Figure 3B,C). These results indicate that IM-93 does not affect either necroptosis or pyroptosis.
Figure 3.
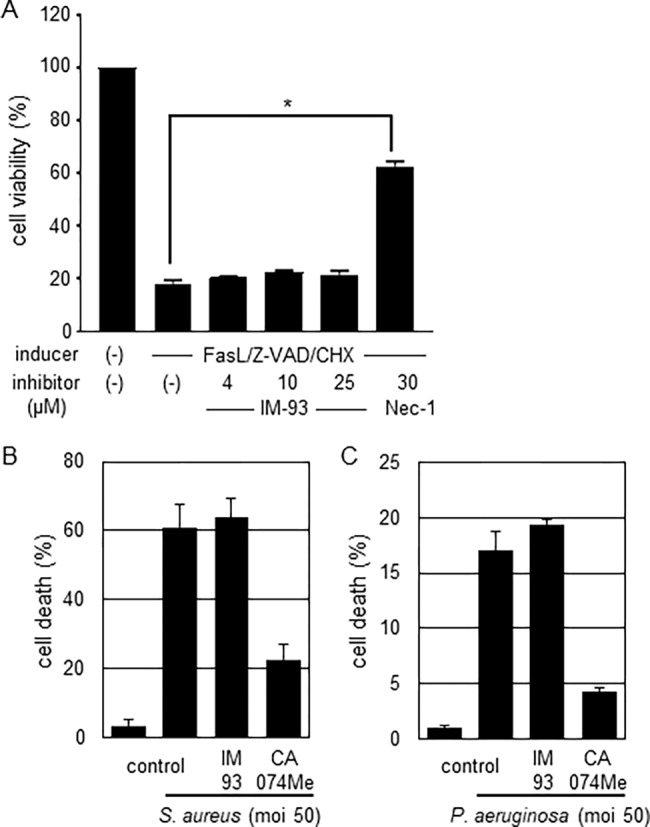
Effects of IM-93 on necroptosis (A) and pyroptosis (B,C). (A) Necroptosis was induced in Jurkat cells by treatment with Fas ligand/cycloheximide/Z-VAD in the presence or absence of IM-93 (4–25 μM) or Nec-1 (30 μM). Cell viability was determined by using a Cell Counting kit. The data shown are mean with SD of triplicate samples. (B,C) Pyroptosis was induced in THP-1 cells by infection with Staphylococcus aureus (B) or Pseudomonas aeruginosa (C) at a multiplicity of infection (moi) of 50 in the presence or absence of IM-93 (10 μM) or CA-074Me (10 μM). Cell death was determined by means of LDH release assay. The data shown are mean with SD of triplicate samples.
Next, we examined the effects of IM-93 on ferroptosis. Erastin was reported to be a chemical inducer of ferroptosis through the inhibition of glutathione synthesis.26 We found that erastin successfully induced ferroptosis in NIH3T3 cells,27 which was inhibited by ferroptosis inhibitors, deferoxamine (DFO), an iron chelator, and ferrostatin-1 (Fer-1, Figure 1), an antioxidant for lipid peroxidation, but not by caspase inhibitor Z-VAD. IM-93 inhibited ferroptosis in a similar manner to DFO or Fer-1 (Figure 4A). However, IM-54 was shown to inhibit TBHP-induced necrotic cell death of HL-60 and H9c2 cells, but not apoptotic cell death induced by various stimuli including hydrogen peroxide.20,23 Although the molecular mechanism of TBHP-induced cell death has not been clarified in detail, we found that ferroptosis inhibitors DFO and Fer-1 inhibited the TBHP-induced cell death of NIH3T3 cells.27 Therefore, we compared the effects of IM-93 with those of DFO and Fer-1 (Figure 4B). Inhibition of cell death by IM-93 was not complete, suggesting that a small portion of cells underwent apoptosis under these conditions. Inhibition profiles of all three compounds on TBHP-induced cell death were similar to those in the case of erastin-induced ferroptosis. These results suggest that TBHP-induced necrosis shares at least a part of its molecular mechanism with erastin-induced ferroptosis in NIH3T3 cells.
Figure 4.

Effects of IM-93 on ferroptosis. Ferroptosis was induced in NIH3T3 cells by treatment with erastin (0.5 μM) for 24 h (A) or TBHP (50 μM) for 12 h (B) in the presence or absence of cell death inhibitors. Cell viability was determined by using a Cell Counting kit. The data shown are mean with SD of triplicate samples.
Finally, we examined the effects of IM-93 on NETosis, necrotic cell death accompanied by NET formation in neutrophils.17,18 NET is extracellular chromosomal DNA coated with histones and proteases, which is released from neutrophils in response to various stimuli, such as microbial molecules, cholesterol crystals, and phorbol-12-myristate-13-acetate (PMA). NET had been thought to work as a protection against microbial infection by trapping microbes.28 However, recent studies indicate that NET formation and NETosis also play important roles in sterile inflammation related to various diseases, such as systemic lupus erythematosus, rheumatoid arthritis, thrombosis, and diabetes.18 NETosis was induced in human peripheral blood neutrophils by treatment with PMA in the presence or absence of IM-93, DFO, and Fer-1 (Figure 5). NET formation was visualized with SYTOX Green (Figure 5A), and NETosis induction was quantified by counting SYTOX Green-positive cells (Figure 5B). We found that NETosis was inhibited by IM-93 but not DFO or Fer-1. Fer-1 did not inhibit NETosis even at 25 μM (Figure S3A). This result shows that IM-93 works in a different manner from these known ferroptosis inhibitors.
Figure 5.

Effects of IM-93 on NETosis. (A) NETosis was induced in human neutrophils by treatment with PMA (0.8 nM) in the presence or absence of IM-93 (25 μM), DFO (100 μM), or Fer-1 (2.5 μM). After 3 h, NETs formed were visualized by fluorescence microscopy. The cells were stained with Hoechest33342 and SYTOX Green. (B) Frequency of NETs-forming neutrophils was determined by counting the number of SYTOX Green positive cells among Hoechst positive cells. The data shown are mean with SD of triplicate samples. *P < 0.01, one-way ANOVA, compared with unstimulated neutrophils.
Although the mechanism of NETosis is not known in detail, reactive oxygen species (ROS), mainly superoxide produced by NADPH oxidase (NOX), are thought to be essential for NETosis induced by PMA.29 After superoxide production, protein-arginine deiminase 4 (PAD4) is activated and induces histone citrullination, leading to DNA decondensation and NET formation. To examine the possibility that IM-93 might inhibit NOX or PAD4, we first examined the effects of IM-93 on NOX activity and ROS production during NETosis.
Superoxide production by NOX was quantified by using luminol lucigenin-enhanced chemiluminescence (Figure 6A). Neither IM-93 nor ferroptosis inhibitors inhibited superoxide production, whereas diphenyleneiodonium (DPI), an inhibitor of NOX, completely suppressed it. In addition, total ROS measured by using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) was also inhibited by DPI, but not by IM-93 and Fer-1 (Figure S4). Next, we examined the in vitro PAD4-inhibitory activity of IM-93 (Figure S5). IM-93 did not inhibit PAD4 even at 200 μM. These results confirm that IM-93 does not suppress NETosis by directly inhibiting NOX or PAD4. Our previous study revealed that lipid peroxidation could enhance the induction of NETosis.27 Therefore, we next detected lipid peroxidation by using C11-Bodipy and quantified it by means of flow cytometry (Figure 6C). DFO did not affect lipid peroxidation, suggesting that ferrous ion was not involved. Both IM-93 and Fer-1 partially suppressed lipid peroxidation to similar extents. Fer-1 directly scavenges lipid radicals and is thought to suppress the lipid peroxidation in NETosis as well as ferroptosis. However, Fer-1 did not inhibit NETosis (Figure 5B), and the more general antioxidant N-acetylcysteine (NAC) also did not show any inhibitory activity (Figure S3B). These results indicate that suppression of lipid peroxidation might not be sufficient for inhibition of NETosis. In contrast, IM-93 inhibited both NET formation and lipid peroxidation, implying that IM-93 is different from other antioxidants such as Fer-1 or NAC. Our previous study indicated that lipid oxides released from neutrophils undergoing NETosis might induce NETosis in neighboring neutrophil sequentially.27 Therefore, IM-93 might inhibit primary NETosis releasing lipid oxides and thus suppress the amplification of lipid peroxidation.
Figure 6.
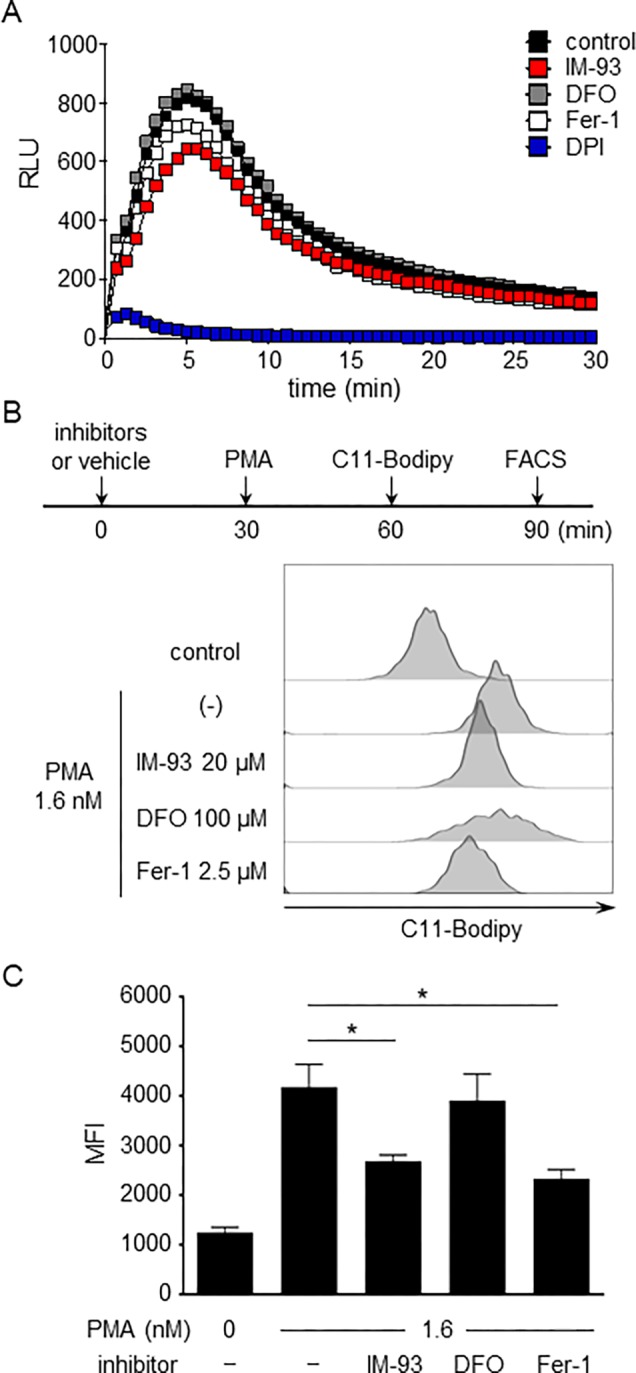
Effects of IM-93 on superoxide production and lipid peroxidation during NETosis. (A) NETosis was induced in mouse neutrophils by treatment with PMA (1 μM) in the presence or absence of IM-93 (10 μM), DFO (100 μM), Fer-1 (2.5 μM), or DPI. After 30 min, superoxide production was measured by using luminol and lucigenin-enhanced chemiluminescence. (B,C) NETosis was induced in human neutrophils by treatment with PMA (1.6 nM) in the presence or absence of IM-93 (20 μM), DFO (100 μM), or Fer-1 (2.5 μM). After 30 min, lipid peroxidation was measured by using C11-bodipy 581/591. (B) Extent of lipid peroxidation was analyzed by flow cytometry. (C) Mean fluorescence intensity (MFI) of C11-bodipy with SD of triplicate samples is shown. *P < 0.01, one-way ANOVA, compared with 1.6 nM PMA without an inhibitor.
Although the mechanism of lipid peroxidation during NETosis remains unclear, the dual inhibitory activities of IM-93 on ferroptosis and NETosis could be favorable for in vivo application. Ferroptosis was recently reported to be involved in heart ischemia-reperfusion injury in which Fer-1 showed protective effects.30 In addition, NET formation was reported to exacerbate myocardial ischemia-reperfusion injury by inducing a no-reflow phenomenon.31 In a rat myocardial ischemia-reperfusion model, cotreatment with DNase I and tissue-type plasminogen activator (t-PA) was reported to reduce the no-reflow area and infarct size by degrading NET.31 Furthermore, long-term analysis indicated beneficial effects of NET degradation on myocardial functional recovery. Therefore, ferroptosis and NETosis appear to contribute to heart ischemia-reperfusion injury in different ways, and so IM-93 might show the protective activity through multiple mechanisms. Indeed, IM-17 has already been shown to have cardioprotective activity in a rat ischemia-reperfusion model22 and might have therapeutic potential for other types of ischemia-reperfusion injury.
In conclusion, we have developed IM-93 as a water-soluble derivative with more potent in vitro activity than the previously reported IM-17. Analysis of cell death inhibitory activities against various types of regulated necrosis revealed dual inhibitory activities of IM-93 toward ferroptosis and NETosis; in this respect, it is distinct from the known ferroptosis inhibitors DFO and Fer-1. The good water solubility and unique inhibitory profile of IM-93 are expected to be favorable for its therapeutic application in ischemia-reperfusion injury, in which both ferroptosis and NETosis are involved. Further in vivo and mechanistic studies of IM derivatives are in progress.
Acknowledgments
We thank K. Kurabayashi and F. Ohnuma for assistance with the cell death inhibition assay in HL-60 cells. We also thank T. Suito and M. Kawana for secretarial assistance.
Glossary
ABBREVIATIONS
- CsA
cyclosporin A
- DCFH-DA
2′,7′-dichlorodihydrofluorescein diacetate
- DFO
deferoxamine
- DPI
diphenyleneiodonium
- Fer-1
ferrostatin-1
- IM
indolylmaleimide
- LDH
lactate dehydrogenase
- NAC
N-acetylcysteine
- NET
neutrophil extracellular trap
- NOX
NADPH oxidase
- PMA
phorbol-12-myristate-13-acetate
- RLU
relative light unit
- ROS
reactive oxygen species
- TBHP
tert-butyl hydroperoxide.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00142.
Details of synthetic procedures, analytical data, and biological studies for IM derivatives (PDF)
Author Present Address
& Advanced Medical Research Center, Hyogo University of Health Science, 1–3–6 Minatojima, Kobe 650-8530, Japan.
Author Contributions
# These authors contributed equally to this work. All authors contributed to writing the manuscript.
This work was supported in part by a Grant-in-Aid for Creation of Biologically Functional Molecules, Scientific Research on Priority Area, a Grant-in-Aid for Young Scientists (B) (18710198 and 20710163), a Grant-in-Aid for Scientific Research (B) (26293089) from Japan Society for the Promotion of Science (JSPS), a Grant-in-Aid for Scientific Research on Innovative Areas (Homeostatic Regulation by Various Types of Cell Death, 26110002, 26110004, and 26110006) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, the Uehara Memorial Foundation, the Takeda Science Foundation, and the Naito Foundation. This work was also supported by AMED-CREST, AMED under Grant Number JP18gm0710004 (to M.S.) and JP19gm1210002 (to M.T.). K.D. was supported by the Special Postdoctoral Researchers’ Program in RIKEN. E.K. was supported by the “Kibou Projects” scholarship for doctoral students from Japanese Society for Immunology. T.S. was supported by a Research Fellowship for Young Scientists from JSPS.
The authors declare no competing financial interest.
This paper was published ASAP on August 2, 2019 with an incorrect Supporting Information file. The revised paper was reposted on August 6, 2019.
Supplementary Material
References
- Kerr J. F. R.; Wyllie A. H.; Currie A. R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis H. M.; Horvitz H. R. Genetic Control of Programmed Cell Death in the Nematode C. Elegans. Cell 1986, 44, 817–829. 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- Thornberry N. A. Caspases: Enemies Within. Science 1998, 281, 1312–1316. 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Kitanaka C.; Kuchino Y. Caspase-Independent Programmed Cell Death with Necrotic Morphology. Cell Death Differ. 1999, 6, 508–515. 10.1038/sj.cdd.4400526. [DOI] [PubMed] [Google Scholar]
- Lockshin R. A.; Zakeri Z. Caspase-Independent Cell Deaths. Curr. Opin. Cell Biol. 2002, 14, 727–733. 10.1016/S0955-0674(02)00383-6. [DOI] [PubMed] [Google Scholar]
- Jäättelä M.; Tschopp J. Caspase-Independent Cell Death in T Lymphocytes. Nat. Immunol. 2003, 4, 416–423. 10.1038/ni0503-416. [DOI] [PubMed] [Google Scholar]
- Lockshin R. A.; Zakeri Z. Caspase-Independent Cell Death?. Oncogene 2004, 23, 2766–2773. 10.1038/sj.onc.1207514. [DOI] [PubMed] [Google Scholar]
- Christofferson D. E.; Yuan J. Necroptosis as an Alternative Form of Programmed Cell Death. Curr. Opin. Cell Biol. 2010, 22, 263–268. 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghe T. V.; Linkermann A.; Jouan-Lanhouet S.; Walczak H.; Vandenabeele P. Regulated Necrosis: The Expanding Network of Non-Apoptotic Cell Death Pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Conrad M.; Angeli J. P. F.; Vandenabeele P.; Stockwell B. R. Regulated Necrosis: Disease Relevance and Therapeutic Opportunities. Nat. Rev. Drug Discovery 2016, 15, 348–366. 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo V.; Bravo-San Pedro J. M.; Sica V.; Kroemer G.; Galluzzi L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016, 26, 655–667. 10.1016/j.tcb.2016.04.006. [DOI] [PubMed] [Google Scholar]
- Frank D.; Vince J. E. Pyroptosis versus Necroptosis: Similarities, Differences, and Crosstalk. Cell Death Differ. 2019, 26, 99–114. 10.1038/s41418-018-0212-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Gao W.; Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Ying Y.; Padanilam B. J. Regulation of Necrotic Cell Death: p53, PARP1 and Cyclophilin D-Overlapping Pathways of Regulated Necrosis?. Cell. Mol. Life Sci. 2016, 73, 2309–2324. 10.1007/s00018-016-2202-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatokun A. A.; Dawson V. L.; Dawson T. M. Parthanatos: Mitochondrial-Linked Mechanisms and Therapeutic Opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell B. R.; Friedmann Angeli J. P.; Bayir H.; Bush A. I.; Conrad M.; Dixon S. J.; Fulda S.; Gascón S.; Hatzios S. K.; Kagan V. E.; Noel K.; Jiang X.; Linkermann A.; Murphy M. E.; Overholtzer M.; Oyagi A.; Pagnussat G. C.; Park J.; Ran Q.; Rosenfeld C. S.; Salnikow K.; Tang D.; Torti F. M.; Torti S. V.; Toyokuni S.; Woerpel K. A.; Zhang D. D. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V.; Zychlinsky A. Neutrophil Extracellular Traps: Is Immunity the Second Function of Chromatin?. J. Cell Biol. 2012, 198, 773–783. 10.1083/jcb.201203170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorch S. K.; Kubes P. An Emerging Role for Neutrophil Extracellular Traps in Noninfectious Disease. Nat. Med. 2017, 23, 279–287. 10.1038/nm.4294. [DOI] [PubMed] [Google Scholar]
- Katoh M.; Dodo K.; Fujita M.; Sodeoka M. Structure-activity relationship of N-methyl-bisindolylmaleimide derivatives as cell death inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 3109–3114. 10.1016/j.bmcl.2005.04.015. [DOI] [PubMed] [Google Scholar]
- Dodo K.; Katoh M.; Shimizu T.; Takahashi M.; Sodeoka M. Inhibition of hydrogen peroxide-induced necrotic cell death with 3-amino-2-indolylmaleimide derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 3114–3118. 10.1016/j.bmcl.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Sodeoka M.; Dodo K. Development of selective inhibitors of necrosis. Chem. Rec. 2010, 10, 308–314. 10.1002/tcr.201000031. [DOI] [PubMed] [Google Scholar]
- Dodo K.; Hayamizu K.; Shimizu T.; Sodeoka M. Structure-activity relationship study of 3-amino-2-indolyllactam derivatives: Development of inhibitors of oxidative stress-induced necrosis. Chem. Pharm. Bull. 2016, 64, 886–898. 10.1248/cpb.c16-00259. [DOI] [PubMed] [Google Scholar]
- Dodo K.; Shimizu T.; Sasamori J.; Aihara K.; Terayama N.; Nakao S.; Iuchi K.; Takahashi M.; Sodeoka M. Indolylmaleimide Derivative IM-17 Shows Cardioprotective Effects in Ischemia-Reperfusion Injury. ACS Med. Chem. Lett. 2018, 9, 182–187. 10.1021/acsmedchemlett.7b00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M.; Buisine E.; Tartar A.; Sergheraert C. Design and Synthesis of New Leads for PKC Bisubstrate Inhibitors. Bioorg. Med. Chem. Lett. 1994, 4, 2845–2850. 10.1016/S0960-894X(01)80826-X. [DOI] [Google Scholar]
- Fujisawa A.; Kambe N.; Saito M.; Nishikomori R.; Tanizaki H.; Kanazawa N.; Adachi S.; Heike T.; Sagara J.; Suda T.; Nakahata T.; Miyachi Y. Disease-Associated Mutations in CIAS1 Induce Cathepsin B-Dependent Rapid Cell Death of Human THP-1 Monocytic Cells. Blood 2006, 109, 2903–2911. 10.1182/blood-2006-07-033597. [DOI] [PubMed] [Google Scholar]
- Dixon S. J.; Lemberg K. M.; Lamprecht M. R.; Skouta R.; Zaitsev E. M.; Gleason C. E.; Patel D. N.; Bauer A. J.; Cantley A. M.; Yang W. S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yotsumoto S.; Muroi Y.; Chiba T.; Ohmura R.; Yoneyama M.; Magarisawa M.; Dodo K.; Terayama N.; Sodeoka M.; Aoyagi R.; Arita M.; Arakawa S.; Shimizu S.; Tanaka M. Hyperoxidation of Ether-Linked Phospholipids Accelerates Neutrophil Extracellular Trap Formation. Sci. Rep. 2017, 7, 16026. 10.1038/s41598-017-15668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V.; Reichard U.; Goosmann C.; Fauler B.; Uhlemann Y.; Weiss D. S.; Weinrauch Y.; Zychlinsky A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- Hakkim A.; Fuchs T. A.; Martinez N. E.; Hess S.; Prinz H.; Zychlinsky A.; Waldmann H. Activation of the Raf-MEK-ERK Pathway Is Required for Neutrophil Extracellular Trap Formation. Nat. Chem. Biol. 2011, 7, 75–77. 10.1038/nchembio.496. [DOI] [PubMed] [Google Scholar]
- Fang X.; Wang H.; Han D.; Xie E.; Yang X.; Wei J.; Gu S.; Gao F.; Zhu N.; Yin X.; et al. Ferroptosis as a Target for Protection against Cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 2672–2680. 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L.; Zhou X.; Ji W.-J.; Lu R.-Y.; Zhang Y.; Zhang Y.-D.; Ma Y.-Q.; Zhao J.-H.; Li Y.-M. Neutrophil Extracellular Traps in Ischemia-Reperfusion Injury-Induced Myocardial No-Reflow: Therapeutic Potential of DNase-Based Reperfusion Strategy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H500–509. 10.1152/ajpheart.00381.2014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.