

Abstract
The nuclear retinoid X receptors (RXRs) are key ligand sensing transcription factors that serve as universal nuclear receptor heterodimer partners and are thus involved in numerous physiological processes. Therapeutic targeting of RXRs holds high potential but available RXR activators suffer from limited safety. Selectivity for RXR subtypes or for certain RXR heterodimers are promising strategies for safer RXR modulation. Here, we report systematic structure–activity relationship studies on biphenyl carboxylates as new RXR ligand chemotype. We discovered specific structural modifications that enhance potency on RXRs, govern subtype preference, and vary modulation of different RXR heterodimers. Fusion of these structural motifs enabled specific tuning of subtype preferential profiles with markedly improved potency. Our results provide further evidence that RXR subtype selective ligands can be designed and present a novel chemotype of RXR modulators that can be tuned for subtype and heterodimer preferences.
Keywords: Nuclear receptor, rexinoids, selectivity, neurodegeneration, cancer
The ligand-sensitive transcriptional regulator retinoid X receptor (RXR)1 has a unique role among nuclear receptors (NRs). As universal partner receptor, RXR forms heterodimers with several NRs such as liver X receptors (LXRs), peroxisome proliferator-activated receptors (PPARs), farnesoid X receptor (FXR), and thyroid hormone receptors (THRs), which respond to ligands of both partners in the dimer.2 Thereby, RXR regulates numerous physiological processes ranging from cell differentiation over metabolic balance to inflammation.2 To date, RXR has only a subordinate therapeutic relevance in cancer treatment with the pan-RXR agonist bexarotene (1a, Scheme 1) as sole drug approved synthetic RXR agonist. According to recent data, RXR may hold enormous potential for the treatment of neurodegenerative diseases.3,4 However, safety issues of available RXR agonists concerning adverse effects on lipid homeostasis5 and thyroid hormone signaling6 prevent further exploration of RXR as therapeutic target in such indications.
Scheme 1. RXR Agonists Bexarotene (1a), 9-cis Retinoic Acid (1b), Selective RXRβ Activator Valerenic Acid (2), and Lead Compound 3.

Ligands with selectivity for the three RXR subtypes (RXRα, RXRβ, and RXRγ), which markedly differ in their expression patterns, and/or for RXR heterodimers are putative strategies to improve the safety of RXR modulators.7,8 However, the ligand binding sites of all three RXRs are identical in all pocket forming residues,7 which renders the design of selective ligands a challenging endeavor. While some heterodimer preferential RXR ligands have been described9−12 demonstrating that heterodimer selectivity can be achieved, little is known about designing RXR subtype selectivity, and only the natural product valerenic acid (2)13 has been reported as a subtype selective RXR ligand, to date. The failure to discover subtype selective RXR activators may in part be due to the limited number of diverse scaffolds14 that have been systematically studied as RXR ligands, which mostly mimic the L-shape of 9-cis configured vitamin A metabolites such as 1b as putative endogenous RXR ligands. Here, we evaluate a new RXR ligand chemotype derived from 3(14) that does not comprise this typical rexinoid geometry for its potential to overcome these issues. We have conducted systematic structure–activity relationship (SAR) studies (compounds 4–28) concerning RXR potency, as well as subtype and heterodimer selectivity. We discovered structural modifications enabling subtype and heterodimer preference and developed markedly improved descendants of 3 with specific RXR modulatory profiles.
Unsubstituted biphenyl-4-carboxylate (4) was commercially available. RXR ligands 5–28 were prepared according to Schemes 2–4 by Suzuki coupling from commercially available aryl halides/aryl boronic acid pairs (Table S1) and biologically characterized in specific Gal4 hybrid reporter gene assays15−17 for the RXRα, RXRβ, and RXRγ subtypes (Table 1). RXR heterodimer modulation was studied using firefly reporter constructs where reporter gene expression is under the control of human response elements for the full-length heterodimers RXR:LXR and RXR:PPAR.10,18,19
Scheme 2. Synthesis of 5–8, 10, 11, 14–18, and 24.

Reagents and conditions: (a) Pd(PPh3)4, dioxane, H2O, rt, 3–42.5 h, 9–69%.
Scheme 4. Synthesis of RXR Ligands 20–23 and 25–27.

Reagents and conditions: (a) Pd(PPh3)4, dioxane, H2O, rt, 14–48 h, 3–72%; (b) LiOH, EtOH, H2O, THF, rt, 15 h, 73%.
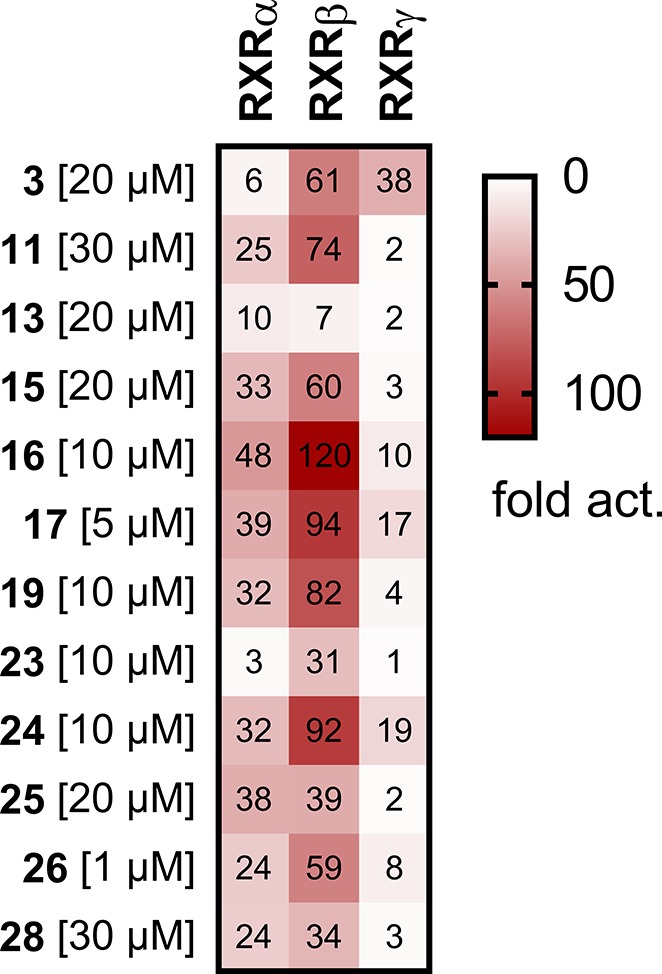
Table 1. Biological Activity of 3 and Derivatives 4–28 on RXRsa.

EC50 and fold activation data are the mean ± SD, n ≥ 3. Fold activation refers to max. fold activation vs. 0.1% DMSO. inactive: no significant activation at the indicated concentration.
Scheme 3. Synthesis of RXR Ligands 9, 12, 13, 19 and 28.

Reagents and conditions: (a) Pd(PPh3)4, dioxane, H2O, rt, 5.5 h, 26%; (b) LiOH, EtOH, H2O, THF, rt, 15–18 h, 74–84%; (c) Pd(PPh3)4, dioxane, H2O, rt, 3.5 h, 24%; (d) Pd(PPh3)4, dioxane, H2O, rt, 24 h, 71%; (e) Pd(PPh3)4, dioxane, H2O, rt, 16 h, 4–16%.
Lead compound 3 has recently been reported as a RXR activator with unusual linear scaffold and preference for the RXRβ and RXRγ subtypes14 (Table 1, Figure S1). To systematically study the SAR of 3 and analogues as RXR ligands (Table 1), we first simplified the structure to the unsubstituted biphenyl-4-carboxylate (4), which was inactive on RXRs up to 30 μM, and the same held true for the 3′-methylbiphenyl-4-carboxylate (5).
With the introduction of an isopropyl (6) or tert-butyl (7) moiety in 3′-position, activity was recovered with a shift in the subtype preference profile toward RXRα and RXRβ, while activity on RXRγ was low. Therein, both 6 and 7 revealed slight relative preference for RXRβ in terms of stronger activation efficacy on this subtype. Consistently, the small 3′-methoxy derivative 8 was inactive up to 30 μM concentration, while RXR agonism was recovered with increasing alkoxy substituent size in 3′-ethoxy analogue 9 and 3′-isopropyloxy derivative 10, which both were RXRα/RXRβ activators. In contrast to its alkyl counterpart 6, 10 had no pronounced RXRβ subtype-preference over RXRα. Notably, none of the monosubstituted biphenylcarboxylates 5–10 achieved an activation efficacy on RXRγ that was comparable to the strong activity of 3 on this subtype, and while some tendencies for RXRβ preference were observed, no structural variation had enhanced potency on RXRs.
When 3′,5′-double substitutions as in 3 were reinstated, small alkyl residues, i.e., 3′,5′-dimethyl (11) and 3′,5′-diethyl (12), achieved considerable potency on RXRs. Both compounds displayed moderate preference for the RXRβ subtype concerning activation efficacy, which was more pronounced for 11, while 12 had higher overall potency in terms of lower EC50 values. With asymmetric 3′-tert-butyl-5′-methyl substitution (13), subtype preference and activation efficacy dropped markedly to a partial RXR agonistic profile. 3′,5′-Dimethoxyderivative 14 was inactive on RXRβ and RXRγ at 30 μM but retained RXRα activation despite weak efficacy. Double chlorine substitution (15) generated strong activity on RXRα and RXRβ but markedly diminished activity on RXRγ. Thereby, 3′,5′-dichlorobiphenyl-4-carboxylate (15) inverted the subtype preferential profile of 3. Double trifluoromethyl substitution (16) was favored by all RXRs and also conserved slight preference for RXRβ concerning activation efficacy.
These observations demonstrate that 3′,5′-substituents affect RXR subtype preference wherein bulky lipophilic residues were most essential for RXRγ activation, while the RXRα and RXRβ subtypes better tolerated smaller moieties. RXRβ preference was most pronounced for the 3′-isopropyl analogue 6, while for dimethoxy derivative 14, activity was only observed on RXRα, and 3′,5′-dichlorosubstitution (15) produced a RXRα/RXRβ preferential profile, while the extraordinarily bulky 3′,5′-di-tert-butyl substitution of 3 seemed sufficient to abrogate activity on RXRα but was favored by RXRγ.
We then focused on the SAR of the benzoic acid part. Chain elongation by one carbon (17) was accompanied by a remarkable gain in potency. Compared to 3, 17 was more active by about a factor of 20. The further elongated propionic acid homologue 18 did not follow this trend and was less active than 17 suggesting that the arylacetic acid motif of 17 comprised an optimal length. Both, 17 and 18, conserved the relative RXRβ preference that was already observed for several analogues of the series but failed to enhance it. When the carboxylic acid was shifted from the 4-position (3) to the 3-position (19), RXRγ agonism dropped markedly, while potency on RXRα was enhanced resulting in RXRα/RXRβ activation, which renders the 1,3-substitution geometry of 19 as another switch toward RXRα/RXRβ preference.
Subsequently, we put our attention on the free positions of the aryl carboxylic acid motif in 3. Introduction of a nitrogen atom (20, 21) to eventually enhance polarity and solubility was not tolerated in either position and a methyl substituent in meta-position (22) to the acidic side chain abrogated activity on all RXRs, as well. Methyl substitution in ortho-position (23), however, had a remarkable impact on subtype selectivity, as 23 exclusively activated RXRβ. Thus, ortho-methylation relative to the carboxylic acid residue as another switch to subtype preference turned out to block RXRγ modulation.
In an attempt to apply the obtained SAR knowledge for structural optimization, we fused both potency supporting modifications of 16 and 17 in trifluoromethyl-substituted arylacetic acid 24, which was indeed endowed with markedly enhanced potency on all RXRs. Compound 24 retained the weak relative preference for RXRβ but was also active on the RXRα and RXRγ subtypes.
We then aimed at designing a preferential RXRα/RXRβ modulator with enhanced potency by fusing the activity promoting 3′,5′-bis(trifuormethyl) substitution (16) with the RXRγ disfavored ortho-methylarylcarboxylate moiety (23). The resulting compound 25 was indeed inactive on RXRγ, while activity on RXRα and RXRβ was conserved, which further confirmed the value of the ortho-methyl substitution to disrupt RXRγ affinity. To improve the potency of this balanced RXRα/RXRβ preferential ligand 25, we then additionally included the activity favored arylacetic acid motif resulting in 26. This triple combination of potency and selectivity promoting structural elements produced high potency on RXRα and RXRβ and retained preference for these subtypes over RXRγ. Aiming to further enhance RXRβ selectivity, we incorporated the RXRγ potency reducing ortho-methylarylcarboxylate motif (23), the RXRα disfavored di-tert-butylphenyl moiety of 3, and the potency promoting acetic acid side chain (17) in 27, which indeed displayed pronounced RXRβ preference. However, 27 exhibited considerable toxicity at concentrations above 10 μM preventing further characterization.
In addition, we combined the RXRα/RXRβ preferential structural elements of 15 (3′,5′-dichlorosubstitution) and 19 (biphenyl-3-carboxylate). The fused compound 28 achieved no improvement in potency or efficacy on RXRα and RXRβ but fully retained preference over RXRγ confirming that the biphenyl-3-carboxylate geometry and the double chlorine substitution are suitable motifs to abrogate RXRγ agonism.
Our SAR studies on this new potent RXR ligand chemotype (summarized in Scheme 5) demonstrate that the modulatory activity of biphenylcarboxylates on RXRs can be tuned to differential subtype preference by minor structural changes that particularly result in remarkable differences in activation efficacy (Figure 1, Figure S1). Therein, the carboxylic acid and its ortho-position as well as the terminal lipophilic substitution pattern turned out to be key motifs of the compound class to govern differential subtype preference profiles. As exemplified in 24–28, these individual selectivity-driving structural changes are compatible for combination in fused molecules to tune subtype preference and promote potency.
Scheme 5. Summarized SAR of Biphenylcarboxylates on RXRs.
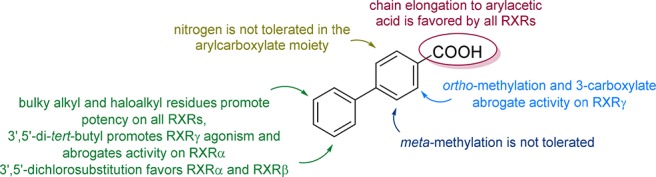
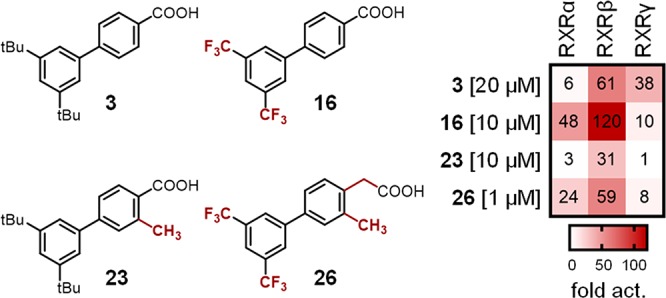
Figure 1.
Subtype preference profiles of selected RXR ligands. Heat map shows mean fold activation at a concentration ≥ EC90 as indicated, n ≥ 3. The corresponding dose–response curves are displayed in Figure S1.
Particularly, compounds 13, 17, 23, 24, 26, and 28 were discovered as potent novel RXR ligands with attractive subtype preference profiles and, therefore, underwent selectivity profiling over NRs related to RXRs in a broad panel of hybrid reporter gene assays (Figure 2). All optimized biphenylacetic acid derivatives 17, 24, 26, and 28 displayed favorable RXR selectivity, which further highlights the acetic acid chain as a favorable motif in this RXR ligand series. The asymmetric partial RXR agonist 3′-tert-butyl-5′-methylbiphenyl-4-carboxylate 13 as well as the biphenyl-3-methyl derivatives 23 and 26 in addition to RXRs activated the fatty acid sensing RARs and PPARs with weak efficacy and turned out less selective. The activity of 17, 24, and 26 on RXRs in the Gal4 hybrid reporter gene assays was blocked by the well characterized RXR antagonist HX531,20 which conclusively confirmed their RXR mediated activity (Figure S2). Exemplary further in vitro characterization of the most active RXR activator 24 demonstrated acceptable solubility for a fatty acid mimetic21 rexinoid (24, 1.15 mg/L; 1a, 0.3 mg/L10) and favorably high stability against microsomal degradation (Figure S3).
Figure 2.

Selectivity profiles of 13, 17, 23, 24, 26, and 28 on nuclear receptors (tested in Gal4 hybrid reporter gene assays). Heat map shows mean fold activation vs DMSO (0.1%), n ≥ 2. RXR activation data is shown for comparison.
RXRs act as universal heterodimer partners for numerous ligand-activated transcription factors, which partly mediate the side effects of RXR agonists such as RXR:LXR induced hyperlipidemia.5,9 Therefore, RXR ligands comprising selectivity for certain RXR heterodimers may hold strong therapeutic potential and provide improved safety, as well. To evaluate whether the biphenylcarboxylate RXR ligand scaffold is also suitable to design RXR heterodimer preferential agents, we studied 13, 17, 24–26, and 28 at concentrations above their EC90s on RXRs for their activity on the human heterodimers RXR:LXR and RXR:PPARγ (Figure 3, Figure S4). The reference RXR agonist bexarotene (1a) strongly activated both heterodimers in this setting and demonstrated marked synergy with LXR and PPARγ agonists observed by superadditive effects.
Figure 3.
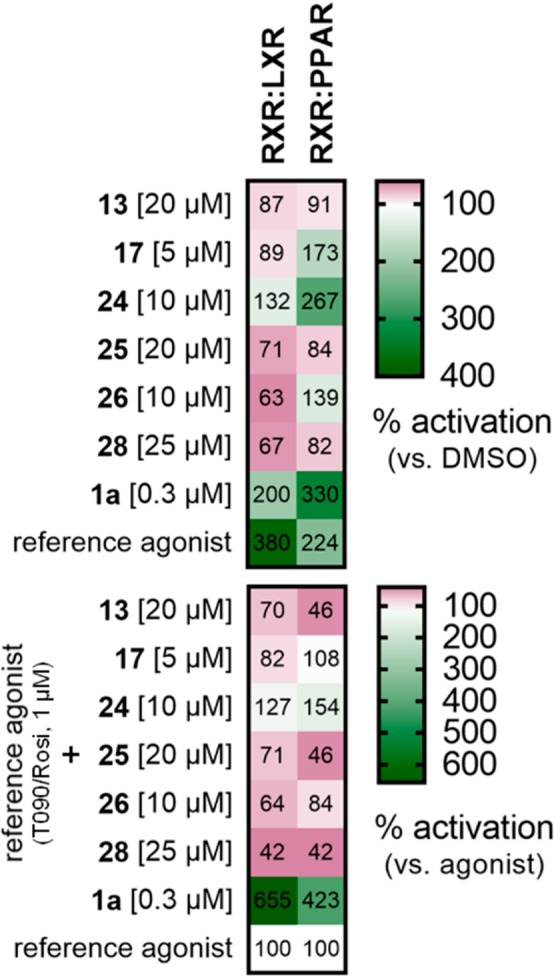
RXR ligands 13, 17, 24–26, and 28 display different activities on the RXR heterodimers RXR:LXR and RXR:PPARγ. Upper panel shows mean relative activation vs DMSO (0.1%), lower panel shows mean relative synergistic activation in the presence of reference agonist T0901317 (1 μM, LXR) or rosiglitazone (1 μM, PPARγ). Heat maps show the mean of three independent experiments conducted in duplicates. See Figure S4 for data distribution.
Partial RXR agonist 13, which is characterized by low activation efficacy on RXRα and RXRβ, failed to activate either heterodimer but blocked activation of both by specific agonists of the partner receptors. Especially RXR:PPARγ activation by rosiglitazone was strongly reduced in the presence of 13. This profile agrees with the compound’s partial agonism, which entails partial antagonism. The markedly more efficacious pan-RXR activator 17, in contrast, strongly activated RXR:PPARγ, while it did not affect rosiglitazone-mediated RXR:PPARγ activation. On RXR:LXR, 17 was indifferent alone and in the presence of LXR agonist T0901317. The optimized pan-RXR agonist 24 also in the heterodimer setting turned out to be the most active RXR agonist and strongly activated the RXR:PPARγ heterodimer with higher efficacy than the PPARγ agonist rosiglitazone and behaved additive with the PPARγ activator. In addition, 24 slightly activated RXR:LXR and enhanced the activity of T0901317, but its efficacy on RXR:PPARγ was markedly higher. Compounds 25 and 28 neither activated RXR:LXR nor RXR:PPARγ but repressed activation of both heterodimers by their respective agonists, particularly in the case of RXR:PPARγ. The ortho-methylbiphenyl-4-acetic acid derivative 26 activated RXR:PPARγ without showing synergy with rosiglitazone but blocked RXR:LXR activity.
These observations on RXR heterodimer modulation also indicate preliminary SAR lessons for heterodimer preference. Compounds 24 and 26, which differ only in the presence of an ortho-methyl substitution in 26, displayed marked difference in RXR activation. While 24 activated both heterodimers and showed synergy with the agonists, 26 displayed a repressor profile on RXR:LXR and only weakly activated RXR:PPARγ without synergy. The ortho-methyl residue, thus, had a remarkable effect on heterodimer preference, too. Similar observations for a single methyl moiety have been reported for other RXR:LXR sparing RXR ligands.9 Shortening the acidic chain from phenylacetic acid in 26 to aryl carboxylate 25 further reduced activating activity on RXR:PPARγ. Moreover, bulky lipophilic residues (17, 24, and 26) appear favored for RXR:PPARγ activation over smaller substituents (13 and 28). Structural modifications on the biphenylcarboxylate scaffold, thus, not only govern RXR subtype preference but can also be employed to tune RXR heterodimer specific activity.
To provide a structural basis for future optimization of biphenyl-based RXR modulators, we solved the X-ray structure of the RXRα LBD in complex with 24 (PDB-ID: 6SJM; Figure 4, Table S2). Compound 24 binds in the classical orthosteric site, which is also addressed by classical rexinoids such as 1a but with distinctive binding properties. The carboxylate moiety of 24 engages the typical anchoring ionic contact with Arg316 at the end of the RXR ligand binding site, and the biphenyl scaffold is entirely surrounded by lipophilic residues with the upper aromatic ring forming an edge-to-face contact to Phe313. The binding site of 24 is defined by helices 2, 6, 7, and 11, but in contrast to known RXR activators, 24 does not extend toward helix 12. This unique binding mode demonstrates that the typical L-shape of most RXR ligands is not essential for RXR activation. This confined binding site occupation provides room for structure-based optimization but may also explain the distinctive biological activity of this new RXR ligand chemotype.
Figure 4.
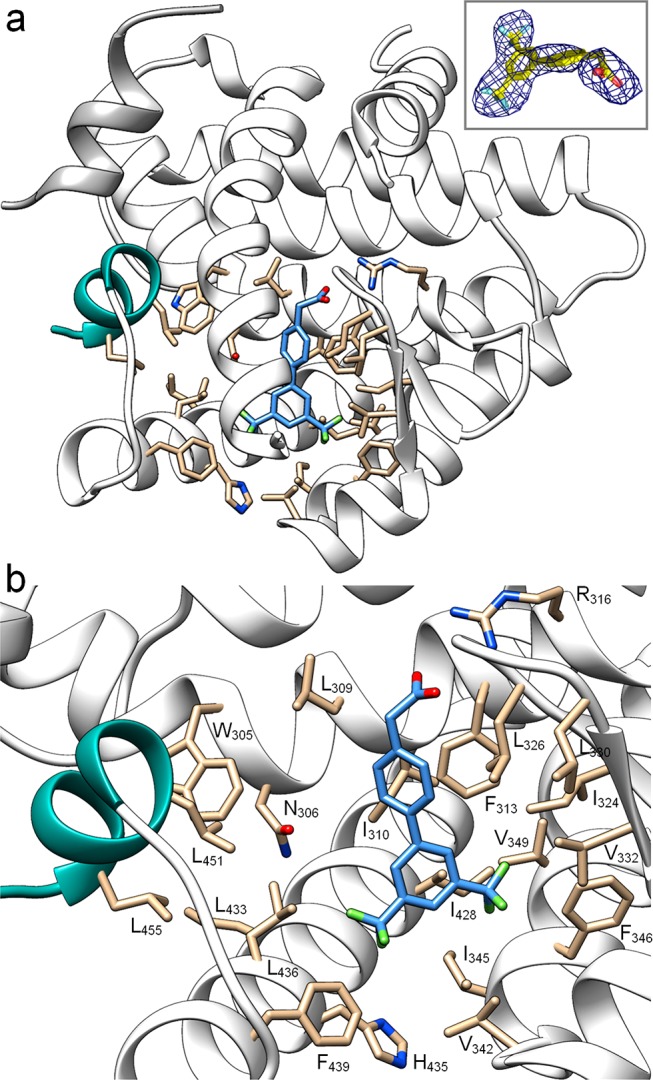
X-ray structure of the RXRα LBD in complex with 24 (blue). Helix 12 (activation function) is depicted in green. PDB-ID: 6SJM. (a) Full RXR LBD; inset shows omitted |FO| – |FC| electron density map contoured at 3σ for the bound ligand. (b) Binding site view. Figures were prepared using UCSF Chimera.22
With the opportunity to structurally control RXR subtype and heterodimer preference, the biphenylcarboxylate chemotype of RXR ligands holds potential to serve as scaffold for the development of innovative RXR ligands to overcome safety and selectivity issues of classical rexinoids. While some heterodimer specific RXR agonists have been developed previously9−12 as essential steps to further explore the therapeutic potential of these essential nuclear receptors, systematic studies on RXR subtype selectivity are rare. Here we demonstrate that minor structural modifications can have remarkable effect on RXR subtype preference. After the recent discovery of valerenic acid13 as selective RXRβ agonist, we thereby provide further evidence that RXRs can be modulated in a subtype selective fashion with small molecules.
Experimental Procedures
General
All final compounds for biological evaluation had a purity of >95% according to HPLC–UV analysis at wavelengths of 245 and 280 nm.
2-(3′,5′-bis(Trifluoromethyl)-(1,1′-biphenyl)-4-yl)acetic Acid (24)
2-(4-Iodophenyl)acetic acid (30b, 0.39 g, 1.5 mmol, 1.0 equiv), 3,5-bis(trifluoromethyl)benzeneboronic acid (29i, 0.46 g, 1.8 mmol, 1.2 equiv), and sodium carbonate (0.57 g, 5.4 mmol, 3.6 equiv) were dissolved in dioxane (10 mL) and water (2.5 mL). The mixture was stirred at room temperature for 10 min, while the solvents were degassed with argon. Then, Pd(PPh3)4 (0.086 g, 0.075 mmol, 0.050 equiv) was added, and the mixture was stirred under reflux for 3 h. The resulting mixture was cleared over Celite 535, 20 mL of EtOAc and 10 mL of HOAc were added, and phases were separated. The organic layer was washed with 2 N HCl (10 mL) and dried over MgSO4, and the solvents were removed in vacuum. Further purification was performed by column chromatography with hexane/EtOAc/HOAc (83:15:2) as mobile phase and recrystallization from EtOAc to obtain 24 as colorless solid (45 mg, 8.6%). Rf (hexane/EtOAc/HOAc = 83:15:2) = 0.26. 1H NMR (500 MHz, acetone-d6): δ = 8.29 (s, 2H), 8.02 (s, 1H), 7.83–7.80 (m, 2H), 7.51 (d, J = 8.4 Hz, 2H), 3.74 (s, 2H). 13C NMR (126 MHz, acetone-d6): δ = 172.51, 144.19, 137.16, 137.04, 132.66, 131.27, 128.23, 125.62, 123.46, 121.56, 40.83. HRMS (MALDI): m/z calculated 348.05795 for C16H10O2F6; found 348.05931 ([M]*).
Glossary
ABBREVIATIONS
- CAR
constitutive androstane receptor
- FXR
farnesoid X receptor
- LXR
liver X receptor
- NR
nuclear receptor
- PPAR
peroxisome proliferator-activated receptor
- RXR
retinoid X receptor
- SAR
structure–activity relationship
- THR
thyroid hormone receptor
- VDR
vitamin D receptor
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00306.
Supplementary Figures and Tables, synthetic methods, in vitro pharmacological methods, protein expression and crystallization, and analytical compound characterization data(PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Germain P.; Chambon P.; Eichele G.; Evans R. M.; Lazar M. A.; Leid M.; De Lera A. R.; Lotan R.; Mangelsdorf D. J.; Gronemeyer H. International Union of Pharmacology. LXIII. Retinoid X Receptors. Pharmacol. Rev. 2006, 58 (4), 760–772. 10.1124/pr.58.4.7. [DOI] [PubMed] [Google Scholar]
- Evans R. M.; Mangelsdorf D. J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157 (1), 255–266. 10.1016/j.cell.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. K.; Jarjour A. A.; Nait Oumesmar B.; Kerninon C.; Williams A.; Krezel W.; Kagechika H.; Bauer J.; Zhao C.; Baron-Van Evercooren A.; et al. Retinoid X Receptor Gamma Signaling Accelerates CNS Remyelination. Nat. Neurosci. 2011, 14 (1), 45–53. 10.1038/nn.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer P. E.; Cirrito J. R.; Wesson D. W.; Lee C. Y. D.; Karlo J. C.; Zinn A. E.; Casali B. T.; Restivo J. L.; Goebel W. D.; James M. J.; et al. ApoE-Directed Therapeutics Rapidly Clear ß-Amyloid and Reverse Deficits in AD Mouse Models. Science 2012, 335 (6075), 1503–1506. 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries-van der Weij J.; de Haan W.; Hu L.; Kuif M.; Oei H. L. D. W.; van der Hoorn J. W. A.; Havekes L. M.; Princen H. M. G.; Romijn J. A.; Smit J. W. A.; et al. Bexarotene Induces Dyslipidemia by Increased Very Low-Density Lipoprotein Production and Cholesteryl Ester Transfer Protein-Mediated Reduction of High-Density Lipoprotein. Endocrinology 2009, 150 (5), 2368–2375. 10.1210/en.2008-1540. [DOI] [PubMed] [Google Scholar]
- Sherman S. I.; Gopal J.; Haugen B. R.; Chiu A. C.; Whaley K.; Nowlakha P.; Duvic M. Central Hypothyroidism Associated with Retinoid X Receptor–Selective Ligands. N. Engl. J. Med. 1999, 340 (14), 1075–1079. 10.1056/NEJM199904083401404. [DOI] [PubMed] [Google Scholar]
- de Lera A. R.; Bourguet W.; Altucci L.; Gronemeyer H. Design of Selective Nuclear Receptor Modulators: RAR and RXR as a Case Study. Nat. Rev. Drug Discovery 2007, 6 (10), 811–820. 10.1038/nrd2398. [DOI] [PubMed] [Google Scholar]
- Merk D. Chances and Challenges of Retinoid X Receptor Gamma Targeting for Regenerative Multiple Sclerosis Treatment. Future Med. Chem. 2015, 7 (18), 2411–2413. 10.4155/fmc.15.163. [DOI] [PubMed] [Google Scholar]
- Atigadda V. R.; Xia G.; Desphande A.; Boerma L. J.; Lobo-Ruppert S.; Grubbs C. J.; Smith C. D.; Brouillette W. J.; Muccio D. D. Methyl Substitution of a Rexinoid Agonist Improves Potency and Reveals Site of Lipid Toxicity. J. Med. Chem. 2014, 57 (12), 5370–5380. 10.1021/jm5004792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollinger J.; Gellrich L.; Schierle S.; Kilu W.; Schmidt J.; Kalinowsky L.; Ohrndorf J.; Kaiser A.; Heering J.; Proschak E.; et al. Tuning Nuclear Receptor Selectivity of Wy14,643 towards Selective Retinoid X Receptor Modulation. J. Med. Chem. 2019, 62 (4), 2112–2126. 10.1021/acs.jmedchem.8b01848. [DOI] [PubMed] [Google Scholar]
- Leibowitz M. D.; Ardecky R. J.; Boehm M. F.; Broderick C. L.; Carfagna M. A.; Crombie D. L.; D’Arrigo J.; Etgen G. J.; Faul M. M.; Grese T. A.; et al. Biological Characterization of a Heterodimer-Selective Retinoid X Receptor Modulator: Potential Benefits for the Treatment of Type 2 Diabetes. Endocrinology 2006, 147 (2), 1044–1053. 10.1210/en.2005-0690. [DOI] [PubMed] [Google Scholar]
- Lagu B.; Lebedev R.; Pio B.; Yang M.; Pelton P. D. Dihydro-[1H]-Quinolin-2-Ones as Retinoid X Receptor (RXR) Agonists for Potential Treatment of Dyslipidemia. Bioorg. Med. Chem. Lett. 2007, 17 (12), 3491–3496. 10.1016/j.bmcl.2007.01.049. [DOI] [PubMed] [Google Scholar]
- Merk D.; Grisoni F.; Friedrich L.; Gelzinyte E.; Schneider G. Computer-Assisted Discovery of Retinoid X Receptor Modulating Natural Products and Isofunctional Mimetics. J. Med. Chem. 2018, 61 (12), 5442–5447. 10.1021/acs.jmedchem.8b00494. [DOI] [PubMed] [Google Scholar]
- Merk D.; Grisoni F.; Friedrich L.; Gelzinyte E.; Schneider G. Scaffold Hopping from Synthetic RXR Modulators by Virtual Screening and de Novo Design. MedChemComm 2018, 9 (8), 1289–1292. 10.1039/C8MD00134K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heering J.; Merk D. Hybrid Reporter Gene Assays: Versatile In Vitro Tools to Characterize Nuclear Receptor Modulators. Methods Mol. Biol. 2019, 1966, 175–192. 10.1007/978-1-4939-9195-2_14. [DOI] [PubMed] [Google Scholar]
- Flesch D.; Cheung S.-Y.; Schmidt J.; Gabler M.; Heitel P.; Kramer J. S.; Kaiser A.; Hartmann M.; Lindner M.; Lüddens-Dämgen K.; et al. Non-Acidic Farnesoid X Receptor Modulators. J. Med. Chem. 2017, 60 (16), 7199–7205. 10.1021/acs.jmedchem.7b00903. [DOI] [PubMed] [Google Scholar]
- Heitel P.; Achenbach J.; Moser D.; Proschak E.; Merk D. DrugBank Screening Revealed Alitretinoin and Bexarotene as Liver X Receptor Modulators. Bioorg. Med. Chem. Lett. 2017, 27 (5), 1193–1198. 10.1016/j.bmcl.2017.01.066. [DOI] [PubMed] [Google Scholar]
- Heitel P.; Gellrich L.; Kalinowsky L.; Heering J.; Kaiser A.; Ohrndorf J.; Proschak E.; Merk D. Computer-Assisted Discovery and Structural Optimization of a Novel Retinoid X Receptor Agonist Chemotype. ACS Med. Chem. Lett. 2019, 10 (2), 203–208. 10.1021/acsmedchemlett.8b00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitel P.; Gellrich L.; Heering J.; Goebel T.; Kahnt A.; Proschak E.; Schubert-Zsilavecz M.; Merk D. Urate Transporter Inhibitor Lesinurad Is a Selective Peroxisome Proliferator-Activated Receptor Gamma Modulator (SPPARγM) in Vitro. Sci. Rep. 2018, 8 (1), 13554. 10.1038/s41598-018-31833-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebisawa M.; Umemiya H.; Ohta K.; Fukasawa H.; Kawachi E.; Christoffel G.; Gronemeyer H.; Tsuji M.; Hashimoto Y.; Shudo K.; et al. Retinoid X Receptor-Antagonistic Diazepinylbenzoic Acids. Chem. Pharm. Bull. 1999, 47 (12), 1778–1786. 10.1248/cpb.47.1778. [DOI] [PubMed] [Google Scholar]
- Proschak E.; Heitel P.; Kalinowsky L.; Merk D. Opportunities and Challenges for Fatty Acid Mimetics in Drug Discovery. J. Med. Chem. 2017, 60 (13), 5235–5266. 10.1021/acs.jmedchem.6b01287. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera--A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25 (13), 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.