

Abstract
Neovascular eye diseases are a major cause of blindness. Excessive angiogenesis is a feature of several conditions, including wet age-related macular degeneration, proliferative diabetic retinopathy, and retinopathy of prematurity. Development of novel anti-angiogenic small molecules for the treatment of neovascular eye disease is essential to provide new therapeutic leads for these diseases. We have previously reported the therapeutic potential of anti-angiogenic homoisoflavanone derivatives with efficacy in retinal and choroidal neovascularization models, although these are racemic compounds due to the C3-stereogenic center in the molecules. This work presents asymmetric synthesis and structural determination of anti-angiogenic homoisoflavanones and pharmacological characterization of the stereoisomers. We describe an enantioselective synthesis of homoisoflavanones by virtue of ruthenium-catalyzed asymmetric transfer hydrogenation accompanying dynamic kinetic resolution, providing a basis for the further development of these compounds into novel experimental therapeutics for neovascular eye diseases.
Keywords: Homoisoflavanone, 3-Benzyl-chroman-4-one, 3-Benzyl-chroman-4-ol, Asymmetric transfer hydrogenation, Dynamic kinetic resolution, Angiogenesis, Ocular neovascularization
Graphical Abstract
INTRODUCTION
Angiogenesis, a physiological process in which new blood vessels are formed, is involved in the progression of several diseases such as cancer and neovascular eye disease.1 These latter diseases, including neovascular (“wet”) age-related macular degeneration (AMD), proliferative diabetic retinopathy (PDR), and retinopathy of prematurity (ROP), are characterized by abnormal angiogenesis in the posterior segment of the eye (retina and choroid), leading to vascular ischemia and leakage that can result in hemorrhage, edema, and retinal detachment. Retinal and choroidal neovascularization (CNV) are major causes of vision loss in eye disease and have an immense effect on quality of life.2 Anti-angiogenic therapies targeting the important, proangiogenic vascular endothelial growth factor (VEGF) pathway have been successful in stabilizing, or even improving central vision in many patients.3 The present anti-VEGF therapies such as ranibizumab, bevacizumab, pegaptanib, and aflibercept are used as standard treatments for neovascular AMD. Although these biologics are now used widely, they have limitations: most target the VEGF pathway only at the level of the VEGF ligand-receptor interaction, approximately 35% of patients with wet AMD are refractive to these biologics4 and they can also be associated with significant side effects.5 There are as yet no approved small molecules for treating retinal or choroidal neovascularization, although some promising small molecules are being developed.6 Thus, novel antiangiogenic small molecules still need to be developed to complement and perhaps combine with existing therapies for wet AMD, PDR and ROP.
Homoisoflavanones consist of a 3-benzylchroman-4-one skeleton and are a small group of natural chromane heterocycles, purified from various plants. Some review articles deal with the plant sources and biological activities of the homoisoflavanones.7 These natural products show a wide range of biological activities such as anti-inflammatory, antioxidant, anti-microbial, and anticancer activities. There is an antiangiogenic homoisoflavanone, 5,7-dihydroxy-3-(3-hydroxy-4-methoxybenzyl)-6-methoxychroman-4-one, called cremastranone (1), that has been isolated from the plants Muscari armeniacum,8 Chionodoxa luciliae,9 Scilla natalensis,10 Merwilla plumbea,11 and Cremastra appendiculata (D. Don).12 Cremastranone was reported to inhibit neovascularization through anti-proliferative activity against human umbilical vein endothelial cells (HUVECs). It had a 50% growth inhibitory concentration (GI50) value of 1.5 μM without cytotoxic effect (Figure 1) and reduced retinal neovascularization in the laser-induced choroidal neovascularization (L-CNV) and oxygen induced retinopathy (OIR) mouse models.13 We successfully synthesized cremastranone (1) as a racemic mixture and evaluated its anti-proliferative activity against HUVECs and also the more tissue-relevant human retinal microvascular endothelial cells (HRECs) with GI50 values of 0.38 and 0.22 μM, respectively, with slightly more potency than the naturally occurring cremastranone.14
Figure 1.

Anti-angiogenic activity of natural and synthetic homoisoflavanones. HREC, target human retinal microvascular endothelial cells; HUVEC, human umbilical vein endothelial cells; 92–1, uveal melanoma cell line; and Y79, retinoblastoma cell line, used as surrogates for non-target ocular cell types. asee references 12 and 14a, bND: not determined
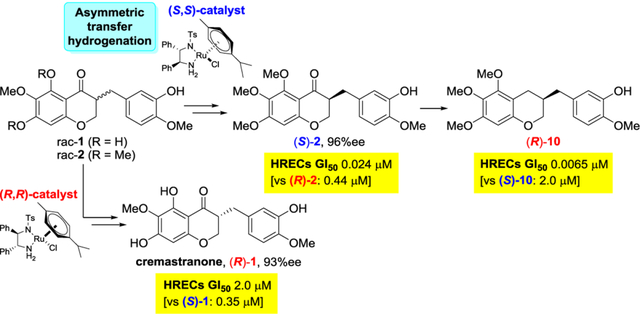
So far, our synthetic efforts toward cremastranone (1) have positively impacted our phenotype-based drug discovery and target identification program. We sought structural modification to increase antiangiogenic activity.15 As synthetic analogs of cremastranone were designed and synthesized by A and B ring modification, 3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one (2) and a phenylalanyl-incorporated analog (3) were identified that inhibit ocular neovascularization with selectivity for HRECs (GI50 of 2.0 and 0.055 μM, respectively), over other cell types as well as promising efficacy in OIR and L-CNV in vivo models.15,16 We also used these compounds as starting points to identify target(s) by chemical proteomics.17,18 Our homoisoflavanone-based photoaffinity probes were exploited for binding protein and target validation for antiangiogenic mechanism, leading to target proteins for two different compounds, ferrochelatase (FECH) and soluble epoxide hydrolase (sEH).19,20
However, the stereochemistry at the C3 position in cremastranone has not been reported, although the (R)-form is known as the configuration of general naturally occurring homoisoflavanones.7 Moreover, the relationship between biological activity and absolute stereochemistry has not yet been confirmed with enantioenriched compounds obtained by chiral separation or asymmetric synthesis. In addition, 5,6,7-trimethoxyhomoisoflavanones 2 and 3 were synthesized and evaluated as mixtures of two stereoisomers.17 In the case of the analog 3, the (S,S)- and (R,S)-isomers were named based on two stereogenic centers at C3 and the α-carbon of the L-phenylalanyl residue (Figure 1). As each isomer potentially exhibited different pharmacological activity, it was necessary to develop an investigational drug for the treatment of wet AMD from the most potent stereoisomer.
There have been many efforts towards the synthesis of homoisoflavonoids including asymmetric synthesis.21 To obtain an enantiopure antipode of homoisoflavanone efficiently, the previous synthetic approaches developed by others were studied as follows (Figure 2): 1) Intramolecular Friedel-Crafts acylation of chiral 2-benzyl-3-aryloxypropanoic acid was adapted for the formation of the 3-benzyl-4-chromanone. Such a chiral carboxylic acid was obtained by lipase-catalyzed reaction of 2-benzyl-1,3-propandiol, followed by Mitsunobu reaction of phenol (Kometani et al.22) and by iridium-catalyzed asymmetric hydrogenation of α-aryloxymethylcinnamic acid (Zhou et al.23), 2) the introduction of a p-methoxybenzyl group (for B ring) by asymmetric aldol reaction with Evans chiral oxazolidinone, followed by the intramolecular etherification to provide chiral 3-benzyl-chromane, which was transformed into the 3-benzyl-4-chromanone by benzylic oxidation (Liu et al.24), 3) enantioselective protonation of enol ether derived from racemic homoisoflavanones by phase transfer catalysts (Levacher et al.25).
Figure 2.

Previous approaches and our plan for asymmetric synthesis of homoisoflavanones
Nevertheless, there are still limitations on asymmetric synthesis of homoisoflavanones with various substituents on the A and B rings. Since we undertook a medicinal chemistry program on antiangiogenic cremastranone, we have been interested in the asymmetric synthesis of cremastranone and the biological effects of chiral homoisoflavanones. Moreover, our synthetic route would be very useful for introducing chemical diversity and for the structural optimization of homoisoflavanone derivatives. In screening the best conditions for asymmetric synthesis of homoisoflavanones, we found that the Metz group,26 Merck group,27 and others28 developed asymmetric syntheses of chroman-4-ones, including flavanone and isoflavanone, by asymmetric transfer hydrogenation using Noyori’s catalysts.29,30 Thus, we envisioned asymmetric transfer hydrogenation of the 3-benzyl-chroman-4-ones 1 and 2 by two Noyori’s ruthenium catalysts (RuCl(p-cymene)[(R,R)-Ts-DPEN], (R,R)-4, and RuCl(p-cymene)[(S,S)-Ts-DPEN], (S,S)-4) and subsequent re-oxidation of the resulting 3-benzyl-chroman-4-ol without racemization to provide each (R)- or (S)-homoisoflavanone (Scheme 1). It was necessary to optimize the reaction conditions through which the reduction of the enolizable ketone of homoisoflavanone would proceed completely by asymmetric transfer hydrogenation to afford enantiomerically pure 3-benzyl-chroman-4-ol. It would be notable if dynamic kinetic resolution accompanied the asymmetric transfer hydrogenation for the enantioselective synthesis of homoisoflavanones, as shown in a previous work.26,27,31
Scheme 1.

Enantioselective synthesis of cremastranone (1) and its analog (2) via asymmetric transfer hydrogenation using Noyori’s ruthenium catalysts 4a and 4b.
Herein, we describe enantioselective synthesis and structural elucidation of the naturally occurring homoisoflavanone cremastranone with (R)-configuration and its (S)-enantiomer in which stereochemistry dictates in vitro efficacy against angiogenesis. It was shown that the stereoisomers with an (S)-configuration on the C3-position are generally superior to the opposite isomers (natural forms) in terms of antiangiogenic efficacy. In addition, we present an efficient method to access several enantioenriched derivatives of homoisoflavanones and reveal the dynamic kinetic resolution mechanism of asymmetric transfer hydrogenation by time-dependent concentration.
RESULTS AND DISCUSSION
Chemistry
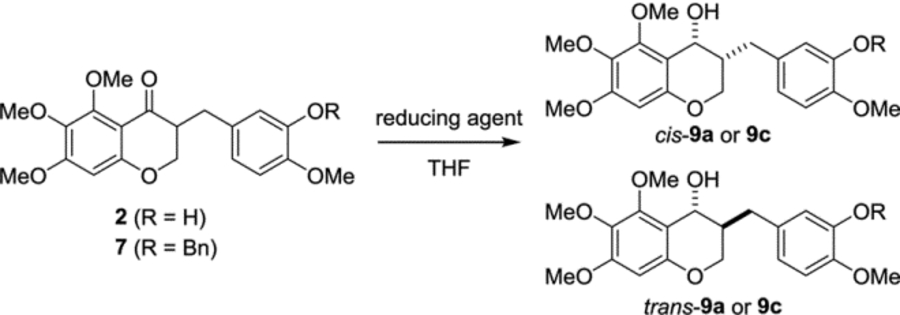
Our initial work was to prepare the ketone substrates for asymmetric transfer hydrogenation (Scheme 2). With two 2-hydroxyacetophenones in hand, 5,6,7-trisubstituted-homoisoflavanones 5a and 5b were prepared via our previous method in which N,N-dimethylformamide dimethyl acetal was used, followed by catalytic hydrogenation or LiAlH4 reduction of 4H-chromen-4-one.17,32 The resulting chroman-4-ones 5a and 5b were treated with isovanillin and p-TsOH, then subsequent catalytic hydrogenation afforded the homoisoflavanones 2 and 6. To screen the substrates for asymmetric transfer hydrogenation, two benzyl-protected analogs of homoisoflavanones (7 and 8) were generated using BnBr and K2CO3 from 2 and 6, respectively. With four homoisoflavanones in hand, we attempted the synthesis of homoisoflavon-4-ol derivatives by diastereoselective (cis or trans) and enantioselective (R- or S-configuration) methods.
Scheme 2.

Preparation of racemic homoisoflavanones for the synthesis of 3-benzyl-chroman-4-ol.
Reagents and conditions: (a) isovanillin or 3-benzyloxy-4-methoxybenzaldehyde, p-TsOH, benzene, reflux, 70% for 2; 76% for 6 and 8; (b) H2, Pd/C, MeOH, rt, 98% for 2; 97% for 6; 56% (97% BORSM) for 8; (c) benzyl bromide, K2CO3, acetone, 99% for 7 and 72% for 8.
First, C4-carbonyl reduction of homoisoflavonoids was carried out using some metal hydride agents to study the chemical yield and the ratio of the cis- to trans-isomer of 3-benzyl-chroman-4-ol (Table 1).33 Homoisoflavanones 2 and 7 were treated with NaBH4 or NaBH4/CeCl3 to provide the resulting rac-9a and rac-9c with cis/trans ratio of 3.3:1 and 3.5:1, respectively. The cis/trans diastereoselectivity was not improved in the case of general procedures using LiAlH4, DIBAL and Super-H®. Unlike the other metal hydride reagents, L-selectride afforded cis-9a and cis-9c, respectively, as a major product selectively with 89% and 99% yield. The structural elucidation of the cis- and trans-isomers of 3-benzyl-chroman-4-ol rac-9a was confirmed by 2D NOESY NMR spectroscopy (Supplementary Figure S1).34
Table 1.
C4-Carbonyl reduction of homoisoflavanones 2 and 7.
| entry | substrate | reducing agent | temp (°C) | yield (%) | cis : transa |
|---|---|---|---|---|---|
| 1 | 2 | NaBH4 | 0 | 68 | 3.3 : 1 |
| 2 | 7 | NaBH4, CeCl3 | 0 | 85 | 3.5 : 1 |
| 3 | 7 | LiAlH4 | 0 | 81 | 1.3 : 1 |
| 4 | 7 | DIBAL | −40 | 98 | 2.2 : 1 |
| 5 | 7 | Super-H® | 0 | 72 | 3.2 : 1 |
| 6 | 7 | Super-H® | −78 | 92 | 3.4 : 1 |
| 7 | 7 | L-selectride | 0 | 61 | 3.8 : 1 |
| 8 | 2 | L-selectride | −78 | 89 | >20 : 1 |
| 9 | 7 | L-selectride | −78 | 99 | >20 : 1 |
The ratio of cis- to trans-isomers was assessed by 1H NMR spectroscopy
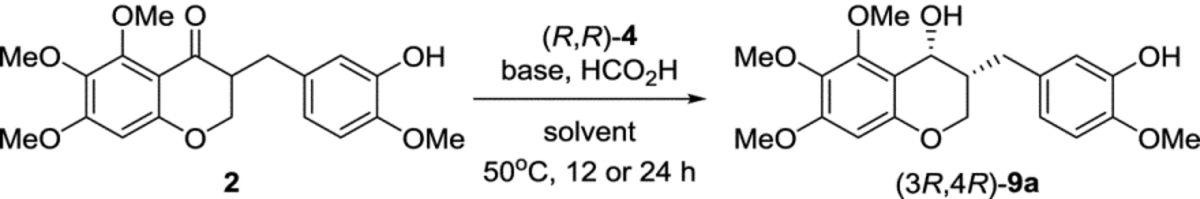
For the enantioselective synthesis of 3-benzyl-chroman-4-ol, the asymmetric transfer hydrogenation was performed with a Noyori’s ruthenium catalyst, RuCl(p-cymene)[(R,R)-Ts-DPEN], (R,R)-4 in order to obtain (3R,4R)-9a selectively (Table 2). Initially, the use of 5 mol% of (R,R)-4 or the mixture of KOH and i-PrOH as a hydrogen source was ineffective. A mixture of triethylamine and formic acid was chosen as a hydrogen source and ethyl acetate was used as a solvent. Low yields were observed without degassing (entry 1 vs 2). Although the yield was not sufficient under these conditions, the ratio of cis/trans and enantioselectivity for cis-9a was promising (entry 2). The cis/trans ratio was calculated with 1H-NMR spectroscopy and enantioselectivity was confirmed by chiral HPLC analysis on Chiralpak AD-3 after the optimization of HPLC conditions with racemic cis-9a. The 3:1 ratio (v/v) of base and formic acid improved yields based on recovered starting material (entries 4 and 5). However, the chemical yield was not improved with 10–20 mol% of (R,R)-4, regardless of screening solvent, base, and the ratio of base and formic acid. The low reactivity under general asymmetric transfer hydrogenation conditions is due to the steric hindrance of the C5-methoxy on the A ring. Given the above results, we postulated that the yield is limited under various conditions, as (R)-homoisoflavanone could react much faster than the (S)-form just by kinetic resolution. Fortunately, the reactions with 30 mol% (R,R)-4 and (S,S)-4 gave rise to excellent yield and enantioselectivity (91–96%, 97%ee). As the carbonyl group in 2 has quite low reactivity due to C5 methoxy group, the chemical yield could increase by loading 30 mol% catalyst and extending the reaction time. Thus, the asymmetric transfer hydrogenation worked with accompanying dynamic kinetic resolution (entries 9–11). The optimized condition was a 3:1 ratio of DBU and formic acid in acetonitrile (entry 10). Similarly, (3S,4S)-9a could be obtained by 30 mol% of (S,S)-4 successfully (entry 11). A high proportion of DBU affected the ratio of the cis/trans isomers and the chemical yield (entry 12). To our knowledge, there are no reports on the asymmetric transfer hydrogenation of chroman-4-ones that contain a C5-substituent on A ring, but we succeeded in the asymmetric transfer hydrogenation of a 5-substituted 3-benzyl-chroman-4-one such as 5,6,7-trimethoxy-homoisoflavanone and the NMR spectra of (3R,4R)-9a and (3S,4S)-9a are identical to those of the racemic cis-9a.
Table 2.
Asymmetric transfer hydrogenation of homoisoflavanone 2 using Noyori’s ruthenium catalysts.
| entry | catalyst (mol%) |
base (equiv.) | base:HCO2H (v/v) | solvent | yield (%)c | cis:trans | cis ee (%)e |
|---|---|---|---|---|---|---|---|
| 1a | (R,R)-4 (10) | Et3N (3.5) | 1:3 | EtOAc | 16 (32) | >20:1 | NDd |
| 2 | (R,R)-4 (10) | Et3N (3.5) | 1:3 | EtOAc | 38 (51) | >20:1 | 98 |
| 3 | (R,R)-4 (10) | Et3N (3.5) | 1:3 | ACN | NR | - | - |
| 4 | (R,R)-4 (10) | Et3N (10) | 3:1 | EtOAc | 45 (80) | >20:1 | ND |
| 5b | (S,S)-4 (10) | Et3N (10) | 3:1 | EtOAc | 56 (92) | >20:1 | 85 |
| 6 | (R,R)-4 (20) | Et3N (10) | 3:1 | ACN | 25 (64) | NDd | ND |
| 7 | (R,R)-4 (20) | DBU (10) | 3:1 | EtOAc | 33 (35) | ND | 97 |
| 8 | (R,R)-4 (20) | DBU (10) | 3:1 | ACN | 23 (26) | ND | 97 |
| 9 | (R,R)-4 (30) | DBU (10) | 3:1 | EtOAc | 91 | ND | 97 |
| 10 | (R,R)-4 (30) | DBU (10) | 3:1 | ACN | 96 | >20:1 | 97 |
| 11b | (S,S)-4 (30) | DBU (10) | 3:1 | ACN | 92 | >20:1 | 97 |
| 12 | (R,R)-4 (10) | DBU (5.0) | 10:1 | ACN | 35 (54) | 1:1 | 97 |
Without degassing;
major product is (3S,4S)-9a;
recovered yield based on starting material in parentheses; NR: no reaction;
ND: not determined;
the enantiomeric excess (%ee) was determined by chiral HPLC analysis using a Chiralpak AD-3.
As a means to elucidate the absolute configuration of 3-benzyl-chroman-4-ol’s C4 secondary alcohol, the ECD spectra of two enantiopure cis-9a isomers synthesized by asymmetric transfer hydrogenation using (R,R)-4 and (S,S)-4 were measured. Following many reports on asymmetric transfer hydrogenation, the two enantiomers of cis-9a were regarded to be (3R,4R)-9a and (3S,4S)-9a, respectively.26 The enantiomers (3R,4R)-9a and (3S,4S)-9a, with 98%ee and 93%ee, as determined by chiral HPLC, showed positive and negative optical rotation, respectively. The experimental ECD confirmed that the two compounds have the opposite configuration as enantiomers (Figure 3; Supplementary Table S1). The theoretical calculations of the ECD spectra were performed for a pair of enantiomers of cis-9a using time-dependent density functional theory (TD-DFT) at the B3LYP functional and the 6–31+G(d) basis set via Gaussian 09. As expected, the calculated ECD spectra of (3R,4R)-9a and (3S,4S)-9a were consistent with their experimental curves. Therefore, the full stereochemistry of two chiral 3-benzyl-chroman-4-ols was obtained by asymmetric transfer hydrogenation was assigned through the ECD and NMR spectra.
Figure 3.


Experimental and calculated ECD spectra of (3R,4R)-9a and (3S,4S)-9a
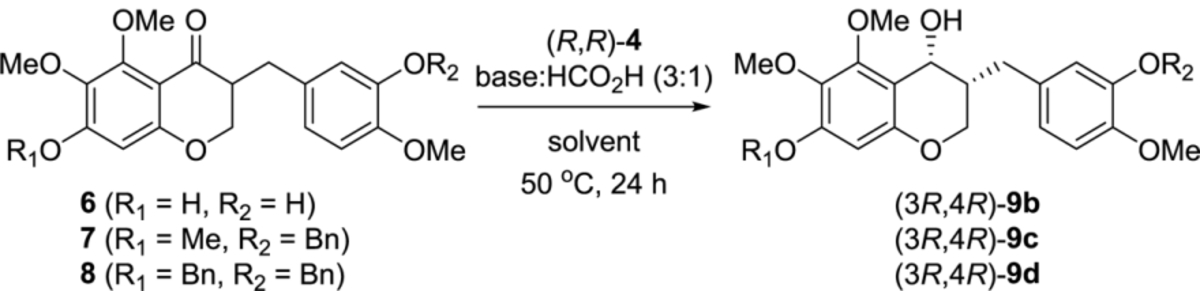
With (3R,4R)-9a in hand, we studied the oxidation of the C4 secondary alcohol without racemization to obtain the chiral homoisoflavanone (R)-2 under various conditions such as TPAP/NMO, PCC, Dess-Martin periodinane and Swern oxidation. Unexpectedly, most oxidation reactions did not proceed due to the phenol group on the C3ʹ position, as was also predicted to affect the synthesis of cremastranone. After introducing a benzyl group to the phenol group at the C3ʹ and C7 positions, asymmetric transfer hydrogenation of the benzyl-protected ketones (7 and 8) as well as the ketone 6 containing two phenol groups was carried out (Table 3). Not surprisingly, the ketone 6 was not converted into the alcohol 9b under two different conditions using Et3N and DBU (entries 1 and 2). The chemical yield was low after the treatment of the ketone 7 with 10 mol% of (R,R)-4 but the enantioselectivity of the cis-isomer was >90%ee (entries 3 and 4). DBU was superior to Et3N as a base in terms of chemical yield and enantioselectivity, and acetonitrile was used as the preferred solvent rather than ethyl acetate. The carbonyl reduction of 7 with 30 mol% of (R,R)-4 and (S,S)-4, DBU and formic acid afforded (3R,4R)-9c and (3S,4S)-9c in 92%–93% yield and 93%ee–97%ee, respectively. The ketone 8, a precursor of cremastranone, was treated with 30 mol% of (R,R)-4 and (S,S)-4 to provide (3R,4R)-9d and (3S,4S)-9d in 87%–92% yield and 96%ee–98%ee, respectively.
Table 3.
Screening conditions for carbonyl reduction of benzyl-protected homoisoflavonoids by Noyori’s chiral ruthenium catalysts.
| entry | substrate | catalyst (mol%) |
base (equiv.) |
solvent | yield (%) | cis:trans | cis ee (%)d |
|---|---|---|---|---|---|---|---|
| 1a | 6 | (R,R)-4 (10) | Et3N (10) | EtOAc | trace | - | - |
| 2 | 6 | (R,R)-4 (30) | DBU (10) | ACN | trace | - | - |
| 3a | 7 | (R,R)-4 (10) | Et3N (10) | EtOAc | 32 | >20:1 | 90 |
| 4 | 7 | (R,R)-4 (10) | DBU (10) | ACN | 55 | NDc | 99 |
| 5 | 7 | (R,R)-4 (30) | DBU (10) | EtOAc | 73 | >20:1 | ND |
| 6 | 7 | (R,R)-4 (30) | DBU (10) | ACN | 93 | ND | 97 |
| 7 | 7 | (S,S)-4 (30) | DBU (10) | ACN | 92 | >20:1 | 93 |
| 8 | 8 | (R,R)-4 (30) | DBU (10) | ACN | 87 | >20:1 | 98 |
| 9 | 8 | (S,S)-4 (30) | DBU (10) | ACN | 92 | >20:1 | 96 |
stirred for 12 h;
major product is (3S,4S)-9a;
ND: not determined;
the enantiomeric excess (%ee) was determined by chiral HPLC analysis using a Chiralpak AD-3.
With the pair of enantiomers of 9c, the oxidation of secondary alcohol under TPAP and NMO conditions, followed by benzyl deprotection provided the desired chiral (R)-2 and (S)-2 in moderate yield (Scheme 3). Fortunately, the two-step reaction did not cause epimerization. Moreover, chiral (R)-2 and (S)-2 were coupled with two different enantiomers of Boc-phenylalanine under EDCI-mediated conditions to provide four stereoisomers of antiangiogenic homoisoflavanone 3: (R,R)-, (S,R)-, (R,S)-, and (S,S)-3, respectively. Chiral 3-benzyl-chromanes were considered as attractive derivatives of homoisoflavonoids; a racemic 3-benzyl-chromane 10 reported recently showed potent activity.35 Thus, (S)-3-benzyl-chromane (S)-10 was obtained from (3R,4R)-9a by treatment with triethylsilane. Similarly, the enantiomer (R)-10 was generated from (3S,4S)-9a. Triethylsilane was more useful to obtain the enantiopure 3-benzyl-chromane compounds than H2 and Pd/C for minimizing racemization. Unfortunately, the catalytic hydrogenation of chiral (R)-2 and (S)-2 afforded (S)-10 and (R)-10 in only 71%ee and 78%ee, respectively.
Scheme 3.

Synthesis of chiral 3-benzyl-chroman-4-one and 3-benzyl-chromane derivatives.
Reagents and conditions: (a) TPAP, NMO, MS 4 Å, CH2Cl2, 66%; (b) H2, Pd/C, EtOAc, 88%; (c) Boc-L-Phe-OH or Boc-D-Phe-OH, EDCI, DMAP, CH2Cl2, 80% for (R,S)-3 and 73% for (R,R)-3; (d) Et3SiH, TFA, THF, 40 °C, 51%.
With (3R,4R)-9d and (3S,4S)-9d in hand, we explored the asymmetric synthesis of the natural product homoisoflavanone, cremastranone (1) for the first time (Scheme 4). Similar to the oxidation issue of (3R,4R)-9a, we observed that the alcohol oxidation of (3R,4R)-9b, did not work, although various oxidizing reagents were screened. Thus, Ley’s oxidation of benzyl-protected (3R,4R)-9d was used, followed by BCl3-mediated deprotection to give the desired cremastranone (1) in a quantitative yield. The synthetic cremastranone obtained stereoselectively exhibited a negative rotation with [α]D20 ‒33.0 (c 0.20, MeOH) and its NMR spectra were identical to those of naturally occurring cremastranone of which the optical rotation was reported with [α]D20 ‒16.0 (c 0.20, MeOH). These experimental data suggested that naturally occurring cremastranone possesses an (R)-configuration at the C3 position of the 3-benzyl-chroman-4-one skeleton. Starting from (3S,4S)-9d, the (S)-enantiomer of cremastranone ((S)-1) were obtained in a similar manner.
Scheme 4.

Asymmetric synthesis of cremastranone.
Reagents and conditions: (a) TPAP, NMO, MS 4 Å, CH2Cl2, 1 h, 94%; (b) BCl3, CH2Cl2, −20 °C, 10 min, 99%.
We also studied the time-concentration of ketone 2 and alcohol 9a in order to prove the mechanism of asymmetric transfer hydrogenation under our developed conditions. To investigate the dynamic kinetic resolution of rac-2, we monitored the progress of (3S,4S)-9a formation via asymmetric transfer hydrogenation of (R)-2 and (S)-2 with (S,S)-4 by chiral HPLC (Figure 4). After 6 h, alcohol (3S,4S)-9a and (S)-2 were generated at 34% and 18%, respectively. We speculated that the racemization on the C3-position occurred to generate (S)-2, which could be converted to (3S,4S)-9a whereas in this reaction using (S,S)-4, starting material, (R)-2 was not directly transformed into the trans-isomer (3S,4R)-9a. The yield of (3S,4S)-9a was increased by 24 h and the enantioselectivity of cis-isomer was improved up to 98%ee. After 24 h, the ratio of cis- to trans-isomer was 10 : 1 and (3R,4R)-9a was no more than 2% ((a), Figure 4). The reaction of starting material (S)-2 using catalyst (S,S)-4 afforded (3S,4S)-9a in 74% yield and 99.9%ee, even after 6 h. As 8%–11% of (R)-2 was found at 6 and 12 h, it was confirmed that racemization occurred partially. Finally, (3S,4S)-9a was provided in 89% yield and 99.9%ee at 24 h.
Figure 4.


Matched/mismatched effects on a pair of homoisoflavanones and catalyst (S,S)-4 in asymmetric transfer hydrogenation. (a) (R)-2; (b) (S)-2. a The concentration of each compound was determined by chiral HPLC analysis using a Chiralpak AD-3.
Antiangiogenic activity of enantiopure homoisoflavanones
As the absolute configuration of natural cremastranone was assigned as the (R)-form and each pair of enantiomers which are 5,6,7-trisubstituted homoisoflavanones were obtained, we considered the antiangiogenic activity of enantiopure homoisoflavanones as (R)- and (S)-isomers compared with that of racemic mixtures. Previously, the GI50 value of cremastranone isolated from C. appendiculata against HUVECs was reported as 1.5 μM (Figure 1).12 In contrast, the growth suppressive activity of racemic cremastranone 1 (synthesized by our group) against HUVECs was GI50 = 377 nM in an alamarBlue fluorescence cell proliferation assay (Figure 1).14a In addition, synthetic 1 showed a dose-dependent, anti-proliferative activity of GI50 = 217 nM against HRECs, a more tissue-relevant, microvascular endothelial cell type. As suggested previously, the somewhat more pronounced inhibitory effect of synthetic compound 1 over the natural product might be due to differences in the biological assays used or purity of the compound. Between the racemic and enantiopure cremastranones, (R)-1 exhibited somewhat weaker inhibitory activity on cell proliferation for HUVECs and HRECs than that of the racemate and (S)-1.
The (S)-isomer of 5,6,7-trimethoxyhomoisoflavanone derivative 2 had 20-fold higher activity than the (R)-isomer, similar to that of cremastranone (Table 4). Among the four stereoisomers of SH-11037 (3), the isomer (S,S)-3 which was incorporated with the coupling of (S)-2 and Boc-L-Phe-OH showed the strongest inhibition of HREC growth. In antiangiogenic activity of 3-benzyl-chroman-4-ol 9, the cis-disubstituted chromane has activity as strong as 3 but the trans-form lost the inhibitory activity in HRECs. In addition, the pair of 3-benzyl-chromanes 10 were evaluated for anti-angiogenic activity. In alamarBlue proliferation assays, racemic and (R)-10 had the most pronounced potency and endothelial-cell selectivity among the antiangiogenic homoisoflavonoids that were made.
Table 4.
Growth inhibitory activity (GI50, μM) of racemic and enantiopure homoisoflavanones (95% confidence interval shown in parentheses)
| compound | structure | HREC | HUVEC | 92–1 | Y79 |
| rac-1 | 0.22 (0.12 – 0.39) |
0.38 (0.24 – 0.59) |
48 (17 – 132) |
9.8 (2.1 – 45) |
|
| (R)-1 | 2.0 (1.0 – 3.8) |
0.98 (0.30 – 3.2) |
4.5 (1.6 – 12) |
3.9 (1.8 – 8.5) |
|
| (S)-1 | 0.35 (0.087 – 1.2) |
0.24 (0.16 – 0.36) |
0.34 (0.15 – 0.72) |
0.36 (0.14 – 0.89) |
|
| rac-2 | 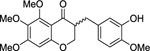 |
2.0 (0.81 – 5.1) |
12 (2.7 – 55) |
>100 | >100 |
| (R)-2 | 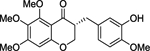 |
0.44 (0.21 – 0.94) |
0.55 (0.16 – 1.9) |
>100 | 0.40 (0.013 – 1.3) |
| (S)-2 | 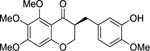 |
0.024 (0.0078 – 0.074) |
0.0027 (0.00066 – 0.011) |
>100 | 0.014 (0.012 – 0.017) |
| rac-3 | 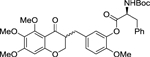 |
0.055 (0.032 – 0.094) |
0.75 (0.37 – 1.5) |
>100 | 12 (5.7 – 25) |
| (R,S)-3 | 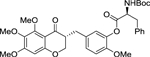 |
0.27 (0.088 – 0.85) |
0.0039 (0.0011 – 0.013) |
>100 | 0.030 (0.0087 – 0.11) |
| (S,S)-3 | 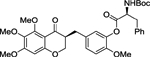 |
0.028 (0.0087 – 0.093) |
0.0030 (0.00067 – 0.013) |
>100 | 0.03 (0.011 – 0.082) |
| (R,R)-3 | 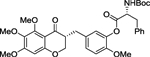 |
4.0 (1.3 – 13) |
1.1 (0.73 – 1.6) |
>100 | 2.5 (1.2 – 5.2) |
| (S,R)-3 | 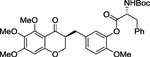 |
0.16 (0.087 – 0.31) |
0.16 (0.040 – 0.64) |
>100 | 0.22 (0.044 – 1.1) |
| cis-9a | 0.034 (0.0081 – 0.15) |
1.1 (0.56 – 2.4) |
>100 | >100 | |
| trans-9a | >100 | 3.9 (0.17 – 130) |
>100 | >100 | |
| (3R,4R)-9a | 0.72 (0.41 – 1.2) |
0.35 (0.087 – 1.4) |
>100 | 0.42 (0.091 – 1.9) |
|
| (3S,4S)-9a | 0.23 (0.049 – 0.95) |
0.036 (0.0067 – 0.19) |
>100 | 0.070 (0.038 – 0.13) |
|
| rac-10 | 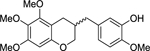 |
0.0015 (0.00056 – 0.0041) |
0.0062 (0.00015 – 0.30) |
28 (3.1 – 246) |
17 (11 – 27) |
| (S)-10 | 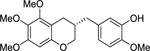 |
2.0 (0.52 – 7.3) |
1.7 (0.41 – 6.9) |
>100 | 3.3 (1.0 – 11) |
| (R)-10 | 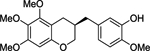 |
0.0065 (0.0046 – 0.0094) |
0.0093 (0.0015 – 0.056) |
>100 | 0.015 (0.0022 – 0.11) |
Previously, we evaluated the cell specificity of synthetic homoisoflavanones by assessing their anti-proliferative effects on non-endothelial ocular cell lines, 92–1 (uveal melanoma cell line) and Y79 (retinoblastoma cell line), as surrogates for non-target ocular tissues. The anti-proliferative activity of racemic mixture 3 on these non-endothelial cell types was considerably less than that on endothelial cells (GI50 = 12 μM and >100 μM for Y79 and 92–1, respectively), suggesting that racemic mixture 3 has pronounced selectivity of antiproliferative effect toward endothelial cells. This trend was also seen for the (R)- or (S)-isomers, although in general in the current work, Y79 cells were more sensitive than in previous reports, likely reflective of a cell batch effect.
Validation of a potent cremastranone derivative in cell-based assays
After demonstrating that enantiopure homoisoflavanones had anti-proliferative activity against HRECs, we explored HREC migration and tube formation in the presence of anti-angiogenic homoisoflavanones and a comparison between more highly anti-proliferative (S)-homoisoflavanones and (R)-antipodes. These assays document key properties of endothelial cells and together provide a good model for the angiogenesis that occurs in vivo. During angiogenesis, cells migrate out from pre-existing capillaries to the location of neovessel formation. We assessed the migration ability of HRECs via the scratch-wound assay, in which a scratch was introduced to a confluent monolayer of HRECs. Migrated cells into the area of the scratch were counted in response to differing concentrations of enantiopure homoisoflavanones (Figure 5). HREC migration was dose-dependently inhibited by the enantiopure homoisoflavanones (Figure 5). The (S)-isomers of 2 and 3 showed more potent effects than their (R)-forms. Similarly, the 3-benzyl-chromane (R)-10, which is the same conformation as the (S)-homoisoflavanones, had stronger inhibitory activity than the enantiomer (S)-10. However, (R)-1 was more potent than (S)-1 in this system.
Figure 5.

Effects of potent enantiomers on HREC migration. Inhibition of HREC migration in a scratch-wound assay. Top, images of control and highest dose treatments, with time zero scratch position marked. Scale bar = 200 μm. Bottom, quantification of migration. Dose ranges chosen based on GI50 data. Mean±SEM, n=4–6; *p<0.05, ***p<0.001, ANOVA with Dunnett’s post hoc tests. Representative data from three independent experiments.
The Matrigel tube formation assay requires most of the events of angiogenesis in vivo to be replicated, including migration, proliferation, and cell-cell adhesion. All enantiopure homoisoflavanones, with the exception of 2, reduced tubule length in a dose-dependent fashion (Figure 6). Again, the (S)-isomer of homoisoflavanone derivative 3 had more favorable activity than the (R)-isomer, with the conformationally similar (R)-10 again showing stronger inhibitory activity than (S)-10.
Figure 6.

Effects of potent enantiomers on HREC tube formation. Inhibition of HREC tube formation in a Matrigel tubule-formation assay Top, images of control and highest dose treatments. Scale bar = 200 μm. Bottom, quantification of tubule length. Dose ranges chosen based on GI50 data. Mean±SEM, n=6; *p<0.05, **p<0.01, ***p<0.001, ANOVA with Dunnett’s post hoc tests. Representative data from three independent experiments ((R)-1 and (S)-1 were assessed in a separate experiment but combined here for ease of visualization).
CONCLUSION
We have developed asymmetric synthesis for the antiangiogenic natural product cremastranone and its homoisoflavanone derivatives. For this, asymmetric transfer hydrogenation of racemic 5,6,7-substituted-homoisoflavanones using Noyori’s ruthenium catalysts afforded the enantiopure 3-benzyl-chroman-4-ol derivatives with excellent yield and enantioselectivity, although the C5-methoxy on the A ring affected the reactivity in the asymmetric transfer hydrogenation. By TPAP oxidation, (R)-3-benzyl-chroman-4-ol was oxidized smoothly into (R)-3-benzyl-chroman-4-one without racemization. During the enantioselective synthesis of cremastranone, a benzyl group was utilized as a protecting group for asymmetric transfer hydrogenation and alcohol oxidation. Two stereoisomers of homoisoflavanones synthesized were evaluated for their antiproliferative activity against HRECs and HUVECs, and the activity of the (S)-isomer was generally more effective than that of the (R)-form and racemate.. Interestingly, the absolute configuration of the natural product cremastranone was confirmed as the (R)-form, whereas the anti-angiogenic effect of the opposite (S)-homoisoflavanone was superior to the natural (R)-form in synthetic derivatives.
EXPERIMENTAL SECTION
Chemistry.
All starting materials and reagents were obtained from commercial sources and used without further purification. Air and moisture sensitive reactions were performed under nitrogen. Flash column chromatography was performed using silica gel 60(230–400 mesh, Merck) with the indicated solvents. Thin-layer chromatography (TLC) was performed using 0.25 mm silica gel plates (Merck). 1H and 13C{1H} NMR spectra were recorded on a Bruker 600 MHz spectrometer as solution in deuterochloroform (CDCl3) or methanol-d4 (CD3OD). 1H NMR data were reported in the order of chemical shift, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet and/or multiplet resonances), number of protons, and coupling constant (J value) in hertz (Hz). Enantiomeric excesses were determined by HPLC on an Agilent 1100 using one chiral column (Chiralpak AD-3 and IC-3). High resolution mass spectra (HRMS) were recorded on a JEOL JMS-700 (EI) and an Agilent 6530 Q-TOF LC/MS/MS system (ESI). The ECD spectra were acquired on an Applied Photophysics Chirascan-Plus circular dichroism spectrometer.
3-(3-Hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one (2).
To a toluene (16 mL) solution of the 5,6,7-trimethoxychroman-4-one (5a) (1.0 g, 4.2 mmol) was added isovanillin (0.70 g, 4.6 mmol) and p-toluenesulfonic acid (82 mg, 0.43 mmol). The reaction mixture was refluxed for 12 h. After cooling to ambient temperature, the reaction mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 2) to afford the (E)-3-(3-hydroxy-4-methoxybenzylidene)-5,6,7-trimethoxychroman-4-one (0.96 g, 60%, BORSM 70%). 1H NMR (600 MHz, CDCl3) δ 7.75 (s, 1H), 6.91 (d, 1H, J = 8.3 Hz), 6.88 (d, 1H, J = 2.0 Hz), 6.86 (t, 1H, J = 4.1 Hz), 6.26 (s, 1H), 5.69 (s, 1H), 5.25 (d, 2H, J = 1.7 Hz), 3.99 (s, 3H), 3.95 (s, 3H), 3.89 (s, 3H), 3.84 (s, 3H). 13C{1H} NMR (150 MHz, CDCl3) δ 179.6, 171.2, 159.4, 159.2, 154.8, 147.5, 145.6, 136.3, 130.2, 128.2, 123.3, 115.8, 110.6, 96.2, 67.7, 61.7, 61.4, 60.4, 56.1, 56.0. HRMS (EI) m/z: [M]+ Calcd for C20H20O7 Calcd for C20H20O7 372.1209; found 372.1208. A methanol solution of the (E)-3-(3-hydroxy-4-methoxybenzylidene)-5,6,7-trimethoxychroman-4-one (0.58 g, 1.6 mmol) and 10% Pd/C (82 mg) was placed under an atmosphere of hydrogen. After stirring for 20 min, the reaction mixture was diluted with ethyl acetate, filtered through a Celite pad, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 1) to afford the 3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one (2) (0.57 g, 97%).1H NMR (600 MHz, CDCl3) δ 6.82 (q, 2H, J = 4.8 Hz), 6.73 (q, 1H, J = 3.4 Hz), 6.27 (s, 1H), 5.63 (d, 1H, J = 1.9 Hz), 4.29 (q, 1H, J = 5.2 Hz), 4.12 (q, 1H, J = 6.3 Hz), 3.95 (s, 3H), 3.90 (d, 6H, J = 1.9 Hz), 3.84 (s, 3H), 3.20 (q, 1H, J = 6.1 Hz), 2.76 (m, 1H), 2.60 (q, 1H, J = 8.3 Hz). 13C{1H} NMR (150 MHz, CDCl3) δ 191.4, 159.8, 159.3, 154.5, 145.7, 145.3, 137.5, 131.7, 120.6, 115.3, 110.8, 108.0, 96.0, 69.1, 61.6, 61.3, 56.1, 56.0, 48.3, 32.2. HRMS (ESI) m/z: [M+H] + Calcd for C20H22O7H 375.1444; found 375.1432.
7-Hydroxy-3-(3-hydroxy-4-methoxybenzyl)-5,6-dimethoxychroman-4-one (6).
To a toluene (3 mL) solution of the 7-benzyloxy-5,6-dimethoxychroman-4-one (5b) (0.27 g, 0.86 mmol) was added isovanillin (0.16 g, 0.99 mmol) and p-toluenesulfonic acid (19 mg, 0.10 mmol) at 0 °C. The reaction mixture was refluxed for 12 h. After cooling to ambient temperature, the reaction mixture was concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 2) to afford the (E)-7-(benzyloxy)-3-(3-hydroxy-4-methoxybenzylidene)-5,6-dimethoxychroman-4-one (0.23 g, 59%, BORSM 76%). 1H NMR (600 MHz, CDCl3) δ 7.74 (s, 1H), 7.44 (d, 2H, J = 7.1 Hz), 7.40 (t, 2H, J = 7.5 Hz), 7.34 (m, 1H), 6.90 (d, 1H, J = 8.3 Hz), 6.87 (d, 1H, J = 2.0 Hz), 6.84 (q, 1H, J = 3.5 Hz), 6.31 (s, 1H), 5.68 (s, 1H), 5.22 (d, 2H, J = 1.7 Hz), 5.14 (s, 2H), 4.00 (s, 3H), 3.94 (s, 3H), 3.85 (s, 3H). 13C{1H} NMR (150 MHz, CDCl3) δ 179.6, 159.2, 158.3, 154.9, 147.5, 145.6, 138.2, 136.3, 135.9, 130.2, 128.7, 128.3, 128.2, 127.3, 123.3, 115.8, 110.9, 110.6, 97.5, 70.7, 67.7, 61.7, 61.4, 56.0. A methanol solution of the (E)-7-(benzyloxy)-3-(3-hydroxy-4-methoxybenzylidene)-5,6-dimethoxychroman-4-one (0.27 g, 0.60 mmol) and 10% Pd/C (64 mg) was placed under an atmosphere of hydrogen. After stirring for 45 min, the reaction mixture was diluted with ethyl acetate, filtered through a Celite pad, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 2) to afford the 7-hydroxy-3-(3-hydroxy-4-methoxybenzyl)-5,6-dimethoxychroman-4-one (6) (0.20 mg, 94%). 1H NMR (600 MHz, CDCl3) δ 6.80 (d, 2H, J = 2.0 Hz), 6.79 (d, 1H, J = 8.2 Hz), 6.71 (dd, 1H, J = 8.2 and 2.0 Hz), 6.33 (d, 1H, J = 5.8 Hz), 6.31 (s, 1H), 5.59 (s, 1H), 4.26 (dd, 1H, J = 11.3 and 4.1 Hz), 4.09 (dd, 1H, J = 11.3 and 7.6 Hz), 3.92 (s, 3H), 3.91 (s, 3H), 3.87 (s, 3H), 3.17 (dd, 1H, J = 14.4 and 4.4 Hz), 2.74 (m, 1H), 2.59 (dd, 1H, J = 14 and 10.7 Hz). 13C{1H} NMR (150 MHz, CDCl3) δ 191.6, 159.9, 155.6, 153.5, 145.7, 145.3, 135.2, 131.7, 120.6, 115.2, 110.8, 108.7, 98.9, 68.9, 61.6, 61.5, 56.0, 48.4, 32.2. HRMS (ESI) m/z: [M+H]+ Calcd for C19H20O7H 361.1287; found 361.1270.
3-(3-(Benzyloxy)-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one (7).
To an acetone (10 mL) solution of 3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one (2) (0.40 g, 1.1 mmol), benzyl bromide (0.2 mL, 1.7 mmol) and K2CO3 (0.3 g, 2.1 mmol) were added. The reaction mixture was refluxed for 4 h. After cooling to ambient temperature, the reaction mixture was diluted with ethyl acetate and the organic phase was washed with water and brine, dried over anhydrous MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 2) to afford the 3-(3-(benzyloxy)-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one (7) (0.50 g, 99%). 1H NMR (600 MHz, CDCl3) δ 7.44 (d, 2H, J = 7.2 Hz), 7.36 (t, 2H, J = 7.3 Hz), 7.29 (t, 1H, J = 7.4 Hz), 6.84 (d, 1H, J = 7.9 Hz), 6.78 (d, 1H, J = 2 Hz), 6.76 (bs, 1H), 6.24 (s, 1H), 5.14 (s, 2H), 4.17 (dd, 1H, J = 11.3 and 4.1 Hz), 3.98 (dd, 1H, J = 11.4 and 7.5 Hz), 3.93 (s, 3H), 3.88 (s, 3H), 3.87 (s, 3H), 3.81 (s, 3H), 3.14 (dd, 1H, J = 13.8 and 4.1 Hz), 2.68 (m, 1H), 2.60 (dd, 1H, J = 13.8 and 10.7 Hz). 13C{1H} NMR (150 MHz, CDCl3) δ 191.4, 159.7, 159.3, 154.5, 148.5, 148.1, 137.5, 137.1, 130.9, 128.5, 127.9, 127.4, 121.9, 115.3, 111.9, 108.7, 95.9, 71.1, 68.9, 61.6, 61.3, 56.1, 56.1, 48.4, 32.3. HRMS (EI) m/z: [M]+ Calcd for C27H28O7 464.1835; found 464.1837.
7-(Benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-one (8).
[2 steps from 5b] An anhydrous ethyl acetate solution of (E)-7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzylidene)-5,6-dimethoxychroman-4-one (88 mg, 0.16 mmol) and 10% Pd/C (8.7 mg) was placed under an atmosphere of hydrogen. After stirring for 30 min, the reaction mixture was diluted with ethyl acetate, filtered through a Celite pad and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 4) to afford the 7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-one (8) (49 mg, 56%, BORSM 97%). 1H NMR (600 MHz, CDCl3) δ 7.44 (m, 6H), 7.37 (m, 3H), 7.29 (t, 1H, J = 7.3 Hz), 6.84 (d, 1H, J = 7.9 Hz), 6.77 (d, 1H, J = 2 Hz), 6.76 (bs, 1H), 6.29 (s, 1H), 5.14 (s, 4H), 4.17 (dd, 1H, J = 11.3 and 4 Hz), 3.97 (dd, 1H, J = 11.3 and 7.5 Hz), 3.94 (s, 3H), 3.87 (s, 3H), 3.83 (s, 3H), 3.15 (dd, 1H, J = 13.7 and 4.1 Hz), 2.67 (m, 1H), 2.60 (dd, 1H, J = 13.8 and 10.7 Hz). 13C{1H} NMR (150 MHz, CDCl3) δ 191. 4, 159.5, 158.4, 154.6, 148.5, 148.1, 137.8, 137.1, 135.8, 130.9, 128.8, 128.5, 128.3, 127.9, 127.4, 127.3, 121.9, 115.3, 111.9, 108.9, 97.30, 71.1, 70.7, 68.9, 61.7, 61.4, 56.1, 48.4, 32.3. HRMS (EI) m/z: [M]+ Calcd for C33H30O7 538.1992; found 538.1988. [2 steps from 6] 1st step: To a N,N-dimethylformamide (2 mL) solution of 7-hydroxy-3-(3-hydroxy-4-methoxybenzyl)-5,6-dimethoxychroman-4-one (6) (22 mg, 0.061 mmol), benzyl bromide (22 μL, 0.18 mmol) and Cs2CO3 (60 mg, 0.18 mmol) were added. After stirring for 2 h at ambient temperature, the reaction mixture was diluted with ethyl acetate and the organic phase was washed with water and brine, dried over anhydrous MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 2) to afford the 7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-one (8) (24 mg, 72%).
cis-3-(3-Hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-ol (cis-9a).
To an anhydrous tetrahydrofuran (1 mL) solution of 3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one (2) (28 mg, 74 μmol), L-Selectride (0.32 mL, 0.15 mmol, 1 M in THF) was added dropwise at −78 °C. After stirring for 30 min, the reaction was quenched with NH4Cl, extracted with ethyl ether, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (ethyl ether : n-hexane = 1 : 2) to afford the 3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-ol (cis-9a) (25 mg, 89%) . For cis-9a, 1H NMR (600 MHz, CDCl3) δ 6.84 (d, 1H, J = 2.0 Hz), 6.79 (d, 1H, J = 8.2 Hz), 6.74 (q, 1H, J = 3.4 Hz), 6.16 (s, 1H), 5.63 (d, 1H, J = 5.0 Hz), 4.69 (t, 1H, J = 3.0 Hz), 3.96 (s, 5H), 3.87 (s, 3H), 3.79 (s, 3H), 3.78 (s, 3H), 2.85 (q, 1H, J = 7.2 Hz), 2.58 (q, 1H, J = 7.3 Hz), 2.15 (m, 2H). 13C{1H} NMR (150 MHz, CDCl3) δ 153.4, 150.8, 149.8, 144.5, 144.0, 134.3, 131.7, 119.4, 114.2, 109.7, 109.5, 94.6, 63.9, 60.3, 59.9, 59.4, 54.9, 54.8, 39.0, 31.3. For trans-9a, 1H NMR (600 MHz, CDCl3) δ 6.78 (dd, 2H, J = 5.3 and 3.1 Hz), 6.66 (dd, 1H, J = 8.2 and 2.1 Hz), 6.23 (s, 1H), 5.57 (s, 1H), 4.59 (s, 1H), 4.11 (dd, 1H, J = 10.9 and 2.3 Hz), 3.99 (s, 3H), 3.95 (m, 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.81 (s, 3H), 2.56 (dd, 1H, J = 10.9 and 7 Hz), 2.51 (d, 1H, J = 2.5 Hz), 2.45 (dd, 1H, J = 13.9 and 9.3 Hz), 2.18 (m, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 154.4, 153.0, 150.8, 146.1, 146.1, 135.7, 132.5, 119.8, 115.7, 111.4, 108.3, 95.6, 63.4, 61.0, 60.5, 60.0, 55.1, 54.9, 41.5, 33.5. HRMS (EI) m/z: [M]+ Calcd for C20H24O7 376.1522; found 376.1519 for cis-9a; HRMS (EI) m/z: [M]+ Calcd for C20H24O7 376.1522; found 376.1524 for trans-9a.
(3R,4R)-3-(3-Hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-ol ((3R,4R)-9a).
To an acetonitrile (1 mL) solution of 3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one (2) (21 mg, 55 μmol) and RuCl(p-cymene)[(R,R)-Ts-DPEN] (R,R)-4 (10 mg, 16 μmol), an acetonitrile (1 mL) solution of DBU/formic acid (75uL : 25uL) was added at ambient temperature. After stirring for 24 h at 50 °C, the reaction was quenched with saturated aqueous NH4Cl solution at ambient temperature. After extraction with diethyl ether, the organic layer was washed with an additional portion of saturated aqueous NaHCO3 solution, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 3) to afford the (3R,4R)-3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-ol ((3R,4R)-9a) (20 mg, 96%). [α]D20= + 100 (c 0.1, CH2Cl2). enantiomeric excess = 98%. 1H NMR (600 MHz, CDCl3) 6.85 (d, 1H, J = 2.1 Hz), 6.81 (d, 1H, J = 8.2 Hz), 6.75 (dd, 1H, J = 8.2 and 2.1 Hz), 6.16 (s, 1H), 5.59 (s, 1H), 4.69 (d, 1H, J = 3.2 Hz), 3.96 (s, 3H), 3.95 (m, 2H), 3.88 (s, 3H), 3.80 (s, 3H), 3.78 (s, 3H), 2.88 (dd, 1H, J =13.9 and 7.7 Hz), 2.60 (dd, 1H, J = 13.9 and 7.9 Hz), 2.17 (m, 2H). 13C{1H} NMR (150 MHz, CDCl3) δ 153.4, 150.8, 149.8, 144.5, 144.0, 134.3, 131.7, 119.4, 114.2, 109.7, 109.5, 94.6, 63.9, 60.3, 59.9, 59.4, 54.9, 54.8, 39.0, 31.3. HRMS (EI) m/z: [M]+ Calcd for C20H24O7 376.1522; found 376.1515. TR = 27 min (chiral AD-3, n-hexane : EtOH = 60 : 40, flow rate = 0.5 mL/min).
(3S,4S)-3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-ol ((3S,4S)-9a).
To an acetonitrile (1 mL) solution of 3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one (2) (20 mg, 53 μmol) and RuCl(p-cymene)[(S,S)-Ts-DPEN] (S,S)-4 (10 mg, 16 μmol), an acetonitrile (1 mL) solution of DBU/formic acid (75 μL : 25 μL) was added at ambient temperature. After stirring for 24 h at 50 °C, the reaction was quenched with saturated aqueous NH4Cl solution at ambient temperature. After extraction with diethyl ether, the organic layer was washed with an additional portion of saturated aqueous NaHCO3 solution, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 3) to afford the (3S,4S)-3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-ol ((3S,4S)-9a) (18 mg, 92%). [α]D20 = –103 (c 0.1, CH2Cl2). enantiomeric excess = 97%. 1H NMR (600 MHz, CDCl3) 6.85 (d, 1H, J = 2.0 Hz), 6.80 (d, 1H, J = 8.2 Hz), 6.74 (dd, 1H, J = 8.2 and 2.1 Hz), 6.16 (s, 1H), 5.61 (s, 1H), 4.69 (d, 1H, J = 3.2 Hz), 3.97 (s, 5H), 3.88 (s, 3H), 3.80 (s, 3H), 3.78 (s, 3H), 2.88 (dd, 1H, J = 14 and 7.7 Hz), 2.60 (dd, 1H, J = 13.9 and 7.9 Hz), 2.15 (m, 2H). 13C{1H} NMR (150 MHz, CDCl3) δ 153.4, 150.8, 149.8, 144.5, 144.0, 134.3, 131.7, 119.4, 114.2, 109.7, 109.5, 94.6, 63.9, 60.3, 59.9, 59.4, 54.9, 54.8, 39.0, 31.3. HRMS (EI) M=m/z: [M]+ Calcd for C20H24O7 376.1522; found 376.1515. TR = 18 min (chiral AD-3, n-hexane : EtOH = 60 : 40, flow rate = 0.5 mL/min).
(3R,4R)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-ol ((3R, 4R)-9c).
To an acetonitrile (1 mL) solution of 3-(3-(benzyloxy)-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one (7) (13 mg, 28 μmol) and (R,R)-4 (5.2 mg, 8.2 μmol), an acetonitrile (1 mL) solution of DBU/formic acid (42 μL:14 μL) was added at ambient temperature. After stirring for 24 h at 50 °C, the reaction was quenched with saturated aqueous NH4Cl solution at ambient temperature. After extraction with diethyl ether, the organic layer was washed with an additional portion of saturated aqueous NaHCO3 solution, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 3) to afford the (3R,4R)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-ol ((3R, 4R)-9c) (12 mg, 93%). [α]D20 = + 60 (c 0.05, CH2Cl2). enantiomeric excess = 97%. 1H NMR (600 MHz, CDCl3) δ 7.44 (d, 2H, J = 7.1 Hz), 7.36 (t, 2H, J = 7.4 Hz), 7.29 (d, 1H, J = 7.4 Hz), 6.85 (d, 1H, J = 8.1 Hz), 6.82 (s, 1H), 6.81 (d, 2H, J = 7.7 Hz), 6.16 (s, 1H), 5.14 (s, 2H), 4.61 (s, 1H), 3.94 (s, 3H), 3.91 (d, 2H, J = 8.5 Hz), 3.87 (s, 3H), 3.79 (s, 3H), 3.77 (s, 3H), 2.83 (dd, 1H, J = 13.9 and 7.9 Hz), 2.57 (dd, 1H, J = 13.9 and 7.8 Hz), 2.11 (m, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 154.4, 151.8, 150.8, 148.2, 148.0, 137.2, 135.3, 131.9, 128.5, 127.8, 127.4, 121.7, 115.4, 112.0, 110.5, 95.6, 71.1, 65.0, 61.3, 60.9, 60.3, 56.1, 55.9, 40.2, 32.4. HRMS (EI) m/z: [M]+ Calcd for C27H30O7 466.1992; found 466.1989. TR = 6.3 min (chiral AD-3, n-hexane : EtOH = 60 : 40, flow rate = 1.0 mL/min).
(3S,4S)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-ol ((3S, 4S)-9c).
To an acetonitrile (1 mL) solution of 3-(3-(benzyloxy)-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one (7) (38 mg, 82 μmol) and (S,S)-4 (15 mg, 24 μmol), an acetonitrile (1 mL) solution of DBU/formic acid (122 μL:40 μL) was added at ambient temperature. After stirring for 24 h at 50 °C, the reaction was quenched with saturated aqueous NH4Cl solution at ambient temperature. After extraction with diethyl ether, the organic layer was washed with an additional portion of saturated aqueous NaHCO3 solution, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 3) to afford the (3S,4S)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-ol ((3S, 4S)-9c) (33 mg, 87%). [α]D20 = + 60 (c 0.05, CH2Cl2). enantiomeric excess = 93%. 1H NMR (600 MHz, CDCl3) δ 7.44 (d, 2H, J = 7.4 Hz), 7.36 (t, 2H, J = 7.4 Hz), 7.29 (d, 1H, J = 7.4 Hz), 6.85 (d, 1H, J = 8.0 Hz), 6.81 (d, 2H, J = 7.8 Hz), 6.16 (s, 1H), 5.14 (s, 2H), 4.61 (d, 1H, J = 3.4 Hz), 3.94 (s, 3H), 3.91 (d, 2H, J = 8.5 Hz), 3.87 (s, 3H), 3.79 (s, 3H), 3.77 (s, 3H), 2.83 (dd, 1H, J = 13.9 and 7.9 Hz), 2.57 (dd, 1H, J = 13.9 and 7.8 Hz), 2.11 (m, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 154.4, 151.9, 150.8, 148.2, 148.0, 137.2, 135.3, 131.9, 128.5, 127.8, 127.4, 121.7, 115.4, 112.0, 110.5, 95.6, 71.1, 65.0, 61.3, 60.9, 60.3, 56.1, 55.9, 40.2, 32.4. HRMS (EI) m/z: [M]+ Calcd for C27H30O7 466.1992; found 466.1999. TR = 5.2 min (chiral AD-3, n-hexane : EtOH = 60 : 40, flow rate = 1.0 mL/min).
(3R,4R)-7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-ol ((3R,4R)-9d).
To an acetonitrile (1 mL) solution of 7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-one (8) (16 mg, 31 μmol) and (R,R)-4 (6.0 mg, 9.4 μmol), an acetonitrile (1 mL) solution of DBU/formic acid (46 μL:15 μL) was added at ambient temperature. After stirring for 24 h at 50 °C, the reaction was quenched with saturated aqueous NH4Cl solution at ambient temperature. After extraction with diethyl ether, the organic layer was washed with an additional portion of saturated aqueous NaHCO3 solution, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 3) to afford (3R,4R)-7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-ol ((3R,4R)-9d) (14 mg, 87%). enantiomeric excess = 98%. [α]D20 = +56 (c 0.11, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 7.45 (dd, 4H, J = 9.7 and 7.9 Hz), 7.37 (dd, 4H, J = 17.8 and 7.4), 7.33 (t, 1H, J = 7.3 Hz), 7.29 (t, 1H, J = 7.4 Hz), 6.86 (d, 1H, J = 8.2 Hz), 6.82 (d, 2H, J = 7.2 Hz), 6.22 (s, 1H), 5.15 (s, 2H), 5.06 (s, 2H), 4.62 (s, 1H), 3.97 (s, 3H), 3.91 (m, 2H), 3.88 (s, 3H), 3.82 (s, 3H), 2.84 (dd, 1H, J = 13.9 and 7.9 Hz), 2.58 (dd, 1H, J = 13.8 and 7.7 Hz), 2.10 (m, 2H). 13C{1H} NMR (150 MHz, CDCl3) δ 153.5, 151.9, 150.7, 148.2, 148.0, 137.2, 136.7, 135.7, 131.9, 128.6, 128.5, 127.9, 127.9, 127.4, 127.2, 121.7, 115.4, 112.0, 110.9, 97.3, 71.1, 70.6, 65.0, 61.4, 61.1, 60.3, 56.1, 40.1, 32.4. HRMS (EI) m/z: [M]+ Calcd for C33H34O7 542.2305; found 542.2303. TR = 15.6 min (chiral AD-3, n-hexane : EtOH = 60 : 40, flow rate = 1.0 mL/min).
(3S,4S)-7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-ol ((3S,4S)-9d).
To an acetonitrile (1 mL) solution of 7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-one (8) (19 mg, 0.04 mmol) and (S,S)-4 (6.5 mg, 0.01 mmol), an acetonitrile (1 mL) solution of DBU/formic acid (60 μL:20 μL) was added at ambient temperature. After stirring for 24 h at 50 °C, the reaction was quenched with saturated aqueous NH4Cl solution at ambient temperature. After extraction with diethyl ether, the organic layer was washed with an additional portion of saturated aqueous NaHCO3 solution, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 3) to afford the (3S,4S)-7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-ol ((3S,4S)-9d) (17 mg, 92%). [α]D20 = – 53 (c 0.15, CH2Cl2). enantiomeric excess = 96%. 1H NMR (600 MHz, CDCl3) δ 7.45 (dd, 4H, J = 10.4 and 7.9 Hz), 7.39 (dd, 4H, J = 17.6 and 7.3 Hz), 7.33 (t, 1H, J = 7.3 Hz), 7.30 (t, 1H, J = 7.4 Hz), 6.86 (d, 1H, J = 7.9 Hz), 6.82 (d, 2H, J = 8.0 Hz), 6.22 (s, 1H), 5.15 (s, 2H), 5.06 (s, 2H), 4.62 (s, 1H), 3.97 (s, 3H), 3.92 (m, 2H), 3.88 (s, 3H), 3.82 (s, 3H), 2.84 (dd, 1H, J = 13.9 and 7.9 Hz), 2.58 (dd, 1H, J = 13.9 and 7.7 Hz), 2.13m, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 153.5, 151.9, 150.7, 148.2, 148.0, 137.2, 136.7, 135.7, 131.9, 128.6, 128.5, 127.9, 127.9, 127.4, 127.2, 121.7, 115.4, 112.0, 110.9, 97.3, 71.1, 70.6, 65.0, 61.4, 61.1, 60.3, 56.1, 40.1, 32.4. HRMS (EI) m/z: [M]+ Calcd for C33H34O7 542.2305; found 542.2302. TR = 10 min (chiral AD-3, n-hexane : EtOH = 60 : 40, flow rate = 1.0 mL/min).
(R)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one ((R)-7).
To a dichloromethane (2 mL) solution of (3R,4R)-9c (21 mg, 42 μmol), tetrapropylammonium perruthenate (4.5 mg, 12 μmol), molecular sieves (4 Å, 50 mg) and N-methylmorpholine N-oxide (7.5 mg, 64 μmol) were added. After stirring for 1 h at ambient temperature. the reaction mixture was filtered through a Celite pad with CH2Cl2 as eluent, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 3 to 1 : 1) to afford the (R)-3-[(3-benzyloxy-4-methoxy-phenyl)methyl]-5,6,7-trimethoxy-chroman-4-one ((R)-7) (20 mg, 66%). [α]D20 = – 25 (c 0.12, CH2Cl2). enantiomeric excess = 94%. 1H NMR (600 MHz, CDCl3) δ 7.45 (d, 2H, J = 7.1 Hz), 7.37 (t, 2H, J = 7.3 Hz), 7.30 (t, 1H, J = 7.3 Hz), 6.83 (d,1H, J = 7.9 Hz), 6.78 (d, 1H, J = 1.9 Hz), 6.76 (s, 1H), 6.24 (s, 1H), 5.14 (s, 2H), 4.19 (dd, 1H, J = 11.3 and 4.1 Hz), 3.97 (dd, 1H, J = 11.3 and 7.4 Hz), 3.93 (s, 3H), 3.89 (s, 3H), 3.87 (s, 3H), 3.81 (s, 3H), 3.15 (dd, 1H, J = 13.8 and 4.1 Hz), 2.66 (m, 1H), 2.60 (dd, 1H, J = 13.8 and 10.7 Hz).13C{1H} NMR (150 MHz, CDCl3) δ 191.4, 159.7, 159.3, 154.5, 148.5, 148.1, 137.5, 137.1, 130.9, 128.5, 127.9, 127.5, 121.9, 115.3, 111.9, 108.7, 95.9, 71.1, 68.9, 61.6, 61.3, 56.1, 56.1, 48.4, 32.3. HRMS (EI) m/z: [M]+ Calcd for C27H28O7 464.1835; found 464.1834. TR = 4.7 min (chiral AD-3, ACN : MeOH = 50 : 50, flow rate = 0.5 mL/min).
(R)-3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one ((R)-2).
An methanol solution of the (R)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one ((R)-7) (11 mg, 24 μmol) and 10% Pd/C (2.6 mg) was placed under an atmosphere of hydrogen. After stirring for 1 h, the reaction mixture was diluted with ethyl acetate, filtered through a Celite pad, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 1) to afford the (R)-3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one ((R)-2) (8.0 mg, 88%). [α]D20 = – 37 (c 0.11, CH2Cl2). enantiomeric excess = 96%. 1H NMR (600 MHz, CDCl3) δ 6.80 (d, 1H, J = 2 Hz), 6.80 (d, 1H, J = 8.2 Hz), 6.71 (dd, 1H, J = 8.2 and 2.0 Hz), 6.25 (s, 1H), 5.60 (s, 1H), 4.28 (dd, 1H, J = 11.3 and 4.2 Hz), 4.10 (dd, 1H, J = 11.3 and 7.7 Hz), 3.93 (s, 3H), 3.88 (d, 6H, J = 2.6 Hz), 3.82 (s, 3H), 3.19 (dd, 1H, J = 14 and 4.3 Hz), 2.76 (m, 1H), 2.60 (dd, 1H, J = 14 and 10.9 Hz). 13C{1H} NMR (150 MHz, CDCl3) δ 191.4, 159.7, 159.3, 154.5, 145.7, 145.3, 137.5, 131.7, 120.6, 115.2, 110.8, 108.7, 96.0, 69.1, 61.6, 61.3, 56.1, 56.0, 48.4, 32.2. HRMS (EI) m/z: [M]+ Calcd for C20H22O7 374.1366; found 374.1366. TR = 5.0 min (chiral AD-3, ACN : MeOH = 50 : 50, flow rate = 1.0 mL/min). For (S)-2, [α]D20 = + 40 (c 0.10, CH2Cl2). enantiomeric excess = 99%. TR = 5.0 min (chiral AD-3, ACN : MeOH = 50 : 50, flow rate = 0.5 mL/min).
2-Methoxy-5-(((R)-5,6,7-trimethoxy-4-oxochroman-3-yl)methyl)phenyl(tert-butoxycarbonyl)-L-phenylalaninate ((R,S)-3).
To a dichloromethane solution (1 mL) of (R)-3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one ((R)-2) (13 mg, 33 μmol) were added Boc-L-Phe-OH (11 mg, 42 μmol), EDCI (10 mg, 52 μmol) and DMAP (1 mg, 8.2 μmol). After stirring for 17 h, the reaction mixture was diluted with dichloromethane and washed with water and brine, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 2) to afford the 2-methoxy-5-(((R)-5,6,7-trimethoxy-4-oxochroman-3-yl)methyl)phenyl(tert-butoxycarbonyl)-D-phenylalaninate ((R,S)-3) (17 mg, 80%). [α]D20 = – 28 (c 0.17, CH2Cl2). enantiomeric excess = >99%. 1H NMR (600 MHz, CDCl3) δ 7.35 (t, 2H, J = 7.5 Hz), 7.30 (t, 3H, J = 7 Hz), 7.18 (d, 1H, J = 6.5 Hz), 7.09 (dd, 1H, J = 8.3 and 1.9 Hz), 6.92 (d, 1H, J = 8.4 Hz), 6.84 (s, 1H), 6.25 (s, 1H), 5.03 (d, 1H, J = 8.4 Hz), 4.86 (dd, 1H, J = 14.2 and 6.2 Hz), 4.30 (dd, 1H, J = 11.3 and 4.1 Hz), 4.10 (dd, 1H, J = 11.3 and 7.6 Hz), 3.93 (s, 3H), 3.89 (s, 3H), 3.82 (s, 3H), 3.81 (s, 3H), 3.35 (dd, 1H, J = 14.0 and 5.7 Hz), 3.24 (dd, 1H, J = 14 and 6.4 Hz), 3.20 (dd, 1H, J = 14 and 4.3 Hz), 2.74 (m, 1H), 2.65 (dd, 1H, J = 13.9 and 10.7 Hz), 1.42 (s, 9H). 13C{1H} NMR (150 MHz, CDCl3) δ 191.1, 170.1, 159.7, 159.4, 155.1, 154.5, 149.7, 139.3, 137.5, 136.0, 131.1, 129.6, 128.6, 127.6, 127.1, 123.4, 112.7, 108.7, 96.0, 80.0, 68.9, 61.6, 61.3, 56.1, 55.9, 54.4, 48.3, 38.3, 31.8, 28.3. HRMS (EI) m/z: [M]+ Calcd for C34H39NO10 621.2574; found 621.2573. TR = 171.9 min (chiral IC, IC-3, TFA in DW : ACN = 50 : 50, flow rate = 0.7 mL/min).
2-Methoxy-5-(((R)-5,6,7-trimethoxy-4-oxochroman-3-yl)methyl)phenyl(tert-butoxycarbonyl)-D-phenylalaninate ((R,R)-3).
To a dichloromethane solution (1 mL) of (R)-3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one ((R)-2) (22 mg, 59 μmol) were added Boc-D-Phe-OH (19 mg, 72 μmol), EDCI (17 mg, 89 μmol) and DMAP (1.5 mg, 12 μmol). After stirring for 17 h, the reaction mixture was diluted with dichloromethane and washed with water and brine, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 2) to afford the 2-methoxy-5-(((R)-5,6,7-trimethoxy-4-oxochroman-3-yl)methyl)phenyl(tert-butoxycarbonyl)-D-phenylalaninate ((R,R)-3) (27 mg, 73%). [α]D20 = – 20 (c 0.27, CH2Cl2). enantiomeric excess = 99%. 1H NMR (600 MHz, CDCl3) δ 7.33 (m, 5H), 7.08 (d, 1H, J = 7.7 Hz), 6.92 (d, 1H, J = 8.1 Hz), 6.84 (s, 1H), 6.26 (s, 1H), 5.03 (bs, 1H), 4.87 (d, 1H, J = 5.6 Hz), 4.28 (d, 1H, J = 8.2 Hz), 4.10 (m, 1H), 3.93 (s, 3H), 3.88 (s, 3H), 3.81 (s, 3H), 3.81 (s, 3H), 3.34 (dd, 1H, J = 13.1 and 4.3 Hz), 3.22 (m, 2H), 2.72 (bs, 1H), 2.65 (t, 1H, J = 12.8 Hz), 1.42 (s, 9H). 13C{1H} NMR (150 MHz, CDCl3) δ 191.1, 170.1, 159.7, 159.4, 155.1, 154.5, 149.7, 139.3, 137.5, 136.0, 131.1, 129.6, 128.6, 127.6, 127.1, 123.4, 112.7, 108.7, 96.0, 80.0, 68.9, 61.6, 61.3, 56.1, 55.9, 54.4, 48.3, 38.3, 31.8, 28.3. HRMS (EI) m/z: [M]+ Calcd for C34H39NO10 621.2574; found 621.2574. TR = 138.8 min (chiral IC, IC-3, TFA in DW : ACN = 50 : 50, flow rate = 0.7 mL/min).
2-Methoxy-5-(((S)-5,6,7-trimethoxy-4-oxochroman-3-yl)methyl)phenyl(tert-butoxycarbonyl)-L-phenylalaninate ((S,S)-3).
To a dichloromethane solution (1 mL) of (S)-3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one ((S)-2) (10 mg, 28 μmol) were added Boc-L-Phe-OH (9 mg, 34 μmol), EDCI (8 mg, 42 μmol) and DMAP (0.6 mg, 4.9 μmol). After stirring for 17 h, the reaction mixture was diluted with dichloromethane and washed with water and brine, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 2) to afford the 2-methoxy-5-(((S)-5,6,7-trimethoxy-4-oxochroman-3-yl)methyl)phenyl(tert-butoxycarbonyl)-L-phenylalaninate ((S,S)-3) (15 mg, 86%). [α]D20 = + 20 (c 0.15, CH2Cl2). enantiomeric excess = 99%. 1H NMR (600 MHz, CDCl3) δ 7.35 (t, 2H, J = 7.4 Hz), 7.30 (t, 3H, J = 6.9 Hz), 7.08 (dd, 1H, J = 8.3 and 1.7 Hz), 6.92 (d, 1H, J = 8.3 Hz), 6.84 (s, 1H), 6.25 (s, 1H), 5.03 (d, 1H, J = 6.8 Hz), 4.87 (d, 1H, J = 7.7 Hz), 4.29 (dd, 1H, J = 11.3 and 4.1 Hz), 4.10 (dd, 1H, J = 11.3 and 7.6 Hz), 3.93 (s, 3H), 3.88 (s, 3H), 3.82 (s, 3H), 3.81 (s, 3H), 3.35 (dd, 1H, J = 14.0 and 5.7 Hz), 3.23 (dd, 1H, J = 13.9 and 6.4 Hz), 3.20 (dd, 1H, J = 14 and 4.3 Hz), 2.74 (m, 1H), 2.64 (dd, 1H, J = 13.8 and 10.7 Hz), 1.42 (s, 9H). 13C{1H} NMR (150 MHz, CDCl3) δ 191.1, 170.1, 159.7, 159.4, 155.1, 154.5, 149.7, 139.3, 137.5, 136.0, 131.1, 129.6, 128.6, 127.6, 127.1, 123.4, 112.7, 108.7, 96.0, 80.0, 68.9, 61.6, 61.3, 56.1, 55.9, 54.4, 48.3, 38.3, 31.8, 28.3. HRMS (EI) m/z: [M]+ Calcd for C34H39NO10 621.2574; found 621.2572. TR = 161.1 min (chiral IC, IC-3, TFA in DW : ACN = 50 : 50, flow rate = 0.7 mL/min).
2-Methoxy-5-(((S)-5,6,7-trimethoxy-4-oxochroman-3-yl)methyl)phenyl(tert-butoxycarbonyl)-D-phenylalaninate ((S,R)-3).
To a dichloromethane solution (1 mL) of (S)-3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one ((S)-2) (14 mg, 37 μmol) were added Boc-D-Phe-OH (12 mg, 45 μmol), EDCI (11 mg, 57 μmol) and DMAP (1 mg, 8.2 μmol). After stirring for 17 h, the reaction mixture was diluted with dichloromethane and washed with water and brine, dried over MgSO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 2) to afford the 2-methoxy-5-(((S)-5,6,7-trimethoxy-4-oxochroman-3-yl)methyl)phenyl(tert-butoxycarbonyl)-D-phenylalaninate ((S,R)-3) (21 mg, 90%). [α]D20 = + 29. (c 0.21, CH2Cl2). enantiomeric excess = 97%. 1H NMR (600 MHz, CDCl3) δ 7.35 (t, 2H, J = 7.5 Hz), 7.30 (t, 3H, J = 7 Hz), 7.09 (dd, 1H, J = 8.3 and 1.8 Hz), 6.93 (d, 1H, J = 8.4 Hz), 6.84 (s, 1H), 6.25 (s, 1H), 5.03 (d, 1H, J = 6.7 Hz), 4.88 (d, 1H, J = 7.9 Hz), 4.29 (dd, 1H, J = 11.3 and 4.1 Hz), 4.10 (dd, 1H, J = 11.3 and 7.4 Hz), 3.94 (s, 3H), 3.88 (s, 3H), 3.82 (s, 3H), 3.81 (s, 3H), 3.36 (dd, 1H, J = 13.9 and 5.6 Hz), 3.24 (dd, 1H, J = 13.9 and 6.4 Hz), 3.20 (dd, 1H, J = 14.1 and 4.3 Hz), 2.75 (m, 1H), 2.65 (dd, 1H, J = 13.9 and 10.7 Hz), 1.43 (s, 9H). 13C{1H} NMR (150 MHz, CDCl3) δ 191.1, 170.1, 159.7, 159.4, 155.1, 154.5, 149.7, 139.3, 137.5, 136.0, 131.1, 129.6, 128.6, 127.6, 127.1, 123.4, 112.7, 108.7, 96.0, 80.0, 68.9, 61.6, 61.3, 56.1, 55.9, 54.4, 48.3, 38.3, 31.8, 28.3. HRMS (EI) m/z: [M]+ Calcd for C34H39NO10 621.2574; found 621.2574. TR = 130.0 min (chiral IC, IC-3, TFA in DW : ACN = 50 : 50, flow rate = 0.7 mL/min).
(S)-2-methoxy-5-((5,6,7-trimethoxychroman-3-yl)methyl)phenol ((S)-10).
[With catalytic hydrogenation] A methanol solution of (R)-3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one ((R)-2) (19 mg, 50 μmol) and 10% Pd/C (5.3 mg) was placed under an atmosphere of hydrogen. After stirring for 1 h, the reaction mixture was diluted with ethyl acetate, filtered through a Celite pad, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 2) to afford the (S)-2-methoxy-5-((5,6,7-trimethoxychroman-3-yl)methyl)phenol ((S)-10) (14 mg, 78%). [α]D20 = + 28 (c 0.14, CH2Cl2). enantiomeric excess = 71%. 1H NMR (600 MHz, CDCl3) δ 6.78 (q, 2H, J = 2.7 Hz), 6.66 (q, 1H, J = 3.4 Hz), 6.18 (s, 1H), 5.58 (d, 1H, J = 2.2 Hz), 4.08 (m, 1H), 3.88 (s, 3H), 3.85 (s, 3H), 3.79 (s, 6H), 3.72 (q, 1H, J = 6.3 Hz), 2.75 (m, 1H), 2.57 (t, 2H, J = 8.0 Hz), 2.32 (q, 1H, J = 8.3 Hz), 2.20 (m, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 152.2, 151.6, 150.7, 145.5, 145.1, 135.8, 132.7, 120.4, 115.1, 110.6, 107.3, 95.9, 69.7, 61.0, 60.6, 56.0, 55.9, 37.6, 33.6, 25.5. [With Et3SiH] To a tetrahydrofuran solution of trifluoroacetic acid (0.16 mL) and triethylsilane (32 μL, 0.2 mmol) was added anhydrous tetrahydrofuran (0.55 mL) solution of (3R,4R)-3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-ol ((3R,4R)-9a) (16 mg, 43 μmol) dropwise over 20 min. After stirring for 1 h at 40 °C, the reaction mixture was diluted with ethyl acetate and the organic phase was washed with water and NaHCO3 and dried over MgSO4. The solvent was removed under reduced pressure and purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 4) to afford the (S)-2-methoxy-5-((5,6,7-trimethoxychroman-3-yl)methyl)phenol ((S)-10) (7.8 mg, 50%). [α]D20= + 55 (c 0.055, CH2Cl2). enantiomeric excess = 99%. 1H NMR (600 MHz, CDCl3) δ 6.79 (d, 1H, J = 4.1 Hz), 6.78 (d, 1H, J =1.9 Hz) 6.67 (dd, 1H, J = 8.2 and 2 Hz), 6.18 (s, 1H), 5.59 (s, 1H), 4.08 (qd, 1H, J = 10.6 and 1.6 Hz), 3.88 (s, 3H), 3.85 (s, 3H), 3.80 (s, 6H), 3.74 (dd, 1H, J = 10.5 and 8.3 Hz), 2.75 (ddd, 1H, J = 16.4, 5.5 and 1.3 Hz), 2.61 (sep, 2H, J = 7.3 Hz), 2.32 (dd, 1H, J = 16.4 and 8.3 Hz), 2.23 (m, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 152.3, 151.8, 150.8, 145.7, 145.2, 135.9, 132.9, 120.6, 115.3, 110.8, 107.4, 96.1, 69.8, 61.2, 60.7, 56.1, 55.9, 37.7, 33.8, 25.6. HRMS (EI) m/z: [M]+ Calcd for C20H24O6 360.1573; found 360.1576. TR = 5.5 min (chiral AD-3, n-hexane : EtOH = 60 : 40, flow rate = 1.0 mL/min).
(R)-2-methoxy-5-((5,6,7-trimethoxychroman-3-yl)methyl)phenol ((R)-10).
[With catalytic hydrogenation] A solution of (S)-3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-one ((S)-2) (20 mg, 0.052 mmol) and 10% Pd/C (5.6 mg) in methanol was placed under an atmosphere of hydrogen. After stirring for 1 h, the reaction mixture was diluted with ethyl acetate, filtered through a Celite pad, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 2) to afford the (R)-2-methoxy-5-((5,6,7-trimethoxychroman-3-yl)methyl)phenol ((R)-10) (15 mg, 78.4%). [α]D20 = – 34 (c 0.15, CH2Cl2). enantiomeric excess = 78%. 1H NMR (600 MHz, CDCl3) δ 6.78 (q, 2H, J = 2.7 Hz), 6.67 (d, 1H, J = 2.0 Hz), 6.18 (s, 2H), 5.60 (s, 1H), 4.08 (m, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.79 (s, 6H), 3.72 (q, 1H, J = 6.3 Hz), 2.75 (m, 1H), 2.57 (t, 1H, J = 8.0 Hz), 2.32 (q, 1H, J = 8.3 Hz), 2.20 (m, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 152.2, 151.6, 150.7, 145.5, 145.1, 135.8, 132.7, 120.4, 115.1, 110.6, 107.3, 95.9, 69.6, 61.0, 60.6, 56.0, 55.9, 37.6, 33.6, 25.5. [With Et3SiH] To a 40 °C mixture of trifluoroacetic acid (0.15 mL) and triethylsilane (32 μL, 0.2 mmol) was added anhydrous tetrahydrofuran (0.5 mL) solution of (3S,4S)-3-(3-hydroxy-4-methoxybenzyl)-5,6,7-trimethoxychroman-4-ol ((3S,4S)-9a) (15 mg, 40 μmol) dropwise over 20 min. After stirring for 1 h, the reaction mixture was diluted with ethyl acetate and the organic phase was washed with water and NaHCO3 and dried over MgSO4. The solvent was removed under reduced pressure and purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 4) to afford the (R)-2-methoxy-5-((5,6,7-trimethoxychroman-3-yl)methyl)phenol ((R)-10) (6.2 mg, 43%). [α]D20= – 32 (c 0.062, CH2Cl2). enantiomeric excess = 98%. 1H NMR (600 MHz, CDCl3) δ 6.79 (d, 1H, J = 4.4 Hz), 6.78 (d, 1H, J =1.6 Hz) 6.67 (dd, 1H, J = 8.2 and 2 Hz), 6.18 (s, 1H), 5.60 (s, 1H), 4.09 (qd, 1H, J = 10.6 and 1.6 Hz), 3.88 (s, 3H), 3.85 (s, 3H), 3.79 (s, 6H), 3.72 (dd, 1H, J = 10.4 and 8.3 Hz), 2.77 (ddd, 1H, J = 16.5, 5.5 and 1.3 Hz), 2.61 (sep, 2H, J = 7.3 Hz), 2.34 (dd, 1H, J = 16.5 and 8.4 Hz), 2.22 (m, 1H). 13C{1H} NMR (150 MHz, CDCl3) δ 152.3, 151.8, 150.8, 145.7, 145.2, 135.9, 132.9, 120.6, 115.3, 110.8, 107.4, 96.1, 69.8, 61.2, 60.7, 56.1, 55.9, 37.7, 33.8, 25.6.. HRMS (EI) m/z: [M]+ Calcd for C20H24O6 360.1573; found 360.1572. TR = 7.6 min (chiral AD-3, n-hexane : EtOH = 60 : 40, flow rate = 1.0 mL/min).
Racemic 2-methoxy-5-((5,6,7-trimethoxychroman-3-yl)methyl)phenol (rac-10).
Racemic 3-benzyl-chromane was prepared by catalytic hydrogenation of 3-benzylidene-chroman-4-one using hydrogen and Pd/C. Particularly, the anhydrous methanol solution of (E)-3-(3-hydroxy-4-methoxybenzylidene)-5,6,7-trimethoxychroman-4-one (22 mg, 59 μmol) and 10% Pd/C (6 mg) was placed under an atmosphere of hydrogen. After stirring for 24 h, the reaction mixture was diluted with ethyl acetate, filtered through a Celite pad and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 2) to afford the rac-10 (19 mg, 88%).
(R)-7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-one ((R)-11).
To a dichloromethane (3 mL) solution of (3R,4R)-9d (11mg, 20 μmol), tetrapropylammonium perruthenate (4 mg, 10 μmol), molecular sieves (4 Å, 8 mg) and N-methylmorpholine N-oxide (8 mg, 70 μmol) were added. After stirring for 1 h at ambient temperature. the reaction mixture was filtered through a Celite pad with dichloromethane as eluent, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 3) to afford the (R)-11 (10 mg, 94%). [α]D20 = – 18 (c 0.056, CH2Cl2). 1H NMR (600 MHz, CDCl3) δ 7.44 (d, 4H, J = 7.3 Hz), 7.42 (t, 2H, J = 7.3 Hz), 7.37 (t, 3H, J = 7.3 Hz), 7.30 (t, 1H, J = 7.3 Hz), 6.84 (d, 1H, J = 7.9 Hz), 6.77 (d, 2H, J = 8.2 Hz), 6.30 (s, 1H), 5.14 (s, 4H), 4.17 (dd, 1H, J = 11.3 and 4.1 Hz), 3.98 (dd, 1H, J = 11.3 and 7.6 Hz), 3.94 (s, 3H), 3.87 (s, 3H), 3.83 (s, 3H), 3.14 (dd, 1H, J = 13.8 and 4.1 Hz), 2.68 (m, 1H), 2.60 (dd, 1H, J = 13.8 and 10.7 Hz). 13C{1H} NMR (150 MHz, CDCl3) δ 191.4, 159.6, 158.4, 154.6, 148.5, 148.1, 137.8, 137.1, 135.8, 130.9, 128.8, 128.5, 128.3, 127.9, 127.5, 127.3, 121.9, 115.3, 112.0, 108.9, 97.3, 71.1, 70.7, 68.9, 61.7, 61.4, 56.1, 48.4, 32.3. HRMS (EI) m/z: [M]+ Calcd for C33H32O7 540.2148; found 540.2153. TR = 19.1 min (chiral AD-3, n-hexane : EtOH = 60 : 40, flow rate = 1.0 mL/min).
(S)-7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-one ((S)-11).
To a dichloromethane (3 mL) solution of (3S,4S)-9d (15 mg, 7.6 μmol), tetrapropylammonium perruthenate (5 mg, 14 μmol), molecular sieves (4 Å, 8 mg) and N-methylmorpholine N-oxide (10 mg, 90 μmol) were added. After stirring for 1 h at ambient temperature. The reaction mixture was filtered through a Celite pad with dichloromethane as eluent, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 3) to afford the (S)-7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-one ((S)-11) (11 mg, 72%). [α]D20 = + 20 (c 0.050, CH2Cl2). enantiomeric excess = 94%. 1H NMR (600 MHz, CDCl3) δ 7.44 (d, 4H, J = 7.4 Hz), 7.42 (t, 2H, J = 7.3 Hz), 7.37 (t, 3H, J = 7.3 Hz), 7.30 (t, 1H, J = 7.3 Hz), 6.84 (d, 1H, J = 7.9 Hz), 6.77 (d, 2H, J = 9.1 Hz), 6.29 (s, 1H), 5.14 (s, 4H), 4.15 (dd, 1H, J = 11.3 and 4.0 Hz), 3.98 (dd, 1H, J = 11.3 and 4 Hz), 3.94 (s, 3H), 3.87 (s, 3H), 3.83 (s, 3H), 3.14 (dd, 1H, J = 13.8 and 4.0 Hz), 2.68 (m, 1H), 2.60 (dd, 1H, J = 13.7 and 10.9 Hz). 13C{1H} NMR (150 MHz, CDCl3) δ 191.4, 159.6, 158.4, 154.6, 148.5, 148.1, 137.8, 137.1, 135.8, 130.9, 128.8, 128.5, 128.3, 127.9, 127.5, 127.3, 121.9, 115.3, 112.0, 108.9, 97.3, 71.1, 70.7, 68.9, 61.7, 61.4, 56.1, 48.4, 32.3. HRMS (EI) m/z: [M]+ Calcd for C33H32O7 540.2148; found 540.2153. TR = 10.8 min (chiral AD-3, ACN : MeOH = 50 : 50, flow rate = 0.5 mL/min).
(R)-Cremastranone ((R)-1).
To a dichloromethane (2 mL) solution of (R)-7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-one ((R)-11) (8.5 mg, 13 μmol) was added boron trichloride (70 μL, 0.07 mmol, 1 M CH2Cl2) at −20 °C. After stirring for 10 min, the reaction mixture was diluted with dichloromethane and washed with NaHCO3 and water, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 3) to afford the (R)-Cremastranone ((R)-1) (4.5 mg, 99%). [α]D20 = – 29 (c 0.034, MeOH). enantiomeric excess = 93%. 1H NMR (600 MHz, CD3OD) δ 6.86 (d, 1H, J = 8.2 Hz), 6.71 (d, 1H, J = 1.9 Hz), 6.68 (d, 1H, J = 8.2 Hz), 5.92 (s, 1H), 4.24 (dd, 1H, J = 11.4 and 4.3 Hz), 4.08 (dd, 1H, J = 11.3 and 7.4 Hz), 3.83 (s, 3H), 3.78 (s, 3H), 3.09 (dd, 1H, J = 13.9 and 4.5 Hz), 2.83 (m, 1H), 2.64 (dd, 1H, J = 13.8 and 10.3 Hz). 13C{1H} NMR (150 MHz, CD3OD) δ 198.7, 159.4, 158.7, 155.5, 146.5, 146.3, 130.9, 129.0, 120.0, 115.6, 111.5, 101.6, 94.4, 68.9, 59.6, 55.0, 46.6, 31.7. HRMS (EI) m/z: [M]+ Calcd for C18H18O7 346.1053; found 346.1057. TR = 11.8 min (chiral AD-3, ACN : MeOH = 50 : 50, flow rate = 0.5 mL/min).
(S)-Cremastranone ((S)-1).
To a dichloromethane (2 mL) solution of (S)-7-(benzyloxy)-3-(3-(benzyloxy)-4-methoxybenzyl)-5,6-dimethoxychroman-4-one ((S)-11) (8.5 mg, 13 μmol) was added boron trichloride (70 μL, 0.07 mmol, 1 M CH2Cl2) at – 20 °C. After stirring for 10 min, the reaction mixture was diluted with dichloromethane and washed with NaHCO3 and water, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (ethyl acetate : n-hexane = 1 : 3) to afford the (S)-Cremastranone ((S)-1) (4.5 mg, 99%). [α]D20 = + 18 (c 0.06, CH3OH). enantiomeric excess = 80%. 1H NMR (600 MHz, CD3OD) δ 6.85 (d, 1H, J = 8.2 Hz), 6.70 (s, 1H), 6.66 (d, 1H, J = 8.2 Hz), 5.90 (s, 1H), 4.23 (qd, 1H, J = 11.4, 4.3 and 2 Hz), 4.06 (m, 1H), 3.82 (s, 3H), 3.77 (s, 3H), 3.07 (dd, 1H, J = 13.9 and 4.5 Hz), 2.81 (m, 1H), 2.63 (dd, 1H, J = 13.7 and 10.8 Hz). 13C{1H} NMR (150 MHz, CD3OD) δ 198.7, 159.3, 158.7, 155.5, 146.5, 146.3, 130.9, 129.0, 120.0, 115.6, 111.5, 101.6, 94.4, 68.9, 59.6, 55.0, 46.6, 31.7. HRMS (EI) m/z: [M]+ Calcd for C18H18O7 346.1053; found 346.1058. TR = 8.4 min (chiral AD-3, ACN : MeOH = 50 : 50, flow rate = 0.5 mL/min).
Biological assay materials.
Human retinal microvascular endothelial cells (HRECs; Cell Systems, Seattle, WA) and human umbilical vein endothelial cells (HUVECs; Lonza, Walkersville, MD), were used between passage 5 and 8. Endothelial Growth Medium (EGM-2) was made by adding an EGM-2 “Bullet Kit” (Cat no. CC-4147) to Endothelial Basal Medium (EBM) (Cat no. CC-3156) (Lonza). 92–1 uveal melanoma cells (a kind gift of Dr. Martine Jager, University of Leiden) were grown in RPMI medium containing 10% FBS and 1% penicillin-streptomycin (pen-strep). Y-79 retinoblastoma cells (a kind gift of Dr. Brenda L. Gallie, Hospital for Sick Children) were grown in RB medium (IMDM + 10% FBS + 55 μM β-mercaptoethanol + 10 μg/mL insulin + 1% pen-strep). AlamarBlue (product code BUF012B) was from AbD Serotec (Raleigh, NC), while Matrigel matrix basement membrane (Cat no.354234) was from Corning (Corning, NY).
Cell proliferation assay.
These compounds’ ability to affect cell proliferation was assessed by an alamarBlue fluorescence assay as previously described.19 Four cell types were used: HRECs, HUVECs, 92–1, and Y79. Briefly, 2,500 cells in 100 μL growth medium were seeded in 96-well clear bottom black plates and grown for 24 hours at 37°C, 5% CO2 Each test compound was then added (concentration range: 0.1 nM to 500 μM) followed by 44 hours’ incubation at 37°C, 5% CO2 (DMSO final concentration = 1%). Then, alamarBlue reagent (11.1 μL) was added and after 4 hours, fluorescence was read on a Synergy H1 plate reader (Biotek, Winooski, VT) with excitation and emission wavelengths of 560 nm and 590 nm, respectively. GraphPad Prism software (v. 7.0) was used for analysis, fitting data to three-parameter log(dose) vs. response curves. Compounds that reduced cell number by ≥50% at the highest concentration tested (relative to DMSO control) were reported as having a GI50 < 100 μM.
In vitro migration assay.
HREC migration was monitored as described before,14,15 with modifications. Briefly, HRECs were grown until confluency in 96‐well plates. A scratch was made across the center of each well with a sterile 10‐μL micropipette tip, and fresh complete medium containing DMSO or indicated concentrations of compounds were added to the wells (DMSO final concentration = 1%). Wells were imaged at 4x using an IncuCyte ZOOM system (Essen BioScience, Ann Arbor, MI) with images taken every 2 hours to monitor cell migration across the scratch. After 8 hours, the cells that migrated into the scratch were counted using the Multipoint tool in ImageJ.
In vitro tube formation assay.
A Matrigel tube formation assay was used to test the ability of HRECs to form tube-like structures when treated with different concentrations of compounds according to our standard protocol.19 Briefly, 15,000 cells in 100 μL EGM-2 medium were grown at 37°C, 5% CO2 in the absence (DMSO treated) and presence of different concentrations of compounds in 96-well clear plates coated with 50 μL of Matrigel basement membrane (DMSO final concentration = 1%). After 8 hours, images were recorded using an EVOS FL microscope and tubule length was measured using Angiogenesis Analyzer macros in ImageJ (http://image.bio.methods.free.fr/ImageJ/?Angiogenesis-Analyzer-for-ImageJ).
Statistical analyses.
Data from tube formation and migration experiments were analyzed by one-way ANOVA with Dunnett’s post hoc tests for comparisons between compound treatments and DMSO control. All analyses were performed using GraphPad Prism software (v.7.0). A p value of <0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGMENT
We thank Emily Sims and the Indiana University AngioBioCore for training on and assistance with the IncuCyte, and Rakshin Kharwadkar for assistance with proliferation assays. This work was supported by grants from the National Eye Institute (Grant number R01EY025641) and the BrightFocus Foundation (Grant number M2015301) to TWC, and grants from the Bio & Medical Technology Development Program of the National Research Foundation funded by the Korean government, MSIP (NRF-2017M3A9C8027781) and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare (Grant number HI14C1135) to S-YS.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website at DOI:.
1H- and 13C{1H} NMR spectral data and HPLC analysis for synthesized compounds. 2D NOESY NMR and ECD spectral data of 9a. (PDF)
BL, SYS, and TWC are named on a patent application disclosing the novel compounds described here. The authors declare no other competing financial interest.
REFERENCES
- 1.(a) Fallah A; Sadeghinia A; Kahroba H; Samadi A; Heidari HR.; Bradaran B; Zeinali S; Molavi O Therapeutic targeting of angiogenesis molecular pathways in angiogenesis-dependent diseases. Biomed. Pharmacother 2019, 110, 775–785. [DOI] [PubMed] [Google Scholar]; (b) Campochiaro PA Ocular neovascularization. J. Mol. Med 2013, 91, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Hodge W; Brown A; Kymes S; Cruess A; Blackhouse G; Hopkins R; McGahan L; Sharma S; Pan I; Blair J; Vollman D; Morrison A, Pharmacologic management of neovascular age-related macular degeneration: systematic review of economic evidence and primary economic evaluation. Can. J. Ophthalmol 2010, 45, 223–230. [DOI] [PubMed] [Google Scholar]; (b) Penn JS; Madan A; Caldwell RB; Bartoli M; Caldwell RW; Hartnett ME, Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res. 2008, 27, 331–371. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brown GC; Brown MM; Sharma S; Stein JD; Roth Z; Campanella J; Beauchamp GR The burden of age-related macular degeneration: a value-based medicine analysis. Trans. Am. Ophthalmol. Soc 2005, 103, 173–186. [PMC free article] [PubMed] [Google Scholar]; (d) Ambati J; Ambati BK; Yoo SH; Ianchulev S; Adamis AP Age-Related Macular Degeneration: Etiology, Pathogenesis, and Therapeutic Strategies. Surv. Ophthalmol 2003, 48, 257–293. [DOI] [PubMed] [Google Scholar]
- 3.(a) Amoaku WM; Chakravarthy U; Gale R; Gavin M; Ghanchi F; Gibson J; Harding S; Johnston RL; Kelly SP; Lotery A; Mahmood S; Menon G; Sivaprasad S; Talks J; Tufail A; Yang Y Defining response to anti-VEGF therapies in neovascular AMD. Eye 2015, 29, 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Folk JC; Stone EM, Ranibizumab Therapy for Neovascular Age-Related Macular Degeneration. New Engl. J. Med 2010, 363, 1648–1655. [DOI] [PubMed] [Google Scholar]; (c) Brown DM; Michels M; Kaiser PK; Heier JS; Sy JP; Ianchulev T, Ranibizumab versus verteporfin photodynamic therapy for neovascular age-related macular degeneration: Two-year results of the ANCHOR study. Ophthalmology 2009, 116, 57–65. [DOI] [PubMed] [Google Scholar]
- 4.Lux A; Llacer H; Heussen FM; Joussen AM Non-responders to bevacizumab (Avastin) therapy of choroidal neovascular lesions. Br. J. Ophthalmol 2007, 91, 1318–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falavarjani KG; Nguyen QD Adverse events and complications associated with intravitreal injection of anti-VEGF agents: a review of literature. Eye 2013, 27, 787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Palanki MSS; Akiyama H; Campochiaro P; Cao J; Chow CP; Dellamary L; Doukas J; Fine R; Gritzen C; Hood JD; Hu S; Kachi S; Kang X; Klebansky B; Kousba A; Lohse D; Mak CC; Martin M; McPherson A; Pathak VP; Renick J; Soll R; Umeda N; Yee S; Yokoi K; Zeng B; Zhu H; Noronha G, Development of Prodrug 4-Chloro-3-(5-methyl-3-{[4-(2-pyrrolidin-1-ylethoxy)phenyl]amino}−1,2,4-benzotriazin-7-yl)phenyl Benzoate (TG100801): A Topically Administered Therapeutic Candidate in Clinical Trials for the Treatment of Age-Related Macular Degeneration. J. Med. Chem 2008, 51, 1546–1559; [DOI] [PubMed] [Google Scholar]; (b) Jo DH; An H; Chang DJ; Baek YY; Cho CS; Jun HO; Park SJ; Kim JH; Lee HY; Kim KW; Lee J; Park HJ; Kim YM; Suh YG; Kim JH, Hypoxia-mediated retinal neovascularization and vascular leakage in diabetic retina is suppressed by HIF-1alpha destabilization by SH-1242 and SH-1280, novel hsp90 inhibitors. J. Mol. Med 2014, 92, 1083–1092. [DOI] [PubMed] [Google Scholar]; (c) An H; Lee S; Lee JM; Jo DH; Kim J; Jeong Y-S; Heo MJ; Cho CS; Choi H; Seo JH; Hwang S; Lim J; Kim T; Jun HO; Sim J; Lim C; Hur J; Ahn J; Kim HS; Seo S-Y; Na Y; Kim S-H; Lee J; Lee J; Chung SJ; Kim Y-M; Kim K-W; Kim SG; Kim JH; Suh Y-G Novel hypoxia-inducible factor 1α (HIF-1α) inhibitors for angiogenesis-related ocular diseases: discovery of a novel scaffold via ring-truncation strategy. J. Med. Chem 2018, 61, 9266–9286. [DOI] [PubMed] [Google Scholar]; (d) Adams CM; Anderson K; Artman III G; Bizec J-C; Cepeda R; Elliott J; Fassbender E; Ghosh M; Hanks S; Hardegger LA; Hosagrahara VP; Jaffee B; Jendza K; Ji N; Johnson L; Lee W; Liu D; Liu F; Long D; Ma F; Mainolfi N; Meredith EL; Miranda K; Peng Y; Poor S; Powers J; Qiu Y; Rao C; Shen S; Sivak JM; Solovay C; Tarsa P; Woolfenden A; Zhang C; Zhang Y The discovery of n-(1-methyl-5-(trifluoromethyl)-1h-pyrazol-3-yl)-5-((6-((methylamino)methyl)pyrimidin-4-yl)oxy)-1h-indole-1-carboxamide (acrizanib), a vegfr-2 inhibitor specifically designed for topical ocular delivery, as a therapy for neovascular age-related macular degeneration. J. Med. Chem 2018, 61, 1622–1635. [DOI] [PubMed] [Google Scholar]
- 7.(a) du Toit K; Drewes SE; Bodenstein J, The chemical structures, plant origins, ethnobotany and biological activities of homoisoflavanones. Nat. Prod. Res 2010, 24, 457–90. [DOI] [PubMed] [Google Scholar]; (b) Mulholland DA; Schwikkard SL; Crouch NR, The chemistry and biological activity of the Hyacinthaceae. Nat. Prod. Rep 2013, 30, 1165–210. [DOI] [PubMed] [Google Scholar]; (c) Li-Gen Lin, Qian-Yu Liu, Yang Ye naturally occurring homoisoflavonoids and their pharmacological activities. Planta Med. 2014, 80, 1053–1066. [DOI] [PubMed] [Google Scholar]; (d) Castelli MV; López SN homoisoflavonoids: occurrence, biosynthesis, and biological activity. Stud. Nat. Prod. Chem 2017, 54, 315–354. [Google Scholar]
- 8.Adinolfi M; Corsaro MM; Lanzetta R; Laonigro G; Mangoni L; Parrilli M Ten homoisoflavanones from two Muscari species. Phytochemistry 1987, 26, 285–290. [Google Scholar]
- 9.Corsaro MM; Lanzetta R; Mancino A; Parrilli M Homoisoflavanones from Chionodoxa luciliae. Phytochemistry 1992, 31, 1395–1397. [Google Scholar]
- 10.Crouch NR; Bangani V; Mulholland DA Homoisoflavanones from Chionodoxa luciliae. Phytochemistry 1999, 51, 943–946. [Google Scholar]
- 11.du Toit K; Elgorashi EE; Malan SF; Drewes SE; van Staden J; Crouch NR; Mulholland DA Anti-inflammatory activity and QSAR studies of compounds isolated from Hyacinthaceae species and Tachiadenus longiflorus Griseb. (Gentianaceae) Bioorg. Med. Chem 2005, 13, 2561–2568. [DOI] [PubMed] [Google Scholar]
- 12.Shim JS; Kim JH; Lee J; Kim SN; Kwon HJ, Anti-angiogenic activity of a homoisoflavanone from Cremastra appendiculata. Planta Med. 2004, 70, 171–173. [DOI] [PubMed] [Google Scholar]
- 13.(a) Kim JH; Yu YS; Jun HO; Kwon HJ; Park KH; Kim KW, Inhibition of choroidal neovascularization by homoisoflavanone, a new angiogenesis inhibitor. Mol. Vis 2008, 14, 556–561. [PMC free article] [PubMed] [Google Scholar]; (b) Kim JH; Kim KH; Yu YS; Kim YM; Kim KW; Kwon HJ Homoisoflavanone inhibits retinal neovascularization through cell cycle arrest with decrease of cdc2 expression. Biochem. Biophys. Res. Commun 2007, 362, 848–852. [DOI] [PubMed] [Google Scholar]
- 14.(a) Lee B; Basavarajappa HD; Sulaiman RS; Fei X; Seo SY; Corson TW The first synthesis of the antiangiogenic homoisoflavanone, cremastranone. Org. Biomol. Chem 2014, 12, 7673–7. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Basavarajappa HD; Lee B; Fei X; Lim D; Callaghan B; Mund JA; Case J; Rajashekhar G; Seo SY; Corson TW, Synthesis and mechanistic studies of a novel homoisoflavanone inhibitor of endothelial cell growth. PLoS One 2014, 9, e95694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basavarajappa HD; Lee B; Lee HJ; Sulaiman RS; An HC; Magaña C; Shadmand M; Vayl A; Rajashekhar G; Kim E-Y; Suh Y-G; Lee K; Seo S-Y; Corson TW Synthesis and biological evaluation of novel homoisoflavonoids for retinal neovascularization. J. Med. Chem 2015, 58, 5015–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sulaiman RS; Merrigan S; Quigley J; Qi X; Lee B; Boulton ME; Kennedy B; Seo S-Y; Corson TW A novel small molecule ameliorates ocular neovascularisation and synergises with anti-VEGF therapy. Sci. Rep 2016, 6, 25509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.For recent reviews of target identification of natural products, see:; (a) Drewes G; Knapp S Chemoproteomics and chemical probes for target discovery. Trends Biotechnol. 2018, 36, 1275–1286. [DOI] [PubMed] [Google Scholar]; (b) Blagg J; Workman P Choose and use your chemical probe wisely to explore cancer biology. Cancer Cell 2017, 32, 9–25. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ziegler S; Pries V; Hedberg C; Waldmann H Target identification for small bioactive molecules: finding the needle in the haystack. Angew. Chem. Int. Ed. Engl 2013, 52, 2744–92. [DOI] [PubMed] [Google Scholar]
- 18.Lee B; Sun W; Lee H; Basavarajappa H; Sulaiman RS; Sishtla K; Fei X; Corson TW; Seo S-Y Design, synthesis and biological evaluation of photoaffinity probes of antiangiogenic homoisoflavonoids. Bioorg. Med. Chem. Lett 2016, 26, 4277–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basavarajappa HD; Sulaiman RS; Qi X; Shetty T; Sheik Pran Babu S; Sishtla KL; Lee B; Quigley J; Alkhairy S; Briggs CM.; Gupta K; Tang B; Shadmand M; Grant MB; Boulton ME; Seo S-Y; Corson TW Ferrochelatase is a therapeutic target for ocular neovascularization. EMBO Mol. Med 2017, 9, 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sulaiman RS; Park B; Sheik Pran Babu SP; Si Y; Kharwadkar R; Mitter SK; Lee B; Sun W; Qi X; Boulton ME; Meroueh SO; Fei X; Seo S-Y; Corson TW Chemical proteomics reveals soluble epoxide hydrolase as a therapeutic target for ocular neovascularization. ACS Chem. Biol 2018, 13, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Gaspar A; Matos MJ; Garrido J; Uriarte E; Borges F Chromone: A Valid Scaffold in Medicinal Chemistry. Chem. Rev 2014, 114, 4960–4992. [DOI] [PubMed] [Google Scholar]; (b) Emami S; Ghanbarimasir Z Recent advances of chroman-4-one derivatives: synthetic approaches and bioactivities. Eur. J. Med. Chem 2015, 93, 539–563. [DOI] [PubMed] [Google Scholar]
- 22.Kawasaki M; Toyooka N; Matsui Y; Tanaka A; Goto M; Kakuda H; Kawabata S; Kometani T Asymmetric synthesis of homoisoflavanone using lipase-catalyzed reaction. Heterocycles 2005, 65, 761–765. [Google Scholar]
- 23.Li Z-Y; Song S; Zhu S-F; Guo N; Wang l.-X.; Zhou Q-L Synthesis of chiral α-benzyl-β2-hydroxy carboxylic acids through iridium-catalyzed asymmetric hydrogenation of α-oxymethylcinnamic acids. Chin. J. Chem 2014, 32, 783–787. [Google Scholar]
- 24.Yu Y-C; Zhu SJ; Lu X-W; Wu Y; Liu B Enantioselective synthesis of four natural homoisoflavonoids. Eur. J. Org. Chem 2015, 22, 4964–4972. [Google Scholar]
- 25.Poisson T; Gembus V; Dalla V; Oudeyer S; Levacher V Organocatalyzed enantioselective protonation of silyl enol ethers: scope, limitations, and application to the preparation of enantioenriched homoisoflavones. J. Org. Chem 2010, 75, 7704–7716. [DOI] [PubMed] [Google Scholar]
- 26.(a) Qin T; Metz P Enantioselective synthesis of isoflavanones by catalytic dynamic kinetic resolution. Org. Lett 2017, 19, 2981–2984. [DOI] [PubMed] [Google Scholar]; (b) Keßberg A; Luübken T; Metz P Enantioselective total synthesis of natural isoflavans: asymmetric transfer hydrogenation/deoxygenation of isoflavanones with dynamic kinetic resolution. Org. Lett 2018, 20, 3006–3009. [DOI] [PubMed] [Google Scholar]; (c) Lemke M-K; Schwab P; Fischer P; Tischer S; Witt M; Noehringer L; Rogachev V; Jäger A; Kataeva O; Frçhlich R; Metz P A practical access to highly enantiomerically pure flavanones by catalytic asymmetric transfer hydrogenation. Angew. Chem. Int. Ed 2013, 52, 11651–11655. [DOI] [PubMed] [Google Scholar]
- 27.Ashley ER; Sherer EC; Pio B; Orr RK; Ruck RT Ruthenium-catalyzed dynamic kinetic resolution asymmetric transfer hydrogenation of β‑chromanones by an elimination-induced racemization mechanism. ACS Catal. 2017, 7, 1446–1451. [Google Scholar]
- 28.(a) Peach P; Cross DJ; Kenny JA; Mann I; Houson I; Campbell L; Walsgroveb T; Wills M Asymmetric transfer hydrogenation of α,β-unsaturated, α-tosyloxy and α-substituted ketones. Tetrahedron 2006, 62, 1864–1876. [Google Scholar]; (b) Fernández R; Ros A; Magriz A; Dietrich H; Lassaletta JM Enantioselective synthesis of cis-α-substituted cycloalkanols and trans-cycloalkyl amines thereof. Tetrahedron 2007, 63, 6755–6763. [Google Scholar]
- 29.For recent reviews of asymmetric transfer hydrogenation, see:; (a) Foubelo F; Najera C; Yus M Catalytic asymmetric transfer hydrogenation of ketones: recent advances. Tetrahedron: Asymmetry 2015, 26, 769–790. [Google Scholar]; (b) Zanotti-Gerosa A; Hems W; Groarke M; Hancock F Ruthenium-catalysed asymmetric reduction of ketones. Platinum Metals Rev. 2005, 49, 158–165. [Google Scholar]
- 30.(a) Fujii A; Hashiguchi S; Uematsu N; Ikariya T; Noyori R Ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation of Ketones Using a Formic Acid-Triethylamine Mixture. J. Am. Chem. Soc 1996, 118, 2521–2522. [Google Scholar]; (b) Ohkuma T; Ishii D; Takeno H; Noyori R Asymmetric hydrogenation of amino ketones using chiral RuCl2(diphophine)(1,2-diamine) complexes. J. Am. Chem. Soc 2000, 122, 6510–6511. [Google Scholar]
- 31.For recent reviews of dynamic kinetic resolution, see:; (a) Mohr JT; Moore JT; Stoltz BM Enantioconvergent catalysis. Beilstein J. Org. Chem 2016, 12, 2038–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huerta FF; Minidis ABE; Bäckvall J-E Racemisation in asymmetric synthesis. Dynamic kinetic resolution and related processes in enzyme and metal catalysis. Chem. Soc. Rev 2001, 30, 321–331. [Google Scholar]
- 32.Lee HJ; Yuan Y; Rhee I; Corson TW; Seo S-Y Synthesis of natural homoisoflavonoids having either 5,7-dihydroxy-6-methoxy or 7-hydroxy-5,6-dimethoxy groups. Molecules 2016, 21, 1058–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salakka AK; Jokela TH; Wähälä K Multiple hydride reduction pathways in isoflavonoids. Beilstein J. Org. Chem 2006, 2, doi: 10.1186/1860-5397-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.(a) Adinolfi M; Barone G; Giordano F; Parrilli RLM Absolute configuration of benzocyclobutene homoisoflavanones from Muscari species. Tetrahedron 1990, 46, 6565–6574. [Google Scholar]; (b) Gomis M; Kirkiacharian BS Reductions par les hydrures et hydroboration etude de la stereoselectivite de nouvelles voies d’acces aux 3-arylmethyl-4-chromanols (Homoisoflavanols). Tetrahedron 1990, 46, 1849–1858. [Google Scholar]
- 35.Schwikkard S; Whitmore H; Sishtla K; Sulaiman RS; Shetty T; Basavarajappa HD; Waller C; Alqahtani A; Frankemoelle L; Chapman A; Crouch N; Wetschnig W; Knirsch W; Andriantiana J; Mas-Claret E; Langat MK; Mulholland D; Corson TW The antiangiogenic activity of naturally occurring and synthetic homoisoflavonoids from the Hyacinthaceae (sensu APGII). J. Nat. Prod 2019, 82, 1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.