

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a deadly disease with a 5-year survival rate of less than 8%. To date, there are no early detection methods or effective treatments available. Many questions remain to be answered in regards to the pathogenesis of PDAC, among which, the controversy over the cell lineage of PDAC demands more attention. Ductal cells were originally thought to be the cell of origin for PDAC due to the ductal morphology of most cases of PDAC. However, recent studies have demonstrated that acinar cells are more sensitive to KRAS mutation and tend to develop to PanIN and PDAC effectively, very likely by undergoing acinar to ductal metaplasia into a transient state that contributes to PDAC initiation. There is also evidence that both ductal and acinar cells can potentially develop to PDAC when exposed to certain genetic settings and stimuli, suggesting that more scrutiny is required for the identification of the true cell lineage of individual cases of PDAC. In this work, we summarize recent findings in the identification of the cellular origin of PDAC, with the goal of advancing our knowledge on the initiation and progression of the disease. We also discuss various models and techniques for investigating early events of PDAC. Better understanding of these cellular events is crucial to identify new methods for the early diagnosis and treatment of PDAC.
Keywords: Pancreatic ductal adenocarcinoma (PDAC), ductal cells, acinar cells, cell lineage, cell of origin
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common form of pancreatic cancer, and the fourth leading cause of cancer deaths in the United States. It is a devastating disease with an overall 5-year survival rate of less than 8%, as currently no early detection methods or effective treatments are available (1). Therefore, understanding the initiation and pathological progression of the disease is crucial to the discovery of novel opportunities for the early diagnosis and treatment of PDAC.
Many questions remain to be answered regarding the pathogenesis of PDAC. In particular, the exact lineage of the cell of origin of PDAC remains unclear. Heterogeneities in morphology, cell surface markers, and drug sensitivity are often noted among different subtypes of tumor cells originating from the same organ (2). The most well-established example would be hematological malignancies, which can be derived from myeloid cell lineage or lymphoid cell lineage and have been classified into more than 100 subtypes according to their cell lineage and clinical features (3,4). Identification of these distinct tumor subtypes thus leads to successful diagnosis and provides guidance for choosing specific therapeutic methods. Recent genomic analysis of PDAC samples identified gene expression patterns which defined several PDAC subtypes that correlate with histopathological characteristics (5–7). Unfortunately, although progress has been made in the identification of the cellular origins of many different cancers, including breast cancer, colorectal cancer, prostate cancer, and brain cancer (2), the cell of origin for PDAC is still controversial, which greatly hinders the diagnosis and treatment of the disease.
Three major precursor lesions for PDAC have been identified and characterized including pancreatic intraepithelial neoplasms (PanINs), intraductal papillary mucinous neoplasms (IPMNs), and mucinous cystic neoplasms (MCNs). It is generally believed that acinar cells may be the cell of origin of PanINs, while IPMNs are derived from ductal cells (8–13). However, these lesions can co-exist with each other (14,15), which adds another layer of complexity to attempts to understand the roles of these precursor lesions in PDAC initiation and progression.
Due to the ductal morphology of most cases of PDAC, ductal cells were originally thought to be the cell of origin for PDAC (16). However, later studies show that acinar cells, the other type of pancreatic exocrine cell, seem to be more sensitive to common pancreatic cancer driver mutations and tend to develop to PanIN and PDAC efficiently, whereas ductal cells are more resistant to mutant KRAS and show limited capacity to develop to PDAC (8,11,17,18). In addition, it has been demonstrated that acinar cells can undergo transdifferentiation to form a population of DCLK1+ cells with pancreatobiliary progenitor phenotype (19,20), which then contribute to PDAC initiation and progression. More recent evidence shows that both ductal and acinar cells can potentially develop to PDAC, but react differently when exposed to certain genetic settings and stimuli (21,22), emphasizing that more scrutiny should be placed in the identification of cell lineages as well as their association with subtypes of PDAC.
In the present article, we summarize recent findings in the identification of the cell of origin for PDAC, aiming to advance our knowledge on the initiation and progression of this disease. We also discuss various models and techniques employed for the investigation of early events of PDAC, as well as their advantages and limitations.
Evidence for the cellular origin for PDAC
PDAC was initially characterized by its ductal, glandular morphology, and so it was conventionally conjectured that PDAC originated from ductal cells (16,23,24). Earlier genetically engineered mouse models (GEMM) of PDAC did not pay attention to cell lineage. The KC (Kras/Cre) model, which utilized Pdx1Cre or p48/Ptf1aCre to activate a conditional knocked-in KrasG12D allele (LSL-KrasG12D) in pancreatic progenitor cells, was the first genetically GEMM to faithfully recapitulate the human PDAC tumorigenesis process in mice (25). Nevertheless, the etiology of this model is still different from that of human PDACs, which more commonly occur in elderly people. More GEMMs have been generated to study gene mutations in PDAC, however, the majority of mouse models use Pdx1Cre or p48/Ptf1aCreKRAS, including the most frequently used KPC (LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre) model (26,27). Pdx1 and p48/Ptf1a are expressed in early progenitor cells during pancreatic development (28). Lineage tracing experiments have shown that both Pdx1 and p48/pft1a expressing cells contribute to all the cell lineages in the pancreas, including both acinar and ductal cells (29–31). The expression of these genes shows relatively restricted pattern in specific cell types in the adult stage; Pdx1 is expressed abundantly in beta cells with lower levels in acinar cells, while Ptf1a is expressed primarily in acinar cells (32,33). Thus, GEMMs of PDAC which use Pdx1Cre or p48/Pf1aCre have unspecific cell lineage. This limitation may explain some of the unexplained GEMM phenotypes. For example, using a Pdx1- and Ptf1a-Cre transgenic mice model, Nabeel Bardeesy et al. observed that while KrasG12D alone led to PanIN formation, the combination of KrasG12D and Smad4 deficiency resulted in the development of IPMN (34). To address this problem, recent efforts have been made in generating GEMMs with Cre or CreER driven by more specific lineage promoters (31).
Unexpectedly, several GEMMs have suggested that, without additional mutations, ductal cells are relatively resistance to oncogenic Kras-induced formation of PDAC precursor lesions. Kopp et al. used transgenic Sox9CreER to activate the expression of a knocked-in KrasG12D allele (LSL- KrasG12D) in ductal cells at postnatal day 10 (11). Although the LSL-KrasG12D allele was effectively recombined by Sox9CreER in about 12% of ductal cells, these mice rarely developed PanIN lesions between the ages of 8 and 17 months. In another model, Ray et al. used a knockin Ck19CreERT to activate KrasG12D expression in larger pancreatic ductal cells between the ages of 6 and 8 weeks (18). In the six mice examined at 4.5 months post-tamoxifen treatment, only two displayed mucinous ductal lesions. Although all five mice exhibited PanIN lesions at the age of 6 months, the absolute numbers of lesions were still low. In addition, it was found that the duct-derived lesions were primarily limited to the large ducts rather than randomly distributed throughout the pancreas.
Nevertheless, several recent studies have revealed that oncogenic Kras could initiate PDAC tumorigenesis in ductal cells in the presence of additional mutations. Kopp et al. used transgenic SOX9CreER to delete the tumor suppressor Pten in ductal cells at the age of 4 weeks (35). These mice developed intraductal papillary lesions resembling human intraductal papillary mucinous neoplasia between the ages of 6 and 14 months. Some of these IPMNS progressed to invasive PDACs in association with the acquisition of spontaneous KRAS mutation. In line with this observation, ductal depletion of Pten in the context of oncogenic KrasG12D resulted in development of IPMN-associated invasive PDACs in one month. Another study used the same transgenic Sox9CreER to simultaneously activate a knockin KrasG12D allele and delete Trp53 in ductal cells between the ages of 3 and 4 weeks (36). These mice developed microscopic tumors as early as 4 weeks post-tamoxifen injection and reached their humane endpoint within 10–13 weeks. It is noteworthy that only small numbers of high grade PanlNs were found in these mice. Ferreira et al. introduced the KrasG12D mutation and Fbw7 deletion into ductal cells by use of knocked-in Ck19CreERT (22). This model developed in situ carcinoma devoid of PanIN lesions within 1 month post-tamoxifen injection. These data imply that either the acquisition of additional mutations in the presence of oncogenic Kras results in ductal cells bypassing the PanIN stage or, more likely, that ductal cells might be responsible for PDAC initiated from a low-grade PanIN-independent route.
In the past decade, the potential role of the pancreatic acinar cell lineage as the cell of origin of PDAC has been extensively studied. Guerra et al. crossed double transgenic Elase-tTA/Tet-O-Cre mice, which express Cre recombinase under the control of the Elastase promoter in a tet-off system, with a line carrying a knockin conditional KrasG12V allele (LSL-KrasG12V) (8). In this model, oncogenic KrasG12V expression was turned on in 20% to 30% of acinar cells as well as a low number of centroacinar cells during late embryonic development if doxycycline was not provided in the drinking water. These mice readily developed full spectrum PanIN lesions, and some progressed to PDACs with about 1-year latency. The similarity of the phenotype of this model and that of KC mice suggests that acinar cells might be the origin of PDACs developed in those models.
Taking advantage of the fact that Ptf1a expression is limited to the acinar cell lineage in the pancreas after birth, Kopp et al. gave Ptf1aCreERKrasG12DR26R7FP mice tamoxifen injections at postnatal day 10 to activate expression of oncogenic KrasGI2D and YFP reporter in acinar cells (11). This lineage tracing model displayed abundant YFP+ PanIN lesions in mice between the ages of 8 and 17 months, clearly demonstrating that these PDAC precursor lesions were derived from acinar cells. Similar phenotypes were also observed in several other postnatal GEMMs using proCPAlCreERT2 or Mist1CreERT2 to turn on oncogenic Kras expression in acinar cells (9,10,37). These data support the notion that at least embryonic or young acinar cells are susceptible to oncogenic Kras-induced PDAC initiation via a PanIN-dependent route.
In contrast to embryonic or young acinar cells, Elase-tTA/Tet-O-Cre/LSL-KrasG12V mice failed to develop even low-grade PanINs if KrasG12V expression in acinar cells was induced at 60 days after birth (8). Under this condition, oncogenic KrasG12V was only expressed in adult acinar cells, suggesting that adult acinar cells were highly refractory to oncogenic Kras-mediated malignant transformation. These mice still developed full spectrum PanIN lesions and invasive PDACs if they were challenged with induction of pancreatitis, suggesting that inflammatory insults restored the susceptibility of adult acinar cells to oncogenic Kras-induced transformation.
The tumor promoting effects of pancreatitis on acinar cells have been observed in several other GEMMs (11,38). These observations have important clinical relevance because chronic pancreatitis is a major risk factor for human PDAC (39). Several clues point to the critical contributions of inflammation induced acinar-to-ductal metaplasia (ADM) to the PDAC promoting effects of pancreatitis (40). Interestingly, Jensen et al. found that acinar cells recapitulate elements normally associated with undifferentiated progenitor cells in pancreatitis (41). It is possible that pancreatitis-induced ADM might alter adult acinar cells to a state similar to that of developing acinar cells, thus restoring their susceptibility to oncogenic Kras-mediated transformation. Liou et al. demonstrated that macrophage-secreted cytokines and tumor necrosis factor α can drive ADM through activation of NF-κB and MMPs, which represents a possible mechanism of pancreatitis-induced ADM (40).
Kopp et al. reported that KrasG12D-driven PanIN formation from acinar cells proceeds through initiation of a gene expression program similar to ductal cells, suggesting that the transition of acinar cells to ductal-like cells is critical for the early development of PDAC (11). Consistently, it was also observed that SOX9, which is primarily expressed in ductal cells, was induced in acinar cells with KrasGI2D mutation, which in turn promoted acinar-to-ductal metaplasia and PanIN formation. In addition to acinar-to-ductal metaplasia, an acinar-to-pancreatobiliary cell conversion was previously proposed to cause the initiation of PDAC. DelGiorno et al. and Bailey et al. identified a group of Dclk1+ and acetylated tubulin+ tuft cells which showed high similarity with pancreatobiliary epithelium cells in PanIN lesions from Kras mutant mouse models (19,20). Further analysis suggested that Soxl7 could induce the conversion of acinar cells to Dclk1+ and acetylated tubulin+ pancreatobiliary cells, which may represent cancer stem cells and contribute to PDAC initiation. More recently, it was reported that Dckl1+ cells are primarily located in the acinar compartment, and low-grade PanINs were infrequently observed in Dclk1CreERTKrasG12D mice after induction with Tamoxifen, while cerulein treatment of Dclk1CreERTKrasG12D mice led to the rapid development of multiple PanINs (42).
Genetic variance in acinar vs ductal-derived PDAC
As discussed above, acinar and ductal lineages possess conspicuously different susceptibilities to oncogenic Kras insult. This raises the possibility that acinar and ductal cells might require distinct genetic/epigenetic abnormalities to promote Kras-induced PDAC tumorigenesis. However, knowledge in this field is still very limited and more questions remain to be answered.
BRG1-SOX9 axis
BRG1 is a component of switch/sucrose non-fermentable (SWI/SNF) chromatin-remodeling complexes. Somatic BRG1-inactivating mutations and deletions were found in human PDACs (43,44). Figura et al. showed that knockout of Brg1 in Ptf1aCre; KrasG12D; Brg1f/f mice resulted in formation of cystic neoplasms resembling human IPMNs (12). However, it is noted that loss of Brg1 in the developing pancreas results in an abnormal pancreatic development, which may account for the observed cystic pancreas. They further generated Hnf1CreERnKrasG12DBrg1f/f mice to simultaneously introduce KrasG12D and Brg1 deletion into adult ductal cells. The authors observed atypical duct cells in a subset of pancreatic ducts from 5 out of 5 experimental mice 6 weeks after tamoxifen induction, only 1 out of 5 developed lesions resembling human IPMNs. In comparison, the mice in the control group (Hnf1CreERT2KrasG12DBrg1f/+) were normal after tamoxifen injection. These data together may suggest a suppressive role of Brg1 in adult ductal cells in PDAC tumorigenesis. However, the non-pancreatic phenotypes in the Hnf1CreERT2KrasG12DBrg1f/f mouse model limited the conclusions that could be drawn from this study regarding cellular origin.
Interestingly, Brg1 seems to play a supporting role in oncogenic KRAS-induced PDAC initiation in acinar cells. Ptf1aCreERKrasG12D mice formed full spectrum of PanINs after tamoxifen injection, while the number of PanINs formed in Ptf1aCreERKrasG12DBrg1f/f mice after tamoxifen injection was significantly lower. Moreover, depletion of Brg1 in established PanINs with an elegant dual recombinase system induced regression of the lesions in vivo, strongly suggesting that Brg1 has a critical role in acinar cells- derived PDAC progression (45).
The different expression levels of SOX9 in acinar and ductal cells may partially explain the seemly paradoxical roles of Brg1 in these two lineages for PDAC tumorigenesis. Adult acinar cells express little to no SOX9 under normal conditions. However, it is ectopically induced to an “intermediate” level in acinar-derived ADM and PanIN lesions. In fact, knockout of Sox9 in adult acinar cells substantially inhibited oncogenic Kras-induced ADM and PanIN formation (11). Tsuda et al. found that BRG1 directly bound to the promoter of SOX9 to induce its expression (45). Depletion of Brg1 in acinar cells prevented SOX9 up-regulation and thus inhibited Kras-induced ADM and PanIN. In line with this observation, overexpression of SOX9 rescued the Kras-driven PanIN phenotype in Brg1 knockout acinar cells, highlighting the importance of the BRG1-SOX9 axis in acinar lineage, PanIN-dependent PDAC progression. On the other hand, a high level of SOX9 expression is shown to play a role in maintaining ductal cell morphology and regulating the expression of duct-specific genes (46–48). It is possible that down-regulation of SOX9 to an “intermediate” level in ductal cells might enhance their susceptibility to oncogenic Kras insult. Indeed, in Hnf1CreERT2KrasG12DBrg1f/f mice, SOX9 expression was reduced in the dysplastic epithelium of the IPMN-like lesions (12), and overexpression of SOX9 reduced IPMN formation in an ex vivo model in a follow-up study (49). Nevertheless, it has not been directly tested whether SOX9-haploinsufficiency in adult ductal cells will increase their susceptibility to oncogenic insults. Whether SOX9 regulates different targets in a dose-dependent manner in adult ductal cells and PDAC precursor lesions also remains unanswered, and any exploration of this question may improve our understanding of PDAC tumorigenesis.
AR1D1A
ARID1A is another frequently mutated component of the SWI/SNF complex in human PDAC. In a recent study, Wang SC et al. demonstrated that Aridla deletion along with oncogenic Kras led to pancreatic mucinous cysts in Ptf1aCreKrasG12DArid1af/f mice which resemble human IPMN (50). They further introduced ductal cell-specific Aridla loss in Sox9CreERKrasG12DArid1af/f mice, which displayed duct dilation 3–4 months after induction and 1 out of 5 mice developed a cystic lesion after 1 year of induction, in association with elevated level of c-MYC expression. In comparison, the authors observed that heterozygous Aridla deletion in acinar cells promoted inflammation, PanIN, and PDAC formation in Pf1aCreERKrasG12DArid1af/+ mice and Ptf1 aCreERKrasG12DTp53f/+Arid1af/+ mice, compared to wild type or null Aridla controls.
In another recent study, Kimura et al. reported that Ptf1aCreKrasG12DArid1af/f mice developed both PanINs and IPMNs after birth (51). More specific lineage tracing analysis showed that acinar specific Ptf1aCTeERr2KrasG12DArid1af/f mice developed PanINs but not IPMNs after tamoxifen injection. In comparison, while oncogenic KrasG12D in adult ductal cells (Hnf1CreEKT2KrasG12D) alone did not induce any sign of ductal atypia, Hnf1CrAERT2KrasG12DArid1af/f mice (1 out of 6) formed IPMN lesions after tamoxifen injection.
Although these two reports seem to suggest that loss of Arid1 promotes PDAC tumorigenesis both in the acinar and ductal cell lineages, the limited number of IPMN formation from ductal cells with Arid1a deletion makes it difficult to identify the role of Arid1a in ductal derived PDAC initiation. In addition, similarly to Brg1 knockout, Arid1a deletion down-regulates SOX9 in adult ductal cells to facilitate a dedifferentiation process, highlighting the critical role of SWI/SNF complex-mediated chromatin modifications to the maintenance of ductal cell identity.
The functions of Arid1a in acinar and ductal derived PDAC initiation were also investigated in several other studies (52,53). For example, Livshits et al. reported that acute Arid1a knockdown in acinar cells in the context of mutant Kras resulted in increased PanIN formation associated with loss of acinar identity (52). Using Ptf1aCreArid1af/f mice, Wang W et al reported that Arid1a deletion led to progressive loss of the acinar cell mass, ADM, and progression of ductal cystic lesions (53). In the context of oncogenic Kras, Arid1a deletion was found to accelerate IPMN progression and PDAC formation in Ptf1aCreKrasG12DArid1af/f mice, compared to Ptf1aCreKrasG12D control. In addition, while PanINs were the most common lesions in Ptf1aCreKrasG12Dp53/+ mice, concurrent Arid1a deletion led to a shift to predominant IPMNs in Ptf1aCreKrasG12Dp53f/+Arid1af/f mice. These observations suggest that Arid1a is important for maintaining acinar homeostasis, while suppressing Kras-driven IPMN formation. Lineage specific Cre lines are needed to dissect the distinct contributions of Arid1a to acinar and ductal derived PDAC formation.
SMAD4
Inactivating-SMAD4 mutations are rarely found in low grade PDAC precursor lesions, but they are common in PDACs as well as high grade PanINs and IPMNs (54,55). Thus, Smad4 mutations have been conventionally considered to associate with late stage progression of PDACs, and its potential roles in early PDAC development have largely been ignored. Both Ptf1aCr’KrasG12DSmad4// and Pdx1CreLSL-KrasG12DSmad4f/f mice showed similar phenotypes to those observed in Ptf1aCreERKrasG12DBrg1f/fmice (34,56). These mice did not develop PanIN lesions, but rapidly formed IPMNs/MCN after birth, raising the possibility that inactivation of Smad4 at an early stage may impair PDAC tumorigenesis from acinar lineage. To date, Smad4 has not been specifically deleted in acinar or ductal cells to investigate its contributions to PDAC tumorigenesis in a lineage specific context. The SMAD proteins have been shown to interact with BRG1 to activate transcription via orchestration of chromatin remodeling. It is possible that SMAD4 is required for BRG1-mediated ADM induction in the early stages of PDAC tumorigenesis of acinar cell lineage, consistent with our previous report that SMAD4 was required for TGFβ induced ADM of human primary acinar cells in vitro (57). The possibility that SMAD4 may play complicated roles in a stage- and cell lineage-dependent manner emphasizes the demand for new models to carefully evaluate its functions in PDAC tumorigenesis.
PTEN
PTEN is a well know tumor suppressor that plays critical roles in controlling the PI3K pathway. Although PTEN inactivation mutations have not been identified in the majority of PDACs, loss and down-regulation of PTEN expression are common (58). Pten loss in adult ductal cells enables oncogenic Kras-induced PDAC development via the IPMN route, similarly to Brg1, but the underlying mechanisms are likely different (35). While Brgl deletion only induced IPMN formation in the presence of oncogenic Kras mutation, depletion of Pten in ductal cells alone was sufficient to induce formation of IPMN lesions. Moreover, some IPMNs subsequently gained spontaneous Kras mutations and progressed to invasive PDACs, suggesting that Pten deficiency might result in chromosomal instability in ductal cells.
The effects of PTEN on PDAC tumorigenesis in acinar cells have not been directly tested. The PDAC progression in Pdx1CreKrasG12DPtenf/+ mice was significantly accelerated when one copy of Pten was deleted in pancreatic progenitor cells (59). Interestingly, the PDACs in this model were mainly developed in a PanIN-dependent manner, suggesting that tumors were most likely initiated from the acinar lineage. In line with this speculation, a large number of ADMs were observed before PanIN formation in this model. Mechanistic analysis revealed that Pten deletion synergized with oncogenic Kras to induce strong stromal response and NF-κB signaling activation, which likely contribute to ADM induction in this model. Nevertheless, the potential involvement of ductal cells in this model cannot be disregarded. The roles of Pten in acinar-derived PDAC tumorigenesis need to be tested more rigorously with appropriate lineage tracing models.
P53
Tumor suppressor p53 is frequently mutated in PDAC. It is not surprising to see a large body of evidence showing inactivation of Trp53 (either heterozygous or complete loss) in acinar or ductal cells in the presence of oncogenic Kras contributed to PDAC formation in genetically engineered mice models (8,12,22,36,50). Interestingly, evidence suggests that gain-of-function mutation of Trp53 along with oncogenic Kras can also promote both acinar- and ductal- derived PDAC formation. For example, Baily et al. reported that oncogenic activation of Kras along with introduction of one allele of mutant Trp53R172H is sufficient to induce transformation of acinar cells in their mouse model (Mist1CreERT2) (21). In comparison, simultaneous activation of oncogenic Kras and two alleles of mutant Trp53R172H in adult ductal cells (Hnf1bCreERT2) are required to generate PDAC as early as 2 months after tamoxifen induction. While the mechanism by which the gain-of-function mutation of Trp53 could promote PDAC development remains unclear, a possible explanation is that the gain-of-function mutation may suppress wild type p53 function through a dominant negative effect (60).
FBW7
The tumor suppressor FBW7 is found to associate with PDAC biology, and its protein expression is downregulated in PDAC patients (61). Ferreria et al. reported that Fbw7 deletion together with oncogenic KrasGI2D in adult acinar cells (Elastase 1CreER) and adult ductal cells (Ckl9CreER) both resulted in formation of carcinoma in mouse model (22). However, the ductal lineage derived PDAC seemed to develop independently of low grade PanlN. Interestingly, the authors also demonstrated that embryonic deletion of Fbw7 in KrasG12DPdx1Cre mice only promoted proliferation and transformation of ductal cells, but not acinar cells.
Taken together, the abovementioned studies make it clear that acinar and ductal cells can respond to certain genetic contexts differently, which adds another layer of complexity for elucidating PDAC initiation (Figure 1). On the other hand, the cell lineage-specific characteristics may provide opportunities for the development of new diagnostic and treatment strategies for the disease.
Figure 1.
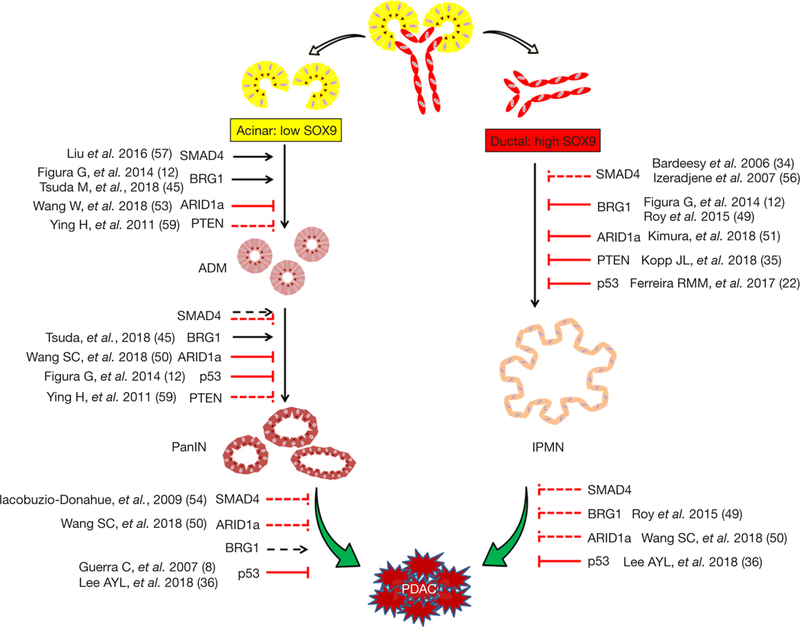
Summary of recent findings on the genetic context in favor of acinar vs. ductal-derived tumorigenesis (8,12,22,34–36,45, 49–51,53,54,56,57,59). Evidence suggest that acinar and ductal cells are capable of transformation and progression to pancreatic ductal adenocarcinoma (PDAC) in response to different genetic abnormalities. For example, BRG1 may suppress ductal-derived PDAC tumorigenesis wile playing supporting roles in acinar-derived PDAC initiation. The effects of SMAD4 on PDAC development are rather complicated, and seem to act in a stage- and cell lineage-dependent manner. At early stages, SMAD4 is believed to inhibit ductal-derived PDAC initiation while being essential for ADM. However, at later stages, SMAD4 may play a tumor suppressive role in both acinar and ductal-derived PDAC formation. In comparison, consistent findings have been reported in regard to the suppressive role of ARID1a, p53 and PTEN in both acinar and ductal-derived tumorigenesis. The solid lines in the figure indicate demonstrated functions of BRG1, SMAD4, PTEN, p53 and ARID1a in PDAC development, while the dashed lines indicate proposed effects from these genes inferred from current evidence.
Clinical implications of PDAC cell of origin
The fact that PDACs can be derived from both acinar and ductal lineages raised the question of whether the cell origin affects the properties of PDAC. Figura et al. compared IPMN-PDAC from Ptf1aCreKrasG12DBrg1f/f mice with PanlN-PDAC from Ptf1aCreKrasG12Dp53f/+ mice (12). Ductal derived IPMN-PDAC developed with shorter latency than acinar derived PanlN-PDAC. However, IPMN-PDAC mice survived longer, and most of them died due to pancreatic exocrine insufficiency without any indications of death from malignant disease. Similar to this study, Kimura et al. compared IPMN-PDAC and PanIN-PDAC derived from Ptf1aCreKrasG12DArid1af/+ and Ptf1aCreKrasG12DArid1af/f mice, respectively (51). They also found that IPMN-PDAC developed faster than PanIN-PDAC, but no significant difference in survival was observed. It needs to be pointed out that these tumors carried different mutations and were not generated from specifically adult acinar or ductal cells, and thus the contributions of cell origin to the cancer phenotype should be interpreted carefully.
Two recent studies showed that PDAC initiation could be triggered in both acinar and ductal cells with an identical combination of oncogenic drivers (KrasGI2D and TP53 deletion) (22,36). The invasive tumors derived from both lineages were histologically indistinguishable, but they developed via two distinct PanIN-dependent and independent manners. Both groups found that the ductal-derived invasive PDACs formed faster than acinar-derived invasive tumors. The mice with ductal-derived PDACs had shorter median survival than mice with acinar-derived tumors. Additionally, Lee et al. reported that distant metastasis only happened with ductal-derived PDACs (36). At the molecular level, a few markers including Keratin 20 and AGR2 were found to be differentially expressed between acinar- and ductal-derived PDACs (22,36). Still, a more comprehensive analysis is essential to evaluate the contributions of cell of origin to the heterogeneity of PDAC.
Together, these data suggest that the cell of origin can affect PDAC development and phenotype, and emphasize the importance of the comprehensive considerations of genetic context when evaluating the contributions of cell origin to PDAC phenotypes.
Different models used to study PDAC cell lineage
Upon careful examination of previous reports, it is evident that different genetic environments are critical to the determination of the fate of acinar and ductal cells. Therefore, it is crucial to develop proper models and methodologies to identify the distinct sets of driver mutations for each cell lineage during PDAC tumorigenesis.
To date, the most compelling data regarding PDAC origination were generated using genetically engineered mouse models (13,31), with the KrasGI2D being the most frequently introduced mutation. The mutation was introduced by using the Cre/LoxP system guided by a cell type-specific gene promoter, a so-called Cre driver. The most commonly used Cre for acinar cells is driven by the promoters of Elastase, Misti, Ptfla, etc., while Sox9, Ckl9, and Hnfl promoters are most frequently used for targeting ductal cells. Despite the advantage of cell type specificity to study of PDAC initiation, engineered mouse models using the Cre/LoxP system may not properly recapitulate the disease mechanisms in humans. For example, SMAD4 is found to be frequently inactivated in human PDAC (~50%), and thus efforts have been made to create Smad4 knockout mice models to mimic PDAC progress in human. However, results showed that Smad4 knockout in mice led to formation of IPMN or MCN-like lesions, which are different from those observed in humans, raising questions about the reliability of mouse models in the study of human PDAC (62).
Given the convincing evidences from lineage tracing studies using GEMM, it would be reasonable to speculate that PDACs can also originate from both acinar and ductal cells in human patients. It is practically infeasible to apply in vivo lineage tracing technique to human beings, and thus robust ex vivo PDAC tumorigenesis models would be valuable tools to investigate this question.
Huang et al. induced the differentiation of human pluripotent stem cells (PSCs) into pancreatic exocrine progenitor organoids (63). These organoids could be further polarized towards acinar and ductal lineages in 3D culture. Expression of mutant KRAS and TP53, a combination which is sufficient to induce mouse PDAC initiation, in these human pancreatic progenitor organoids only induced abnormal ductal architecture and nuclear morphology consistent with neoplastic transformation both in culture and in vivo. Thus, more work is required to recapitulate the full spectrum of human PDAC tumorigenesis with this model. It is noteworthy that pancreatic progenitor organoids could be polarized towards acinar and ductal lineages in 3D ex vivo culture, as it indicates that this model has potential to investigate human PDAC tumorigenesis in specific exocrine lineages.
Boj et al. recently developed a Wnt signaling-dependent 3D culture method for primary PDACs from human patients (64). Importantly, these PDAC organoids retained similar phenotypes to the original PDACs in patients, suggesting that a 3D organoid culture system might be a robust ex vivo system for study of human PDAC. Importantly, primary normal exocrine pancreatic cells, probably ductal cells, from human organ donors could also be propagated with the same method. This system is compatible with genetic engineering techniques, highlighting its potential to model human PDAC tumorigenesis. Indeed, Seino et al. showed that normal human exocrine pancreatic cells could be engineered to malignant cells which recapitulated human PDACs by introducing oncogenic KRASG12D and deletion of TP53, SMAD4 and P16 (65). Thus, genetically engineering human primary exocrine pancreatic cells represents another appealing strategy to investigate the mechanisms of human PDAC tumorigenesis.
Efforts have been made to use flow-cytometry to sort primary human pancreatic ductal cells and genetically modify them to carry the four most common PDAC driver mutations (KRASG12V and deletions of TP53, SMAD4 and P16) (66). Unexpectedly, these cells failed to give rise to invasive PDAC when transplanted into the pancreases of NSG (NOD/SCID/gamma, NOD.Cg-Prkdcscid IL2R tm1Wjl/SzJ) immune-deficient mice. These data raised the possibility that additional abnormalities might be required for human PDAC initiation from ductal lineage. It should be noticed that these cells were expanded without Wnt protein. Given the fact that KRAS, TP53, SMAD4, and P16 mutations did not confer Wnt-independency to PDACs, the ex vivo expansion under Wnt-free condition might have impaired the tumorigenicity of these genetically modified cells. Thus, well-controlled experiments are still required to draw a solid conclusion.
To further examine the roles of cellular origin in human PDAC tumorigenesis, our lab has developed flow cytometry-based methods to separate and isolate viable primary acinar and ductal cells from normal human exocrine pancreatic tissues (57,67). We found that a combination of UEA/CD133/CLA surface markers can be used to separate viable acinar and ductal cells from the adult human exocrine pancreas by flow cytometry, which can be used in ex vivo lineage tracing experiments. Transient TGF-β exposure accelerated the ADM process in sorted primary human acinar cells, which synergized with oncogenic KRAS to promote the growth of acinar-derived ductal-like cells. These data suggest the importance of ADM in human PDAC initiation from acinar lineage, but the underlying mechanisms need to be further investigated. More recently, we found that both primary acinar and ductal cells could be expanded in 3D culture (unpublished data), which enabled us to directly compare human PDAC tumorigenesis in these two lineages. We have generated invasive PDAC from both acinar and ductal cells (unpublished data). Our ongoing studies are focused on identifying lineage-specific oncogenic drivers for human PDAC and evaluating their effects on PDAC phenotypes such as drug resistance and metastatic potential.
Despite recent promising findings from genetically engineered primary human primary pancreatic cells, theses ex vivo models are also associated with several concerns. For example, transplantation of primary cells to immunodeficient mice does not allow us to investigate cell fate in the context of the immune system, and so it does not fully recapitulate human disease. Due to cellular plasticity, the in vivo cultured exocrine cells may change their cell fate after isolation from endogenous content. Therefore, caution should be taken when interpret the data generated from these models.
Conclusion and summary
PDAC remains one of the deadliest diseases in the world, however, its pathogenesis is still unclear and the cell lineage of PDAC is controversial. Identification of the cellular origins and the associated driver mutations of PDAC will offer opportunities for cancer prevention and provide insight into the development of diagnostic methods and new therapeutic treatments.
Due to technical advances in genetic engineering, researchers have been extensively interrogating the cell of origin for PDAC using in vivo murine models. It has now become clear that acinar cells are more sensitive to KRAS mutation than ductal cells, and can undergo acinar-ductal metaplasia to effectively develop PanIN and eventually PDAC. By comparison, although ductal cells seem refractory to single KRAS mutation, they still can transform to PDAC by other mechanisms when exposed to certain genetic settings. These findings suggest that more scrutiny should be placed on the identification the actual cell lineage of individual cases of PDAC.
A major obstacle that remains is that current in vitro and in vivo models suffer from critical drawbacks in the identification of PDAC cell lineage, which questions the reliability of the research outcomes. Therefore, there is an urgent need to develop new methodologies to investigate PDAC initiation. Models employing normal human pancreatic acinar and ductal cells allow for identification of the cell lineage of human PDAC. In addition, Shen et al. recently developed a sensitive tumor detection and classification method using plasma cell-free DNA methylomes (68), suggesting the possibility of identifying cancer cell lineage using lineage specific epigenetic alterations. The development of new models and techniques for investigating the early events of PDAC will help to advance our knowledge on the initiation and progress of this disease.
Acknowledgments
We apologize to colleagues whose work we could not cite because of space constraints.
Funding: The P Wang group was supported by Cancer Prevention and Research Institute of Texas, the William and Ella Owens Medical Research Foundation, National Institute of Diabetes and Digestive and Kidney Diseases (R01DK115696), and National Cancer Institute (1R21CA21896801). P Wang was a CPRIT scholar.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Stark AP, Sacks GD, Rochefort MM, et al. Long-term Survival in Patients with Pancreatic Ductal Adenocarcinoma. Surgery 2016;159:1520–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Visvader JE. Cells of origin in cancer. Nature 2011;469:314–22. [DOI] [PubMed] [Google Scholar]
- 3.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–405. [DOI] [PubMed] [Google Scholar]
- 4.Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016;127:2375–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collisson EA, Sadanandam A, Olson P, et al. Subtypes of Pancreatic Ductal Adenocarcinoma and Their Differing Responses to Therapy. Nat Med 2011;17:500–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moffitt RA, Marayati R, Flate EL, et al. Virtual microdissection identifies distinct tumor- and stroma- specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 2015;47:1168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 8.Guerra C, Schuhmacher AJ, Cañamero M, et al. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 2007;11:291–302. [DOI] [PubMed] [Google Scholar]
- 9.De La O, Emerson LL, Goodman JL, et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A 2008;105:18907–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Habbe N, Shi G, Meguid RA, et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A 2008;105:18913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kopp JL, von Figura G, Mayes E, et al. Identification of Sox9-Dependent Acinar-to-Ductal Reprogramming as the Principal Mechanism for Initiation of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012;22:737–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.von Figura G, Fukuda A, Roy N, et al. The chromatin regulator Brg1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nat Cell Biol 2014;16:255–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamaguchi J, Yokoyama Y, Kokuryo T, et al. Cells of origin of pancreatic neoplasms. Surg Today 2018;48:9–17. [DOI] [PubMed] [Google Scholar]
- 14.Verbeke CS. Intraductal papillary-mucinous neoplasia of the pancreas: Histopathology and molecular biology. World J Gastrointest Surg 2010;2:306–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brosens LAA, Hackeng WM, Offerhaus GJ, Hruban RH, Wood LD. Pancreatic adenocarcinoma pathology: changing “landscape”. J Gastrointest Oncol 2015;6:358–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Busnardo AC, DiDio LJ, Tidrick RT, Thomford NR. History of the pancreas. Am J Surg 1983;146:539–50. [DOI] [PubMed] [Google Scholar]
- 17.Brembeck FH, Schreiber FS, Deramaudt TB, et al. The Mutant K-ras Oncogene Causes Pancreatic Periductal Lymphocytic Infiltration and Gastric Mucous Neck Cell Hyperplasia in Transgenic Mice. Cancer Res 2003;63:2005–9. [PubMed] [Google Scholar]
- 18.Ray KC, Bell KM, Yan J, et al. Epithelial Tissues Have Varying Degrees of Susceptibility to KrasG12D- Initiated Tumorigenesis in a Mouse Model. PLoS One 2011;6:e16786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delgiorno KE, Hall JC, Takeuchi KK, et al. Identification and Manipulation of Biliary Metaplasia in Pancreatic Tumors. Gastroenterology 2014;146:233–44e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bailey JM, Alsina J, Rasheed ZA, et al. DCLK1 Marks a Morphologically Distinct Subpopulation of Cells with Stem Cell Properties in Pre-invasive Pancreatic Cancer. Gastroenterology 2014;146:245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bailey JM, Hendley AM, Lafaro KJ, et al. p53 mutations cooperate with oncogenic Kras to promote adenocarcinoma from pancreatic ductal cells. Oncogene 2016;35:4282–8. [DOI] [PubMed] [Google Scholar]
- 22.Ferreira RMM, Sancho R, Messal HA, et al. Duct- and Acinar-Derived Pancreatic Ductal Adenocarcinomas Show Distinct Tumor Progression and Marker Expression. Cell Rep 2017;21:966–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hezel AF, Kimmelman AC, Stanger BZ, et al. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev 2006;20:1218–49. [DOI] [PubMed] [Google Scholar]
- 24.Bailey JM, DelGiorno KE, Crawford HC. The secret origins and surprising fates of pancreas tumors. Carcinogenesis 2014;35:1436–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437–50. [DOI] [PubMed] [Google Scholar]
- 26.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7:469–83. [DOI] [PubMed] [Google Scholar]
- 27.Guerra C, Barbacid M. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol Oncol 2013;7:232–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burlison JS, Long Q, Fujitani Y, et al. Pdx-1 and Ptf1a concurrently determine fate specification of pancreatic multipotent progenitor cells. Dev Biol 2008;316:74–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002;129:2447–57. [DOI] [PubMed] [Google Scholar]
- 30.Kawaguchi Y, Cooper B, Gannon M, et al. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet 2002;32:128–34. [DOI] [PubMed] [Google Scholar]
- 31.Magnuson MA, Osipovich AB. Pancreas-specific Cre driver lines and considerations for their prudent use. Cell Metab 2013;18:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roy N, Takeuchi KK, Ruggeri JM, et al. PDX1 dynamically regulates pancreatic ductal adenocarcinoma initiation and maintenance. Genes Dev 2016;30:2669–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoang CQ, Hale MA, Azevedo-Pouly AC, et al. Transcriptional Maintenance of Pancreatic Acinar Identity, Differentiation, and Homeostasis by PTF1A. Mol Cell Biol 2016;36:3033–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bardeesy N, Cheng KH, Berger JH, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 2006;20:3130–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kopp JL, Dubois CL, Schaeffer DF, et al. Loss of Pten and Activation of Kras Synergistically Induce Formation of Intraductal Papillary Mucinous Neoplasia From Pancreatic Ductal Cells in Mice. Gastroenterology 2018;154:1509–1523.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee AYL, Dubois CL, Sarai K, et al. Cell of origin affects tumour development and phenotype in pancreatic ductal adenocarcinoma. Gut 2018. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 37.Gidekel Friedlander SY, Chu GC, Snyder EL, et al. Context-Dependent Transformation of Adult Pancreatic Cells by Oncogenic K-Ras. Cancer Cell 2009;16:379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guerra C, Collado M, Navas C, et al. Pancreatitis-induced Inflammation Contributes to Pancreatic Cancer by Inhibiting Oncogene-Induced Senescence. Cancer Cell 2011;19:728–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raimondi S, Lowenfels AB, Morselli-Labate AM, et al. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol 2010;24:349–58. [DOI] [PubMed] [Google Scholar]
- 40.Liou GY, Doppler H, Necela B, et al. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-κB and MMPs. J Cell Biol 2013;202:563–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jensen JN, Cameron E, Garay MVR, et al. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology 2005;128:728–41. [DOI] [PubMed] [Google Scholar]
- 42.Westphalen CB, Takemoto Y, Tanaka T, et al. Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell 2016;18:441–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shain AH, Giacomini CP, Matsukuma K, et al. Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci U S A 2012;109:E252–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsuda M, Fukuda A, Roy N, et al. The BRG1/SOX9 axis is critical for acinar cell-derived pancreatic tumorigenesis. J Clin Invest 2018;128:3475–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manfroid I, Ghaye A, Naye F, et al. Zebrafish sox9b is crucial for hepatopancreatic duct development and pancreatic endocrine cell regeneration. Dev Biol 2012;366:268–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shih HP, Kopp JL, Sandhu M, et al. A Notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development 2012;139:2488–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delous M, Yin C, Shin D, et al. sox9b Is a Key Regulator of Pancreaticobiliary Ductal System Development. PLOS Genet 2012;8:e1002754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roy N, Malik S, Villanueva KE, et al. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev 2015;29:658–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang SC, Nassour I, Xiao S, et al. SWI/SNF component ARID1A restrains pancreatic neoplasia formation. Gut 2019;68:1259–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kimura Y, Fukuda A, Ogawa S, et al. ARID1A Maintains Differentiation of Pancreatic Ductal Cells and Inhibits Development of Pancreatic Ductal Adenocarcinoma in Mice. Gastroenterology 2018;155:194–209.e2. [DOI] [PubMed] [Google Scholar]
- 52.Livshits G, Alonso-Curbelo D, Morris JP IV et al. Arid1a restrains Kras-dependent changes in acinar cell identity. van Lohuizen M, editor. eLife 2018;7:e35216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang W, Friedland SC, Guo B, et al. ARID1A, a SWI/SNF subunit, is critical to acinar cell homeostasis and regeneration and is a barrier to transformation and epithelial-mesenchymal transition in the pancreas. Gut 2019;68:1245–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iacobuzio-Donahue CA, Fu B, Yachida S, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol 2009;27:1806–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yonezawa S, Higashi M, Yamada N, et al. Precursor Lesions of Pancreatic Cancer. Gut Liver 2008;2:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Izeradjene K, Combs C, Best M, et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 2007;11:229–43. [DOI] [PubMed] [Google Scholar]
- 57.Liu J, Akanuma N, Liu C, et al. TGF-β1 promotes acinar to ductal metaplasia of human pancreatic acinar cells. Sci Rep 2016;6:30904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Asano T, Yao Y, Zhu J, et al. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene 2004;23:8571–80. [DOI] [PubMed] [Google Scholar]
- 59.Ying H, Elpek KG, Vinjamoori A, et al. Pten is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-κB-cytokine network. Cancer Discov 2011;1:158–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Willis A, Jung EJ, Wakefield T, et al. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004;23:2330. [DOI] [PubMed] [Google Scholar]
- 61.Ji S, Qin Y, Shi S, et al. ERK kinase phosphorylates and destabilizes the tumor suppressor FBW7 in pancreatic cancer. Cell Res 2015;25:561–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ijichi H Genetically-engineered mouse models for pancreatic cancer: Advances and current limitations. World J Clin Oncol 2011;2:195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang L, Holtzinger A, Jagan I, et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell and patient-derived tumor organoids. Nat Med 2015;21:1364–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boj SF, Hwang CI, Baker LA, et al. Organoid Models of Human and Mouse Ductal Pancreatic Cancer. Cell 2015;160:324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seino T, Kawasaki S, Shimokawa M, et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018;22:454–67.e6. [DOI] [PubMed] [Google Scholar]
- 66.Lee J, Snyder ER, Liu Y, et al. Reconstituting development of pancreatic intraepithelial neoplasia from primary human pancreas duct cells. Nat Commun 2017;8:14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Akanuma N, Liu J, Liou GY, et al. Paracrine Secretion of Transforming Growth Factor ß by Ductal Cells Promotes Acinar-to-Ductal Metaplasia in Cultured Human Exocrine Pancreas Tissues. Pancreas 2017;46:1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen SY, Singhania R, Fehringer G, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018;563:579. [DOI] [PubMed] [Google Scholar]