

Abstract
Osteoarthritis (OA) is a progressive degenerative disease of the joints caused in part by a change in the phenotype of resident chondrocytes within affected joints. This altered phenotype, often termed proinflammatory or procatabolic, features enhanced production of endoproteinases and matrix metallo-proteinases (MMPs) as well as secretion of endogenous inflammatory mediators. Degradation and reduced retention of the proteoglycan aggrecan is an early event in OA. Enhanced turnover of hyaluronan (HA) is closely associated with changes in aggrecan. Here, to determine whether experimentally increased HA production promotes aggrecan retention and generates a positive feedback response, we overexpressed HA synthase-2 (HAS2) in chondrocytes via an inducible adenovirus construct (HA synthase-2 viral overexpression; HAS2-OE). HAS2-OE incrementally increased high-molecular-mass HA >100-fold within the cell-associated and growth medium pools. More importantly, our results indicated that the HAS2-OE expression system inhibits MMP3, MMP13, and other markers of the procatabolic phenotype (such as TNF-stimulated gene 6 protein (TSG6)) and also enhances aggrecan retention. These markers were inhibited in OA-associated chondrocytes and in chondrocytes activated by interleukin-1β (IL1β), but also chondrocytes activated by lipopolysaccharide (LPS), tumor necrosis factor α (TNFα), or HA oligosaccharides. However, the enhanced extracellular HA resulting from HAS2-OE did not reduce the procatabolic phenotype of neighboring nontransduced chondrocytes as we had expected. Rather, HA-mediated inhibition of the phenotype occurred only in transduced cells. In addition, high HA biosynthesis rates, especially in transduced procatabolic chondrocytes, resulted in marked changes in chondrocyte dependence on glycolysis versus oxidative phosphorylation for their metabolic energy needs.
Keywords: hyaluronan, chondrocyte, matrix metalloproteinase (MMP), metabolomics, osteoarthritis, inflammation, cartilage catabolism, hyaluronan synthase 2 (HAS2), aggrecan
Introduction
The loss of aggrecan from cartilage is an early event in osteoarthritis (OA)2 (1–3). Aggrecan monomers are retained extracellularly by the interaction of their G1 protein domains with long filaments of hyaluronan (HA). Whereas HA serves to retain aggrecan, the presence of aggrecan (bound to HA), blocks CD44-mediated endocytosis and clearance of HA (4). In reverse, when aggrecan is cleaved, HA endocytosis is enhanced. In this way, aggrecan and HA turnover is tightly coordinated, with both macromolecules being lost from cartilage during OA (5, 6) or IL1 treatment (7–14). HA in cartilage is synthesized primarily by the hyaluronan synthase-2 (HAS2) isoform (15). Conditional knockout of Has2 in mice gives rise to limbs that are severely shortened, and the mutant phenotype includes reduced retention of aggrecan within the growth plate extracellular matrix (16). Aggrecan is also not retained by rat chondrosarcoma chondrocytes following CRISPR/Cas9-mediated silencing of Has2 (17). HAS2-OE applied to these Has2−/− cells rescues aggrecan retention necessary for pericellular coat formation.
We have long posited that the loss of cell-associated HA and aggrecan (as occurs in OA) contributes to the development of a procatabolic phenotype in chondrocytes. This is based on our observations that experimental release or displacement of HA (and bound aggrecan) from the chondrocyte cell surface via HA oligosaccharide or hyaluronidase treatment results in the activation of the cells (18, 19). This includes activation of several matrix metalloproteinases, such as MMP3, MMP13 (18–21), endoproteinases ADAMTS4 and ADAMTS5 (21, 22), and other mediators such as TSG6 (21, 23), nitric oxide, and nitric oxide synthase (24). Thus, the removal of an HA/aggrecan pericellular matrix enhances the same markers as those seen in early OA. The next question raised is whether forced repair of the chondrocyte pericellular matrix chondrocytes reverses or diminishes the procatabolic phenotype. Such a rescue of matrix has long been the goal of many investigators supporting the application of exogenous high-molecular-mass HA both in vitro and in patients, a process termed viscosupplementation (25, 26).
In this study, we explored an alternative approach to replenish HA at the surface of chondrocytes and within the extracellular medium environment that is shared with other chondrocytes. We have subcloned two different HAS2-coding sequences, one human HAS2 and one murine mycHAS2, into adenovirus plasmids. One of these viral plasmids incorporates a GFP (ZsGreen) for visualization purposes; the other incorporates a Tet-On doxycycline-inducible promoter to selectively drive HAS2 protein transcription. We have optimized the transduction conditions to the point where we consistently obtain ∼50% transduction efficiency in both normal bovine and human OA chondrocytes (17, 21). Given that the chondrocytes have a very low cell proliferation rate, we have observed continuous viral transduction extending from weeks to months. With these tools, we sought to determine the effects of enhancing HA production by the chondrocytes at the local level, with variable HA synthesis allowed to occur in parallel with experimental challenge by cytokines or other pro-inflammatory mediators. Such data will address whether chondrocytes can sense that HA repair has occurred and signal chondrocytes to return to quiescent homeostasis.
Results
HAS2-OE results in a diminution of MMP13 and TSG6 mRNA expression in chondrocytes
Chondrocytes in vitro respond to treatment with IL1β by altering their phenotype and expressing markers associated with OA, two of which include MMP13 and TSG-6. Fig. 1A depicts three examples of human OA chondrocyte cultures transduced with Ad-Tet-murine-mycHAS2 and subsequently treated with 1 ng/ml IL1β. All three exhibited an up-regulation of MMP13 mRNA (dark bars), the magnitude of which varies from patient to patient. Nonetheless, when co-treated with doxycycline (Dox) to turn on HAS2 transgene expression, MMP13 stimulation was reduced nearly by half. Normalization of the IL1β-stimulated values to 100%, to allow for better quantification of experiments conducted over many months, showed that Dox addition resulted in a 59 and 55% decrease in MMP13 and TSG6 mRNA, respectively (Fig. 1A, right panels). Similar results were obtained when OA chondrocyte cultures were transduced with Ad-ZsGreen-human-HAS2 (Fig. 1B). For these experiments, Ad-ZsGreen-LacZ was used as the negative control to rule out potential alternative effects of strong viral expression of a transgene protein on MMP13; no effect on MMP13 was observed. Also, in the experiments shown in Fig. 1B, cells were exposed to IL1β at 10 ng/ml. Nonetheless, HAS2-OE still resulted in a 54% reduction in MMP13 mRNA. Thus, whether the transgene was human or murine, driven by a Tet or strong CMV promoter, consistent knockdown of these catabolic markers was observed.
Figure 1.

A, three examples of independent human OA chondrocyte cultures transduced with Ad-Tet-murine-mycHAS2 and subsequently treated with 1 ng/ml IL1β and Dox as labeled, analyzed for changes in MMP13 mRNA. The right-hand bar graphs depict the mean ± 95% confidence interval (error bars) of independent experiments showing the -fold change in MMP13 (n = 6) and TSG6 (n = 5) mRNA due to HAS2-OE (percent inhibition) relative to values with IL1β treatment (without Dox) set to 100% (dotted line). The actual mean value is shown beside the bar. B, a representative example of human OA chondrocytes transduced with Ad-ZsGreen-human-HAS2 (bars 2 and 4) or Ad-ZsGreen-LacZ (bars 1 and 3) and subsequently treated with 10 ng/ml IL1β (bars 3 and 4). The adjacent bar graph depicts the mean ± 95% confidence interval of independent experiments showing the -fold change in MMP13 mRNA (n = 10) due to HAS2-OE (percent inhibition) relative to values with IL1β treatment set to 100% (dotted line). C, a representative example of bovine articular chondrocytes transduced with varying concentrations (IFU/cell) of Ad-ZsGreen-human-HAS2, followed by treatment with 10 ng/ml IL1β and then analyzed for changes in ZsGreen, human HAS2, and bovine MMP13 mRNA. In the adjacent panel, aliquots of proteinase K–treated media and cell lysates from human chondrocytes transduced with Ad-ZsGreen-human-HAS2 (ZsGreen) or Ad-Tet-murine-mycHAS2 (Tet) were analyzed on a 1.0% agarose electrophoresis sizing gel stained with DMMB. Lane markers denote a 1.0 mg/ml aliquot of HA of 1200–1800 kDa (H); HA of 180–350 kDa (M), and HA of <5 kDa (L).
In Fig. 1C, bovine chondrocytes were exposed to varying infectious units (IFU) of Ad-ZsGreen-human-HAS2, exposure that resulted in the progressive up-regulation of both ZsGreen and human HAS2 transgene mRNAs as well as a ∼55% knockdown of IL1β-stimulated bovine MMP13 mRNA, at doses above 12.5 IFU/cell. In the far-right panel of Fig. 1C is a representative agarose gel electrophoresis run, used to visualize the size of HA being synthesized (compared with samples of purified high-, middle-, and low-molecular-mass HA, shown as markers in the first three lanes). Chondrocytes transduced with either varying IFU of Ad-ZsGreen-human-HAS2 or Dox-activated Tet-murine-mycHAS2 resulted in the enhanced synthesis of high-molecular-mass HA—namely HA that does not enter the agarose gel, present in both the cell and medium fractions.
The effect of HAS2-OE on IL1β-stimulated chondrocytes was also studied at the protein level. Fig. 2, A–C depicts Western blot analyses of MMP13 protein expression of three representative cultures of Ad-Tet-mycHAS2–transduced human OA chondrocytes exposed to 1 ng/ml IL1β. Untreated control lanes (lanes 1) illustrate how MMP13 protein levels varied in these cultures of chondrocytes derived from different patients. Nonetheless, all exhibited a substantial increase in the ∼54-kDa MMP13 protein with IL1β treatment (lanes 2) in both the cell-associated and medium fractions. Co-treatment with 100 ng/ml Dox and IL1β resulted in a knockdown of MMP13 protein accumulation. Digitizing several Western blotting experiments (Fig. 2D) demonstrated the range of knockdown due to HAS2-OE with mean values that closely mimic the percent knockdown of MMP13 seen at the mRNA level shown in Fig. 1.
Figure 2.

A–C, Western blot analyses of cell lysate and medium aliquots from three representative experiments with human OA chondrocytes. Chondrocytes were transduced with Ad-Tet-mycHAS2 and subsequently treated without (lane 1) or with (lanes 2 and 3) 1.0 ng/ml IL1β in the absence (lanes 1 and 2) or presence (lane 3) of 100 ng/ml Dox. Blots were probed for detection of MMP13 protein (∼54 kDa), and the cell lysate blots were reprobed for β-actin. The bar graph in D depicts the mean ± 95% confidence interval (error bars) of 10 independent experiments showing the changes in cell-associated and medium accumulation of MMP13 protein due to HAS2-OE (percent inhibition) relative to values with IL1β treatment (without Dox) set to 100% (dotted line). Mean values are shown beside each bar. E, Western blot analyses for CD44 from two representative experiments on bovine chondrocytes transduced with Ad-Tet-human-CD44 and treated with 100 ng/ml Dox (lanes 3 and 4), without IL1β (lanes 1 and 3), or with 1.0 ng/ml IL1β (lanes 2 and 4). Shown is RT-PCR analysis for MMP13 (F) and MMP3 (G) from no Dox (bars 1 and 2) or CD44-OE chondrocytes (bars 3 and 4) treated without (bars 1 and 3) or with 1.0 ng/ml IL1β (bars 2 and 4). Shown is a representative experiment (n = 3) of three independent experiments with similar results. No change in MMP13 or MMP3 mRNA was observed with CD44-OE.
As another control, bovine chondrocytes were transduced with Ad-Tet-human-CD44, as an irrelevant transgene driven by same Tet-Dox adenoviral system. Fig. 2E illustrates robust protein expression of human CD44 in transduced bovine chondrocytes. Under these conditions, other cultures were examined for changes in IL1β-induced expression of MMP13 or MMP3 mRNA. As shown in Fig. 2, F and G, the addition of Dox (CD44-OE) exerted no inhibitory effect on these two procatabolic markers in chondrocytes activated by IL1β.
Effect of Dox concentration and varying HAS2-OE
To determine the dose dependence effects of HAS2-OE (distinct from varying the IFU/cell as shown in Fig. 1C), chondrocytes were transduced with Ad-Tet-mycHAS2 and subsequently treated with various concentrations of Dox. Human chondrocytes, all transduced as a common pool with the same 10 IFU/cell of virus, were plated as monolayers and then exposed to varying Dox concentrations. In Fig. 3A, murine HAS2 mRNA (in human chondrocytes) increased progressively with Dox dose, resulting in the progressive increase in high-molecular-mass HA (Fig. 3D) and increased accumulation of HA in the cell layer (Fig. 3F, gray bars) and medium (Fig. 3F, red bars) as measured by HA ELISA. As the HA levels increased, these same cells showed a coordinate inhibition of MMP13 mRNA (Fig. 3B) and TSG6 mRNA (Fig. 3G), both with a substantial knockdown even observed at low Dox doses. These results were mirrored by changes to MMP13 at the protein level (Fig. 3C). Numbers shown above the MMP13 bands depict the pixel intensity of the MMP13 band, setting the intensity of the IL1β-positive control to 1.0 (and all values normalized to intensities of respective β-actin bands). An ∼50% knockdown of MMP13 protein was observed at all Dox doses tested.
Figure 3.

Human chondrocytes were transduced with 100 IFU/cell Ad-Tet-mycHAS2; untreated control is labeled C. Following transduction, the chondrocytes were treated with 1 ng/ml IL1β (bars 2–6 or labeled with a plus sign), exposed to varying Dox concentrations (as labeled, ng/ml) and analyzed for changes in mycHAS2 (A), MMP13 (B), and TSG6 mRNA (G). Chondrocyte lysates were analyzed by Western blot analysis of MMP13 protein (C; blots reprobed for β-actin). Media from these cultures were analyzed on an agarose electrophoresis sizing gel (D) and by HA ELISA (F; red, medium; gray, cell-associated HA in units of ng/ml × 103). Western blot analyses were performed on three additional human OA chondrocyte cultures (E, H, and K) exposed to Dox (as labeled, ng/ml). Blots were probed for MMP13 protein, β-actin, or mycHAS2 as labeled. Numbers shown above MMP13 bands indicate band intensity (normalized to β-actin) relative to the IL1β-induced MMP13 band set to 1.0. H and K, two experiments in which nontransduced (no virus as labeled) and Ad-Tet-mycHAS2–transduced human OA chondrocytes were exposed to Dox (as labeled, ng/ml). I and J, the effect of Dox concentrations on transduced bovine chondrocytes without (C) or with 1 ng/ml IL1β (+) on expression for mycHAS2 (I) or MMP13 (J) mRNA; results from independent bovine cultures n = 3 for each condition (n = 12 for 100 ng/ml Dox comparison). I, -fold change relative to control set to 1.0; J, -fold change relative to values with IL1β treatment (without Dox) set to 100. An unpaired t test was used for statistical analysis: **, p < 0.01. Error bars, S.D.
In another experiment (Fig. 3E), MMP13 protein was inhibited at 20, 200, and 1000 ng/ml Dox. This experiment also illustrates the progressive Dox-induced accumulation of the mycHAS2 protein, a monomer at ∼64 kDa (arrow) that forms a series of dimers, tetramers, and multimers necessary for HA biosynthesis (27) (Fig. 3E, bottom blot). Another experiment shown in Fig. 3H again demonstrated inhibition of MMP13 protein in transduced human chondrocytes exposed to 100 or 1000 ng/ml Dox (right four lanes). However, the addition of 0, 100, or 1000 ng/ml Dox to chondrocytes that were not transduced with Ad-Tet-mycHAS2 exhibited no change in the level of IL1β-stimulated MMP13 protein (Fig. 3H, left four lanes). This demonstrates that Dox alone has no inhibitory, stimulatory, or otherwise detrimental effect on chondrocyte MMP13 protein accumulation (or MMP13 mRNA expression; data not shown). Another example of this same experimental approach on human OA chondrocytes is shown in Fig. 3K. Again, whereas substantial inhibition of MMP13 protein was observed by Dox addition to transduced chondrocytes, no change in MMP13 was observed following Dox addition to nontransduced chondrocytes.
Ad-Tet-mycHAS2 transduction in bovine chondrocytes mimicked results obtained in human OA chondrocytes. MycHAS2 mRNA was elevated with increase in the concentration of Dox (Fig. 3I). This increase in mycHAS2 resulted in a knockdown of IL1β-stimulated MMP13 mRNA but was only significant at higher Dox concentrations (Fig. 3J).
Biological effects of HAS2-OE on chondrocytes and proteoglycan retention
To determine the effects of IL1β and HAS2-OE at a functional level, high-density cultures of bovine chondrocytes were transduced with Ad-Tet-murine-mycHAS2 or Ad-ZsGreen-murine-HAS2 and then treated for 1 week with IL1β and varying concentrations of Dox. The cultures were then fixed and stained with dimethylmethylene blue (DMMB), a dye that provides pink staining of accumulated sulfated glycosaminoglycan (sGAG; indicative of proteoglycan) in monolayers (as well as a bluish counterstaining of nuclei). It should be also noted that HA alone does not stain with DMMB; however, Streptomyces hyaluronidase treatment results in the release of retained sGAG (4, 28). Control, untreated chondrocytes shown in Fig. 4A appeared as rounded chondrocytes with many (but not all) surrounded with intense pink dye staining. In some areas, the DMMB staining can be seen extending away and between cells. Following treatment with 1 ng/ml IL1β for 1 week (Fig. 4B), the pink staining surrounding the chondrocytes was now absent from most cells, revealing only the counterstaining of the nuclei. Transduced chondrocytes treated with 50 or 100 ng/ml Dox alone (Fig. 4, C and E, respectively) exhibited staining and cell shape patterns similar to control chondrocytes. However, the addition of 50 ng/ml Dox alone to IL1β-treated chondrocytes (with no transduction) did not result in sGAG accumulation (Fig. 4B, inset).
Figure 4.

To determine the effects of IL1β and HAS2-OE at a functional level, control bovine chondrocytes (A and B), chondrocytes transduced with Ad-Tet-murine-mycHAS2 (C–G), or Ad-ZsGreen-human-HAS2 (H–J) were then treated without or with IL1β (1 ng/ml) and varying concentrations of Dox (as labeled) for 1 week. B (inset), nontransduced chondrocytes co-treated with IL1β and 50 ng/ml Dox. The cultures were then fixed and stained with DMMB, a dye that provides pink staining of accumulated proteoglycan (as well as a bluish counterstaining of nuclei). Images shown are representative fields of four independent experiments. J, overlay image of green fluorescence (due to Ad-ZsGreen-mycHAS2 transduction) and bright field. For this image, the green fluorescence intensity has been digitally enhanced to better illustrate successfully transduced cells. Bars in each image, 50 μm.
Interestingly, co-treatment with 50 or 100 ng/ml Dox together with IL1β (Fig. 4, D and F, respectively) revealed substantially improved sGAG retention as compared with IL1β treatment alone (Fig. 4B). This suggests that HAS2-OE provides a degree of functional protection of chondrocytes during periods of procatabolic activation. Other control experiments included transduced cells with no Dox added (Fig. 4G).
Fig. 4, H–J depicts bovine chondrocytes transduced with Ad-ZsGreen-murine-mycHAS2. Again, transduced cells alone exhibited DMMB staining (Fig. 4H) similar to control chondrocytes as well as improved retention in transduced chondrocytes treated with IL1β (Fig. 4I). Interestingly, an overlay of green fluorescence onto Fig. 4I shown in Fig. 3J illustrates that many but not all ZsGreen-positive cells displayed pink sGAG staining. In sum, sGAG accumulation and retention in chondrocytes overexpressing HAS2 appears as robust as in control chondrocytes. Moreover, the effects of HAS2-OE appear to protect chondrocytes from negative features associated with IL1β treatment.
Mechanism responsible for HAS2-OE effects: Comparison with 4MU
To determine whether HA biosynthesis was necessary for the HAS2-OE effects observed in chondrocytes, a typical next step would be to introduce the chemical HA inhibitor, 4MU. However, we have previously determined that 4MU alone, by a still unknown mechanism, blocks the expression of procatabolic markers in chondrocytes, including MMP13 and TSG6 (21). Nonetheless, control and HAS2-OE–transduced chondrocytes were exposed to IL1β in the absence or presence of 4MU (Fig. 5). Western blot analysis of human chondrocyte cultures demonstrated again that HAS2-OE affected an inhibition of MMP13 protein in both the cell layer and medium fractions (Fig. 5, A–C). Treatment with 4-MU alone also blocked the accumulation of MMP13 protein in these two fractions. Co-incubation with 4-MU and Dox generated a combined effect, clearly evident in Fig. 5, A and B. Interestingly, background MMP13 levels were high in the sample of human OA cultures shown in Fig. 5C (control (Ctr)). HAS2-OE and 4-MU both reduced MMP13 protein accumulation by these cells even in the absence of IL1β stimulation. Similar results were obtained when human chondrocytes (Fig. 5D) and bovine chondrocytes (Fig. 5E) were examined for changes in MMP13 mRNA. At the mRNA level, although both treatment conditions were inhibitory, 4-MU appeared more effective than HAS2-OE at reducing MMP13 mRNA, opposite to observations at the protein level in Fig. 5, A–C. Nonetheless, co-treatment with 4MU and Dox resulted in enhanced knockdown. The mechanism of 4-MU–inhibitory activity includes reducing availability of cytoplasmic UDP-GlcUA necessary for HA biosynthesis and blocking transcription of HAS2 itself (29–31). This is illustrated here by a 4MU-mediated reduction of HA present in the medium of chondrocytes as detected by HA ELISA (Fig. 5D) and a marked reduction in mycHAS2 mRNA (Fig. 5E) as well as mycHAS2 protein (Fig. 5C, lanes 4 and 8). Nonetheless, there is likely still sufficient mycHAS2 and HA synthesized to exert an HAS2-OE effect in the presence of 4MU. This suggests that the effects of HAS2-OE and 4MU are similar, as both knock down MMP13 mRNA and MMP13 protein.
Figure 5.

To determine the independent and combined effects of HAS2-OE and 4MU, chondrocytes were transduced with Ad-Tet-mycHAS2 and subsequently incubated with or without IL1β (1 ng/ml) and co-treated in the absence (control (C)) or presence of 100 ng/ml Dox (labeled HAS2 and both), and/or 0.5 mm 4MU (labeled 4MU and both). The effects of these treatments on MMP13 protein are shown in A–C, representing Western blot analysis of three independent preparations of human OA chondrocytes. A and B, MMP13 present in cell lysates and medium (as labeled); C includes visualization of the mycHAS2 protein and a representative blot of β-actin (β-actin for studies in A and B not shown). Numbers shown above MMP13 bands indicate the relative band intensity (normalized to β-actin) as compared with the intensity of the IL1β-induced MMP13 band set to 1.0. D, a representative example of human OA chondrocytes analyzed for MMP13 mRNA as well as HA content present in the medium (units = ng/ml × 102). Chondrocytes were treated with IL1β (bars 2–5) and co-treated with DOC (0 or 100 as labeled) and 4MU (depicted as − or +). MMP13 data depict the relative -fold change in mRNA relative to control (C) set to 1.0. E, changes in relative expression of MMP13 and mycHAS2 mRNA in bovine chondrocytes. Chondrocytes were treated with IL1β (bars 2–6) and co-treated with DOC (0 or 100 as labeled) and 4MU (depicted as − or +). MMP13 data depict the relative -fold change in mRNA relative to control (C) set to 1.0. E summarizes results from three independent bovine cultures for each condition. An unpaired t test was used for statistical analysis: *, p < 0.05; **, p < 0.01. Error bars, S.D.
HAS2-OE inhibition in co-cultures and conditioned medium
Our original hypothesis was that enhanced accumulation of extracellular HA (by HAS2-OE) would bind to receptors on chondrocytes, such as CD44, and the occupied receptors would signal a return to quiescence or steady state. To determine whether increased extracellular HA was indeed necessary for HAS2-OE inhibition of MMPs, we designed experiments to test these conditions on nontransduced cells. In one approach, Ad-HAS2–transduced human chondrocytes were co-cultured with nontransduced bovine chondrocytes at a 1:1 ratio, as depicted in Fig. 6A (phase-contrast overlay image using Ad-ZsGreen-mycHAS2). For biochemical analysis, co-cultures were treated for 24 h without or with Dox and IL1β. As shown in Fig. 6B, using specific primer pairs for human MMP13, a knockdown of human MMP13 mRNA in the presence of IL1β and Dox was observed. However, no knockdown of bovine MMP13 mRNA was observed in the co-cultures (Fig. 6C).
Figure 6.

Ad-Tet-mycHAS2–transduced human chondrocytes were co-cultured with nontransduced bovine chondrocytes. A, example of a coculture, except Ad-ZsGreen-mycHAS2 was used. The co-cultures (B and C) were treated for 24 h with 0 or 100 ng/ml Dox (as labeled) and without (−) or with (+) 1 ng/ml IL1β. B and C, qRT-PCR using human-specific (B) or bovine-specific (C) primers for MMP13. Data represent duplicate experiments and depict relative -fold change with control lysates set to 1.0. In a second series of experiments (E–L), bovine chondrocytes were transduced with Ad-Tet-mycHAS2, wherein >50% were successfully transduced (D; coat image using Ad-ZsGreen-mycHAS2 for illustration). Transduced chondrocyte cultures were incubated with 0, 50, or 100 ng/ml Dox (as labeled) and without (−) or with (+) 1 ng/ml IL1β. After 24 h of treatment, the conditioned medium from each of these cultures was added to fresh, nontransduced bovine chondrocytes (F and I) and allowed to incubate for an additional 24 h. The conditioned medium was also analyzed for HA content via an HA ELISA (G; units of ng of HA/ml). Analysis by qRT-PCR used bovine-specific primers for MMP13 (E and F), MMP3 (H and I), COL2A1 (K), and ACAN (L). The -fold changes in bovine mRNA are relative to values with IL1β treatment (without Dox) set to 100. E–I, results from six independent bovine cultures for each condition. An unpaired t test was used for statistical analysis; ***, p < 0.001. J, -fold change expression of human COL2A1 mRNA using lysates of the human OA chondrocyte experiment shown in Fig. 3B. Error bars, S.D.
Another approach was to test the conditioned medium of transduced cells. Chondrocytes exhibited a transfection efficiency of ∼50%, and in many cases, transfected and nontransfected cells share a pericellular matrix or coat, as illustrated in Fig. 6D. When Ad-Tet-mycHAS2–transduced bovine chondrocytes were co-treated with IL1β and Dox, there was a highly significant decrease in expression of bovine MMP13 mRNA (Fig. 6E) as well as MMP3 mRNA (Fig. 6H), another procatabolic marker of activated chondrocytes. This inhibition correlated with a dose-dependent increase in newly synthesized HA released into the media (Fig. 6G).
Next, the conditioned medium from each of these cultures (representatives shown in Fig. 6G) was removed, added directly to fresh naive chondrocytes, and incubated on these new cells for an additional 24 h. The addition of conditioned medium from IL1β-treated cells (e.g. from a culture depicted in Fig. 6G, bar 2) resulted in a stimulation of both MMP13 (Fig. 6F, bar 2) and MMP3 (Fig. 6I, bar 2) mRNA. This suggests that there were sufficient levels of active IL1β still present in the conditioned medium to activate new target cultures of chondrocytes. However, the addition of conditioned medium from IL1β plus 50 or 100 ng/ml Dox-treated cultures to fresh cells did not inhibit the IL1β stimulation of MMP13 or MMP3 mRNA (Fig. 6, F and I, bars 3 and 4). The fact that this 24-h conditioned medium contained substantial levels of extracellular HA (Fig. 6G, bars 3 and 4) indicated that the inhibitory activity is not contained in the conditioned medium rich in soluble extracellular HA. This suggests that the HAS2-OE inhibitory effects only occurred in transduced cells and transduced cells that are actively synthesizing HA.
Bovine cDNA from experiments shown in Fig. 6, E–I as well as cDNA from human chondrocyte experiments shown in Fig. 3B were also assayed for the effects of HAS2-OE on type II collagen and aggrecan expression. Treatment with 1 ng/ml IL1β significantly reduced the expression of human COL2A1 (Fig. 6J) and bovine COL2A1 (Fig. 6K) as well as bovine aggrecan mRNA (Fig. 6L, ACAN). However, unlike the effects observed on MMPs, HAS2-OE did not exhibit a capacity to rescue this inhibition of collagen or aggrecan mRNA (at least not at the 24-h time point). No statistically significant changes in expression of COL2A1 or ACAN were observed.
HAS2-OE inhibition of catabolic markers induced by other pro-inflammatory agents
To determine whether HAS2-OE–mediated inhibition of MMPs was specific to IL1β activation, bovine or human chondrocytes were activated by other known pro-inflammatory/damage-associated molecular pattern (DAMP) agents, namely TNFα, LPS, or HA oligosaccharides (HAo). Treatment of Ad-Tet-mycHAS2–transduced human chondrocytes with 100 ng/ml Dox resulted in enhanced synthesis of high-molecular-mass HA (Fig. 7A) as well as mycHAS2 protein (Fig. 7C) under both control (Ctr) and agent-treated conditions. Fig. 7B depicts a representative Western blot analysis of lysates from these same treated cultures probed for MMP13. Similar to IL1β treatment, TNFα, LPS, or HA oligosaccharides all affected an increase in MMP13 protein production. As well, co-treatment with Dox reduced accumulation of MMP13 protein in both the cell layer and medium fractions of agent-activated chondrocytes as well as MMP13 present in untreated control cultures. In a similar fashion, co-incubation of transduced human chondrocytes with either Dox plus LPS (Fig. 7D) or Dox plus HA oligosaccharides (Fig. 7E) resulted in the inhibition of DAMP-stimulated MMP13 mRNA expression.
Figure 7.

Bovine or human chondrocytes were activated by other known pro-inflammatory/DAMP agents: TNFα, LPS, or HAo. Ad-Tet-mycHAS2-transduced human chondrocytes, treated with 100 ng/ml Dox as labeled, exhibited enhanced synthesis of high-molecular-mass HA (A) under control (Ctr) and agent-treated conditions. B, representative Western blot analysis of lysates from these same treated cultures probed for MMP13 (blots reprobed for β-actin) and for expression of mycHAS2 protein (C). Next, human chondrocytes were transduced with Ad-Tet-mycHAS2 and then incubated with or without Dox (as labeled 0 and 100); without (bar 1) or with (bars 2 and 3) 10 ng/ml LPS (D) or 250 μg/ml HAo (E). D and E, relative -fold change in MMP13 mRNA, assayed in duplicate, with untreated control values set to 1.0. Bovine chondrocytes transduced with Ad-Tet-mycHAS2 (F–J) were incubated with 0, 50, or 100 ng/ml Dox (as labeled) and without (bar 1) or with (bars 2–4) co-treatment with LPS (F and G), HAo (H and I), or 10 ng/ml TNFα (J). After cultures were exposed to LPS and HAo, the 24-h conditioned medium (LPS C.M. or HAo C.M.) was collected and added to fresh bovine monolayer cultures for an additional 24-h incubation, similar to conditions described in the legend to Fig. 6. Changes in mycHAS2, MMP13, or MMP3 mRNA were quantified. F–J, results from three independent bovine chondrocyte cultures for each condition (each assayed in duplicate). The -fold changes in mRNA are relative to control values set to 1.0. An unpaired t test was used for statistical analysis: **, p < 0.01. Error bars, S.D.
These results were replicated in cultures of bovine chondrocytes (Fig. 7, F–J). Similar levels of mycHAS2 mRNA following Dox treatment were observed in each of these cultures (Fig. 7, F, H, and J, left panels), indicating the likelihood that substantial extracellular HA was being produced. LPS treatment provided a strong stimulation of MMP13 (Fig. 7F, middle) as well as MMP3 (Fig. 7G) mRNA. Stimulation of both MMP13 and MMP3 were significantly blocked by co-treatment with 50 or 100 ng/ml Dox. Similar stimulation of bovine chondrocyte MMP13 or MMP3 was obtained by treatment with HA oligosaccharides (Fig. 7, H and I, middle panels) or TNFα (Fig. 7J, right). In each case, the expression of both of these activation markers was blocked by Dox-mediated activation of HAS2-OE.
As such, it was of interest to test the effects of the 24-h conditioned medium of the Ad-Tet-mycHAS2–transduced cultures treated without or with LPS and HAo with or without Dox, on naive chondrocytes. As shown in the right-hand panels (Fig. 7, F–I), sufficient levels of active LPS and HA oligosaccharides were present to stimulate the new expression of MMP13 or MMP3 mRNA in cultures of fresh, nontransduced target chondrocytes. However, the conditioned medium transferred from Dox co-treated (transduced) cultures exhibited no transferrable capacity to inhibit the stimulation of MMP13 (Fig. 7, F and H) or MMP3 (Fig. 7, G and I) mRNA in the fresh cultures. Again, although substantial levels of newly synthesized HA are present in the 50 and 100 ng/ml Dox conditioned medium (Fig. 6G), this HA-enriched medium does not offer inhibitory activity.
The effect of HAS2-OE on chondrocyte energy metabolism
To determine whether the inhibitory effects of HAS2-OE on MMPs were due to changes in intracellular metabolism, bovine chondrocytes were transduced with Ad-Tet-mycHAS2; plated into 24-well or 96-well Seahorse XF Cell Culture Microplates as confluent monolayers; and incubated with 0, 50, or 100 ng/ml Dox without or with 1.0 ng/ml IL1β, as labeled, for 24 h. The cells were then tested live, in real time, for rate changes in medium accumulation of +H protons (indicative of lactic acid accumulation) and simultaneously for O2 consumption (indicative of mitochondrial respiration). After establishing values for baseline metabolism, the cells are put under stress by injection of select metabolic mediators to parse out sub-pathways associated with the glycolysis or TCA pathways. Fig. 8A shows a representative cell energy phenotype test wherein averaged values for extracellular acidification rate (ECAR, mpH/min) are plotted versus oxygen consumption rate (OCR, pmol/min). The primary observation is that IL1β-treated chondrocytes (red squares) were more glycolytic as compared with control chondrocytes (blue squares). HAS2-OE chondrocytes had reduced glycolytic activity (purple squares), but of more importance, co-treatment with Dox and IL1β (green squares) resulted in reducing the ECAR metabolism of IL1β-activated chondrocytes back toward that of control cells. In other words, HAS2-OE did drive chondrocytes toward a more “quiescent” phenotype. These changes in ECAR were quantified in Fig. 8B. IL1β treatment resulted in a prominent increase in ECAR values (black bar) indicative of enhanced cellular usage of glycolysis. HAS2-OE significantly reversed IL1β-induced increase in glycolysis back to levels similar to control chondrocytes.
Figure 8.
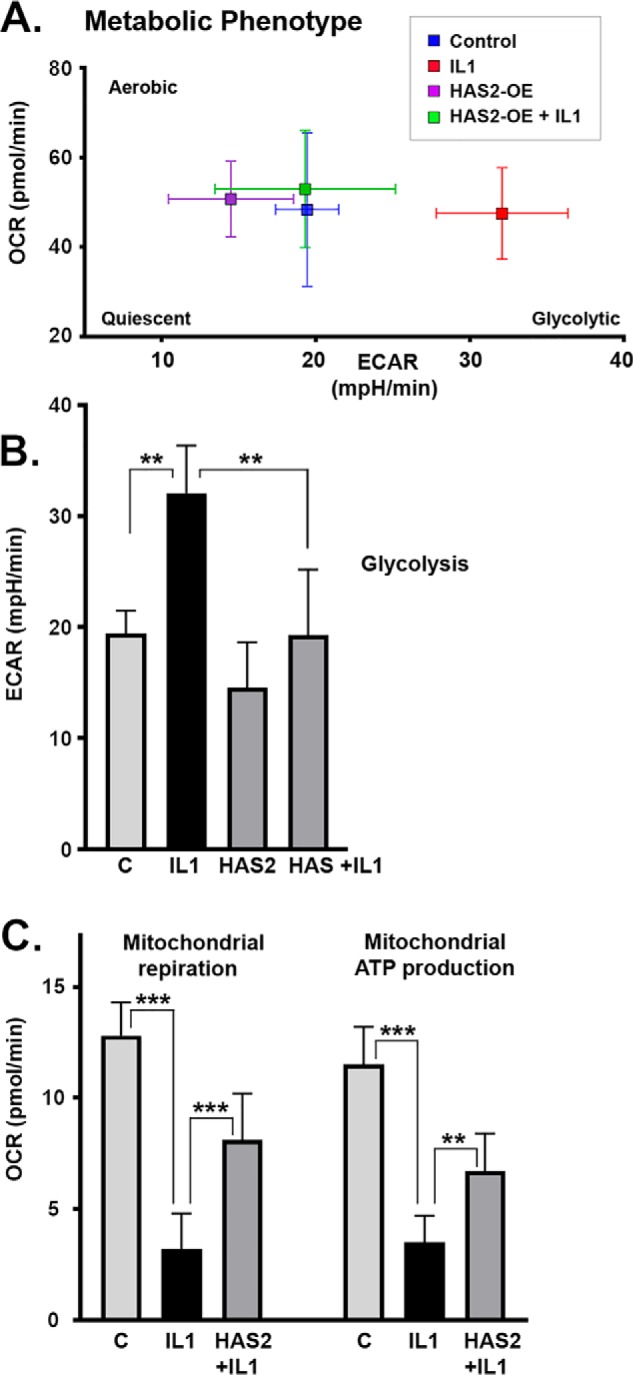
To determine whether the inhibitory effects of HAS2-OE on MMPs was due to changes in intracellular metabolism, bovine chondrocytes were transduced with Ad-Tet-mycHAS2 and then plated into 24-well (A) or 96-well (B and C) Seahorse XF or XFe cell culture microplates. The confluent monolayers were then incubated with 0, 50, or 200 ng/ml Dox, without or with 1 ng/ml IL1β as labeled. The cells were then mated with a sensor cartridge and analyzed in a Seahorse XF or XFe Flux Analyzer for real-time changes in proton accumulation and oxygen consumption within the specialized DMEM culture media. A, representative cell energy phenotype test wherein values for ECAR (mpH/min) under baseline conditions are plotted versus OCR (pmol/min). B, summary of ECAR data (average ± S.D. (error bars), n = 3) representative of basal glycolysis rates. C, results of a representative Mito Stress Test of mitochondrial function, wherein bars depict mitochondrial respiration as a corrected OCR value (average ± S.D., n = 9) as well as the ATP production rates (pmol/min) that can be calculated from the results of this test. Each panel of Seahorse data depicts a representative experiment of 3–4 independent experiments. An unpaired t test was used for statistical analysis: **, p < 0.01; ***, p < 0.001.
Less change in OCR was noted in the phenotype test as compared with ECAR (Fig. 8A). Nonetheless, a second assay (mitochondrial stress test) was used to examine the effects on mitochondrial respiration more closely. In Fig. 8C, mitochondrial respiration (measured by OCR) revealed that IL1β-treated chondrocytes (black bars) displayed a substantial reduction in mitochondrial respiration as compared with control cells (gray bars labeled C). Responses of these cells to stressed conditions (namely oligomycin followed by carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) injections) can be used to show changes in mitochondrial contribution to ATP production. Again, IL1β-treated chondrocytes show a substantial deficit in ATP production by mitochondria, as compared with controls (black bars). However, co-treatment of chondrocytes with IL1β and Dox (labeled HAS2+IL1) rescued both overall mitochondrial respiration and mitochondrial contribution to ATP production. Mitochondrial spare capacity displayed a similar profile, and mitochondrial proton leak was minimal but slightly elevated in IL1β conditions (data not shown). These data suggest that a metabolic shift had occurred following IL1β treatment, a shift that was rescued by HAS2-OE.
Discussion
We demonstrated previously that chondrocytes and cartilage explants, treated with the inflammatory cytokine IL1β, exhibit a substantial loss of HA and proteoglycan (8, 21). This occurs even though IL1β enhances the synthesis of endogenous chondrocyte HAS2 (8). We subsequently determined that HA and bound aggrecan G1 domains were rapidly internalized by CD44 after IL1β activation, in close coordination with the ADAMTS/MMP-mediated cleavage and release of degraded aggrecan monomers (4, 32, 33). Our hypothesis was that this matrix loss would result in a negative feedback loop wherein deficiencies in the cell-associated HA/proteoglycan pericellular matrix would further promote the procatabolic phenotype of activated chondrocytes. For example, we observed enhanced MMP3, MMP13, and ADAMTS4 activation in chondrocytes after the HA/proteoglycan-rich pericellular matrix was experimentally removed by treatment with HA oligosaccharides (18, 20, 22) or hyaluronidase (21). We proposed that if extracellular HA could be restored, more proteoglycan would be retained, chondrocytes would sense these changes (possibly through CD44 signaling), and the cells would return to a more quiescent, steady-state phenotype. In the current study, we examined the effects of HAS2-OE as an approach to test these hypotheses. HAS2-OE did allow chondrocytes to locally synthesize high levels of high-molecular-mass HA (Fig. 1C) well beyond what these cells could produce with their endogenous HAS2. For example, in the HA ELISA shown in Fig. 3F, total HA content changed from 110 ng/ml in basal chondrocytes to 16,600 ng/ml with HAS2-OE. This included a more than 100-fold increase in cell-associated HA. However, it should be noted that this level of HA (16.6 μg/ml) is far less than levels used in studies testing the addition of exogenous HA to cells (50–1000 μg/ml) (25, 34–36).
Under these conditions, we demonstrated that HAS2-OE exerted a profound effect on chondrocytes. Induced procatabolic markers, such as MMP13, MMP3, and TSG6, were diminished, and sGAG, indicative of proteoglycan, was retained even in the continued presence of IL1β.
Nonetheless, it has been suggested in recent years that IL1β does not appropriately reflect the phenotype of OA (37). Other stressors contribute to the altered OA phenotype in chondrocytes, including chronic exposure to excessive loading, aging, and obesity (38, 39). More recent reports have proposed the role of activation of the innate immune responses of chondrocytes in the pathogenesis and early inflammation associated with OA (38, 40, 41). Toll-like receptors (TLRs) constitute one receptor family that gives rise to these innate immunity responses. These receptors can be activated by agents such as LPS as well as by ligands derived from the products of cartilage breakdown known collectively as DAMPs. HA oligosaccharides are an example of an extracellular matrix breakdown product that is viewed by some as a DAMP (40, 42, 43). Activation of an innate immunity response results in the enhanced production of MMPs as well as endogenous IL1β, IL6, and TNFα. Given the activation of MMPs, these output events conspire to generate another wave of DAMPs and establishment of a self-amplifying loop that leads to progressive cartilage degeneration. Many damaged fragments of cartilage matrix are thought to have the capacity to serve as DAMPs. The relative contribution of DAMP-mediated activation of chondrocytes in OA patients remains unknown. Nonetheless, the addition of exogenous HA was found to reduce NF-κB activation and diminish TNFα, IL1β, MMP13, and iNOS expression in LPS-stimulated chondrocytes (44). HA binding to CD44 decreased the release of IL6 and TNFα by macrophages following LPS exposure (45) and decreased TLR4 signaling induced by LPS treatment of fibroblasts (46). Based on these reports, we tested the effects of HAS2-OE on chondrocytes activated by three representatives of this pathway—namely TNFα, LPS, and HA oligosaccharides. All three agents affected a robust activation of MMP3 and MMP13, both in human and bovine chondrocytes. Nonetheless, the combination of HAS2-OE treatment with any of these agents resulted in a significant knockdown of the procatabolic markers (Fig. 7). Even the elevated MMP13 level of expression in human OA chondrocytes at baseline (without any cytokine or DAMP enhancement) was reduced by HAS2-OE. This suggests that the mechanism for the HAS2-OE inhibitory effect is likely downstream of procatabolic initiating events that impinge on chondrocytes, including proinflammatory cytokines, DAMPs, excessive loading, etc.
The mechanism for this inhibition by HAS2-OE is more difficult to address. Our original hypothesis included the occupation of CD44 receptors by extracellular HA to promote homeostasis (7, 47). Others have provided data indicating that extracellular HA directly blocks the access of ligands to TLR4 (34, 44) or blocks required interactions of TLR4 and CD44 (35, 45, 46).
To address the mechanism, we tested whether the enhanced HA due to HAS2-OE, present within the extracellular matrix or medium, was sufficient and effective to knock down markers of adjacent, nontransduced but otherwise cytokine-activated chondrocytes. At high density, co-cultures of transduced and nontransduced chondrocytes are in close association with each other and sometimes even share the same pericellular matrix (Fig. 6D). Moreover, all cells (transduced and nontransduced) are exposed to the elevated HA present in the medium fraction. However, nontransduced bovine chondrocytes in co-culture with human HAS2-OE chondrocytes did not display a knockdown of markers. In a second approach, conditioned medium from transduced bovine cultures, ones that had exhibited HAS2-OE–mediated knockdown of MMP13 and MMP3, failed to transfer this inhibitory activity to naive nontransduced chondrocytes. A similar observation was made of conditioned medium from cultures with successful HAS2-OE–mediated inhibition of LPS or HA oligosaccharide-stimulated MMP13 or MMP3. One conclusion from these results is that HAS2-OE activity occurs only in transduced cells and that extracellular HA (or another unknown soluble factor) is not directly responsible. This may be the reason that the average level of knockdown of markers by HAS2-OE of ∼50% is similar to our typical transduction efficiency.
When testing the comparative effectiveness of 4MU and HAS2-OE, we noted that 4MU was more effective at knockdown of MMP13 mRNA (Fig. 5). One explanation is that 4MU and HAS2-OE function via different mechanisms. Alternatively, 4MU may be more effective because it affects all cells within the cultures, whereas HAS2-OE is restricted to only the transduced cells. That 4MU also effectively blocks the procatabolic phenotype of chondrocytes and, moreover, enhances aggrecan retention in intact bovine or human cartilage explants exposed to IL1β or HA oligosaccharides (21) is difficult to fit with our original hypothesis. One of the major effects of 4MU is as a selective chemical inhibitor of HA biosynthesis (21, 31). However, 4MU also blocks HAS2 mRNA expression (21, 30, 31, 48), and, like HAS2-OE in this study, 4MU also blocks the expression of MMP13, ADAMTS4, and TSG6 (21). The mechanism for the 4MU transcriptional effects is still unknown, but it is not dependent on HA biosynthesis, alteration of early cell signaling events, message stability, or GlcNAcylation (21). This leaves open the difficult question: How can 4MU, an inhibitor of HA production, and HAS2-OE enhancement of HA production both act as anti-catabolic agents? One possibility is that both apply stress on the availability of intracellular UDP-sugar pools (49).
Chanmee et al. (50, 51) observed that HAS2-OE in breast cancer cells resulted in a phenotype change of these cells into cancer stem cells, a phenotype switch that was based on a metabolic reprogramming event. Their observations suggested that HAS2-OE gave rise to a flux in the hexosamine biosynthetic pathway and subsequent alterations in energy metabolism, due to the large quantity of UDP-sugar substrates (namely UDP-GlcUA and UDP-GlcNAc) necessary for enhanced HA production. One intriguing possibility of these results is that HAS2-OE can drive metabolic reprogramming in cells, reprogramming that can subsequently drive changes in phenotype. Moreover, such changes in metabolism represent a mechanism that would only occur in successfully HAS2-transduced cells. Enhanced HA must still be produced and secreted to drive cellular responses to UDP-sugar depletion.
Whether HAS2-OE affected a metabolic reprogramming in chondrocytes was addressed by measuring changes in their use of glycolysis or mitochondrial respiration to satisfy their metabolic needs. In this current study, IL1β-activated chondrocytes exhibited enhanced utilization of glycolysis and less reliance on mitochondrial respiration than control, quiescent chondrocytes. A deficit in mitochondrial activity associated with OA chondrocytes is the central message in a recent review (39). HAS2-OE exerted a pronounced effect on this OA-like metabolic signature, blocking the dependence on glycolysis (ECAR) and simultaneously enhancing mitochondrial respiration and ATP production. This is the opposite result as observed in cancer cells by Chanmee et al. (50), wherein HAS2-OE reduced glycolysis and enhanced mitochondrial function. In another study, HAS2-OE in epithelial cells, resulting in an epithelial-mesenchymal transition; this transition included enhancement of a mesenchymal pericellular matrix but was also associated with increased production of MMP9 (52). Thus, it is likely that HAS2-OE exerts differing effects on different cell types. Chondrocytes represent a more quiescent, nonproliferating cell type, residing in a nonvascularized environment with a unique function to synthesize large quantities of extracellular matrix.
Additional studies will be required to determine whether HAS2-OE–associated changes in the metabolic signature (reported in Fig. 8) are directly responsible for inhibition of the procatabolic phenotype. Nonetheless, a close linkage of these two events is consistent with observations of this study that MMP inhibition by HAS2-OE occurs only in successfully transduced cells and is similar to the effects of 4MU.
Experimental procedures
Materials
Ham's F-12 and DMEM were obtained from Mediatech; FBS was from Hyclone; and TNFα and IL1β were from R&D Systems, Inc. 4-MU was from Alfa Aesar (A10337). Pronase (53702; EMD Millipore Calbiochem), collagenase P (11249002001; Roche Applied Science), and collagenase D (11088882001; Roche Applied Science) were used in dissociation of tissues. Cell lysis buffer was from Cell Signaling Technologies, and Clear Blue X-ray film was from Genesee Scientific. All other reagents were from Sigma-Aldrich.
Specific primers for real-time RT-PCR were custom-made by Integrated DNA Technologies (Coralville, IA). The iScriptTM cDNA synthesis kit was obtained from Bio-Rad, and RT2 Real TimeTM SYBR Green reagents were from SA Biosciences. The DuoSet HA ELISA kit for hyaluronan (DY3614-05) was purchased from R&D Systems, Inc., and used following the manufacturer's instructions.
Specific antibodies used for analysis included rabbit polyclonal anti-MMP13 (sc-30073, clone H-230, lot F1312; Santa Cruz Biotechnology, Inc.). However, given that this antibody is no longer commercially available, we verified that identical results could be obtained using rabbit polyclonal anti-MMP13 (ab39012, lot CR1575 14-27, Abcam) or mouse monoclonal anti-MMP13 (sc515284, lot C1617, Santa Cruz Biotechnology). Other antibodies included β-actin (A1978, clone AC-15, lot 065M4837V; Sigma-Aldrich). Anti-CD44 (BU52; 193–020, lot 254902) was from Ancell. Mouse anti-Myc was obtained from Cell Signaling Technology (catalog no. 2276, clone 9B11, lot 24). The secondary antibodies horseradish peroxidase–conjugated donkey anti-rabbit (SA1-200) and horseradish peroxidase–conjugated donkey anti-mouse (SA1-100) were from Thermo Fisher Scientific.
HA oligosaccharides were prepared and purified as described previously (18) and derived from rooster comb HA (Sigma). The HA oligosaccharides are present as a mixture of HA hexa-, octa-, and decasaccharides. We recently reviewed information on the activity and comparative use of these and other HA oligosaccharide preparations (7).
Cell culture
Primary bovine articular chondrocytes were isolated from the metacarpophalangeal joints of 18–24-month-old adult steers as described previously (21, 22). Primary human articular chondrocytes were isolated from knee cartilage obtained following joint replacement surgery, within 24 h after surgery and with institutional approval. Human cartilage samples were from patients (∼60% female, 40% male) with an average age of 66.8 ± 10.0 years and taken from normal-looking cartilage present on the remnants of medial and lateral femoral condyles. Bovine and human chondrocytes were liberated from full-thickness slices of articular cartilage by sequential Pronase/collagenase P digestion. After harvest and before plating, chondrocytes in suspension were incubated with Ad-HAS2 constructs (10 IFU/cell) for 2 h at 37 °C with occasional gentle mixing, in a 1:1 mixture of DMEM/Ham's F-12 medium, 50 units/ml penicillin, l-glutamate, and ascorbic acid but without serum. Chondrocytes were then plated as high-density monolayers (0.5–1.0 × 106 cells/cm2) in medium still containing virus and allowed to attach overnight at 37 °C. Medium was then changed to fresh medium containing 10% FBS and doxycycline (Dox) if the Ad-Tet-On virus was being used. After 24 h in culture, the medium was changed to 1% FBS (with Dox if appropriate) for 12 h and then incubated in serum-free medium for 1 h prior to the addition of 10 ng/ml TNFα, 1–10 ng/ml IL1β, 250 μg/ml HA oligosaccharides, or 10 ng/ml LPS, with or without 0.5–2.0 mm 4-MU and with or without varying concentrations of Dox in fresh serum-free culture medium treatment for varying times. In experiments with 4-MU, the reagent was dissolved in DMSO and then added to the culture medium with a final concentration of 0.1% DMSO; DMSO only at the same concentration was used as a control.
Particle exclusion assay
To visualize the pericellular matrix, the medium of Ad-ZsGreen1-mycHas2–transduced bovine chondrocytes was replaced with a suspension of formalin-fixed erythrocytes in PBS plus 0.1% BSA (53). Cells were photographed using a Nikon TE2000 inverted phase-contrast microscope, and images were captured digitally in real time using a Retiga 2000R digital camera (QImaging). The presence of the cell-associated matrix is seen as the particle-excluded zone surrounding the chondrocytes.
DMMB staining of monolayers
Ad-Tet-mycHAS2–transduced or Ad-ZsGreen-mycHAS2–transduced bovine chondrocytes were plated into 12-well plates at 1 × 106 cells/well. Following overnight attachment, cells were treated without or with varying concentrations of Dox and without or with 1.0 ng/ml IL1β for 4 days. Cell medium was removed, and the cells were washed with PBS followed by 15-min treatment with 4% paraformaldehyde solution at room temperature. The cells were washed again with PBS and then incubated overnight in a solution of DMMB in the dark with no rocking. The DMMB was prepared as described previously (4, 28, 54). Cells were examined in plates using a Nikon TE2000 inverted phase-contrast microscope, and images were captured digitally in real time using a Retiga 2000R digital camera (QImaging).
Generation of Adeno-Has2 virus constructs
The human HAS2 ORF (NM_005328) in pCR3.1 (a gift from Dr. Tim Bowen, Cardiff University School of Medicine, Cardiff, UK) (55) was PCR-amplified using AccuPrime Pfx DNA Polymerase (Life Technologies) and primers designed specifically for use with the Adeno-X Adenoviral System 3 (Clontech). The amplified human HAS2 sequence was ligated into the pAdenoX-CMV-ZsGreen1 linearized vector (Clontech) to form Ad-ZsGreen-hHAS2. All PCR-amplified regions were verified by DNA sequencing. In the adeno-ZsGreen-HAS2 vector, HAS2 and ZsGreen1 sequence are present on the same plasmid but driven by separate CMV-1E promoters. The murine Has2 coding sequence (NM_008216), containing an NH2-terminal 6× Myc tag in pCDNA3, was kindly provided by Drs. Davide Vigetti and Alberto Passi (27). The murine Has2 coding sequence was PCR-amplified and subcloned into the pAdenoX-CMV-ZsGreen1 linearized vector to form Ad-ZsGreen-mycHas2 as described previously (17). The murine Has2 coding sequence was subcloned into the pAdenoX-Tet3G linearized vector (Clontech) as described previously (21) to form Ad-Tet-mycHas2. Primers and plasmid for LacZ, included in the Adeno-X Adenoviral System 3 kit (Clontech) were used according to the manufacturer's protocol to generate Ad-ZsGreen-LacZ. In addition, the human CD44-coding sequence that we have described previously (56–58) was also subcloned into the pAdenoX-Tet3G linearized vector (Clontech) to generate the Ad-Tet-human-CD44. Each of these viral constructs was amplified and packaged in HEK293 cells (ATCC). Viral particles were purified using the Adeno-X Purification Kit (Clontech) and titered using the Adeno-X Rapid Titer Kit (Clontech) to obtain 3–5 × 109 IFU/ml purified viral stock.
Western blotting
Total protein was extracted using cell lysis buffer–containing protease and phosphatase inhibitor mixtures. Equivalent protein concentrations were loaded into 4–12% NuPAGE® Novex® Tris acetate gradient minigels (Thermo Fisher Scientific). In some experiments, the conditioned culture medium was also collected and processed for Western blotting by loading aliquots of equivalent volume to minigels. Following electrophoresis, proteins within the acrylamide gel were transferred to a nitrocellulose membrane using a Criterion blotter apparatus (Bio-Rad), and the nitrocellulose membrane was then blocked in TBS containing 0.1% Tween 20 and 5% nonfat dry milk (TBS-T-NFDM) for 1 h. Immunoblots were incubated overnight with primary antibody in TBS-T-NFDM at 4 °C, rinsed three times in TBS-T, and incubated with secondary antibody in TBS-T-NFDM for 1 h at room temperature. Detection of immunoreactive bands was performed using chemiluminescence (Novex ECL, Invitrogen). In some cases, the blots were stripped using Restore Plus Western Stripping Buffer (Thermo Fisher Scientific) for 30 min at room temperature and reprobed using another primary antibody. Developed X-ray films were imaged and digitized using a Bio-Rad GelDoc with ImageLab software. Pixel intensities for MMP13 bands were used for quantification after normalization to loading control bands (β-actin or GAPDH). All other experimental details not mentioned here are described in the figure legends.
Real-time quantitative RT-PCR (qRT-PCR)
Total RNA was isolated from the bovine and human chondrocyte cultures according to the manufacturer's instructions for the use of TRIzol® reagent (Thermo Fisher Scientific). Total RNA was reverse-transcribed to cDNA using the iScript cDNA synthesis kit (Bio-Rad). Quantitative PCR was performed using Sso-Advanced SYBR-Green Supermix (Bio-Rad) and amplified on a StepOnePlus real-time PCR system (Applied Biosystems) to obtain cycle threshold (Ct) values for target and internal reference cDNA levels.
The human-specific primer sequences are as follows: ACAN, forward (5′-TCT GTA ACC CAG GCT CCA AC-3′) and reverse (5′-CTG GCA AAA TCC CCA CTA AA-3′); COL2A1 (59), forward (5′-GGC AAT AGC AGG TTC ACG TAC A-3′) and reverse (5′-CGA TAA CAG TCT TGC CCC ACT T-3′); GAPDH, forward (5′-GAA TTT GGC TAC AGC AAC AGG-3′) and reverse (5′-AGT GAG GGT CTC TCT CTT CC-3′); human-specific MMP13, forward (5′-CAG TGG TGG TGA TGA AGA TGA T-3′) and reverse (5′-CGC GAG ATT TGT AGG ATG GTA G-3′); HAS2, forward (5′-CTG GAA GAA CAA CTT CCA CGA A-3′) and reverse (5′-GAC CAA TTG CGT TAC GTG TTGC-3′); TSG6, forward (5′-GTG GCG TCT TTA CAG ATC CAA AGC-3′) and reverse (5′-CAA CAT AAT CAG CCA AGC AAC-3′).
The bovine-specific primer sequences are follows: ACAN, forward (5′-AAA TAT CAC TGA GGG TGA AGC CCG-3′) and reverse (5′-ACT TCA GGG ACA AAC GTG AAA GGC-3′); COL2A1 (60), forward (5′-TGC AGG ACG GGC AGA GGT AT-3′) and reverse (5′-CAC AGA CAC AGA TCC GGC AG-3′); GAPDH, forward (5′-ATT CTG GCA AAG TGG ACA TCG TCG-3′) and reverse (5′-ATG GCC TTT CCA TTG ATG ACG AGC-3′); MMP3, forward (5′-CTC ACA GAC CTG ACT CGG TT-3′) and reverse (5′-CAC GCC TGA AGG AAG AGA TG-3′); bovine-specific MP13, forward (5′-CCT GCT GGA ATC CTG AAG AAA-3′) and reverse (5′-AGT CTG CCA GTC ACC TCT AA-3′); 18S rRNA, forward (5′-GTA ACC CGT TGA ACC CCA TT-3′) and reverse (5′-CCA TCC AAT CGG TAG TAG CG-3′). The mouse primer sequence for mycHAS2 was as follows: forward (5′-GCA TGA ATT TGT GGA AGA CTG G-3′) and reverse (5′-GCC GTG TAT TTA GTT GCA TAG C-3′). Also used were primers for ZsGreen: forward (5′-AGA AGA TGA CCG ACA ACT GG-3′) and reverse (5′-GTA CAC GGT GTC GAA CTG G-3′).Real-time RT-PCR efficiency (E) was calculated as E = 10(−1/slope) (61). The -fold increase in copy numbers of mRNA was calculated as a relative ratio of target gene to GAPDH (ΔΔCt), following the mathematical model introduced by Pfaffl (62) as described previously (21, 28).
Agarose gel electrophoresis
HA and chondroitin sulfate chains remaining after papain digestion of proteoglycans were separated on 1% agarose gels prepared in Tris acetate–EDTA buffer and cast into 10 × 15-cm trays of an MP-1015 horizontal electrophoresis apparatus (ISI Scientific) as described previously (4). Briefly, samples (15 μl) were loaded into each well. Electrophoresis was carried out for 30 min at 150 V. Gels were fixed by rinsing in 70% ethanol for 30 min and stained overnight with Stains-all or followed by destaining in 70% ethanol. Stained bands were imaged by trans-white light on the GelDoc Imager.
Metabolomic studies using Seahorse flux analyzer
Transduced bovine chondrocytes were plated at 8.0 × 104 cells/well into specially designed 96-well (or at 2.5 × 105 cells/well into 24-well) Seahorse XF cell culture microplates. The confluent monolayers were preincubated with 0 and 50–200 ng/ml Dox in 1% FBS for 12 h and then for 24 h with or without 2 ng/ml IL1β and with or without the same level of Dox. The medium was changed to serum-free Seahorse XF Base Medium (without phenol red but 10 mm glucose, 1.0 mm pyruvate, and 2.0 mm glutamine added) or Seahorse XF DMEM, pH 7.4, cells, depending on the assay. Assay medium also contained fresh IL1β. The cells were then mated with a sensor cartridge and analyzed in a Seahorse XF 24 or XFe 96 Flux Analyzer (Agilent Tech) for real-time detection of changes in proton accumulation and oxygen consumption following the manufacturer's guidelines. For the Agilent XF cell energy phenotype test, a combined injection of oligomycin (2 μm final) and FCCP (0.25 μm final) were applied after the instrument completed measurement of basal values. When performing an Agilent XF Mito Stress Test, timed sequential injections of oligomycin (2 μm final) followed by FCCP (0.25 μm final) and, last, a 1:1 mixture of antimycin A with rotenone (0.50 μm final) were applied after measurement of basal values. Algorithms provided in Agilent assay report generator Excel files were used to generate blots and bar graphs. Mitochondrial ATP production derived from the Mito Stress test is expressed as OCR (pmol of O2/min) in their software.
Statistical analysis
All data except as noted were obtained from at least three independent experiments performed in duplicate or triplicate. A two-tailed unpaired Student's t test was used for direct comparison of treatment group with control. A p value of <0.05 was considered significant (*, p value ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001).
Author contributions
W. K., C. B. K., and S. I. designed this study and wrote the paper; W. K. prepared the figures. E. B. A. contributed to the writing of the paper and designed, subcloned, and constructed all of the adenoviral vectors used for expression of HAS2. S. I., N. I., K. T., S. T., and Y. O. designed, performed, and analyzed all of the experiments shown in Figs. 1–8. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Larry J. Dobbs, Jr. (Department of Pathology and Laboratory Medicine, East Carolina University) for assistance in the appropriation of human osteoarthritic tissue and Michelle Cobb and Joani Zary Oswald for technical assistance with this project. We thank Drs. Alberto Passi and Davide Vigetti (Universita degli Studi dell'Insurbria) and Dr. Paraskevi (Uppsala University) for graciously providing the mycHAS2 plasmid and Dr. Tim Bowen (Cardiff University School of Medicine, Cardiff, UK) for the human HAS2 plasmid.
The work was supported in part by National Institutes of Health Grants R01-AR039507 (to C. B. K.), R21-AR066581 (to W. K.), and R21-AR072682 (to W. K.) as well as a grant from the Mizutani Foundation (to W. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- OA
- osteoarthritis
- ACAN
- aggrecan
- DMMB
- dimethylmethylene blue
- Dox
- doxycycline
- ECAR
- extracellular acidification rate
- FCCP
- carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- HA
- hyaluronan
- HAS
- hyaluronan synthase
- HAS2-OE
- hyaluronan synthase-2 viral overexpression
- MMP
- matrix metalloproteinase
- 4MU
- 4-methylumbelliferone
- OCR
- oxygen consumption rate
- TSG6
- TNF-stimulated gene 6 protein
- GlcNAc
- N-acetylglucosamine
- GlcUA
- glucuronic acid
- IL
- interleukin
- CMV
- cytomegalovirus
- IFU
- infectious units
- sGAG
- sulfated glycosaminoglycan
- HAo
- HA oligosaccharides
- TLR
- Toll-like receptor
- DAMP
- damage-associated molecular pattern
- DMEM
- Dulbecco's modified Eagle's medium
- qRT-PCR
- real-time quantitative RT-PCR
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- FCCP
- carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone.
References
- 1. Rizkalla G., Reiner A., Bogoch E., and Poole A. R. (1992) Studies of the articular cartilage proteoglycan aggrecan in health and osteoarthritis: evidence for molecular heterogeneity and extensive molecular changes in disease. J. Clin. Invest. 90, 2268–2277 10.1172/JCI116113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hardingham T. E., and Fosang A. J. (1992) Proteoglycans: many forms and many functions. FASEB J. 6, 861–870 10.1096/fasebj.6.3.1740236 [DOI] [PubMed] [Google Scholar]
- 3. Knudson C. B., and Knudson W. (2001) Cartilage proteoglycans. Semin. Cell Dev. Biol. 12, 69–78 10.1006/scdb.2000.0243 [DOI] [PubMed] [Google Scholar]
- 4. Danielson B. T., Knudson C. B., and Knudson W. (2015) Extracellular processing of the cartilage proteoglycan aggregate and its effect on CD44-mediated internalization of hyaluronan. J. Biol. Chem. 290, 9555–9570 10.1074/jbc.M115.643171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Holmes M. W., Bayliss M. T., and Muir H. (1988) Hyaluronic acid in human articular cartilage: age-related changes in content and size. Biochem. J. 250, 435–441 10.1042/bj2500435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roughley P. J., and Mort J. S. (2014) The role of aggrecan in normal and osteoarthritic cartilage. J. Exp. Orthop. 1, 8 10.1186/s40634-014-0008-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Knudson W., Ishizuka S., Terabe K., Askew E. B., and Knudson C. B. (2019) The pericellular hyaluronan of articular chondrocytes. Matrix Biol. 78–79, 32–46 10.1016/j.matbio.2018.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nishida Y., D'Souza A. L., Thonar E. J., and Knudson W. (2000) Stimulation of hyaluronan metabolism by interleukin-1α in human articular cartilage. Arthritis Rheum. 43, 1315–1326 [DOI] [PubMed] [Google Scholar]
- 9. Morales T. I., and Hascall V. C. (1988) Correlated metabolism of proteoglycans and hyaluronic acid in bovine cartilage organ cultures. J. Biol. Chem. 263, 3632–3638 [PubMed] [Google Scholar]
- 10. Ng C. K., Handley C. J., Preston B. N., and Robinson H. C. (1992) The extracellular processing and catabolism of hyaluronan in cultured adult articular cartilage explants. Arch. Biochem. Biophys. 298, 70–79 10.1016/0003-9861(92)90095-E [DOI] [PubMed] [Google Scholar]
- 11. Durigova M., Roughley P. J., and Mort J. S. (2008) Mechanism of proteoglycan aggregate degradation in cartilage stimulated with oncostatin M. Osteoarthr. Cartil. 16, 98–104 10.1016/j.joca.2007.05.002 [DOI] [PubMed] [Google Scholar]
- 12. Durigova M., Troeberg L., Nagase H., Roughley P. J., and Mort J. S. (2011) Involvement of ADAMTS5 and hyaluronidase in aggrecan degradation and release from OSM-stimulated cartilage. Eur. Cell Mater. 21, 31–45 10.22203/eCM.v021a03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fosang A. J., Tyler J. A., and Hardingham T. E. (1991) Effect of interleukin-1 and insulin like growth factor-1 on the release of proteoglycan components and hyaluronan from pig articular cartilage in explant culture. Matrix 11, 17–24 10.1016/S0934-8832(11)80223-4 [DOI] [PubMed] [Google Scholar]
- 14. Sztrolovics R., Recklies A. D., Roughley P. J., and Mort J. S. (2002) Hyaluronate degradation as an alternative mechanism for proteoglycan release from cartilage during interleukin-1β-stimulated catabolism. Biochem. J. 362, 473–479 10.1042/0264-6021:3620473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nishida Y., Knudson C. B., Nietfeld J. J., Margulis A., and Knudson W. (1999) Antisense inhibition of hyaluronan synthase-2 in human articular chondrocytes inhibits proteoglycan retention and matrix assembly. J. Biol. Chem. 274, 21893–21899 10.1074/jbc.274.31.21893 [DOI] [PubMed] [Google Scholar]
- 16. Matsumoto K., Li Y., Jakuba C., Sugiyama Y., Sayo T., Okuno M., Dealy C. N., Toole B. P., Takeda J., Yamaguchi Y., and Kosher R. A. (2009) Conditional inactivation of Has2 reveals a crucial role for hyaluronan in skeletal growth, patterning, chondrocyte maturation and joint formation in the developing limb. Development 136, 2825–2835 10.1242/dev.038505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang Y., Askew E. B., Knudson C. B., and Knudson W. (2016) CRISPR/Cas9 knockout of HAS2 in rat chondrosarcoma chondrocytes demonstrates the requirement of hyaluronan for aggrecan retention. Matrix Biol. 56, 74–94 10.1016/j.matbio.2016.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Knudson W., Casey B., Nishida Y., Eger W., Kuettner K. E., and Knudson C. B. (2000) Hyaluronan oligosaccharides perturb cartilage matrix homeostasis and induce chondrogenic chondrolysis. Arthritis Rheum. 43, 1165–1174 [DOI] [PubMed] [Google Scholar]
- 19. Schmitz I., Ariyoshi W., Takahashi N., Knudson C. B., and Knudson W. (2010) Hyaluronan oligosaccharide treatment of chondrocytes stimulates expression of both HAS-2 and MMP-3, but by different signaling pathways. Osteoarthr. Cartil. 18, 447–454 10.1016/j.joca.2009.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ohno S., Im H. J., Knudson C. B., and Knudson W. (2006) Hyaluronan oligosaccharides induce matrix metalloproteinase 13 via transcriptional activation of NFκB and p38 MAP kinase in articular chondrocytes. J. Biol. Chem. 281, 17952–17960 10.1074/jbc.M602750200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ishizuka S., Askew E. B., Ishizuka N., Knudson C. B., and Knudson W. (2016) 4-Methylumbelliferone diminishes catabolically activated articular chondrocytes and cartilage explants via a mechanism independent of hyaluronan inhibition. J. Biol. Chem. 291, 12087–12104 10.1074/jbc.M115.709683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ariyoshi W., Takahashi N., Hida D., Knudson C. B., and Knudson W. (2012) Mechanisms involved in enhancement of the expression and function of aggrecanases by hyaluronan oligosaccharides. Arthritis Rheum. 64, 187–197 10.1002/art.33329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chou C. H., Attarian D. E., Wisniewski H. G., Band P. A., and Kraus V. B. (2018) TSG-6: a double-edged sword for osteoarthritis (OA). Osteoarthr. Cartil. 26, 245–254 10.1016/j.joca.2017.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iacob S., and Knudson C. B. (2006) Hyaluronan fragments activate nitric oxide synthase and the production of nitric oxide by articular chondrocytes. Int. J. Biochem. Cell Biol. 38, 123–133 10.1016/j.biocel.2005.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goldberg V. M., and Buckwalter J. A. (2005) Hyaluronans in the treatment of osteoarthritis of the knee: evidence for disease-modifying activity. Osteoarthr. Cartil. 13, 216–224 10.1016/j.joca.2004.11.010 [DOI] [PubMed] [Google Scholar]
- 26. Strand V., McIntyre L. F., Beach W. R., Miller L. E., and Block J. E. (2015) Safety and efficacy of US-approved viscosupplements for knee osteoarthritis: a systematic review and meta-analysis of randomized, saline-controlled trials. J. Pain Res. 8, 217–228 10.2147/JPR.S83076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karousou E., Kamiryo M., Skandalis S. S., Ruusala A., Asteriou T., Passi A., Yamashita H., Hellman U., Heldin C. H., and Heldin P. (2010) The activity of hyaluronan synthase 2 is regulated by dimerization and ubiquitination. J. Biol. Chem. 285, 23647–23654 10.1074/jbc.M110.127050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Terabe K., Takahashi N., Cobb M., Askew E. B., Knudson C. B., and Knudson W. (2019) Simvastatin promotes restoration of chondrocyte morphology and phenotype. Arch. Biochem. Biophys. 665, 1–11 10.1016/j.abb.2019.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vigetti D., Rizzi M., Viola M., Karousou E., Genasetti A., Clerici M., Bartolini B., Hascall V. C., De Luca G., and Passi A. (2009) The effects of 4-methylumbelliferone on hyaluronan synthesis, MMP2 activity, proliferation, and motility of human aortic smooth muscle cells. Glycobiology 19, 537–546 10.1093/glycob/cwp022 [DOI] [PubMed] [Google Scholar]
- 30. Kultti A., Pasonen-Seppänen S., Jauhiainen M., Rilla K. J., Kärnä R., Pyöriä E., Tammi R. H., and Tammi M. I. (2009) 4-Methylumbelliferone inhibits hyaluronan synthesis by depletion of cellular UDP-glucuronic acid and downregulation of hyaluronan synthase 2 and 3. Exp. Cell Res. 315, 1914–1923 10.1016/j.yexcr.2009.03.002 [DOI] [PubMed] [Google Scholar]
- 31. Kakizaki I., Kojima K., Takagaki K., Endo M., Kannagi R., Ito M., Maruo Y., Sato H., Yasuda T., Mita S., Kimata K., and Itano N. (2004) A novel mechanism for the inhibition of hyaluronan biosynthesis by 4-methylumbelliferone. J. Biol. Chem. 279, 33281–33289 10.1074/jbc.M405918200 [DOI] [PubMed] [Google Scholar]
- 32. Embry Flory J. J., Fosang A. J., and Knudson W. (2006) The accumulation of intracellular ITEGE and DIPEN neoepitopes in bovine articular chondrocytes is mediated by CD44 internalization of hyaluronan. Arthritis Rheum. 54, 443–454 10.1002/art.21623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ariyoshi W., Knudson C. B., Luo N., Fosang A. J., and Knudson W. (2010) Internalization of aggrecan G1 domain neoepitope ITEGE in chondrocytes requires CD44. J. Biol. Chem. 285, 36216–36224 10.1074/jbc.M110.129270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Campo G. M., Avenoso A., Nastasi G., Micali A., Prestipino V., Vaccaro M., D'Ascola A., Calatroni A., and Campo S. (2011) Hyaluronan reduces inflammation in experimental arthritis by modulating TLR-2 and TLR-4 cartilage expression. Biochim. Biophys. Acta 1812, 1170–1181 10.1016/j.bbadis.2011.06.006 [DOI] [PubMed] [Google Scholar]
- 35. Ruppert S. M., Hawn T. R., Arrigoni A., Wight T. N., and Bollyky P. L. (2014) Tissue integrity signals communicated by high-molecular weight hyaluronan and the resolution of inflammation. Immunol. Res. 58, 186–192 10.1007/s12026-014-8495-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Furuta J., Ariyoshi W., Okinaga T., Takeuchi J., Mitsugi S., Tominaga K., and Nishihara T. (2017) High molecular weight hyaluronic acid regulates MMP13 expression in chondrocytes via DUSP10/MKP5. J. Orthop. Res. 35, 331–339 10.1002/jor.23266 [DOI] [PubMed] [Google Scholar]
- 37. Sandy J. D., Chan D. D., Trevino R. L., Wimmer M. A., and Plaas A. (2015) Human genome-wide expression analysis reorients the study of inflammatory mediators and biomechanics in osteoarthritis. Osteoarthr. Cartil. 23, 1939–1945 10.1016/j.joca.2015.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goldring M. B., and Berenbaum F. (2015) Emerging targets in osteoarthritis therapy. Curr. Opin. Pharmacol. 22, 51–63 10.1016/j.coph.2015.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mobasheri A., Rayman M. P., Gualillo O., Sellam J., van der Kraan P., and Fearon U. (2017) The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 13, 302–311 10.1038/nrrheum.2017.50 [DOI] [PubMed] [Google Scholar]
- 40. Orlowsky E. W., and Kraus V. B. (2015) The role of innate immunity in osteoarthritis: when our first line of defense goes on the offensive. J. Rheumatol. 42, 363–371 10.3899/jrheum.140382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu-Bryan R., and Terkeltaub R. (2015) Emerging regulators of the inflammatory process in osteoarthritis. Nat. Rev. Rheumatol. 11, 35–44 10.1038/nrrheum.2014.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Campo G. M., Avenoso A., Campo S., D'Ascola A., Nastasi G., and Calatroni A. (2010) Small hyaluronan oligosaccharides induce inflammation by engaging both toll-like-4 and CD44 receptors in human chondrocytes. Biochem. Pharmacol. 80, 480–490 10.1016/j.bcp.2010.04.024 [DOI] [PubMed] [Google Scholar]
- 43. Liu-Bryan R., and Terkeltaub R. (2010) Chondrocyte innate immune myeloid differentiation factor 88-dependent signaling drives procatabolic effects of the endogenous Toll-like receptor 2/Toll-like receptor 4 ligands low molecular weight hyaluronan and high mobility group box chromosomal protein 1 in mice. Arthritis Rheum. 62, 2004–2012 10.1002/art.27475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Campo G. M., Avenoso A., Campo S., D'Ascola A., Nastasi G., and Calatroni A. (2010) Molecular size hyaluronan differently modulates toll-like receptor-4 in LPS-induced inflammation in mouse chondrocytes. Biochimie 92, 204–215 10.1016/j.biochi.2009.10.006 [DOI] [PubMed] [Google Scholar]
- 45. Muto J., Yamasaki K., Taylor K. R., and Gallo R. L. (2009) Engagement of CD44 by hyaluronan suppresses TLR4 signaling and the septic response to LPS. Mol. Immunol. 47, 449–456 10.1016/j.molimm.2009.08.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hirabara S., Kojima T., Takahashi N., Hanabayashi M., and Ishiguro N. (2013) Hyaluronan inhibits TLR-4 dependent cathepsin K and matrix metalloproteinase 1 expression in human fibroblasts. Biochem. Biophys. Res. Commun. 430, 519–522 10.1016/j.bbrc.2012.12.003 [DOI] [PubMed] [Google Scholar]
- 47. Knudson C. B., and Knudson W. (1993) Hyaluronan-binding proteins in development, tissue homeostasis and disease. FASEB J. 7, 1233–1241 10.1096/fasebj.7.13.7691670 [DOI] [PubMed] [Google Scholar]
- 48. Vigetti D., Genasetti A., Karousou E., Viola M., Clerici M., Bartolini B., Moretto P., De Luca G., Hascall V. C., and Passi A. (2009) Modulation of hyaluronan synthase activity in cellular membrane fractions. J. Biol. Chem. 284, 30684–30694 10.1074/jbc.M109.040386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hascall V. C., Wang A., Tammi M., Oikari S., Tammi R., Passi A., Vigetti D., Hanson R. W., and Hart G. W. (2014) The dynamic metabolism of hyaluronan regulates the cytosolic concentration of UDP-GlcNAc. Matrix Biol. 35, 14–17 10.1016/j.matbio.2014.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chanmee T., Ontong P., Izumikawa T., Higashide M., Mochizuki N., Chokchaitaweesuk C., Khansai M., Nakajima K., Kakizaki I., Kongtawelert P., Taniguchi N., and Itano N. (2016) Hyaluronan production regulates metabolic and cancer stem-like properties of breast cancer cells via hexosamine biosynthetic pathway-coupled HIF-1 signaling. J. Biol. Chem. 291, 24105–24120 10.1074/jbc.M116.751263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chanmee T., Ontong P., Mochizuki N., Kongtawelert P., Konno K., and Itano N. (2014) Excessive hyaluronan production promotes acquisition of cancer stem cell signatures through the coordinated regulation of Twist and the transforming growth factor β (TGF-β)-Snail signaling axis. J. Biol. Chem. 289, 26038–26056 10.1074/jbc.M114.564120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zoltan-Jones A., Huang L., Ghatak S., and Toole B. P. (2003) Elevated hyaluronan production induces mesenchymal and transformed properties in epithelial cells. J. Biol. Chem. 278, 45801–45810 10.1074/jbc.M308168200 [DOI] [PubMed] [Google Scholar]
- 53. Knudson C. B. (1993) Hyaluronan receptor-directed assembly of chondrocyte pericellular matrix. J. Cell Biol. 120, 825–834 10.1083/jcb.120.3.825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Farndale R. W., Sayers C. A., and Barrett A. J. (1982) A direct spectrophotometric microassay for sulfated glycosaminoglycans in cartilage cultures. Connect. Tissue Res. 9, 247–248 10.3109/03008208209160269 [DOI] [PubMed] [Google Scholar]
- 55. Simpson R. M., Meran S., Thomas D., Stephens P., Bowen T., Steadman R., and Phillips A. (2009) Age-related changes in pericellular hyaluronan organization leads to impaired dermal fibroblast to myofibroblast differentiation. Am. J. Pathol. 175, 1915–1928 10.2353/ajpath.2009.090045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jiang H., Peterson R. S., Wang W., Bartnik E., Knudson C. B., and Knudson W. (2002) A requirement for the CD44 cytoplasmic domain for hyaluronan binding, pericellular matrix assembly and receptor mediated endocytosis in COS-7 cells. J. Biol. Chem. 277, 10531–10538 10.1074/jbc.M108654200 [DOI] [PubMed] [Google Scholar]
- 57. Thankamony S. P., and Knudson W. (2006) Acylation of CD44 and its association with lipid rafts are required for receptor and hyaluronan endocytosis. J. Biol. Chem. 281, 34601–34609 10.1074/jbc.M601530200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mellor L., Knudson C. B., Hida D., Askew E. B., and Knudson W. (2013) Intracellular domain fragment of CD44 alters CD44 function in chondrocytes. J. Biol. Chem. 288, 25838–25850 10.1074/jbc.M113.494872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Indrawattana N., Chen G., Tadokoro M., Shann L. H., Ohgushi H., Tateishi T., Tanaka J., and Bunyaratvej A. (2004) Growth factor combination for chondrogenic induction from human mesenchymal stem cell. Biochem. Biophys. Res. Commun. 320, 914–919 10.1016/j.bbrc.2004.06.029 [DOI] [PubMed] [Google Scholar]
- 60. Mehlhorn A. T., Niemeyer P., Kaiser S., Finkenzeller G., Stark G. B., Südkamp N. P., and Schmal H. (2006) Differential expression pattern of extracellular matrix molecules during chondrogenesis of mesenchymal stem cells from bone marrow and adipose tissue. Tissue Eng. 12, 2853–2862 10.1089/ten.2006.12.2853 [DOI] [PubMed] [Google Scholar]
- 61. Rasmussen T. B., Uttenthal A., de Stricker K., Belák S., and Storgaard T. (2003) Development of a novel quantitative real-time RT-PCR assay for the simultaneous detection of all serotypes of foot-and-mouth disease virus. Arch. Virol. 148, 2005–2021 10.1007/s00705-003-0145-2 [DOI] [PubMed] [Google Scholar]
- 62. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45 10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]