

Abstract
Hydrogen sulfide (H2S) participates in prokaryotic metabolism and is associated with several physiological functions in mammals. H2S reacts with oxidized thiol derivatives (i.e. disulfides and sulfenic acids) and thereby forms persulfides, which are plausible transducers of the H2S-mediated signaling effects. The one-cysteine peroxiredoxin alkyl hydroperoxide reductase E from Mycobacterium tuberculosis (MtAhpE–SH) reacts fast with hydroperoxides, forming a stable sulfenic acid (MtAhpE–SOH), which we chose here as a model to study the interactions between H2S and peroxiredoxins (Prx). MtAhpE–SOH reacted with H2S, forming a persulfide (MtAhpE–SSH) detectable by mass spectrometry. The rate constant for this reaction was (1.4 ± 0.2) × 103 m−1 s−1 (pH 7.4, 25 °C), six times higher than that reported for the reaction with the main low-molecular-weight thiol in M. tuberculosis, mycothiol. H2S was able to complete the catalytic cycle of MtAhpE and, according to kinetic considerations, it could represent an alternative substrate in M. tuberculosis. MtAhpE–SSH reacted 43 times faster than did MtAhpE–SH with the unspecific electrophile 4,4′-dithiodipyridine, a disulfide that exhibits no preferential reactivity with peroxidatic cysteines, but MtAhpE–SSH was less reactive toward specific Prx substrates such as hydrogen peroxide and peroxynitrite. According to molecular dynamics simulations, this loss of specific reactivity could be explained by alterations in the MtAhpE active site. MtAhpE–SSH could transfer its sulfane sulfur to a low-molecular-weight thiol, a process likely facilitated by the low pKa of the leaving thiol MtAhpE–SH, highlighting the possibility that Prx participates in transpersulfidation. The findings of our study contribute to the understanding of persulfide formation and reactivity.
Keywords: hydrogen sulfide, Mycobacterium tuberculosis, peroxiredoxin, antioxidant, enzyme kinetics, alkyl hydroperoxide reductase E, hydrodisulfide, persulfide, signaling compound, sulfenic acid
Introduction
Hydrogen sulfide (H2S)5,6 has been related to the origin and evolution of life on our planet, and several organisms can produce or utilize H2S in various metabolic processes. In mammals, beyond its toxicological relevance, H2S has been associated with a variety of physiological functions, including vasodilation, neuromodulation and immunoregulation (1–3).
The pathogen Mycobacterium tuberculosis is the causative agent of tuberculosis disease. M. tuberculosis proliferates inside the phagosomes of activated macrophages, its main host cells, where it is exposed to oxidants, including hydrogen peroxide (H2O2), organic hydroperoxides (ROOH) and peroxynitrite (ONOO−/ONOOH) (4–7). Its antioxidant defense battery includes several enzymes as well as mycothiol, the main low-molecular-weight thiol in the bacterium, with functions analogous to those of GSH. Supplementation with H2S was shown to complement the growth defects of M. tuberculosis strains with impaired ability to recycle mycothiol, either in cellular or animal models of disease (8). H2S produced by host cells could potentially reach the interior of M. tuberculosis, because it can easily cross membranes (9). Furthermore, M. tuberculosis produces H2S by different enzymatic mechanisms (10–13).
Among the possible reactions of H2S, those with oxidized thiol derivatives have received attention as sources of persulfides (RSSH/RSS−).7 Indeed, hydrosulfide (HS−, the conjugate base in equilibria with H2S, pKa = 6.9 (14)), can react with sulfenic acids (RSOH) and disulfides (RSSR) to produce persulfides, also referred to as hydropersulfides, hydrodisulfides or disulfanes (Equations 1 and 2) (15).
| (Eq. 1) |
| (Eq. 2) |
Persulfides are important intermediates in sulfur metabolism in bacteria, where they are produced in enzymatic catalytic cycles (16, 17). Several enzymes, some also present in mammals, produce or transfer these functional groups; these include cystathionine γ-lyase, cystathionine β-synthase (18), mercaptopyruvate sulfurtransferase (19), sulfide:quinone oxidoreductase (20, 21) and thiosulfate sulfurtransferases (22, 23). Other enzymes are able to react with persulfides; a dioxygenase can use GSH persulfide as substrate and is encoded by the ethe1 gene, which is mutated in ethylmalonic encephalopathy, a severe infantile metabolic disorder (24). Recently proposed as intermediates in the transduction of the signaling effects observed after the administration of H2S (25, 26), persulfides have been generating increased interest. According to the hypothesis of persulfide-mediated signaling, the formation of a persulfide in certain cysteines could unleash changes in the activity of effector proteins, like the inhibition of papain, a cysteine-dependent protease (27), PTEN, a lipid phosphatase (28), and aquaporin-8, a membrane channel (29), among others. Possible roles in regulation and catalysis are still being explored, and the reactivity and physicochemical features of these species are poorly understood. Lately, some molecular models have been proposed and analytical methods have been developed to study persulfides both in vivo and in vitro (15, 30–38). When thiols are modified to persulfides, nucleophilicity is maintained and probably increased due to two factors: (a) increased acidity with respect to thiols (39), which results in increased availability of the deprotonated, more nucleophilic form at neutral pH; and (b) the α effect, i.e. the enhanced reactivity of a nucleophilic atom when it is adjacent to an atom containing one or more unshared pairs of electrons (40). In addition, a new property is acquired: electrophilicity. The reduction and the oxidation of persulfides are also possible; either H2S and thiols or perthiosulfenic acids (RSSOH) and polysulfides are produced, respectively. The high reactivity of persulfides determines the instability of these compounds in aqueous solutions (35), limiting their study and highlighting the importance of developing suitable models.
Peroxiredoxins (Prxs) are a family of antioxidant enzymes that play crucial roles in redox signaling (41–43). These enzymes are thiol-dependent peroxidases with ping-pong kinetic mechanisms. The oxidizing substrate (H2O2, organic hydroperoxide or peroxynitrite) reacts with the thiolate at the peroxidatic cysteine in the reduced enzyme to form a sulfenic acid (44). The reactivities of the thiolates in peroxidatic cysteines of Prx with hydroperoxides are several orders of magnitude faster than those of typical low- or high-molecular-weight thiols. This can be explained by the decrease in the energy of activation of the reaction by an exquisite network of electrostatic and hydrogen-bonding interactions involving the functional groups of an arginine and a threonine among others (45–47). Besides, the environment of the peroxidatic cysteine lowers the pKa of the thiol by several units relative to free cysteine (48). Once oxidized, the sulfenic acid is then reduced back to thiol by the reducing substrate(s), either directly or after a resolution step that involves the formation of a disulfide bond with a second cysteine residue (resolving cysteine), depending on the Prx subfamily (44, 49). Often, a thioredoxin/thioredoxin reductase system reduces the disulfide bond to complete the catalytic cycle (44). The direct reduction of the sulfenic acid occurs in the so-called one-cysteine Prxs, such as alkyl hydroperoxide reductase E of M. tuberculosis (MtAhpE). This Prx catalyzes the reduction of several hydroperoxides, being most active with peroxynitrite and fatty acid hydroperoxides (50, 51). The sulfenic acid of MtAhpE (MtAhpE–SOH) is reduced by the glutaredoxin-like protein mycoredoxin-1, either directly or after formation of a mixed disulfide with mycothiol (52–54). H2S is another possible reducing substrate for MtAhpE–SOH; however, it is not clear how effective its contribution could be. Moreover, both the ability of the resulting persulfide (MtAhpE–SSH) to react with typical Prx substrates or, alternatively, the capacity to be transferred to acceptor thiols remain unexplored.
In this work, MtAhpE was chosen as a model for persulfidation studies because this one-cysteine Prx presents the advantage that its sulfenic acid is relatively stable (52). We focused on the kinetic characterization of the reaction between MtAhpE–SOH and H2S to form a persulfide. Kinetic methods were employed to assess the possibility that H2S could act as a reducing substrate of the sulfenic acid and the relative contribution with respect to the better characterized mycobacterial reducing systems (mycothiol and mycoredoxin-1) is discussed. To compare the reactivity of the persulfide in the peroxidatic cysteine to that of the thiol, we evaluated the kinetics with specific substrates and unspecific reactants of Prxs. Additionally, we performed computational simulations to analyze the structural basis of the effects observed. Furthermore, the possibility of Prx assistance in persulfidation reactions (transpersulfidation) was explored.
Results
Detection of the persulfide in MtAhpE
The formation of the persulfide from the reaction of H2S with MtAhpE–SOH was revealed by the detection of its alkylation product after treatment with iodoacetamide and by the reduction of this product to MtAhpE–SH with DTT (Fig. 1A). Cysteine modifications to MtAhpE–SSH and sulfinic acid (MtAhpE–SO2H) involve similar mass shifts. The use of an alkylating agent allows us to distinguish unequivocally the nature of the product, because only persulfides will be alkylated due to their high nucleophilicity in opposition to the poor one of sulfinic acids. Furthermore, alkylated persulfides are characteristically reduced by thiol-containing compounds like DTT (55). A species with a molecular mass of 19,408 Da, consistent with the S-carbamidomethyl derivative of the peroxidatic cysteine persulfide (MtAhpE–SS–CAM), was detected in equimolar mixtures of MtAhpE–SH (19,319 Da), H2S and H2O2 at different incubation times. The species was already present when iodoacetamide was added 30 s after mixing, and maximum yields were obtained after 2–5 min (Fig. 1B). MtAhpE–SSH was relatively stable, because it could still be detected after incubation times before alkylation of 15 and 30 min (Fig. S1), although the signal of the MtAhpE–SS–CAM derivative decreased while that corresponding to reduced MtAhpE (MtAhpE–S–CAM, 19,376 Da) increased. No alkylated MtAhpE polysulfide derivatives (i.e. MtAhpE–SSnS–CAM, with n ≥1) were detected under these experimental conditions. MtAhpE–SS–CAM, an unsymmetrical disulfide, was reduced by treatment with DTT to form the original thiol (19,319 Da) (Fig. 1C). Additionally, a peak corresponding to 19,352 Da was detected in all samples and was particularly evident in those where MtAhpE–SH was incubated with H2S and H2O2. Because this species remained after DTT addition, it most probably reflects the presence of protein over-oxidized to MtAhpE–SO2H.
Figure 1.

Detection of persulfide in MtAhpE. A, persulfide formation and derivatization with iodoacetamide. The species were represented in their protonated state for simplicity. B, deconvoluted mass spectra for the reaction mixtures of MtAhpE–SH (10 μm), H2O2 (10 μm) and Na2S (10 μm) in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C). After the indicated incubation times, samples were treated with iodoacetamide (40 mm) during 30 min and gel-filtered. C, mass spectra of MtAhpE–SH alone (black) or incubated with H2O2 and Na2S for 5 min and treated with iodoacetamide and gel-filtered as in A, before (red) or after treatment with DTT (2 mm) (blue).
Kinetics of the reaction of H2S with MtAhpE–SOH
As shown above, H2S is able to react with MtAhpE–SOH forming a persulfide. With the aim of evaluating the viability of this reaction among alternative reducing systems, the kinetics of the reaction was studied. Because direct measurements of concentration changes are not straightforward in this time scale, determinations were performed by two competition assays. The first one was a competition between H2S and 5-thio-2-nitrobenzoic acid (TNB) for MtAhpE–SOH (Fig. 2A). The incubation of MtAhpE–SOH with the colored thiol TNB under pseudo-first–order conditions in the absence of H2S (Fig. 2B, blue trace) reproduced previous observations and confirmed the second-order rate constant of the direct reaction between MtAhpE–SOH and TNB to be (2.2 ± 0.1) × 103 m−1 s−1 (50). The reaction yielded a mixed disulfide (MtAhpE–S–TNB) that slowly reacted with a second molecule of TNB regenerating the reduced enzyme, as reported previously (50). When MtAhpE–SOH was mixed with TNB in the presence of H2S, the observed exponential rate constants of TNB decay (kobs) increased linearly with the concentration of H2S while the amplitudes decreased (Fig. 2, B and C), as expected for competition kinetics (56). A second-order rate constant of (1.4 ± 0.2) × 103 m−1 s−1 for the reaction of H2S with MtAhpE–SOH (pH 7.4, 25 °C) was obtained from this competition assay. The MtAhpE–S–TNB product reacted with excess H2S and produced TNB, explaining the linear increases in absorbance after the exponential decays of TNB in Fig. 2B.
Figure 2.
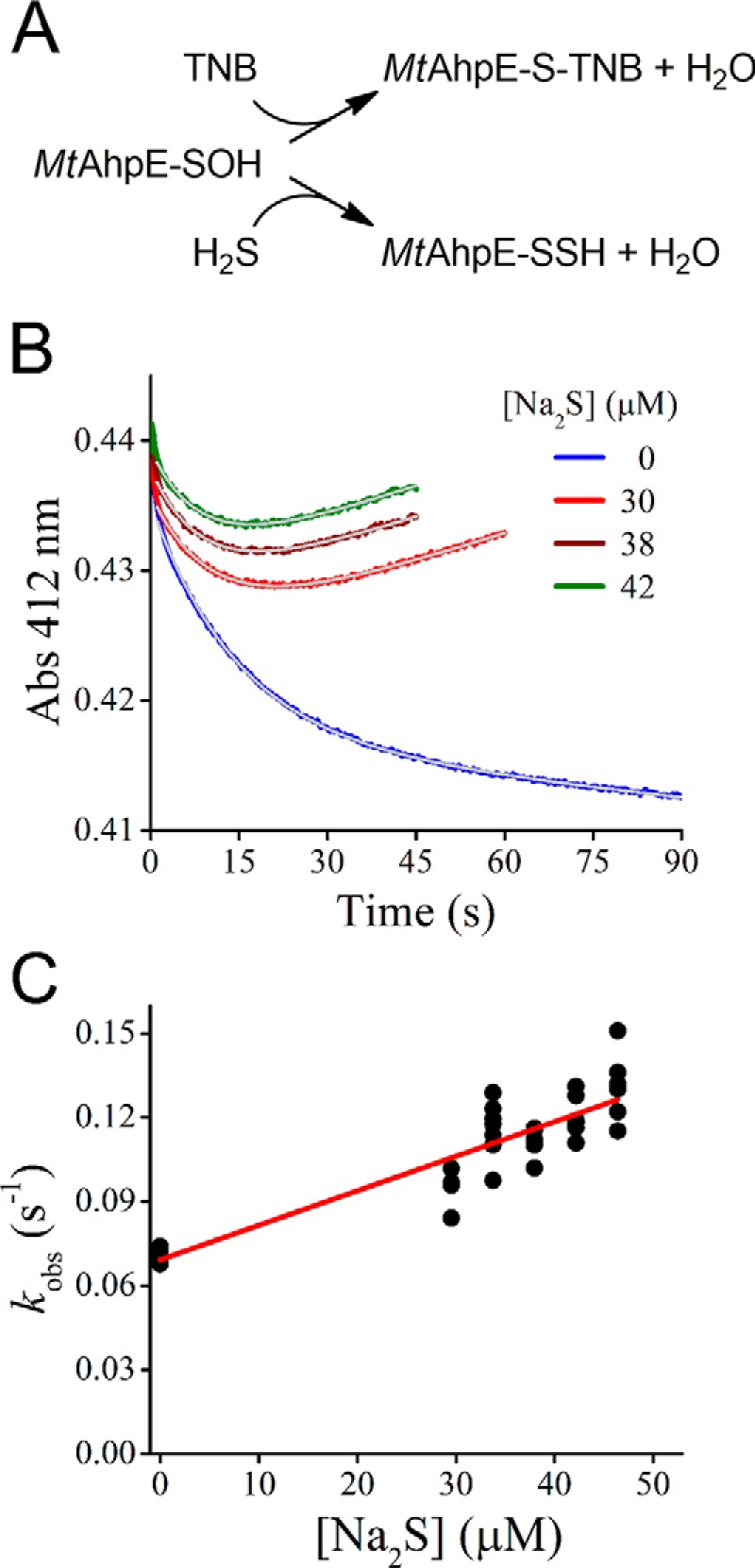
Competition assay of H2S and TNB toward MtAhpE–SOH. A, reactions of the competition assay with TNB. The species are represented in their protonated state for simplicity. B, time courses for the absorbance of TNB (30 μm) when mixed with MtAhpE–SOH (1.4 μm) and different concentrations of Na2S in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C). The gray trace represents the best fit to an exponential plus straight-line function (Abs412 nm = A·exp(−kobs·t) + b·t + c, where A is the amplitude of the exponential contribution; kobs is the observed exponential rate constant; b is the slope of the linear contribution; and c is the offset). The data used for the fits corresponded to 10 half-lives minus the first second. C, exponential rate constants obtained from the exponential phase of absorbance decay versus Na2S concentration. Representative results of an experiment performed three independent times are shown.
A second approach for determining the kinetics of the reaction of H2S with MtAhpE–SOH consisted of a competition assay with the over-oxidation reaction of MtAhpE–SOH to MtAhpE–SO2H following the changes in the protein intrinsic fluorescence emission that occur when the enzyme is exposed to excess H2O2 (50, 52). The fast oxidation of MtAhpE–SH produced a rapid decrease in its fluorescence emission due to the formation of the MtAhpE–SOH during the first s, which is consistent with the reported rate constant of 8.2 × 104 m−1 s−1 (50). A second phase of the reaction showed a slow recovery of the emission due to enzyme over-oxidation to MtAhpE–SO2H, as shown previously (50–52). The presence of H2S produced changes in the amplitude and in the observed rate constants of this second phase (Fig. 3). The global fitting of the fluorescence changes according to a simple competition model (Fig. 3A) yielded a rate constant of (1.0 ± 0.4) × 103 m−1 s−1 for the reaction of H2S with MtAhpE–SOH, which is similar to the value determined using TNB described above. Also, the rate constant of over-oxidation yielded by the global fitting was 60 m−1 s−1, in agreement with our previous report (50).
Figure 3.
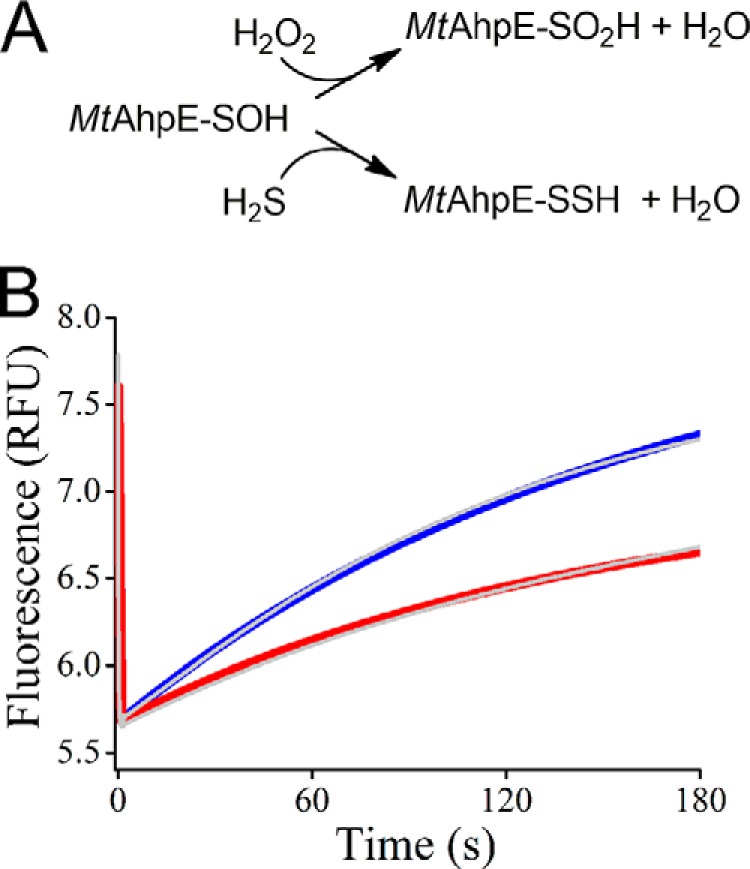
Competition assay of H2S and H2O2 toward MtAhpE–SOH. A, reactions of the competition assay with H2O2. The species are represented in their protonated state for simplicity. B, time courses of intrinsic fluorescence changes of MtAhpE–SH (1 μm) when mixed with H2O2 (100 μm) to produce MtAhpE–SOH and MtAhpE–SO2H in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C) (blue). The red trace shows the effect of Na2S (1 μm) in the reaction mixture. Data obtained from time course fits to determine the rate constant are shown as gray traces. Representative results of an experiment performed two independent times are shown.
Reactivity of MtAhpE–SSH with an unspecific target, the disulfide DTDPy
The relative stability of the persulfide derivative of this Prx facilitated the study of its reactivity. The kinetics of the reaction of the persulfide toward the electrophile DTDPy was evaluated. DTDPy is a synthetic disulfude used as a reporter, which allows the study of the reactivity of both thiol and persulfide and is not expected to establish specific interactions with the active site. A mixture of MtAhpE–SOH and H2S was aged for increasing time periods and mixed with DTDPy under pseudo-first–order conditions (Fig. 4). The 4-thiopyridone released by reaction of DTDPy with H2S and MtAhpE–SSH was followed at 324 nm, and the kinetics showed a biphasic profile. The fast phase, attributable to the reaction of MtAhpE–SSH with DTDPy, increased its amplitude during the first 5 min while MtAhpE–SSH was being slowly produced, as expected from the concentrations of MtAhpE–SOH and H2S, and the rate constant of (1.4 ± 0.2) × 103 m−1 s−1 reported in the previous section. After ∼400 s, the amplitude of the fast phase became constant, showing that all persulfide had been formed and remained stable during at least 30 min under these experimental conditions (Fig. 4B). From the amplitude of the fast phase, the amount of MtAhpE–SSH formed was calculated as 1.1 μm, representing 81% of total MtAhpE. The slow phase that decreased in amplitude with time was attributed to the reaction of H2S with DTDPy and had a rate constant of 545 ± 1 m−1 s−1 at pH 7.4 and 25 °C. Given that at ∼900 s the formation of MtAhpE–S–SH had reached a plateau while H2S had been depleted, experiments with varying concentrations of DTDPy were performed after this aging period. The observed rate constants for the fast phase increased linearly with DTDPy concentration (Fig. 4C). From the slope, the second-order rate constant for the reaction of MtAhpE–SSH with DTDPy was calculated to be (1.8 ± 0.1) × 103 m−1 s−1 at pH 7.4 and 25 °C. In comparison, the rate constant for the reaction of the thiol MtAhpE–SH with DTDPy was 42 ± 8 m−1 s−1 under the same conditions (Fig. 4C).
Figure 4.
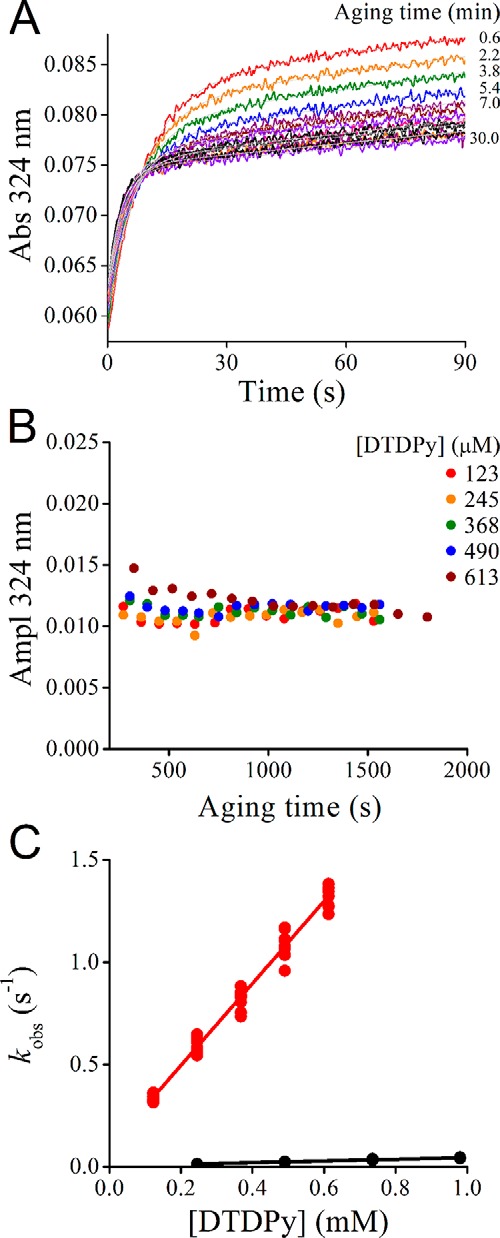
Reactivity of MtAhpE–SSH toward the electrophile DTDPy. A, persulfide was prepared by incubation of MtAhpE–SH (1.30 μm) with H2O2 (1.29 μm) in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C) during 3 min. Then, Na2S (1.29 μm) was added and mixed with DTDPy (123 μm) every 1.5 min in a stopped-flow instrument. The gray trace represents the best fit to an exponential plus straight-line function (Abs324 nm = A·exp(−kobs·t) + b·t + c, where A is the amplitude of the exponential contribution; kobs is the observed rate constant; b is the slope of the linear contribution; and c is the offset). B, amplitude of experiments performed as in A but with increasing concentrations of DTDPy. The variations in the amplitude of the fast phase reveal the timing for the formation of MtAhpE–SSH. C, after aging mixtures of MtAhpE–SH, H2O2 and Na2S for ∼900 s, samples were mixed with varying concentrations of DTDPy, and the observed rate constants of the fast phase were determined. From the linear correlation with the concentration of DTDPy, a second-order rate constant of (1.8 ± 0.1) × 103 m−1 s−1 was obtained for the persulfide (red circles). The observed rate constants for the reaction of MtAhpE–SH with DTDPy are shown in black circles (42 ± 8 m−1 s−1). The rate constant values are the mean and standard deviation of three independent experiments.
Reactivity of MtAhpE–SSH toward the specific Prx substrates, peroxynitrite and H2O2
Despite the increased intrinsic reactivity of the persulfide with respect to the thiol, the ability of MtAhpE–SSH to consume specific substrates of the enzyme, which implies additional factors, remained to be evaluated. The decay of peroxynitrite (12 μm) was highly accelerated in the presence of 15 μm reduced MtAhpE–SH (Fig. 5A, black trace) and occurred mostly in the mixing time of the apparatus (1.1 ms), in agreement with the fast reaction previously reported (1.7 × 107 m−1 s−1 at pH 7.4 and 25 °C (50)). On the contrary, both MtAhpE–SOH and MtAhpE–SSH caused only modest increases in peroxynitrite decay rate (Fig. 5A, blue and red traces, respectively). Considering ∼80% MtAhpE–SSH formation yield, as determined above (Fig. 4A), the rate constant of the reaction with peroxynitrite was estimated as ∼104 m−1 s−1 at pH 7.4 and 25 °C.
Figure 5.
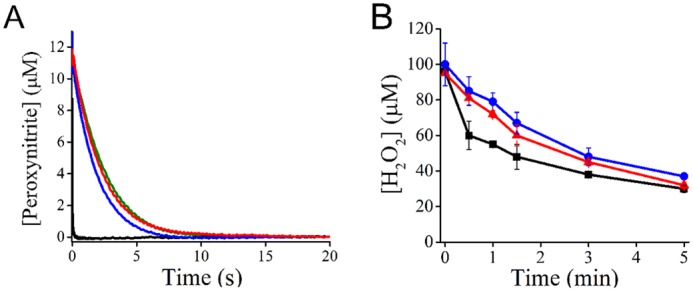
Reactivity of MtAhpE–SSH with peroxynitrite and H2O2. A, peroxynitrite (12 μm) decay in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C) was followed at 310 nm with no further addition (green trace) or in the presence of 15 μm MtAhpE–SH (black), MtAhpE–SOH (blue) or MtAhpE–SSH (red). B, H2O2 decay in 0.05 m phosphate buffer (pH 7.4, 25 °C) in the presence of 45 μm MtAhpE–SH (black squares), MtAhpE–SOH (blue circles) or MtAhpE–SSH (red triangles). H2O2 concentration was determined using the ferrous oxidation–xylenol orange method. Representative results of experiments performed two independent times are shown.
In the case of H2O2, its consumption by MtAhpE–SH (45 μm) was almost 1:1 during the first 30 s, as already described (51), and in agreement with the reported rate constant of 8.2 × 104 m−1 s−1 at pH 7.4 (Fig. 5B, black squares) (50). In the presence of MtAhpE–SOH (Fig. 5B, blue circles), the decay of H2O2 was slower, and from the initial rate of H2O2 decay a rate constant of 78 m−1 s−1 was estimated, which agrees with the low rate constant of the reaction between MtAhpE–SOH and H2O2 previously determined (40 m−1 s−1) (50). The rate of H2O2 consumption by MtAhpE–SSH (Fig. 5B, red triangles) was similar to that of MtAhpE–SOH. From the initial rate of H2O2 decay and considering persulfide formation yields of ∼80%, the rate constant of MtAhpE–SSH oxidation by H2O2 was estimated to be 109 m−1 s−1 at pH 7.4 and 25 °C.
Catalytic consumption of H2S and H2O2 by MtAhpE
To study whether H2S is able to complete the catalytic cycle of the enzyme, MtAhpE–SH was added to a solution containing H2S and H2O2, and aliquots were removed to determine H2S as well as H2O2 consumption during the incubation. Although H2O2 is able to consume H2S in the absence of catalysts, in agreement with the slow uncatalyzed reaction already reported (0.35 m−1 s−1 at pH 7.4 and 25 °C (57)), increased rates of H2S and H2O2 consumption were observed when substoichiometric amounts of the enzyme were included (Fig. 6, A and B). The initial rates of H2S decay showed a linear dependence on the concentration of MtAhpE–SH with a slope 2.2 × 10−2 s−1 at the assayed substrate concentrations, confirming the H2S-consuming activity in the presence of H2O2 (Fig. 6C).
Figure 6.
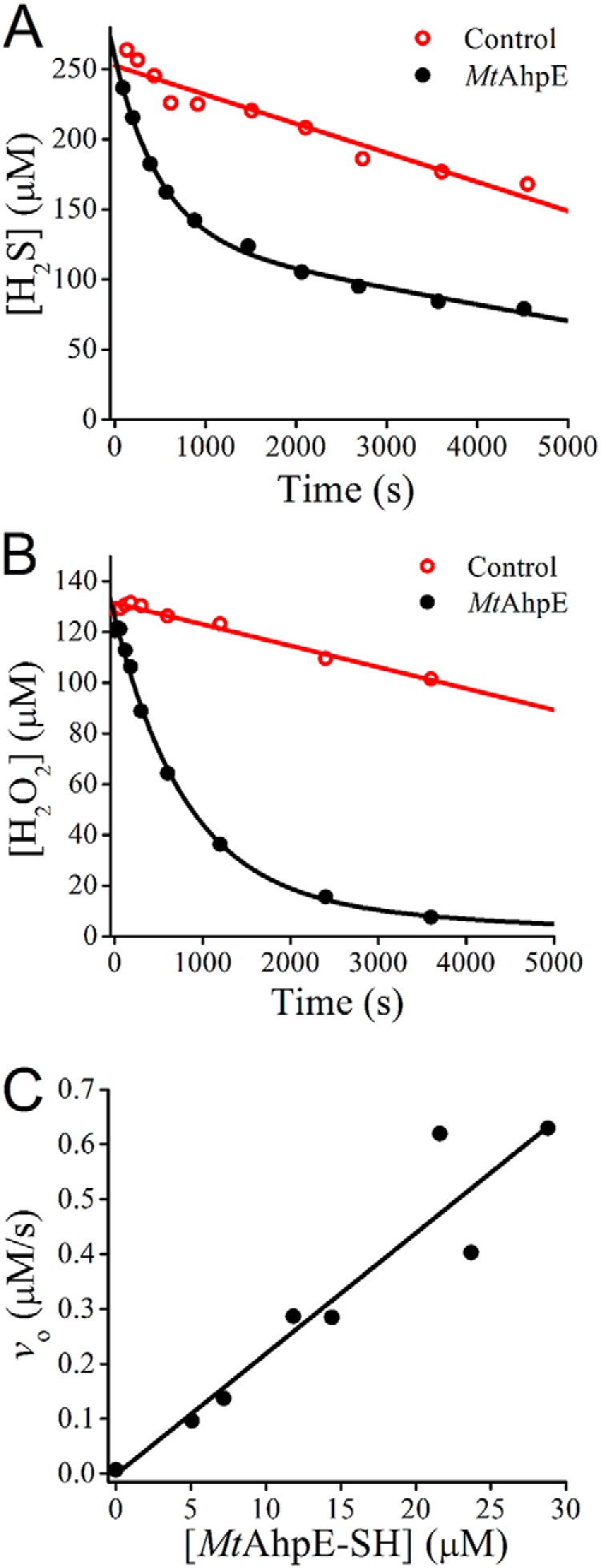
MtAhpE catalysis of the reaction between H2S and H2O2. A, mixtures of Na2S (250 μm) and H2O2 (125 μm) were incubated in the presence (black) or absence (red) of MtAhpE–SH (2.9 μm) in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C). The concentration of H2S was determined using the methylene blue method. B, same experiment as in A, but the mixtures were done in 0.05 m phosphate buffer (pH 7.4, 25 °C), and the H2O2 concentration was determined using the ferrous oxidation–xylenol orange method. C, initial rates of H2S consumption obtained at different concentrations of MtAhpE in solutions of Na2S (125 μm) and H2O2 (127 μm). Data from two independent experiments are shown.
Molecular dynamics of MtAhpE–SSH
Molecular dynamics (MD) simulations of the thiolate (MtAhpE–S−) and the persulfide forms (MtAhpE–SS−) of the peroxidatic cysteine (Cys45) were performed to evaluate the structural and dynamic effect of persulfidation. As shown in Fig. 7, A and C, upon 700 ns of MD simulations, neither global structural rearrangements nor big conformational changes were detected when comparing the thiolate and the persulfide forms on the MtAhpE dimer (Fig. 7C). However, when taking a closer look at the active sites, significant disruptions in the interactions of Cys45 with the active-site residues Thr42 and Arg116 were identified. These interactions were previously highlighted as essential for hydroperoxide recognition and reduction, not only for this particular Prx (45, 58) but also for the entire Prx family (46–48, 59, 60). Specifically, a gain in the Thr42 region's flexibility upon persulfidation was observed (Fig. 7D). Furthermore, neither Sγ nor Sδ atoms of the persulfide in Cys45 interacted strongly with Thr42 and Arg116 (Fig. 7, B and D), whereas strong interactions are established with the sulfur of the corresponding thiolate (45). In summary, MD simulations suggest that persulfidation of MtAhpE leads to a significant disruption in the topology of the active site that alters key interactions involved in catalysis.
Figure 7.

Structural changes upon persulfidation in MtAhpE's active site. A, structural alignment of representative snapshots of the MtAhpE dimer obtained from MD simulations with Cys45 as thiolate (black) or as persulfide (red). B, comparison of thiolate and persulfide in Cys45 in the active site of MtAhpE depicting residues Thr42, Cys45 and Arg116. C, distribution of α-helix and β-sheet content (%) for both MD simulations. D, comparison of root mean square fluctuations (Å) in a per-residue basis calculated from thiolate in Cys45 (black) and persulfide in Cys45 (red) MD simulations. E, distribution of relevant active-site interactions. Selected distances are the same as highlighted in B.
Transfer of the sulfane sulfur of MtAhpE–SSH to a thiol probe
With the aim of determining whether MtAhpE–SSH is able to transfer its sulfane sulfur to other thiols, MtAhpE–SSH was incubated with sulfane sulfur probe 4 (SSP4), a nonfluorescent thiol-containing compound used as a probe for sulfane sulfurs. The transfer of a sulfane sulfur to a thiol of the probe leads to the formation of a persulfide, which undergoes spontaneous cyclization to release a fluorophore (61, 62). Incubation of SSP4 with MtAhpE–SSH led to an increase in the fluorescence emission (Fig. 8). This increase occurred to a much larger extent than in controls with MtAhpE–SH or MtAhpE–SOH. According to calibration curves using Na2S2, the yield of the transsulfuration process was 95 ± 5%.
Figure 8.
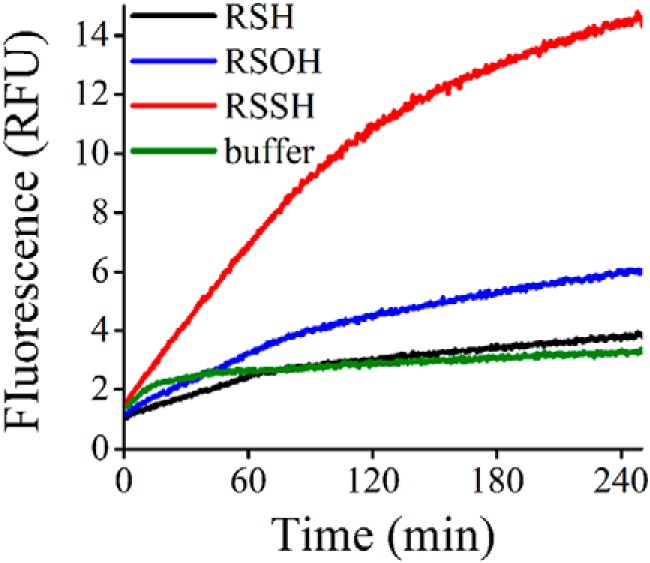
Transfer of the sulfane sulfur of MtAhpE–SSH to the thiol in the SSP4 probe. MtAhpE–SOH was prepared by incubation of MtAhpE–SH (4 μm) with H2O2 (4 μm) in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C) during 2 min. The persulfide was formed by adding Na2S (4 μm) to sulfenic acid preparations and aging during 15 min, time in which the reaction is expected to be completed. Then, samples were diluted 2-fold; SSP4 (20 μm) was added, and fluorescence emission was followed for MtAhpE–SH (black), MtAhpE–SOH (blue), MtAhpE–SSH (red) and buffer (green). Traces are the average of four runs from two independent experiments performed in duplicates.
Discussion
A plausible fate of H2S in cells is represented by its reactions with cysteine sulfenic acids (15, 36). The peroxidatic cysteines in Prxs constitute preferential targets for hydroperoxides due to the high reactivity and cellular abundance (63, 64). Their reaction constitutes a source of sulfenic acids, which are then reduced by several pathways. The feasibility of the reaction of H2S with a sulfenic acid is determined by kinetic aspects and is favored when the latter is long-lived. We particularly focused on MtAhpE–SH, which produces a relatively stable sulfenic acid due to the absence of thiols in the vicinity of the active site (52).
The sulfenic acid form of MtAhpE, MtAhpE–SOH, reacts with H2S to form a persulfide (MtAhpE–SSH). The rate constant for this reaction was determined to be (1.4 ± 0.2) × 103 m−1 s−1 at pH 7.4 and 25 °C. Considering that the reactive species are protonated MtAhpE–SOH and ionized HS− (65) and because the reported pKa values are 6.6 for MtAhpE–SOH (50) and 6.9 for H2S (14), the pH-independent rate constant can be calculated to be 1.4 × 104 m−1 s−1.8
The values obtained for MtAhpE–SOH can be compared with those obtained for the reaction of the sulfenic acid in human serum albumin (HSA–SOH) with H2S (15). The rate constant for HSA–SOH was (2.7 ± 0.8) × 102 m−1 s−1 at pH 7.4 and 25 °C, whereas the pH-independent rate constant was ∼4 × 102 m−1 s−1 (assuming that most HSA–SOH was protonated at pH 7.4). Thus, it can be concluded that MtAhpE–SOH is 30 times more reactive than HSA–SOH with HS−.
Substoichiometric concentrations of MtAhpE were able to consume H2S and H2O2 catalytically, suggesting that MtAhpE–SH can be regenerated and that the enzyme can initiate a new catalytic cycle. As precedent, it was proposed that bovine Prx6, another one-cysteine Prx from a different subfamily, is able to consume H2O2 using H2S as a reducing substrate via the formation of a persulfide in its peroxidatic cysteine (66). In contrast, in Prx6 from Arenicola marina, no evidence could be obtained for H2S participation in the catalytic cycle (67). In the case of MtAhpE, the catalysis in the presence of excess H2S and H2O2 could proceed by a variety of pathways. It surely starts with the fast oxidation of MtAhpE–SH by H2O2 (8.2 × 104 m−1 s−1, pH 7.4 (50), Equation 3) to produce MtAhpE–SOH, followed by the reaction with H2S to form MtAhpE–SSH ((1.4 ± 0.2) × 103 m−1 s−1, pH 7.4, Equation 4). Then, MtAhpE–SSH can react with H2S to recover the original thiol and produce HSSH and other polysulfide by-products (Equation 5). Alternatively, MtAhpE–SSH can react with H2O2 to form a perthiosulfenic acid (RSSOH) and other higher oxidation states (RSSO2H and RSSO3H), which can then be reduced to thiol or persulfide by H2S. The favored pathway is determined by kinetic aspects that remain to be elucidated. Nevertheless, the slope of the plot of rate versus enzyme concentration indicated a turnover of 2.2 × 10−2 s−1 at the used H2S and H2O2 concentrations (Fig. 6C). Assuming that the rate-limiting step is a second-order reaction, either the reaction of MtAhpE–SSH with H2S or with H2O2, and considering that the concentrations of H2S and H2O2 were 125 and 127 μm, respectively, it can be calculated that the rate constant for the rate-limiting second-order reaction is ∼170 m−1 s−1. Clearly, the reaction of MtAhpE–SOH with H2S, which has an 8-fold higher rate constant ((1.4 ± 0.2) × 103 m−1 s−1, pH 7.4), is not rate-limiting in the catalytic process. Besides, the rate of the reaction of MtAhpE–SSH with H2O2 (∼109 m−1 s−1) is below the expected rate-limiting step, leading to the reaction in Equation 5 as the most likely to participate in the catalytic cycling.
| (Eq. 3) |
| (Eq. 4) |
| (Eq. 5) |
In cellular contexts, the reaction of H2S with sulfenic acids could be of relevance in one-cysteine Prxs, where the resolving cysteine is absent and the sulfenic acid could be long-lived. It could also be relevant in eukaryotic two-cysteine Prx, particularly in those cases where the reaction of the resolving cysteine with the sulfenic acid to form a disulfide is relatively slow so that the sulfenic acid would have a significant half-life, and that is the case of eukaryotic typical two-cysteine Prxs in opposition to bacterial counterparts (68).
The second-order rate constant of the reaction of MtAhpE–SOH with H2S ((1.4 ± 0.2) × 103 m−1 s−1) is six times higher than that reported for the reaction of MtAhpE–SOH with mycothiol and is similar to that reported for mycoredoxin-1 (237 and 1.6 × 103 m−1 s−1, respectively (52)), which are the endogenous substrates in M. tuberculosis known up to date. The main fate of MtAhpE–SOH in cells is dictated not only by kinetic constants but also by the concentration of the targets. Although reports on the steady-state concentrations of mycoredoxin-1 in M. tuberculosis are still lacking, levels of both mycothiol (1–8 mm (69)) and H2S (∼370 μm (69, 70)) have been estimated. Thus, H2S could represent an effective substrate in M. tuberculosis, an alternative to the mycothiol and mycoredoxin-1. The mechanisms of regulation of mycothiol and H2S synthesis in the bacterium are only starting to be unraveled (71, 72); therefore, further work is required to establish their relative contribution for MtAhpE reduction during different metabolic conditions. Furthermore, the roles of H2S and mycothiol/mycoredoxin-1 as electron donors for AhpE and AhpE-like proteins expressed in other Actinomycetes (csb.wfu.edu/prex.test/prxInfo.php?subfamily=6) (94), which differ in mycothiol content (70) and can be exposed, depending on their habitat, to high H2S concentrations, deserve further investigation. Indeed, H2S supplementation was shown to complement the growth defect of bacterial strains with decreased ability to regenerate the reduced form of mycothiol (8). In addition, it was shown that Rv2238c, the gene encoding MtAhpE, is transcriptionally up-regulated in a cellular model of intraocular tuberculosis (73). Our study gives insights into possible mechanisms of cross-talk between the pathogen and its host at a junction between H2S signaling and the antioxidant defense systems.
The reactivity of MtAhpE–SSH toward an unspecific electrophilic target was probed using DTDPy. This synthetic disulfide was chosen because it has high intrinsic reactivity, because the reaction can be followed through the absorbance of 4-thiopyridone, because it can be used in pseudo-first–order excess so that the concentration of persulfide does not need to be exactly known, and because it has been used with HSA before (15). In addition, it constitutes an unspecific “substrate” for MtAhpE, which would allow us to interrogate thiol and persulfide reactivity in the absence of specificity aspects. Apparent second–order rate constants of (1.8 ± 0.1) × 103 m−1 s−1 and 42 ± 8 m−1 s−1 were obtained at pH 7.4 for MtAhpE–SSH and for MtAhpE–SH, respectively. The value obtained for the persulfide was 43 times higher than that for the thiol. Considering that the reactive species are ionized, because the pKa of MtAhpE–SH is 5.2 (50), and the pKa of MtAhpE–SSH is also likely to be much lower than 7.4 (39), the values obtained at pH 7.4 are likely to reflect pH-independent values. Thus, the increased reactivity of MtAhpE–SSH with respect to MtAhpE–SH cannot be ascribed to changes in availability of the ionized species. Rather, they can be ascribed to an increase in intrinsic reactivity. A previous publication (15) reported rate constants for the reaction of DTDPy with the persulfide (HSA–SSH) and the thiol (HSA–SH) in human serum albumin as (1.7 ± 0.1) × 104 m−1 s−1 and (7.6 ± 0.4) × 102 m−1 s−1, respectively, at pH 7.4 and 25 °C (Table 1). These values translate into pH-independent rate constants of ∼2 × 104 m−1 s−1 for HSA–SSH and 7 × 103 m−1 s−1 for HSA–SH, which has a pKa of 8.1 (74). Thus, the formation of a persulfide produced a 20-fold increase in the reactivity at pH 7.4 and just a 3-fold increase in pH-independent rate constants. The increase in intrinsic reactivity with DTDPy of the persulfide relative to the thiolate can be due to the α-effect, to changes in solvation, to alterations in weak interactions in the environment of the cysteine or to combinations of these effects.
Table 1.
Rate constants for thiols and persulfides
|
k (m−1 s−1) |
|||
|---|---|---|---|
| DTDPy | ONOOH | H2O2 | |
| MtAhpE–SH | 42 a,b | 1.9 × 107 a,b (50) | 8.2 × 104 a (50) |
| MtAhpE–SSH | 1.8 × 103 a,b | ∼104 a,b | ∼109 a,b |
| HSA–SH | 7.6 × 102 a (15) | 2.7 × 103 c (15) | 2.1 d (74) |
| HSA–SSH | 1.7 × 104 a (15) | 1.2 × 104 c (15) | ND e |
a Data were determined at 25 °C and pH 7.4.
b Data were determined in this work.
c Data were determined at 20 °C and pH 7.4.
d Data were determined at 37 °C and pH 7.4.
e ND means not determined.
Remarkably, the reactivity of MtAhpE–SH toward DTDPy at pH 7.4 was 1 order of magnitude lower than that of HSA–SH. In contrast, the reactivity toward hydroperoxides, the specific substrates of Prxs, is several orders of magnitude higher for MtAhpE–SH than for HSA–SH (Table 1). This is another example of the low reactivity of Prxs toward nonspecific compounds and contributes to the concept that there is no such thing as a general reactive cysteine (46, 75). The peroxidatic cysteine microenvironment in Prxs specifically accelerates the reaction with their hydroperoxide substrates.
In contrast to the increased reactivity toward the synthetic disulfide DTDPy of MtAhpE–SSH versus MtAhpE–SH, the reactivity of MtAhpE–SSH toward H2O2 and peroxynitrite was several orders of magnitude lower than that of MtAhpE–SH. The mild reactivity of MtAhpE–SSH with these specific Prx substrates appears to be an effect of geometrical distortion of the catalytic site, which seems to fit the requirements for the correct interaction of hydroperoxides with the peroxidatic cysteine in the thiolate but not the persulfide state, through hydrogen bonds with Arg116 and Thr42 during both the formation of the substrate complex and the transition state (45). In addition to changing the reactivity of this site due to shifts in distances, the formation of a persulfide could also change the value of the pKa. Although the acidity appears to be lower in low-molecular-weight persulfides with respect to their analogous thiols (39), it is not easy to predict the persulfide pKa in the case of a Prx because of the special environment of the active site. Furthermore, persulfides are expected to improve the reactivity as soft bases (15), which make them more likely to react with disulfides than with hard peroxides.
Once formed in a Prx, what fate could the persulfide have? Although further reaction with H2S or H2O2 can occur in vitro, in vivo it is likely that reactions with thiols predominate, considering the high cellular concentrations of low- and high-molecular-weight thiols. In fact, proteins of the thioredoxin and glutaredoxin families have been shown to react with persulfidated proteins (36, 37). The possibility of direct attack of a protein thiolate in the outer sulfur of a Prx persulfide would be promoted by the relatively high acidity of the leaving group thiol. The result would be the formation of a persulfide in the attacking protein. This would constitute a mechanism for transpersulfidation that could contribute to the relatively high levels of persulfidation that have been detected (36–38). As proof of concept, MtAhpE–SSH was able to transfer the persulfide to a low-molecular-weight thiol in high yield (Fig. 8). Thus, the reaction of H2S with Prx sulfenic acids shown in this study opens up the possibility of Prx participation in the persulfidation of proteins.
Experimental procedures
Chemicals
Sodium sulfide (Na2S·9H2O) was obtained from either J. T. Baker or Carlo Erba. H2O2 was obtained from Mallinckrodt Chemicals. 5,5′-Dithiobis-(2-nitrobenzoic acid) (DTNB), 1,4-dithiothreitol (DTT), iodoacetamide and diethylenetriaminepentaacetic acid (DTPA) were purchased from Sigma. 4,4′-Dithiodipyridine (DTDPy) was purchased from Acros Organics. SSP4 and Na2S2 were obtained from Dojindo Molecular Technologies.
Preparation of reagents
H2S solutions were prepared by dissolving Na2S·9H2O in extensively degassed distilled water plus 0.1 mm DTPA in sealed vials and used immediately. H2O2 was prepared by dilution of stock solutions in ultrapure water and quantified by measuring absorbance at 240 nm (ϵ240 = 39.4 m−1 cm−1) (76). Peroxynitrite was synthesized from H2O2 and nitrous acid as described previously and treated with granular manganese dioxide to eliminate residual H2O2 (77). Nitrite contamination was typically <30% of peroxynitrite concentration. Peroxynitrite concentrations were determined spectrophotometrically at 302 nm (1705 m−1 cm−1 (78)). Solutions of Na2S2 were prepared in ultrapure water immediately before use. Solutions of 5-thio-2-nitrobenzoic acid (TNB) were prepared as described previously (79). DTDPy stock solutions were prepared in 95% ethanol. SSP4 was diluted in DMSO.
Protein expression and purification
MtAhpE (encoded by the gene Rv2238c, https://mycobrowser.epfl.ch) (95) was expressed in Escherichia coli BL21(DE3) (expression vector pDEST17) as a recombinant His-tagged protein and purified as described previously (80). The concentration of the protein subunits of this homodimeric enzyme was determined spectrophotometrically at 280 nm, ϵ280 = 23,950 m−1 cm−1 calculated according to protein sequence using the ProtParam tool in ExPASy, http://web.expasy.org/protparam (81).
Protein thiol reduction and quantitation
MtAhpE was reduced immediately before use by incubation with 2 mm DTT for 30 min at 4 °C. Excess reductant was removed by gel filtration using a HiTrap desalting column (Amersham Biosciences) and UV detection at 280 nm. Protein thiol content was measured by reaction with either DTNB (ϵ412 = 14,150 m−1 cm−1) (82) or DTDPy (ϵ324 = 21,400 m−1 cm−1) (83). As expected, the reduced enzyme contained one thiol per protein subunit.
Preparation of MtAhpE derivatives
MtAhpE–SOH was obtained by treatment of the reduced enzyme with an equivalent amount of H2O2 in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C). Incubation times required for completion of the reaction under the experimental conditions employed were determined by computational modeling using Gepasi (84). MtAhpE–SSH was prepared by mixing equimolar concentrations of both H2O2 and Na2S with MtAhpE–SH. Because H2O2 reacts several orders of magnitude faster with MtAhpE–SH than with H2S (8.2 × 104 (50) versus 0.35 m−1 s−1 (57) at pH 7.4 and 25 °C, respectively), MtAhpE–SOH is formed, which in turn reacts with H2S to form MtAhpE–SSH.
Detection of MtAhpE–SSH by ESI-Q MS
The detection of persulfides was carried out in samples of MtAhpE–SH (10 μm) and Na2S (10 μm) treated with H2O2 (10 μm) in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C). At different incubation times, iodoacetamide (40 mm) was added for 30 min at 25 °C. The excess alkylating agent was removed by gel filtration using PD SpinTrap G-25 (GE Healthcare) in 20 mm ammonium bicarbonate buffer (pH 7.4, 25 °C). After this gel filtration step, some samples were further treated with DTT (2 mm) before analysis. All samples were loaded into a C4 column (GraceVydac 214MS5115) for HPLC separation. Mobile phase consisted of 0.1% formic acid in nanopure water (solvent A) and 0.1% formic acid in CH3CN (solvent B), and elution of the protein was performed with a 10-min linear gradient of solvent B (5–50%) followed by an additional 10 min at 50% solvent B at 100 μl/min. An ESI-triple quadrupole mass spectrometer (QTrap4500, ABSciex) was employed for detection. The spectrometer was set in Q1 positive mode in the 500–2000 m/z range with a scan rate of 200 Da/s and Q1 resolution in UNIT. Parameters used were as follows: IS, 5000; TEM, 300; DP, 120; EP, 10; CUR, 20; GS1, 30; GS2, 20. Data acquisition was done using Analyst 1.6.2 (ABSciex) and PeakView 2.2 (ABSciex) software was used for data analysis and deconvolution of all spectra.
Kinetics of H2S reaction with MtAhpE–SOH
The kinetics of the reaction of MtAhpE–SOH with H2S was determined by two competition approaches, using a SX20 Applied Photophysics stopped-flow spectrophotometer and either absorbance or fluorescence detectors. In the competition of H2S and TNB for MtAhpE–SOH, the latter (1.4 μm) was mixed with solutions of Na2S (0–50 μm) and TNB (30 μm) in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C), and the absorbance was recorded at 412 nm (ϵ412 = 14,150 m−1 cm−1) (82). The exponential rate constants were obtained from the best fits to exponential plus straight line functions. The second-order rate constant of the reaction between H2S and MtAhpE–SOH was obtained from the slope of the plot of kobs versus Na2S concentration.
The competition of H2S and H2O2 for MtAhpE–SOH was studied following total fluorescence emission (λex 295 nm), taking advantage of the changes in the protein intrinsic fluorescence that occur during MtAhpE–SH oxidation to MtAhpE–SOH and over-oxidation to sulfinic acid (MtAhpE–SO2H) as described before (50). MtAhpE–SH (1 μm) was mixed with solutions of Na2S (0 or 1 μm) and H2O2 (10, 100, 250 μm) in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C). Results were fitted and modeled using DynaFit (85) to determine the rate constant for the reaction of H2S toward MtAhpE–SOH. The initial concentration of the reagents was considered as well as the previously reported rate constant for the oxidation of MtAhpE–SH to MtAhpE–SOH and that of MtAhpE–SOH to MtAhpE–SO2H (50).
Reactivity of MtAhpE–SSH toward DTDPy
MtAhpE–SOH (1.3 μm) was prepared as described above in a syringe of the SX20 Applied Photophysics stopped-flow spectrophotometer. Then, Na2S (1.3 μm) was added to initiate the formation of MtAhpE–SSH. This solution was mixed at different aging times with DTDPy (100–700 μm), and the absorbance at 324 nm (ϵ324 = 21,400 m−1 cm−1) was recorded. Data were fitted to double-exponential functions to obtain the kobs and amplitudes of the phases. The rate constant of the reaction of MtAhpE–SSH with DTDPy was calculated from the slope of the plot of the observed rate constants for the fast phase versus DTDPy concentration obtained for mixtures of MtAhpE–SH, H2O2 and Na2S aged for at least 900 s.
Reactivity of MtAhpE–SSH toward peroxynitrite and H2O2
The decay of peroxynitrite (12 μm) in the absence or presence of MtAhpE–SH, MtAhpE–SOH or MtAhpE–SSH (15 μm) in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C) was followed at 310 nm using an SX20 Applied Photophysics stopped-flow spectrophotometer. The reduction of H2O2 by the different forms of MtAhpE was followed using the ferrous oxidation–xylenol orange method (FOX assay) (51, 86). Briefly, MtAhpE–SH, MtAhpE–SOH or MtAhpE–SSH (45 μm) were mixed with H2O2 (100 μm) in 0.05 m phosphate buffer (pH 7.4, 25 °C). Aliquots (100 μl) were taken at different times, mixed with 900 μl of the FOX reagent, and further incubated for 30 min at room temperature followed by absorbance measurement at 560 nm. The extinction coefficient for H2O2 using this assay was determined (ϵ560 = 51,520 m−1 cm−1) and was in close agreement with previously reported values (51). The second-order rate constants of the reduction of these oxidants by the different forms of MtAhpE were estimated by initial velocity kinetics.
Catalytic consumption of H2O2 and H2S by MtAhpE
H2O2 (125 μm) and Na2S (250 μm) were mixed in the absence or presence of increasing concentrations of MtAhpE–SH (5–29 μm) in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C) using sealed vials. Samples were removed using gas-tight syringes and the remaining H2S was determined by the methylene blue method (87). Alternatively, the same concentrations of H2O2 and Na2S used in the previous experiment were mixed in the absence or presence of MtAhpE–SH (2.9 μm) in 0.05 m phosphate buffer (pH 7.4, 25 °C), and the remaining H2O2 was determined by the FOX assay as described above.
Molecular dynamics of MtAhpE–SH and MtAhpE–SSH
Classical MD of the MtAhpE dimer were performed for the thiolate form of MtAhpE–SH (MtAhpE–S−) and for the persulfide form (MtAhpE–SS−). For the MtAhpE–S− MD, the recently reviewed crystal structure of MtAhpE (PDB code 4X0X) (88) was used as the starting structure. The persulfide initial model was generated by a modification of the oxidized structure of MtAhpE (PDB code 4X1U) (88) in which the sulfenic acid was in silico transformed to the persulfide form (MtAhpE–SS−). Classical parameters for simulating cysteine persulfide were developed using standard protocols (89).
Both initial models were submitted to the same MD protocol as we have previously performed for this and other related enzymes (45, 58, 90). Briefly, the system was solvated with an octahedral box 12 Å in radius with TIP3P water molecules (91). With the exception of cysteine persulfide in the MtAhpE–SS− model, residue parameters correspond to the parm14SB AMBER force field (92). Simulations were performed using periodic boundary conditions with a 10-Å cutoff and a particle mesh Ewald summation method for treating the electrostatic interactions. The hydrogen bond lengths were kept at their equilibrium distance by using the SHAKE algorithm, whereas temperature and pressure were kept constant with a Langevin thermostat and barostat, respectively, as implemented in the AMBER14 program. The system was minimized in 1000 steps (10 with steep gradient and the rest with the conjugate gradient). Then, it was heated from 0 to 300 K for 20 ps at constant pressure with a Berendsen thermostat, and pressure was equilibrated at 1 bar for 5 ps. After these two steps, a 10-ns MD long simulation at a constant temperature (300 K) and a constant volume was performed. An unrestrained 700-ns-long production MD at the NPT ensemble was performed. All dynamics visualizations and molecular drawings were performed with VMD 1.9.1 (93).
Transfer of the sulfane sulfur of MtAhpE–SSH to the thiol in SSP4
MtAhpE–SOH was prepared by incubation of MtAhpE–SH (4 μm) with H2O2 (4 μm) in 0.1 m phosphate buffer with 0.1 mm DTPA (pH 7.4, 25 °C) during 2 min. MtAhpE–SSH was formed by adding Na2S (4 μm) to sulfenic acid preparations and aging during 15 min. Then, samples were diluted 2-fold in phosphate buffer and mixed with SSP4 (20 μM and 4% DMSO, final concentrations). Fluorescence emission at 515 nm (λex = 482 nm) was recorded in a Varioskan Flash plate reader (Thermo Fisher Scientific). Calibration curves were performed with Na2S2 assuming equimolarity of sulfane sulfur and Na2S2 in reference solutions. The yield of sulfane sulfur transfer to the probe was estimated based on the amplitude of the fit of the fluorescence increase to an exponential function.
Data processing
Data were plotted and analyzed using OriginPro 8.0 (OriginLab). Unless specified, results are expressed as the mean ± S.D. of independent experiments.
Author contributions
E. C., A. M. R., B. A., and M. T. conceptualization; E. C., A. M. R., A. Z., M. M., R. R., B. A., and M. T. resources; E. C., A. M. R., A. Z., M. M., R. R., B. A., and M. T. formal analysis; E. C., A. M. R., R. R., B. A., and M. T. funding acquisition; E. C., A. M. R., B. A., and M. T. validation; E. C., A. M. R., A. Z., M. M., and M. I. D. A. investigation; E. C., A. M. R., and A. Z. visualization; E. C., A. M. R., A. Z., M. M., B. A., and M. T. writing-original draft; E. C., A. M. R., B. A., and M. T. project administration; E. C., A. M. R., A. Z., M. M., M. I. D. A., R. R., B. A., and M. T. writing-review and editing; B. A. and M. T. supervision.
Supplementary Material
This work was supported in part by grants from Comisión Sectorial de Investigación Científica (CSIC, Universidad de la República) CSIC-Iniciación 2015 (to E. C. and A. M. R.), CSIC I+D 2016 (to B. A.), CSIC I+D 2016 (to M. T.), CSIC Grupos 2014 and 2018 (to E. C. and B. A.), CSIC Grupos 2014 and 2018 (to R. R.), CSIC Centros Interdisciplinarios 2015 (to R. R.), Fondo Vaz Ferreira MEC, I/FVF2017/069 (to E. C.), and I/FVF2017/185 (to A. Z.), and by funding from PEDECIBA. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Fig. S1.
The term “H2S” is used throughout the text to refer to the mixture of H2S (sulfane or hydrogen sulfide) and HS− (sulfanide or hydrogen(sulfide)(1−)) in rapid equilibrium at the pH of the solution, unless otherwise specified.
In this text, “persulfide” is used for the mixture of RSSH and RSS− in rapid equilibrium at the pH of the solution, unless otherwise specified. RSSH, usually referred to as hydropersulfide or hydrodisulfide in bibliography, is named hydridodisulfide, disulfanyl or dithiohydroperoxide by IUPAC.
kpH = kpH-ind × (Ka H2S/(Ka H2S + [H+])) × ([H+]/(Ka MtAhpE–SOH + [H+])).
- Prx
- peroxiredoxin
- MtAhpE
- alkyl hydroperoxide reductase E of M. tuberculosis
- DTT
- 1,4-dithiothreitol
- TNB
- 5-thio-2-nitrobenzoic acid
- DTDPy
- 4,4′-dithiodipyridine
- MD
- molecular dynamics
- DTNB
- 5,5′-dithiobis-(2-nitrobenzoic acid)
- DTPA
- diethylenetriaminepentaacetic acid
- PDB
- Protein Data Bank.
References
- 1. Abe K., and Kimura H. (1996) The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 16, 1066–1071 10.1523/JNEUROSCI.16-03-01066.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hosoki R., Matsuki N., and Kimura H. (1997) The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 237, 527–531 10.1006/bbrc.1997.6878 [DOI] [PubMed] [Google Scholar]
- 3. Wang R. (2012) Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 92, 791–896 10.1152/physrev.00017.2011 [DOI] [PubMed] [Google Scholar]
- 4. Nathan C., and Shiloh M. U. (2000) Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. U.S.A. 97, 8841–8848 10.1073/pnas.97.16.8841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shiloh M. U., and Nathan C. F. (2000) Reactive nitrogen intermediates and the pathogenesis of Salmonella and mycobacteria. Curr. Opin. Microbiol. 3, 35–42 10.1016/S1369-5274(99)00048-X [DOI] [PubMed] [Google Scholar]
- 6. Alvarez M. N., Peluffo G., Piacenza L., and Radi R. (2011) Intraphagosomal peroxynitrite as a macrophage-derived cytotoxin against internalized Trypanosoma cruzi: consequences for oxidative killing and role of microbial peroxiredoxins in infectivity. J. Biol. Chem. 286, 6627–6640 10.1074/jbc.M110.167247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Piacenza L., Trujillo M., and Radi R. (2019) Reactive species and pathogen antioxidant networks during phagocytosis. J. Exp. Med. 216, 501–516 10.1084/jem.20181886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nambi S., Long J. E., Mishra B. B., Baker R., Murphy K. C., Olive A. J., Nguyen H. P., Shaffer S. A., and Sassetti C. M. (2015) The oxidative stress network of Mycobacterium tuberculosis reveals coordination between radical detoxification systems. Cell Host Microbe 17, 829–837 10.1016/j.chom.2015.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cuevasanta E., Denicola A., Alvarez B., and Möller M. N. (2012) Solubility and permeation of hydrogen sulfide in lipid membranes. PLoS ONE 7, e34562 10.1371/journal.pone.0034562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hatzios S. K., and Bertozzi C. R. (2011) The regulation of sulfur metabolism in Mycobacterium tuberculosis. PLoS Pathog. 7, e1002036 10.1371/journal.ppat.1002036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhave D. P., Muse W. B. 3rd, and Carroll K. S. (2007) Drug targets in mycobacterial sulfur metabolism. Infect. Disord. Drug Targets 7, 140–158 10.2174/187152607781001772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wheeler P. R., Coldham N. G., Keating L., Gordon S. V., Wooff E. E., Parish T., and Hewinson R. G. (2005) Functional demonstration of reverse transsulfuration in the Mycobacterium tuberculosis complex reveals that methionine is the preferred sulfur source for pathogenic mycobacteria. J. Biol. Chem. 280, 8069–8078 10.1074/jbc.M412540200 [DOI] [PubMed] [Google Scholar]
- 13. Nzungize L., Ali M. K., Wang X., Huang X., Yang W., Duan X., Yan S., Li C., Abdalla A. E., Jeyakkumar P., and Xie J. (2019) Mycobacterium tuberculosis metC (Rv3340) derived hydrogen sulphide conferring bacteria stress survival. J. Drug Target 2019, 1–13 10.1080/1061186X.2019.1579820 [DOI] [PubMed] [Google Scholar]
- 14. Hughes M. N., Centelles M. N., and Moore K. P. (2009) Making and working with hydrogen sulfide: the chemistry and generation of hydrogen sulfide in vitro and its measurement in vivo: a review. Free Radic. Biol. Med. 47, 1346–1353 10.1016/j.freeradbiomed.2009.09.018 [DOI] [PubMed] [Google Scholar]
- 15. Cuevasanta E., Lange M., Bonanata J., Coitiño E. L., Ferrer-Sueta G., Filipovic M. R., and Alvarez B. (2015) Reaction of hydrogen sulfide with disulfide and sulfenic acid to form the strongly nucleophilic persulfide. J. Biol. Chem. 290, 26866–26880 10.1074/jbc.M115.672816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wright C. M., Christman G. D., Snellinger A. M., Johnston M. V., and Mueller E. G. (2006) Direct evidence for enzyme persulfide and disulfide intermediates during 4-thiouridine biosynthesis. Chem. Commun. 2006, 3104–3106 10.1039/b604040c [DOI] [PubMed] [Google Scholar]
- 17. Mueller E. G. (2006) Trafficking in persulfides: delivering sulfur in biosynthetic pathways. Nat. Chem. Biol. 2, 185–194 10.1038/nchembio779 [DOI] [PubMed] [Google Scholar]
- 18. Ida T., Sawa T., Ihara H., Tsuchiya Y., Watanabe Y., Kumagai Y., Suematsu M., Motohashi H., Fujii S., Matsunaga T., Yamamoto M., Ono K., Devarie-Baez N. O., Xian M., Fukuto J. M., and Akaike T. (2014) Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 7606–7611 10.1073/pnas.1321232111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yadav P. K., Yamada K., Chiku T., Koutmos M., and Banerjee R. (2013) Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 288, 20002–20013 10.1074/jbc.M113.466177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jackson M. R., Melideo S. L., and Jorns M. S. (2012) Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 51, 6804–6815 10.1021/bi300778t [DOI] [PubMed] [Google Scholar]
- 21. Libiad M., Yadav P. K., Vitvitsky V., Martinov M., and Banerjee R. (2014) Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 289, 30901–30910 10.1074/jbc.M114.602664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Melideo S. L., Jackson M. R., and Jorns M. S. (2014) Biosynthesis of a central intermediate in hydrogen sulfide metabolism by a novel human sulfurtransferase and its yeast ortholog. Biochemistry 53, 4739–4753 10.1021/bi500650h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Libiad M., Motl N., Akey D. L., Sakamoto N., Fearon E. R., Smith J. L., and Banerjee R. (2018) Thiosulfate sulfurtransferase-like domain-containing 1 protein interacts with thioredoxin. J. Biol. Chem. 293, 2675–2686 10.1074/jbc.RA117.000826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hildebrandt T. M., and Grieshaber M. K. (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 275, 3352–3361 10.1111/j.1742-4658.2008.06482.x [DOI] [PubMed] [Google Scholar]
- 25. Toohey J. I. (2011) Sulfur signaling: is the agent sulfide or sulfane? Anal. Biochem. 413, 1–7 10.1016/j.ab.2011.01.044 [DOI] [PubMed] [Google Scholar]
- 26. Filipovic M. R., Zivanovic J., Alvarez B., and Banerjee R. (2018) Chemical biology of H2S signaling through persulfidation. Chem. Rev. 118, 1253–1337 10.1021/acs.chemrev.7b00205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Francoleon N. E., Carrington S. J., and Fukuto J. M. (2011) The reaction of H(2)S with oxidized thiols: generation of persulfides and implications to H(2)S biology. Arch. Biochem. Biophys. 516, 146–153 10.1016/j.abb.2011.09.015 [DOI] [PubMed] [Google Scholar]
- 28. Greiner R., Pálinkás Z., Bäsell K., Becher D., Antelmann H., Nagy P., and Dick T. P. (2013) Polysulfides link H2S to protein thiol oxidation. Antioxid. Redox Signal. 19, 1749–1765 10.1089/ars.2012.5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bestetti S., Medraño-Fernandez I., Galli M., Ghitti M., Bienert G. P., Musco G., Orsi A., Rubartelli A., and Sitia R. (2018) A persulfidation-based mechanism controls aquaporin-8 conductance. Sci. Adv. 4, eaar5770 10.1126/sciadv.aar5770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Artaud I., and Galardon E. (2014) A persulfide analogue of the nitrosothiol SNAP: formation, characterization and reactivity. Chembiochem 15, 2361–2364 10.1002/cbic.201402312 [DOI] [PubMed] [Google Scholar]
- 31. Ubuka T. (2002) Assay methods and biological roles of labile sulfur in animal tissues. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 781, 227–249 10.1016/S1570-0232(02)00623-2 [DOI] [PubMed] [Google Scholar]
- 32. Pan J., and Carroll K. S. (2013) Persulfide reactivity in the detection of protein S-sulfhydration. ACS Chem. Biol. 8, 1110–1116 10.1021/cb4001052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang D., Macinkovic I., Devarie-Baez N. O., Pan J., Park C. M., Carroll K. S., Filipovic M. R., and Xian M. (2014) Detection of protein S-sulfhydration by a tag-switch technique. Angew. Chem. Int. Ed. Engl. 53, 575–581 10.1002/anie.201305876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagahara N., Nirasawa T., Yoshii T., and Niimura Y. (2012) Is novel signal transducer sulfur oxide involved in the redox cycle of persulfide at the catalytic site cysteine in a stable reaction intermediate of mercaptopyruvate sulfurtransferase? Antioxid. Redox Signal. 16, 747–753 10.1089/ars.2011.4468 [DOI] [PubMed] [Google Scholar]
- 35. Bailey T. S., Zakharov L. N., and Pluth M. D. (2014) Understanding hydrogen sulfide storage: probing conditions for sulfide release from hydrodisulfides. J. Am. Chem. Soc. 136, 10573–10576 10.1021/ja505371z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wedmann R., Onderka C., Wei S., Szijártó I. A., Miljkovic J. L., Mitrovic A., Lange M., Savitsky S., Yadav P. K., Torregrossa R., Harrer E. G., Harrer T., Ishii I., Gollasch M., Wood M. E., et al. (2016) Improved tag-switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem. Sci. 7, 3414–3426 10.1039/C5SC04818D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dóka É., Pader I., Bíró A., Johansson K., Cheng Q., Ballagó K., Prigge J. R., Pastor-Flores D., Dick T. P., Schmidt E. E., Arnér E. S., and Nagy P. (2016) A novel persulfide detection method reveals protein persulfide- and polysulfide-reducing functions of thioredoxin and glutathione systems. Sci. Adv. 2, e1500968 10.1126/sciadv.1500968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gao X. H., Krokowski D., Guan B. J., Bederman I., Majumder M., Parisien M., Diatchenko L., Kabil O., Willard B., Banerjee R., Wang B., Bebek G., Evans C. R., Fox P. L., Gerson S. L., et al. (2015) Quantitative H2S-mediated protein sulfhydration reveals metabolic reprogramming during the integrated stress response. Elife 4, e10067 10.7554/eLife.10067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Everett S. A., Folkes L. K., Wardman P., and Asmus K. D. (1994) Free-radical repair by a novel perthiol: reversible hydrogen transfer and perthiyl radical formation. Free Radic. Res. 20, 387–400 10.3109/10715769409145638 [DOI] [PubMed] [Google Scholar]
- 40. Jencks W. P., Cordes S., and Carriuolo J. (1960) The free energy of thiol ester hydrolysis. J. Biol. Chem. 235, 3608–3614 [PubMed] [Google Scholar]
- 41. Rhee S. G., Chae H. Z., and Kim K. (2005) Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 38, 1543–1552 10.1016/j.freeradbiomed.2005.02.026 [DOI] [PubMed] [Google Scholar]
- 42. Randall L. M., Ferrer-Sueta G., and Denicola A. (2013) Peroxiredoxins as preferential targets in H2O2-induced signaling. Methods Enzymol. 527, 41–63 10.1016/B978-0-12-405882-8.00003-9 [DOI] [PubMed] [Google Scholar]
- 43. Brigelius-Flohé R., and Flohé L. (2011) Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 15, 2335–2381 10.1089/ars.2010.3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Poole L. B. (2007) The catalytic mechanism of peroxiredoxins. Subcell. Biochem. 44, 61–81 10.1007/978-1-4020-6051-9_4 [DOI] [PubMed] [Google Scholar]
- 45. Zeida A., Reyes A. M., Lebrero M. C., Radi R., Trujillo M., and Estrin D. A. (2014) The extraordinary catalytic ability of peroxiredoxins: a combined experimental and QM/MM study on the fast thiol oxidation step. Chem. Commun. 50, 10070–10073 10.1039/C4CC02899F [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Portillo-Ledesma S., Sardi F., Manta B., Tourn M. V., Clippe A., Knoops B., Alvarez B., Coitiño E. L., and Ferrer-Sueta G. (2014) Deconstructing the catalytic efficiency of peroxiredoxin-5 peroxidatic cysteine. Biochemistry 53, 6113–6125 10.1021/bi500389m [DOI] [PubMed] [Google Scholar]
- 47. Hall A., Parsonage D., Poole L. B., and Karplus P. A. (2010) Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. J. Mol. Biol. 402, 194–209 10.1016/j.jmb.2010.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Flohé L., Budde H., Bruns K., Castro H., Clos J., Hofmann B., Kansal-Kalavar S., Krumme D., Menge U., Plank-Schumacher K., Sztajer H., Wissing J., Wylegalla C., and Hecht H. J. (2002) Tryparedoxin peroxidase of Leishmania donovani: molecular cloning, heterologous expression, specificity, and catalytic mechanism. Arch. Biochem. Biophys. 397, 324–335 10.1006/abbi.2001.2688 [DOI] [PubMed] [Google Scholar]
- 49. Soito L., Williamson C., Knutson S. T., Fetrow J. S., Poole L. B., and Nelson K. J. (2011) PREX: PeroxiRedoxin classification indEX, a database of subfamily assignments across the diverse peroxiredoxin family. Nucleic Acids Res. 39, D332–D337 10.1093/nar/gkq1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hugo M., Turell L., Manta B., Botti H., Monteiro G., Netto L. E., Alvarez B., Radi R., and Trujillo M. (2009) Thiol and sulfenic acid oxidation of AhpE, the one-cysteine peroxiredoxin from Mycobacterium tuberculosis: kinetics, acidity constants, and conformational dynamics. Biochemistry 48, 9416–9426 10.1021/bi901221s [DOI] [PubMed] [Google Scholar]
- 51. Reyes A. M., Hugo M., Trostchansky A., Capece L., Radi R., and Trujillo M. (2011) Oxidizing substrate specificity of Mycobacterium tuberculosis alkyl hydroperoxide reductase E: kinetics and mechanisms of oxidation and over-oxidation. Free Radic. Biol. Med. 51, 464–473 10.1016/j.freeradbiomed.2011.04.023 [DOI] [PubMed] [Google Scholar]
- 52. Hugo M., Van Laer K., Reyes A. M., Vertommen D., Messens J., Radi R., and Trujillo M. (2014) Mycothiol/mycoredoxin 1-dependent reduction of the peroxiredoxin AhpE from Mycobacterium tuberculosis. J. Biol. Chem. 289, 5228–5239 10.1074/jbc.M113.510248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kumar A., Balakrishna A. M., Nartey W., Manimekalai M. S. S., and Grüber G. (2016) Redox chemistry of Mycobacterium tuberculosis alkylhydroperoxide reductase E (AhpE): structural and mechanistic insight into a mycoredoxin-1 independent reductive pathway of AhpE via mycothiol. Free Radic. Biol. Med. 97, 588–601 10.1016/j.freeradbiomed.2016.07.007 [DOI] [PubMed] [Google Scholar]
- 54. Kumar A., Nartey W., Shin J., Manimekalai M. S. S., and Gruber G. (2017) Structural and mechanistic insights into mycothiol disulphide reductase and the Mycoredoxin-1–alkylhydroperoxide reductase E assembly of Mycobacterium tuberculosis. Biochim. Biophys. Acta 1861, 2354–2366 10.1016/j.bbagen.2017.05.007 [DOI] [PubMed] [Google Scholar]
- 55. Cuevasanta E., Möller M. N., and Alvarez B. (2017) Biological chemistry of hydrogen sulfide and persulfides. Arch. Biochem. Biophys. 617, 9–25 10.1016/j.abb.2016.09.018 [DOI] [PubMed] [Google Scholar]
- 56. Espenson J. H. (1995) Chemical Kinetics and Reaction Mechanisms, 2nd Ed., Grall S. ed., pp. 46–69, The McGraw-Hill Companies, Inc., New York [Google Scholar]
- 57. Hoffmann M. R. (1977) Kinetics and mechanism of oxidation of hydrogen sulfide by hydrogen peroxide in acidic solution. Environ. Sci. Technol. 11, 61–66 10.1021/es60124a004 [DOI] [Google Scholar]
- 58. Zeida A., Reyes A. M., Lichtig P., Hugo M., Vazquez D. S., Santos J., González Flecha F. L., Radi R., Estrin D. A., and Trujillo M. (2015) Molecular basis of hydroperoxide specificity in peroxiredoxins: the case of AhpE from Mycobacterium tuberculosis. Biochemistry 54, 7237–7247 10.1021/acs.biochem.5b00758 [DOI] [PubMed] [Google Scholar]
- 59. Hall A., Nelson K., Poole L. B., and Karplus P. A. (2011) Structure-based insights into the catalytic power and conformational dexterity of peroxiredoxins. Antioxid. Redox Signal. 15, 795–815 10.1089/ars.2010.3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nagy P., Karton A., Betz A., Peskin A. V., Pace P., O'Reilly R. J., Hampton M. B., Radom L., and Winterbourn C. C. (2011) Model for the exceptional reactivity of peroxiredoxins 2 and 3 with hydrogen peroxide: a kinetic and computational study. J. Biol. Chem. 286, 18048–18055 10.1074/jbc.M111.232355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bibli S. I., Luck B., Zukunft S., Wittig J., Chen W., Xian M., Papapetropoulos A., Hu J., and Fleming I. (2018) A selective and sensitive method for quantification of endogenous polysulfide production in biological samples. Redox Biol. 18, 295–304 10.1016/j.redox.2018.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen W., Liu C., Peng B., Zhao Y., Pacheco A., and Xian M. (2013) New fluorescent probes for sulfane sulfurs and the application in bioimaging. Chem. Sci. 4, 2892–2896 10.1039/c3sc50754h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cox A. G., Winterbourn C. C., and Hampton M. B. (2009) Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 425, 313–325 10.1042/BJ20091541 [DOI] [PubMed] [Google Scholar]
- 64. Trujillo M., Ferrer-Sueta G., and Radi R. (2008) Peroxynitrite detoxification and its biologic implications. Antioxid. Redox Signal. 10, 1607–1620 10.1089/ars.2008.2060 [DOI] [PubMed] [Google Scholar]
- 65. Portillo-Ledesma S., Randall L. M., Parsonage D., Dalla Rizza J., Karplus P. A., Poole L. B., Denicola A., and Ferrer-Sueta G. (2018) Differential kinetics of two-cysteine peroxiredoxin disulfide formation reveal a novel model for peroxide sensing. Biochemistry 57, 3416–3424 10.1021/acs.biochem.8b00188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Peshenko I. V., and Shichi H. (2001) Oxidation of active center cysteine of bovine 1-Cys peroxiredoxin to the cysteine sulfenic acid form by peroxide and peroxynitrite. Free Radic. Biol. Med. 31, 292–303 10.1016/S0891-5849(01)00579-2 [DOI] [PubMed] [Google Scholar]
- 67. Loumaye E., Ferrer-Sueta G., Alvarez B., Rees J. F., Clippe A., Knoops B., Radi R., and Trujillo M. (2011) Kinetic studies of peroxiredoxin 6 from Arenicola marina: rapid oxidation by hydrogen peroxide and peroxynitrite but lack of reduction by hydrogen sulfide. Arch. Biochem. Biophys. 514, 1–7 10.1016/j.abb.2011.07.002 [DOI] [PubMed] [Google Scholar]
- 68. Wood Z. A., Poole L. B., and Karplus P. A. (2003) Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 300, 650–653 10.1126/science.1080405 [DOI] [PubMed] [Google Scholar]
- 69. Newton G. L., and Fahey R. C. (2002) Mycothiol biochemistry. Arch. Microbiol. 178, 388–394 10.1007/s00203-002-0469-4 [DOI] [PubMed] [Google Scholar]
- 70. Newton G. L., Arnold K., Price M. S., Sherrill C., Delcardayre S. B., Aharonowitz Y., Cohen G., Davies J., Fahey R. C., and Davis C. (1996) Distribution of thiols in microorganisms: mycothiol is a major thiol in most actinomycetes. J. Bacteriol. 178, 1990–1995 10.1128/jb.178.7.1990-1995.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cumming B. M., Lamprecht D. A., Wells R. M., Saini V., Mazorodze J. H., and Steyn A. J. C. (2014) The physiology and genetics of oxidative stress in mycobacteria. Microbiol. Spectr. 2, 10.1128/microbiolspec.MGM2-0019-2013 [DOI] [PubMed] [Google Scholar]
- 72. Pal V. K., Bandyopadhyay P., and Singh A. (2018) Hydrogen sulfide in physiology and pathogenesis of bacteria and viruses. IUBMB Life 70, 393–410 10.1002/iub.1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Abhishek S., Saikia U. N., Gupta A., Bansal R., Gupta V., Singh N., Laal S., and Verma I. (2018) Transcriptional profile of Mycobacterium tuberculosis in an in vitro model of intraocular tuberculosis. Front. Cell. Infect. Microbiol. 8, 330 10.3389/fcimb.2018.00330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bonanata J., Turell L., Antmann L., Ferrer-Sueta G., Botasini S., Méndez E., Alvarez B., and Coitiño E. L. (2017) The thiol of human serum albumin: acidity, microenvironment and mechanistic insights on its oxidation to sulfenic acid. Free Radic. Biol. Med. 108, 952–962 10.1016/j.freeradbiomed.2017.04.021 [DOI] [PubMed] [Google Scholar]
- 75. Peskin A. V., Low F. M., Paton L. N., Maghzal G. J., Hampton M. B., and Winterbourn C. C. (2007) The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 282, 11885–11892 10.1074/jbc.M700339200 [DOI] [PubMed] [Google Scholar]
- 76. Nelson D. P., and Kiesow L. A. (1972) Enthalpy of decomposition of hydrogen peroxide by catalase at 25 °C (with molar extinction coefficients of H2O2 solutions in the UV). Anal. Biochem. 49, 474–478 10.1016/0003-2697(72)90451-4 [DOI] [PubMed] [Google Scholar]
- 77. Alvarez B., Demicheli V., Durán R., Trujillo M., Cerveñansky C., Freeman B. A., and Radi R. (2004) Inactivation of human Cu,Zn superoxide dismutase by peroxynitrite and formation of histidinyl radical. Free Radic. Biol. Med. 37, 813–822 10.1016/j.freeradbiomed.2004.06.006 [DOI] [PubMed] [Google Scholar]
- 78. Bohle D. S., Hansert B., Paulson S. C., and Smith B. D. (1994) Biomimetic synthesis of the putative cytotoxin peroxynitrite, ONOO−, and its characterization as a tetramethylammonium salt. J. Am. Chem. Soc. 116, 7423–7424 10.1021/ja00095a062 [DOI] [Google Scholar]
- 79. Turell L., Botti H., Carballal S., Ferrer-Sueta G., Souza J. M., Durán R., Freeman B. A., Radi R., and Alvarez B. (2008) Reactivity of sulfenic acid in human serum albumin. Biochemistry 47, 358–367 10.1021/bi701520y [DOI] [PubMed] [Google Scholar]
- 80. Li S., Peterson N. A., Kim M. Y., Kim C. Y., Hung L. W., Yu M., Lekin T., Segelke B. W., Lott J. S., and Baker E. N. (2005) Crystal structure of AhpE from Mycobacterium tuberculosis, a 1-Cys peroxiredoxin. J. Mol. Biol. 346, 1035–1046 10.1016/j.jmb.2004.12.046 [DOI] [PubMed] [Google Scholar]
- 81. Wilkins M. R., Gasteiger E., Bairoch A., Sanchez J. C., Williams K. L., Appel R. D., and Hochstrasser D. F. (1999) Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112, 531–552 [DOI] [PubMed] [Google Scholar]
- 82. Riener C. K., Kada G., and Gruber H. J. (2002) Quick measurement of protein sulfhydryls with Ellman's reagent and with 4,4′-dithiodipyridine. Anal. Bioanal. Chem. 373, 266–276 10.1007/s00216-002-1347-2 [DOI] [PubMed] [Google Scholar]
- 83. Grassetti D. R., and Murray J. F. Jr. (1967) Determination of sulfhydryl groups with 2,2′- or 4,4′-dithiodipyridine. Arch. Biochem. Biophys. 119, 41–49 10.1016/0003-9861(67)90426-2 [DOI] [PubMed] [Google Scholar]
- 84. Mendes P. (1993) GEPASI: a software package for modelling the dynamics, steady states and control of biochemical and other systems. Comput. Appl. Biosci. 9, 563–571 [DOI] [PubMed] [Google Scholar]
- 85. Kuzmic P. (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 237, 260–273 10.1006/abio.1996.0238 [DOI] [PubMed] [Google Scholar]
- 86. Jiang Z. Y., Hunt J. V., and Wolff S. P. (1992) Ferrous ion oxidation in the presence of xylenol orange for detection of lipid hydroperoxide in low density lipoprotein. Anal. Biochem. 202, 384–389 10.1016/0003-2697(92)90122-N [DOI] [PubMed] [Google Scholar]
- 87. Siegel L. M. (1965) A direct microdetermination for sulfide. Anal. Biochem. 11, 126–132 10.1016/0003-2697(65)90051-5 [DOI] [PubMed] [Google Scholar]
- 88. van Bergen L. A., Alonso M., Palló A., Nilsson L., De Proft F., and Messens J. (2016) Revisiting sulfur H-bonds in proteins: the example of peroxiredoxin AhpE. Sci. Rep. 6, 30369 10.1038/srep30369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wang J., Wolf R. M., Caldwell J. W., Kollman P. A., and Case D. A. (2004) Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 10.1002/jcc.20035 [DOI] [PubMed] [Google Scholar]
- 90. Reyes A. M., Vazquez D. S., Zeida A., Hugo M., Piñeyro M. D., De Armas M. I., Estrin D., Radi R., Santos J., and Trujillo M. (2016) PrxQ B from Mycobacterium tuberculosis is a monomeric, thioredoxin-dependent and highly efficient fatty acid hydroperoxide reductase. Free Radic. Biol. Med. 101, 249–260 10.1016/j.freeradbiomed.2016.10.005 [DOI] [PubMed] [Google Scholar]
- 91. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
- 92. Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., and Simmerling C. (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 94. Nelson L. J., Knutson S. T., Soito L., Klomsiri C., Poole L. B., and Fetrow J. S. (2011) Analysis of the peroxiredoxin family: using active site structure and sequence information for global classification and residue analysis. Proteins 79, 947–964 10.1002/prot.22936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kapopoulou A., Lew J. M., and Cole S. T. (2011) The MycoBrowser portal: a comprehensive and manually annotated resource for mycobacterial genomes. Tuberculosis (Edinb) 91, 8–13 10.1016/j.tube.2010.09.006 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.