

Abstract
A 33-year-old woman presented with a productive cough from childhood. She had suffered from repeated bacterial pneumonia. Her clinical and imaging findings revealed chronic sinusitis, bronchiectasis and situs inversus. We suspected primary ciliary dyskinesia (PCD) and performed a bronchial mucosal biopsy. The ciliary beat pattern according to high-speed video microscopy was complete loss. Electron microscopic findings of cilia showed defect of outer dynein arm (ODA). A genetic examination detected compound heterozygous mutations of DNAH5 that encode ODA components. There are few reports of genetic mutation analyses in Japanese PCD patients. We herein report a PCD patient with DNAH5 mutations and review the related literature.
Keywords: primary ciliary dyskinesia, DNAH5, gene, outer dynein arm
Introduction
Primary ciliary dyskinesia (PCD) is a genetically heterogeneous disorder of motile cilia caused by structural and functional defects of cilia. Most cases of PCD involve autosomal recessive inheritance. In recent years, the utility of genetic examinations has rapidly increased with the discovery of a number of novel cilia-related genes. Approximately 39 cilia-related genes have been identified, and mutations in DNAH5, which encodes the outer dynein arm (ODA) components, are the most common in Caucasians (1, 2). However, few reports have performed genetic mutation analyses in Japanese PCD cases (3, 4), and all that have been evaluated thus far have been children and teenagers.
We herein report an adult PCD case with compound heterozygous mutations of DNAH5.
Case Report
A 33-year-old non-smoking woman had a chronic cough and purulent sputum. She had been found to have situs inversus at five years old. She had had a productive cough since childhood and had experienced bacterial pneumonia on repeated occasions for the past five years. She had been married for three years and had no children.
On a physical examination, she was afebrile. Her blood pressure was 126/90 mmHg, pulse rate 112/min, respiratory rate of 15 times/min and O2 saturation of 98% with room air. On a cardiovascular examination, heart beats were audible at the right side of the chest. A respiratory examination revealed coarse crackles in both lungs. An initial investigation revealed a white cell count of 8,670 /μL and a C-reactive protein level of 1.10 mg/dL.
The saccharin test, which measures the duration until saccharin is tasted in the mouth after its deposition on the inferior turbinate, took longer than 60 minutes. In sputum culture, Escherichia coli was identified. Chest X-ray (posteroanterior view) showed a cardiac shadow at the right side and bronchiectasis with fibrotic bands in lower field of both sides (Fig. 1). Chest computed tomography (CT) showed bronchiectasis and bronchial wall thickening changes prominently in the lower lobes of both lungs (Fig. 2). Abdominal CT confirmed situs inversus totalis, paranasal sinus CT showed bilateral mucosal thickening of both maxillary antra, suggesting chronic sinusitis. Spirometry showed a forced vital capacity (FVC) of 1.40 L (43.5% of predicted), forced expiratory volume in 1 second (FEV1) of 0.91 L (32.5% of predicted) and an FEV1/FVC ratio of 63.6%. She was found to have otitis media with effusion on an otolaryngological examination. Based on these findings, she was diagnosed with Kartagener's syndrome.
Figure 1.
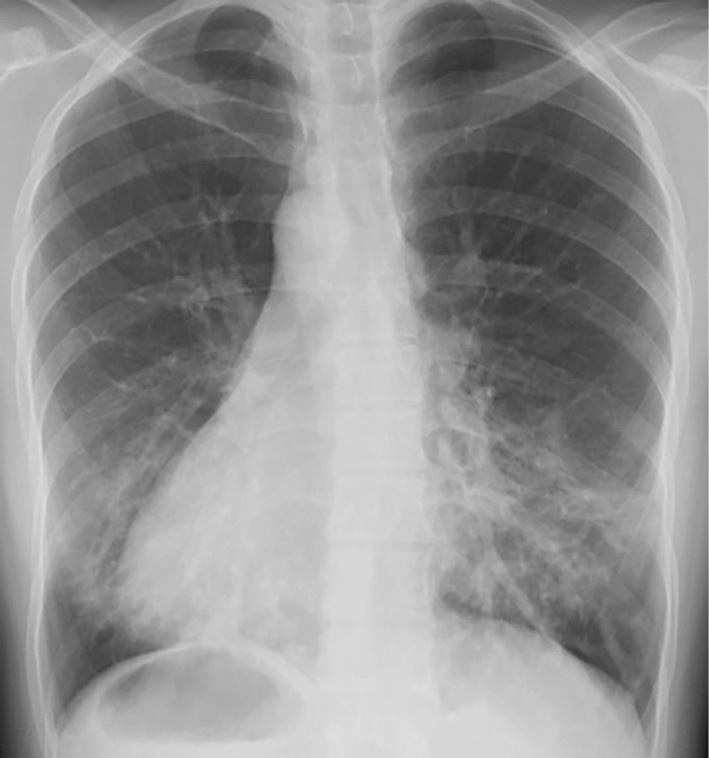
Chest X-ray shows dextrocardia in a patient with situs inversus and bronchiectasis changes in the bilateral lower areas.
Figure 2.
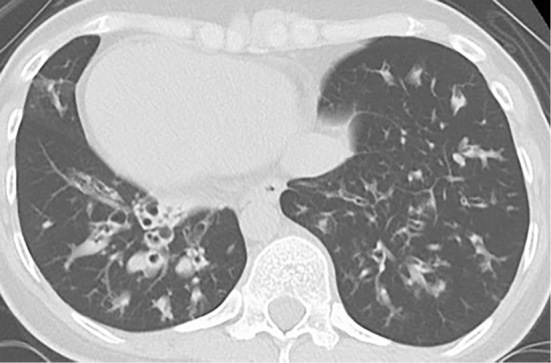
Chest computed tomography shows bronchiectasis and bronchial wall thickening changes prominently in both lower fields.
Given her history of repeated respiratory infections, situs inversus and physical examination findings, we suspected her of having primary ciliary dyskinesia and decided to proceed with the diagnosis according to certain guidelines. We used the European Respiratory Society (ERS) guidelines for the diagnosis of PCD and an international registry for PCD as a reference (5, 6). However, the ERS guidelines algorithm required several further evaluations in addition to a clinical symptom assessment. First, we needed to evaluate the nasal nitric oxide (nNO) and perform a high-speed video analysis (HSVA). Unfortunately, we were unable to examine the nNO level, but the fractional exhaled nitric oxide was under 5 ppb. An HSVA showed a loss of ciliary motion. We then had to perform transmission electron microscopy (TEM). This examination of a respiratory epithelial biopsy sample revealed a defect of ODA in the cilia (Fig. 3A). We then performed genetic testing of her peripheral blood by the method reported by Takeuchi et al. (4). We analyzed her genes using Sanger sequencing and a next-generation sequencing panel of 32 genes. She had compound heterozygous mutations in exon 55 of DNAH5 (NM_001369.2 c.9365delT p.Leu3122Ter) and in exon 34 of DNAH5 (NM_001369.2 c.5563_5564insA p.Ile1855fs) (Fig. 4). No mutations were found in the other 31 genes. It was impossible to obtain blood for a gene analysis from her parents, but they had no clinical findings suggestive of PCD. Of note, her sister also had situs inversus and a history of repeated episodes of respiratory tract infection. We therefore suspected that the patient was compound heterozygous. The international registry required a positive clinical presentation and consistent findings for PCD by at least two different assessments (e.g. HSVA, TEM, immunofluorescence microscopy, nNO, genetic test). Based on the above results, we diagnosed the patient with PCD according to two guidelines. While the saccharine test is not advocated by the ERS guideline, it is said to reflect the mucociliary clearance. Our patient's saccharine test result was quite long, and we considered this result to support the diagnosis.
Figure 3.

Electron microscopy of cilia from bronchial mucosa. A: A transmission electron microscopic analysis shows a defect in the outer dynein arm in the cilia (arrow). B: Normal structure of cilia, characterized by a typical ’9+2’ structure with nine outer microtubule doublets and a central pair of microtubules.
Figure 4.

The results of Sanger sequencing in this case. Compound heterozygous mutations in the DNAH5 gene are shown. Regarding the mutations of this case, c.5563_5564insA is a frame-shift mutation caused by the insertion of A between nucleotide positions 5563 and 5564, which leads to the frame-shift of an isoleucine codon at position 1855 and a stop codon at position 6. c.9365delT is a nonsense mutation caused by a defect in T at nucleotide position 9365 of the coding sequence of the gene, resulting in the leucine codon at position 3122 being changed to a stop codon. Green: Adenine (A). Black: Guanine (G). Red: Thymine (T). Blue: Cytosine (C)
Primary ciliary dyskinesia rule (PICADAR) is a diagnostic prediction tool for PCD (7), and our patient was positive for six of the parameters (chest symptoms, situs inversus, chronic rhinitis and otitis media). PICDAR is the first validated tool for diagnosing PCD, and we believe it will prove useful for specialists in various departments.
Discussion
PCD is a rare genetically heterogeneous disorder that is predominantly an autosomal-recessive inherited disorder. It is associated with structural and functional abnormalities of motile cilia and characterized by recurrent upper and lower airway infection.
Cilia have microtubule-based cytoskeletons called cilia axonemes and are characterized by a typical ‘9+2’ structure, consisting of nine outer microtubule doublets and a central pair of microtubules (Fig. 3B). The cilia proteome has been determined to contain more than 1,000 different proteins. In PCD, genetic defects in ciliary proteins have been found in over 250 species. Several recent studies have been conducted on PCD-associated genes. A total of 39 PCD-associated genes have been identified, and around 65% PCD patients have been found to have biallelic mutations (1, 2). The earliest reported PCD-associated genes were DNAI1 and DNAH5, which encode a component of ODA, and several dysmotile strains of Chlamydomonas reinhardtii with axonemal structures similar to human cilia have been reported (8, 9). DNAH5 is located in the genomic region with 79 exons and alternative exon 1, spanning 250 kb, and it encodes an axonemal heavy dynein chain of ODA. DNAH5 is associated with the activity of ATPase and microtubule motor proteins. Mutations in DNAH5 genes can be found in approximately 15-24% of all PCD cases among Caucasians. However, there are few reports of genetic mutation analyses in Japanese cases of PCD. Our patient had PCD with DNAH5 compound heterozygous mutations. One of the mutations of this case, c.9365delT, is a nonsense mutation caused by a defect in T at nucleotide position 9365 of the coding sequence of the gene, resulting in the leucine codon at position 3122 being changed to a stop codon. c.5563_5564insA is a frame-shift mutation caused by the insertion of A between nucleotide positions 5563 and 5564, which leads to the frame-shift of the isoleucine codon at position 1855 and the stop codon at position 6. c.5563_5564insA has previously been reported to be pathogenic (4), and the c.9365delT mutation is mentioned in ClinVar.
The present case was found to have PCD with an ODA defect. PCD with an ODA defect is easy to detect by electron microscopy, but the detection of PCD with an inner dynein arm (IDA) defect or others is difficult. There are few reported cases of an isolated IDA defect, and disease-causing mutations resulting in an isolated IDA defect have yet to be identified (10). O'Callaghan found that a patient whose initial results suggested PCD with an IDA defect or radial spoke defect had no abnormality after retesting (11). We must therefore interpret the results of electron microscopy carefully and conduct diagnostic tests multiple times if possible.
Treatment of PCD has not been standardized due to the lack of randomized controlled trials. The goal of treatment is the early detection of the disease and maintenance of the lung function by the prevention of exacerbation. However, it is difficult to diagnose PCD by reliable non-invasive diagnostic tests in some cases, so genetic tests can be useful. Recently, whole-exon sequencing was performed in a case of PCD to diagnose and identify new PCD-associated genes (3, 4, 12, 13). Genetic tests are expected to become the preferred diagnosis approach for PCD in the future. If gene analyses become more widely performed, then the need for gene-based treatment would thus be expected to increase.
The authors state that they have no Conflict of Interest (COI).
Fainacial Support
This study was supported by Takeda Science Foundation and a JSPS KAKENHI Grant Number JP 16K11210.
Acknowledgement
The authors thank Makoto Ikejiri and Kaname Nakatani (Mie University Graduate School of Medicine) for performing the gene analysis.
References
- 1.Mirra V, Werner C, Santamaria F. Primary ciliary dyskinesia: an update on clinical aspects, genetics, diagnosis, and future treatment strategies. Front Pediatr 5: 135, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Damseh N, Quercia N, Rumman N, Dell SD, Kim RH. Primary ciliary dyskinesia: mechanisms and management. Appl Clin Gene 10: 67-74, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kano G, Tsujii H, Takeuchi K, et al. Whole-exome sequencing identification of novel DNAH5 mutations in a young patient with primary ciliary dyskinesia. Mol Med Rep 14: 5077-5083, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeuchi K, Kitano M, Kiyotoshi H, et al. A targeted next-generation sequencing panel reveals novel mutations in Japanese patients with primary ciliary dyskinesia. Auris Nasus Larynx 45: 585-591, 2018. [DOI] [PubMed] [Google Scholar]
- 5.Lucas JS, Barbato A, Collins SA, et al. European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia. Eur Respir J 4: 49, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Werner C, Lablans M, Ataian M, et al. An international registry for primary ciliary dyskinesia. Eur Respir J 47: 849-859, 2016. [DOI] [PubMed] [Google Scholar]
- 7.Behan L, Dimitrov BD, Kuehni CE, et al. PICADAR: a diagnostic predictive tool for primary ciliary dyskinesia. Eur Respir J 47: 1103-1112, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rupp G, O'Toole E, Gardner LC, Mitchell BF, Porter ME. The sup-pf-2 mutations of Chlamydomonas alter the activity of the outer dynein arms by modification of the gamma-dynein heavy chain. J Cell Biol 135: 1853-1865, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Omran H, Häffner K, Völkel A, et al. Homozygosity mapping of a gene locus for primary ciliary dyskinesia on chromosome 5p and identification of the heavy dynein chain DNAH5 as a candidate gene. Am J Respir Cell Mol Biol 23: 696-702, 2000. [DOI] [PubMed] [Google Scholar]
- 10.Shoemark A, Ives A, Becker-Heck A, et al. Inner dynein arm defects in primary ciliary dyskinesia. J Genet Syndr Gene Ther 4: 7, 2013. [Google Scholar]
- 11.O'Callaghan C, Rutman A, Williams GM, Hirst RA. Inner dynein arm defects causing primary ciliary dyskinesia: repeat testing required. Eur Respir J 38: 603-607, 2011. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, Luo H. Whole-exome sequencing identified a novel compound heterozygous mutation of LRRC6 in a Chinese primary ciliary dyskinesia patient. Biomed Res Int 2018: 1854269, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boycott K, Hartley T, Adam S, et al. The clinical application of genome-wide sequencing for monogenic diseases in Canada: position statement of the Canadian college of medical geneticists. J Med Genet 52: 431-437, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]