

SUMMARY
Cells respond to nutrient stress by trafficking cytosolic contents to lysosomes for degradation via macroautophagy. The endoplasmic reticulum (ER) serves as an initiation site for autophagosomes and is also remodeled in response to nutrient stress through ER-phagy, a form of selective autophagy. Quantitative proteome analysis during nutrient stress identified an unstudied single-pass transmembrane ER protein TEX264 as a ER-phagy receptor. TEX264 uses an LC3-interacting region (LIR) to traffic into ATG8-positive puncta that often initiate from 3-way ER tubule junctions and subsequently fuse with lysosomes. Interaction and proximity biotinylation proteomics identified a cohort of autophagy regulatory proteins and cargo adaptors located nearby TEX264 in a LIR-dependent manner. Global proteomics and ER-phagy flux analysis revealed stabilization of a cohort of ER proteins in TEX264−/− cells during nutrient stress. This work reveals TEX264 as an unrecognized ER-phagy receptor that acts independently of other candidate ER-phagy receptors to remodel the ER during nutrient stress.
Graphical Abstract
eTOC blurb
TEX264, an endoplasmic reticulum (ER) resident protein, remodels sub-domains of the ER into ring-like structures in association with ATG8 proteins upon nutrient stress, which then fuse with lysosomes for ER turnover. TEX264 accounts for ~50% of the ER-phagic flux during starvation.
INTRODUCTION
Macroautophagy is a process wherein cellular proteins and organelles are captured within a double membrane vesicle called an autophagosome prior to fusion with a lysosome and degradation of the captured material by lysosomal proteases (Anding and Baehrecke, 2017). As such, autophagy contributes to the availability of amino acids and other cellular building blocks in times of need. Autophagy is a highly regulated process involving the ULK1-RB1CC1 protein kinase complex, the VPS34 phosphatidylinositol-3-phosphate (PI3P) lipid kinase complex (PIK3C3, PIK3R4, BECN1, ATG14), and an ATG7-driven conjugation system that links the ubiquitin-like ATG8 proteins to phosphatidylethanolamine on autophagosomal membranes (Hurley and Young, 2017; Wirth et al., 2013). Continuous supply of nutrients suppresses autophagy, in part, through molecular target of rapamycin (MTOR)-mediated phosphorylation of the ULK1 complex, which blocks its ability to activate autophagy (Hurley and Young, 2017; Wirth et al., 2013). In response to nutrient deprivation or inhibition of MTOR, these systems promote a burst of autophagosome production in a process referred to as macroautophagy. ATG8 conjugation promotes expansion and maturation of autophagosomes and is critical for fusion with lysosomes through a poorly understood mechanism (Mizushima et al., 2011). Selective autophagy is used to capture damaged organelles or protein aggregates for delivery to the lysosome. Here, ATG8 proteins on growing autophagosomal membranes are thought to directly bind cargo receptors, including SQSTM1 (also called p62), OPTN, TAX1BP1, and CALCOCO2 (also called NDP52), that are assembled on cargo through a variety of mechanisms (Harper et al., 2018; Khaminets et al., 2016). The interaction of ATG8 proteins, 6 family members of which exist in humans (MAP1LC3A, B, C, GABARAP, GABARAPL1, L2), with cargo receptors involves a short motif in the cargo receptor referred to as the LC3 interaction region (LIR) and a hydrophobic pocket in ATG8 proteins (Khaminets et al., 2016).
A major question concerns the extent to which individual cellular proteins or organelles are degraded upon activation of macroautophagy via starvation signals – in other words, how selective is macroautophagy? Previous studies have used Stable Isotope Labeling with Amino acids in Cell culture (SILAC)-based proteomics in cell lines displaying increased basal autophagic flux as a result of KRAS mutation, leading to the finding that acute starvation promotes dramatic changes in the proteome and that defects in autophagy suppress innate immune signaling through stabilization of IRF3 (Mathew et al., 2014). Additional experiments suggest that autophagy receptors are rapidly degraded by endosomal microautophagy (Mejlvang et al., 2018). At the same time, evidence is accumulating that even in the absence of the ATG8 conjugation, turnover of cytosolic contents may occur through a lysosomal pathway that still requires the kinase-driven arms of the pathway (An and Harper, 2018; Nguyen et al., 2016; Nishida et al., 2009; Pontano Vaites et al., 2017; Tsuboyama et al., 2016). In order to address specificity of the autophagy system, we performed a global analysis of proteome abundance employing Tandem Mass Tagging (TMT) quantitative proteomics in HEK293T (293T) cells with or without the ATG8-activating enzyme ATG7 or the ULK1 complex subunit RB1CC1 in response to either amino acid withdrawal (−AA) or MTOR inhibition. This approach allowed us to quantify >8000 proteins in triplicate or quadruplicate analyses in each genotype, revealing a cohort of primarily cell cycle proteins whose abundance is decreased independent of autophagy, as well as a set of proteins that appear to undergo degradation via autophagy in an ATG8-conjugation dependent manner. Among the ATG8-conjugation dependent cohort were proteins linked with general and selective autophagy, as well as proteins linked with the endoplasmic reticulum (ER).
The ER has dual links with the autophagy system. First, the initiation of autophagosome production, for example in response to nutrient deprivation, involves the formation of an omegasome, a membrane structure that emerges from ER and is marked by PI3P via VPS34. PI3P serves to recruit WIPI proteins, DFCP1 and the ATG8 conjugation machinery, ultimately leading to the expansion of the omegasome into the isolation membrane (IM)/phagophore (Hayashi-Nishino et al., 2009; Hurley and Young, 2017; Ktistakis and Tooze, 2016). During this process, lipidated ATG8 becomes embedded in the growing phagophore. Second, the ER itself is subject to remodeling through a form of selective autophagy called ER-phagy (Anding and Baehrecke, 2017; Dikic, 2018; Smith and Wilkinson, 2017). ER-phagy in mammalian cells has primarily been studied in response to nutrient deprivation, or upon overexpression of LIR motif containing ER-phagy regulators that can promote removal of particular regions of the ER network. The LIR containing proteins FAM134B and RTN3L contain reticulon helical domains that are inserted into bent ER membranes (primarily curved sheets in the case of FAM134B and tubules in the case of RTN3L) (Grumati et al., 2017; Khaminets et al., 2015). These proteins appear to participate in distinct types of ER-phagy. FAM134B overexpression leads to fragmented ER and enhanced degradation of SEC61B, a protein that is concentrated in ER sheets (Khaminets et al., 2015). Similarly, overexpression of RTN3L promotes fragmentation and turnover of ER tubules (Grumati et al., 2017). To date, there is no evidence that FAM134B or RTN3L interact either physically or genetically to promote ER-phagy, but ER turnover by starvation is reduced by deletion of either FAM134B or RTN3L, indicating that these pathways are independently contributing to ER homeostasis (Grumati et al., 2017). Previous studies have also suggested that SEC62, a protein that functions as part of the SEC61 ER-translocon for co-translational insertion of ER-resident and secreted proteins into the ER lumen, functions as a selective ER-phagy receptor during recovery from ER stress (Fumagalli et al., 2016). Moreover, the ER transmembrane protein CCPG1 has also been shown to be recruited into autophagosomal puncta in response to starvation, and interacts with LC3 through a LIR motif (Smith et al., 2018). Analogous systems have also been reported in budding yeast (Mochida et al., 2015). The extent to which any of these candidate ER-phagy regulators are linked with mobilization of ER membranes to promote phagophore formation in response to starvation is unclear, as is whether additional pathways for regulation of ER turnover exist (Dikic, 2018). Given that ER constitutes over 50% of total cellular membrane and ~10% of total cellular volume, it is plausible that multiple ER membrane resident proteins may function together to mediate efficient and timely turnover of ER in response to diverse signals (Ktistakis and Tooze, 2016).
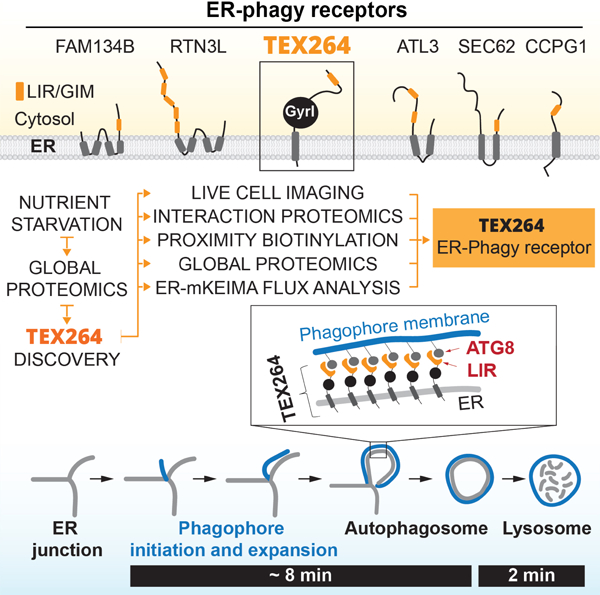
Through our global proteomic analysis of cells after AA withdrawal or MTOR inhibition, we identified a previously unstudied protein TEX264 (testis-expressed protein 264) as a candidate selective autophagy substrate linked with ER-phagy. TEX264 contains an N-terminal hydrophobic segment and a cytosolic gyrase inhibitor (Gyrl)-like domain followed by an unstructured C-terminal region. Interaction proteomics, proximity biotinylation, live-cell imaging, global protein abundance measurements, and ER-phagy flux experiments demonstrate that: 1) TEX264 is localized throughout the ER network, 2) TEX264 uses a C-terminal LIR motif to interact selectively with a subset of ATG8 family members in a lipidation-dependent manner, 3) TEX264 is degraded through autophagy in a LIR-dependent manner, a property reminiscent of autophagic cargo adaptors, 4) during starvation, TEX264 is trafficked into LC3A/B-positive phagophores that are tethered to mobile ER tubule 3-way junctions and then fuses with lysosomes, 5) deletion of TEX264 leads to partial stabilization of a large cohort of ER proteins in response to starvation, and 6) increasing or decreasing TEX264 abundance can alter ER-phagy flux as assessed by multiple ER-phagy flux reporters. We conclude that TEX264 is a critical determinant of ER-phagy in response to starvation and propose a working model for how TEX264 is trafficked from tubular ER to growing autophagosomes via its cytosolic LIR motif.
RESULTS
Quantitative analysis of proteome abundance in response to AA withdrawal and MTOR inhibition
We examined proteome abundance in 293T cells with or without ATG7 or RB1CC1 using TMT proteomics (see STAR METHODS). Triplicate or quadruplicate cultures were either left untreated or incubated in media lacking AAs (−AA) or containing the MTOR inhibitor Torin1 for 10 h, and total cell extracts processed for TMT and analyzed by synchronous precursor selection-MS3 methods (McAlister et al., 2014) (Figure 1A). Immunoblotting of cell extracts demonstrated that phosphorylation of T389 in S6K, a nutrient regulated event, was fully inhibited by AA withdrawal or MTOR inhibition and the abundance of SQSTM1 was also reduced only in WT cells, as expected for this cargo receptor (Figure 1B, S1A). More than 8000 proteins were quantified across all conditions (Figure 1C, S1C–E, Table S1). In response to AA withdrawal, we identified 340 and 17 proteins in WT cells, respectively, whose abundance was reduced or increased by > 1.4-fold (0.5 in Log2 scale) (p<0.01, q<0.05) (Figure 1C, Table S1). This compares with 353 and 17 proteins in ATG7−/− cells, and 226 and 16 proteins in RB1CC1−/− cells, respectively (Figure 1C). Among 340 proteins decreased in WT cells, 240 proteins were detected in all three genotypes, passed p < 0.01 filter, and had more than 1 peptide quantified (Figure 1D, F). In total, the abundance of 82 proteins was reduced by more than 40% regardless of the absence of ATG7 or RB1CC1 upon AA withdrawal (Figure 1D). These proteins were enriched in cell cycle and cytoskeletal regulators (Figure 1E), consistent with reduced rates of cell proliferation upon AA withdrawal, as measured in parallel (Figure S1B). A similar analysis of proteins whose abundance was decreased specifically in WT cells (>30% total abundance difference between WT and ATG7−/−) identified 22 proteins, including autophagy adaptors/receptors (SQSTM1, GABARAPL2, and CCPG1) (Khaminets et al., 2016; Smith et al., 2018), several proteins known to be targeted by selective forms of autophagy (centriolar satellites) (Tang et al., 2013), and 9 proteins not previously linked with autophagy-dependent turnover (Figure 1F,G) (p<0.01, q<0.05). Broadly similar results were obtained with Torin1 treated cells (Figure S1F–H, Table S1). The abundance of 20 proteins was found to be reduced in an ATG7 and RB1CC1-dependent manner 10h after Torin1 treatment (Figure S1F,H). Overall, there was a 0.72 correlation coefficient (Pearson’s) when comparing changes with Torin1 versus AA withdrawal (Figure S1I). Interestingly, the abundance of SLC38A2, a glutamine transporter, and SESN3, the cytosolic receptor for leucine, were among the most significantly changing proteins only upon AA withdrawal and not affected by Torin1 treatment. These data provide a resource for the identification of proteins whose abundance is under nutrient and/or MTOR control through the autophagy system.
Figure 1. Quantitative analysis of cellular proteome remodeling in response to nutrient stress.

(A) Overview of quantitative proteomics approach to examine proteome remodeling. The indicated cells were left untreated (UT), or incubated for 10h in media either lacking AAs (−AA) or containing Torin1 (150 nM). Total cell extracts were processed for TMT-MS3 analysis.
(B) Immunoblotting of extracts used for TMT proteomics with the indicated antibodies. Lower panel represents the total protein abundance present on immunoblots.
(C) Volcano plots (−Log10 p-value versus Log2 ratio of -AA/UT for 293T WT, ATG7−/−, or RB1CC1−/− cells as described in panel A. Proteins with Log2(−AA/UT) < −0.5 or > 0.5 (p-value <0.01) are indicated as colored empty circles, and filled colored circles indicate statistically significant hits (Welch’s t-test (S0 = 0.585), corrected for multiple comparison by permutation-based FDR (5%)).
(D) Among 240 proteins with Log2(−AA/UT) value lower than −0.5 in WT HEK293T cells and quantified in all three genotypes with more than one peptide (p < 0.01 in all three), Log2(−AA/UT) values for 82 individual proteins (top) or the average value (lower) whose abundance was reduced (p<0.01) by log2(−AA/UT) < −0.5 across WT, ATG7−/− and RB1CC1−/− cells are indicated. Data are represented as mean ± SEM for triplicate or quadruplicate measurements.
(E) Gene ontology (GO) analysis of proteins from panel D. The lower portion of the panel shows the results of Functional Annotation Clustering using the DAVID Bioinformatics Resource V6.8 (https://david.ncifcrf.gov/home.jsp).
(F) Log2(−AA/UT) values for 22 individual proteins (top) or the average value (lower) whose abundance was reduced (p<0.01) by log2(−AA/UT) <−0.5 in WT cells but not in ATG7−/− and RB1CC1−/− cells. These 22 proteins showed more than 30% abundance difference between WT and ATG7−/−. Data for RB1CC1−/− is indicated as grey because proteins with p value higher than 0.01 are included in RB1CC1−/− cells. Data are represented as mean ± SEM.
(G) Heat map of Log2(−AA/UT) values for 22 individual proteins from panel F. See also Figure S1 and Table S1.
TEX264 is an ER protein that accumulates in LC3B-positive puncta during nutrient stress
Among the proteins whose abundance was reduced by both AA withdrawal and MTOR inhibition in an ATG7 and RB1CC1-dependent manner was a previously unstudied protein, TEX264 (Figure 1C,G, S1D–E,H). The magnitude of reduction found by TMT for TEX264 was comparable to that of SQSTM1 measured in parallel (Figure 2A). As such, we selected TEX264 for further validation as a candidate selective autophagy substrate. Immunoblotting of cell extracts from 293T, HEK293 (293), or HCT116 cells with AA withdrawal (10 or 14h) confirmed a reduction in TEX264 protein and this reduction was blocked upon removal of ATG7, ATG5, or RB1CC1 (Figure 2B–D, S2A). TEX264 turnover in 293T cells in response to AA withdrawal was blocked by addition of BafA, indicating lysosomal turnover (Figure S2A). This result was independently verified in 293 cells with AA withdrawal (10h) or MTOR inhibition (Torin1, 24h) using TMT-based proteomics (Figure S2B).
Figure 2. TEX264 is a resident ER protein that is degraded by autophagy in response to nutrient deprivation.

(A) TMT based quantification of TEX264 and SQSTM1 abundance in 293T cells in response to AA withdrawal (10h) or MTOR inhibition with Torin1 (10h). Data are derived from Table S1. Data are represented as mean ± SD for triplicate or quadruplicate measurements.
(B, C) The indicated 293T cells were either left untreated of subjected to AA withdrawal (10h) prior to immunoblotting of cell extracts using the indicated antibodies (panel B). Equal loading of extracts is demonstrated by Ponceau S staining. Data are represented as mean ± SD for triplicate or quadruplicate measurements.
(D) The indicated 293T, 293, or HCT116 cells were either left untreated of subjected to AA withdrawal (14h) prior to immunoblotting of cell extracts using the indicated antibodies.
(E, F) COS7 cells expressing TEX264-eGFP and RFP-KDEL (panel E) or the indicated cells gene edited to express endogenous TEX264-eGFP (panel F) were either left untreated or subjected to nutrient deprivation (EBSS and BafA, 3h) prior to confocal microscopy. Scale bar in panel E = 20 µm, in panel F = 10 µm.
(G) Schematic displaying of the properties of Keima that allow it to be used to monitor flux into the lysosome, where the acidic environment causes an increase in the ratio of 561 nm/488 nm excitation. Once in the lysosome, the Keima fusion protein is “processed” to degrade the fusion protein, but the Keima fragment is resistant to lysosomal proteases and maintains its fluorescence within the lysosome.
(H) TEX264-Keima was stably expressed in HCT116 cells with or without ATG5 and sorted by flow-cytometry for equal expression. The cells left untreated or subjected to AA withdrawal (5h) prior to live cell imaging by confocal microscopy. Scale bar = 10 µm.
(I,J) Stable HCT116 cells prepared as in panel H were left untreated or subjected to Torin1 (18h) with or without BafA (added 1h before analysis) prior to analysis by flow cytometry to measure the ratio of 561 nm/488 nm excitation in single cells (panel I). A plot of cell count versus fluorescence ration for 561/488 nm is shown in panel J. Data are represented as mean ± SD for triplicate measurements.
(K) TEX264-Keima was stably expressed in HCT116 cells with or without ATG5 and cells left untreated or subjected to SAR405, Torin1 or AA withdrawal (15h). Cell extracts were subjected to immunoblotting with the indicated antibodies. The position of processed Keima is shown. Asterisk (red) indicates a band resulting from N-acyl group hydrolysis in Keima chromophore during denaturation, leading to a loss of C-terminus amino acids (18.3 kDa) from intact TEX264-Keima as reported in (An and Harper, 2018). See also Figure S2.
Although annotated as a secreted protein in UniProt (www.uniprot.org), TEX264 has a candidate N-terminal transmembrane segment [probability 0.94 using the transmembrane-hidden Markov model (TMHMM) program, see STAR METHODS], with a short N-terminal sequence predicted to be luminal and the larger C-terminal domain predicted to be cytosolic. Consistent with membrane localization, we found extensive co-localization between TEX264-eGFP and the pan-ER marker RFP-KDEL in COS7 cells, which have a flat morphology facilitating microscopic analysis of reticular ER, as well as in HCT116 cells (ER-tracker red) (Figure 2E, S2C). To verify ER localization, we used CRISPR-Cas9 (Ran et al., 2013) to create HCT116, 293 and 293T cells containing a homozygous C-terminal eGFP fusion with TEX264, which was also shown to localize in ER in all cell lines (Figure 2F, S2D,E).
Nutrient deprivation promotes autophagic removal of ER (Grumati et al., 2017). In order to examine TEX264 trafficking and dynamics in response to nutrient stress, we first incubated endogenously tagged TEX264-eGFP cells (HCT116, 293T, 293), HCT116 cells stably expressing exogenous TEX264-eGFP (2.4-fold over endogenous TEX264), or COS7 cells stably expressing TEX264-eGFP in nutrient stress conditions (Earle’s balanced salt solution, EBSS) with or without BafA, and examined cells by confocal microscopy (Figure 2E,F, S2F,G). In response to AA withdrawal, TEX264-GFP accumulated in cytosolic puncta in all cell lines tested, with puncta easily detected within 1h in HCT116 cells stably expressing TEX264-eGFP (Figure S2F). Image analysis of TEX264-eGFP knock-in cells revealed a mean of ~4 and ~24 puncta/cell, without and with 3h nutrient stress, respectively, for HCT116 and 293 cells (>0.7 micron puncta size), although 293T cells displayed ~3-fold fewer puncta under both conditions (Figure 2F, S2G). Analogous results were observed in COS7 cells (Figure 2E). Importantly, endogenous TEX264-eGFP puncta were frequently co-incident with mCherry-LC3B-positive autophagosomal structures (Figure S2H) (Pearson’s correlation = 0.65), consistent with their incorporation into autophagosomes.
TEX264 is targeted to the lysosome for degradation
The accumulation of TEX264-eGFP in LC3B-positive puncta is consistent with turnover of TEX264 via autophagy. To directly examine autophagic flux of TEX264, we fused TEX264 with Keima, a pH-responsive reporter that undergoes a chromophore resting charge state change upon trafficking to the lysosome (pH of ~4.5), allowing flux measurements by flow cytometry or microscopy to be performed by measuring the ratio of 561 nm/488 nm excitation (Figure 2G) (Katayama et al., 2011). Keima itself is also stable to lysosomal proteases in mammals, and the appearance of a “processed” Keima fusion protein (wherein the cellular fusion protein is cleaved) therefore reveals lysosomal trafficking (Figure 2G) (An and Harper, 2018). TEX264-Keima was stably expressed in WT or ATG5−/− HCT116 cells at 2.7-fold higher levels than endogenous TEX264. Upon AA withdrawal (5h), we observed an increase of 561 nm excitation TEX264-Keima puncta in ATG5+/+ cells by confocal microscopy, consistent with trafficking of TEX264-Keima to the lysosome, and this was greatly diminished in ATG5−/− cells (Figure 2H). Parallel flow cytometry (Figure 2I,J) demonstrated that control cells displayed 561 nm/488 nm TEX264-Keima signal that was reduced by addition of BafA, indicating ongoing targeting of TEX264 to the lysosome under basal conditions (Katayama et al., 2011). This result is consistent with the immunoblot analysis of endogenous TEX264 using 293T cells (Figure S2A). The flux of TEX264-Keima through the autophagy system was increased 3.2-fold upon incubation with Torin1 (18h) compared with the basal flux, and this was blocked by BafA and was absent in cells lacking ATG5 (Figure 2I,J). Finally, we examined “processing” of TEX264-Keima (Figure 2G) using immunoblotting. In WT untreated cells, a fraction of α-Keima signal was observed at ~25 KDa (“processed” Keima) and this was markedly reduced upon treatment with SAR405, a VPS34 inhibitor that blocks the autophagy system (Ronan et al., 2014), consistent with ongoing basal turnover of TEX264-Keima through autophagy (Figure 2K). This signal was increased upon either AA withdrawal (2.7-fold) or addition of Torin1 (2-fold), and was abolished in ATG5−/− cells (Figure 2K). Taken together, these data reveal that TEX264 is an ER protein that undergoes basal autophagic flux, and this process is accelerated in response to nutrient deprivation or MTOR inhibition.
TEX264 associates with ATG8 family members through its C-terminal LIR motif
Given the accumulation of TEX264 in LC3B-positive puncta, we examined whether TEX264 may interact directly with ATG8 modifiers through a LIR motif. In addition to its N-terminal transmembrane segment, we identified a GyrI-like domain related to several prokaryotic proteins including the bacterial Gyrase Inhibitor with a sheet-helix-sheet-sheet architecture (Anantharaman and Aravind, 2004), located in the central region of TEX264’s cytosolic domain. Moreover, we found a candidate LIR motif (F273EEL) in TEX264’s predicted unstructured C-terminus (residues 185–313) and an additional candidate sequence at the end of the presumptive transmembrane segment (Y27SGL), both of which are conserved from zebrafish to human (Figure 3A). We expressed TEX264-eGFP Y27A and F273A mutant constructs in HCT116 cells, then we sorted the cells using flow-cytometry to equalize expression levels to near endogenous levels. Both proteins localized to reticular ER structures, as expected (Figure 3B left panel). In response to nutrient deprivation in the presence of BafA (1h), we found that both WT and Y27A TEX264-eGFP proteins mobilized into ring-shaped structures with equal efficiency (Figure 3B). In contrast, TEX264F273A-eGFP failed to form puncta to an extent similar to HCT116 ATG5−/− cells expressing WT TEX264-eGFP, indicating that the C-terminal LIR motif is required to mobilize TEX264 into autophagic structures (Figure 3B). Moreover, α-GFP immune complexes from WT TEX264-GFP cells contained LC3B and the amount of LC3B associated with TEX264 increased (~3-fold) in the context of AA withdrawal and BafA (3h) (Figure 3C). However, this interaction was dramatically reduced with either TEX264F273A-eGFP or with WT TEX264-eGFP in cells lacking ATG5 (Figure 3C). As expected, GFP control constructs failed to associate with LC3B.
Figure 3. TEX264 associates with specific ATG8 family members and is targeted for autophagy through a C-terminal LIR motif.

(A) Domain structure of TEX264 showing the transmembrane segment, the GyrI-like domain, and the position of two candidate LIR motifs. The sequences on individual LIR motifs are shown below.
(B) The indicated TEX264-eGFP proteins were expressed in HCT116 cells with or without ATG5 and cells either left untreated or treated with EBSS and BafA (1h) prior to imaging by confocal microscopy. Quantification of TEX264-GFP puncta number per cell after starvation is shown in the right panel. Data are represented as mean ± SEM. Scale bar = 10 µm.
(C) Lysates from the indicated HCT116 cells with or without ATG5 stably expressing the indicated TEX264-eGFP protein or HA-FLAG-eGFP as a control were subjected to immunoprecipitation with α-GFP antibodies. Immune complexes or whole cell lysates were subjected to immunoblotting with the indicated antibodies.
(D) HCT116 TEX264−/− cells were reconstituted with WT or mutant TEX264-eGFP at near endogenous levels followed by clonal selection. Cells were left untreated or subjected to AA withdrawal in the presence of BafA (3h) in duplicate or triplicate and extracts subjected to immunoblotting with the indicated antibodies.
(E) Cells from panel D were subjected to α-GFP immunoprecipitation prior to analysis by TMT-MS3. Volcano plots of -Log10(p-value) versus Log2(TEX264F273A/WT) in either untreated conditions or upon AA withdrawal in the presence of BafA (3h) are shown. Proteins with Log2(TEX264F273A/WT ) < −0.5 or > 0.5 (p-value <0.05) are indicated as colored empty circles, and filled colored circles indicate statistically significant hits (Welch’s t-test (S0 = 0.585), corrected for multiple comparison by permutation-based FDR (5%))
(F) Label-free quantification of MS1 precursor ions present in α-GFP immune complexes from panel D (relative intensity). Data are represented as mean ± SD (n=2 or 3, as shown in panel D).
(G) The indicated cells were subjected to SAR405 treatment, AA withdrawal or Torin1 treatment for the indicated times and cell extracts subjected to immunoblotting with the indicated antibodies. See also Figure S3 and Table S2.
In order to systematically examine both the association of TEX264 with the 6 ATG8 family members and to look for associated proteins, we performed unbiased affinity purification-mass spectrometry (AP-MS) using HCT116 TEX264−/− cells reconstituted with either WT TEX264-eGFP or TEX264F273A-eGFP at near endogenous levels (Figure S3A,B). Cells were either left untreated or starved of AAs for 3h in the presence of BafA in duplicate or triplicate (Figure 3D). Immunoblotting of cell extracts revealed loss of S6K pT389 and 4EBP1 phosphorylation, and conversion of LC3B-I to LC3B-II, as expected (Figure 3D). α-GFP immune complexes were subjected to TMT-MS3 analysis and proteins associated with TEX264 visualized using a Volcano plot of p-value versus the Log2-Ratio for the F273A mutant compared with WT in either untreated cells or after AA withdrawal (Figure 3E, Table S2). In untreated and starved cells, we observed strong LIR-dependent association between LC3B (MAP1LC3B), as well as the 3 GABARAP isoforms. Because the TMT experiment doesn’t provide an indication of relative abundance across the ATG8 isoforms, we extracted label-free quantification of precursor ions (MS1) for individual ATG8 isoforms to estimate the relative abundance of ATG8 isoforms in association with TEX264-eGFP (Figure 3F) in the different 11 TMT channels. The most abundant interaction with WT TEX264-eGFP in HCT116 was with LC3B and GABARAP, with very low levels of GABARAPL1 and L2 detected. Among these, LC3B and GABARAPL2 increased 3-fold upon AA withdrawal, whereas association with other GABARAPs was unchanged (Figure 3F). We were not able to examine LC3A independently in this experiment as the only peptides found for LC3A were shared with LC3B, and we did not detect LC3C in this experiment. The absence of endogenous LC3A association with TEX264-eGFP likely reflects the very low levels of LC3A relative to LC3B in HCT116 cells (Figure S3C). As an alternative approach for examining association with ATG8 proteins, we stably expressed all 6 ATG8 proteins as GFP fusions at equal levels in HCT116 cells. α-GFP immune complexes revealed association of endogenous TEX264 with LC3A and LC3B, both of which increased ~3-fold upon nutrient stress, as well as association with GABARAPL1, which did not undergo detectable increase in association upon AA withdrawal (Figure S3D). In addition to ATG8 proteins, we also observed LIR-dependent association of TEX264-eGFP with components of the VPS34 lipid kinase important for initiation of autophagy (PIK3C3/VPS34, PIK3R4/VPS15, BECN1, and ATG14). Like GABARAP proteins, interaction with the VPS34 complex occurred in both untreated and starvation conditions but was enriched in WT versus F273A mutant TEX264 (1.6~1.9-fold enrichment) (Figure 3E). These results were validated by immunoprecipitation followed by immunoblotting (Figure S3E). Moreover, we observed LIR-dependent association with TMEM97 (also called sigma-2 receptor), a multi-pass transmembrane protein known to localize in the ER, as well as RGS3 (Figure 3E).
TEX264 turnover through autophagy requires its LIR motif
The association of TEX264 with ATG8 family members through a LIR motif together with ATG5-dependent turnover of TEX264 led us to examine the effect of the F273A mutant on autophagic flux in HCT116 cells. We found that, unlike TEX264WT-Keima, TEX264F273A-Keima did not undergo lysosomal processing in response to either AA withdrawal or MTOR inhibition (Figure 3G). This, together with the ATG5-dependent targeting of TEX264 to the lysosome, indicates that the C-terminal LIR motif in TEX264 targets the protein to the autophagy system through direct interaction with ATG8 proteins.
TEX264 accumulates at ER tubule junctions previously marked by ATG8
To visualize TEX264 trafficking in relation to ER and autophagosomal structure, we expressed TEX264-eGFP in COS7 cells together with mCherry-LC3A and performed live-cell imaging. We frequently observed the emergence of small mCherry-LC3A puncta near 3-way junctions of ER tubules, followed by the appearance of TEX264 puncta co-localizing with LC3A (Figure 4A). These TEX264/LC3A puncta remain tethered at or nearby 3-way junctions over the next several minutes as ER tubules undergo significant sliding through the junction architecture (Figure 4A, Movie S1). This behavior was observed with multiple independent LC3A as well as LC3B-positive/TEX264-positive puncta in multiple experiments (Figure 4A, S4A,B). We monitored the increase in TEX264-eGFP signal intensity and puncta size in time lapse images, finding that the average time from the initial detection of TEX264 concentration to maximal size (0.8~1.1 microns, ± 0.109 µm resolution limit) was ~8 min (Figure 4B,C).
Figure 4. TEX264 accumulates in ATG8-positive punctate structures at ER tubule 3-way junctions followed by fusion with lysosomes.

(A) COS7 cells stably expressing TEX264-eGFP and mCherry-LC3A were subjected to confocal imaging during nutrient withdrawal. Time-lapse images of individual regions of the cell cortex containing an LC3B-positive punctate structure located near a ER tubule 3-way junction are shown. Scale bar = 1 µm. Also see Movie S1.
(B, C) Examples of time-lapse images showing the accumulation of TEX264-eGFP into puncta over an ~8 min time period (panel B). Quantification of 10 such events is provided in panel C. Data are represented as mean ± SEM (n=10).
(D) COS7 cells stably expressing TEX264-eGFP and treated with lysotracker red were subjected to confocal imaging during starvation. Time-lapse images of individual regions of the cell cortex containing a lysotracker red-positive lysosome fusing with a TEX264-eGFP-positive ring-structure are shown. Scale bar = 1 µm. Also see Movie S2.
(E) HCT116 TEX264−/− cells expressing TEX264-APEX2 were starved (EBSS) in the presence of BafA for 3h prior to H2O2 and DAB treatment. Cell thin sections were examined by electron microscopy. A region of DAB-positive perinuclear ER is indicated by the yellow arrow. Red arrows indicate DAB staining in autophagosome/autolysosome-like structures. Case 1 employs near endogenous levels of TEX264-APEX2 and Case 2 employs cells transiently expressing TEX264-APEX2. Scale bar = 1 µm.
(F) COS7 cells stably expressing TEX264-eGFP and mCherry-LC3B were subjected to confocal imaging during starvation in the presence of BafA (3h). Scale bar in the enlarged images = 1 µm. See also Figure S4 and Movies S1 and S2.
We next sought to examine the integration of TEX264 containing puncta into lysosomes by following TEX264-eGFP puncta in the context of lysotracker-red, which identifies acidic lysosomal vesicles. The initial encounter of an acidic lysosome with a TEX264-eGFP-positive puncta (120 sec in Figure 4D, Movie S2) was followed by partial acidification of the entire TEX264-eGFP-positive puncta at 180 sec and full acidification by 360 sec. Thus, in COS7 cells, TEX264 trafficking from the ER to autophagosomes and subsequent fusion with a lysosome can occur on the order of ~15 min.
Sub-structural analysis of TEX264 targeting to autophagosomes
The topology of TEX264 on the intact ER membrane and presumed engagement of autophagosomal membranes harboring LC3A/B places specific constraints on the location of TEX264 during capture within autophagosomes. To examine autophagosomal engagement, we reconstituted HCT116 TEX264−/− cells with TEX264-APEX2 (Figure S4C) (and when indicated overexpression of TEX264-APEX2) and performed electron microscopy (EM) of cell thin sections after treatment of cells first treated under starvation conditions (EBSS + BafA, 3h) followed by DAB (diaminobenzidine) and H2O2. APEX2 reacts with H2O2 to catalyze the generation of mono-DAB radical at the heme-containing active center, which leads to the osmiophilic polymer production that result in intensified electron density in EM within ~10–20nm of the APEX2 molecule (Hung et al., 2017). We observed dense staining in ER structures including the nuclear periphery (Figure 4E, case 1 yellow arrow), as well as within autophagosome- and autolysosome-like structures (Figure 4E, case 1 and 2 red arrows and Figure S4D). Interestingly, TEX264-APEX2 staining appears juxtaposed to the membrane of autophagosome-like structures, as expected for proteins that associate with ATG8.
We next examined COS7 cells expressing TEX264-eGFP and mCherry-LC3B 3h post starvation in the presence of BafA (Figure 4F). We observed numerous vesicular organelles that contained one or more TEX264-positive ring structures surrounded by diffuse mCherry-LC3B (lysosomal contents) (Figure 4F). This morphology is similar to the electron dense structure observed in DAB staining (Figure 4E case 2, S4D). This places certain constraints on the capture of TEX264 through autophagy, as described in the DISCUSSION. In accordance with this, fluorescence intensity of a cross section of 442ex Keima positive vesicles in starved HCT116 cells expressing TEX264-Keima showed hollow circle structures (Figure S4E,F). However, once acidified after fusion with a lysosome, the cross-section signal intensity of 561ex positive puncta was highest at the center of individual vesicles, suggesting that TEX264 was degraded by the lysosomal hydrolases, which in turn released Keima from the ER-membranes juxtaposed to autophagosomal inner membranes (Figure S4E,F).
Proximity biotinylation identifies proteins nearby TEX264 in autophagosomes
We next sought to elucidate proteins that are nearby TEX264-APEX2, but not TEX264F273A-APEX2, in response to AA withdrawal in the presence of BafA (3h) using TMT-MS3 (Figure 5A). We expected that proximity biotinylation-MS should detect a distinct group of proteins than IP-MS as it detects targets within ~10nm of the APEX2 domain fused to TEX264 regardless of the binding affinity toward TEX264 (Hung et al., 2017), thus providing a complementary picture of LIR dependent TEX264 interactors. Extracts from HCT116 TEX264−/− cells reconstituted with near endogenous level of WT or LIR mutant TEX264-APEX2 (Figure S4C) were subjected to purification on streptavidin beads with stringent washing (Hung et al., 2017) prior to trypsinization and proteomics. Under basal conditions, we identified multiple ER membrane proteins (CANX, CISD2, TMEM43, TMX1), autophagy regulators (ATG14, WIPI2, RAB7A), and autophagy cargo receptors (SQSTM1, CALCOCO2/NDP52, TAX1BP1) enriched in TEX264WT-APEX2 (Figure 5B), as expected given basal turnover of TEX264 via autophagy (Figure 2I). Three hours post AA withdrawal, we identified many of the same ER membrane proteins even further statistically enriched with TEX264WT-APEX2 as well as additional ER localized proteins including LRRC59, CALR, CALU, SCD, NOMO2, and BCAP31 (Figure 5C,D). We validated endogenous CALCOCO2, SQSTM1, CANX biotinylation using immunoblotting of streptavidin purified proteins. Interactions were increased in response to AA withdrawal in the presence of BafA and were largely dependent upon the F273 in TEX264 (Figure S4G). This suggests that, upon starvation, TEX264WT-APEX2 is incorporated into autophagosomes such that it is adjacent to autophagy machinery known to be localized on the concave face of autophagosomal membranes, which includes cargo receptors, while maintaining association with ER proteins. In stark contrast, tubulin biotinylation (a known target of ER-APEX2 probes) was not altered by starvation or in the absence of the LIR motif (Figure S4G).
Figure 5. Proximity biotinylation identifies autophagy machinery nearby TEX264-APEX2 during capture in autophagosomes.

(A) Schematic of proximity biotinylation approach for identification of proteins nearby TEX264-APEX2 during capture in autophagosomes.
(B,C) Lysates from cells from panel A were subjected to streptavidin affinity enrichment under denaturating conditions prior to analysis by TMT-MS3. Volcano plots of -Log10(p-value) versus Log2(TEX264F273A-APEX2/TEX264-APEX2WT) in either untreated conditions or upon AA withdrawal in the presence of BafA (3h). Proteins with Log2-Ratio < −0.5 or > 0.5 (p-value <0.05) are indicated as colored empty circles, and filled colored circles indicate statistically significant hits (Welch’s t-test (S0 = 0.585), corrected for multiple comparison by permutation-based FDR (1%))
(D) Comparison of LIR dependent proximity biotinylation targets for TEX264. A plot of Log2(F273A/WT) in untreated cells versus Log2(F273A/WT) in cells upon AA withdrawal in the presence of BafA (3h). See also Figure S4 and Table S2.
A role for TEX264 in ER-phagy revealed by quantitative proteomics
The data presented thus far indicates that TEX264 traffics from the ER to autophagosomes in response to nutrient stress in a manner that depends on ATG8 conjugation and is degraded in lysosomes. This led us to ask whether TEX264 might contribute to the turnover of ER proteins either generally or in a selective manner. We initially examined whether deletion of TEX264 altered the response of cells to ER stress or increased ER stress under basal conditions. WT and TEX264−/− cells displayed similar sensitivities to DTT and similar activation of ER-stress markers upon treatment with DTT or Thapsigargin (Figure S5A–C). Moreover, we did not observe obvious alterations in ER area in WT versus TEX264−/− cells either in untreated conditions or after treatment with EBSS, DTT, or Thapsigargin, as examined using ER-tracker and an image analysis method described previously with modification (Smith et al., 2018) (Figure S5D).
To examine how AA withdrawal affects global proteome abundance in the absence of TEX264, we performed a TMT-MS3 experiment. 293T WT or TEX264−/− cells were either left untreated or subjected to AA withdrawal (10h) prior to immunoblotting of total cell extracts and TMT-MS3 proteomics (Figure 6A,B, Table S3). S6K, S6 and 4EBP1 phosphorylation was decreased and LC3B-II was increased in both WT and TEX264−/− cells in response to AA withdrawal, indicating that the absence of TEX264 does not affect starvation signals (Figure 6B). We initially compared the proteomes of WT and TEX264−/− cells in untreated conditions (Figure 6C left panel). The abundance of 11 proteins were reduced more than 2-fold and 3 proteins were increased upon loss of TEX264, indicating only minor proteome changes upon TEX264 deletion. As expected, starvation led to a more than 40% reduction in 391 proteins in WT cells and 469 in cells lacking TEX264 (Figure S6A, B). In untreated conditions, 310 ER-localized proteins (based on (Itzhak et al., 2016)) were largely distributed symmetrically within the volcano plot (Figure 6C left panel). However, the distribution of ER proteins changed dramatically and skewed to the right in the volcano plot upon starvation (Figure 6C right panel). Comparison of proteome changes in response to AA withdrawal in WT versus TEX264−/− cells using correlation plots (Figure 6D,E) revealed that: 1) the majority of the proteome is localized along the diagonal (p<0.0001), indicating that the bulk of the proteome change is unaffected by loss of TEX264 (Figure 6D), 2) ER membrane and lumenal proteins are primarily found below the diagonal, indicating that loss of TEX264 leads to stabilization of a broad range ER proteins upon starvation when compared with WT cells (Figure 6D,E), 3) the abundance of other organelles were largely unaffected by loss of TEX264 (p>0.36), indicating selectivity for ER (Figure 6E), 4) degradation of two ER-phagy receptor proteins (RTN3 and SEC62) detected in the TMT analysis were intermediate among ER proteins in terms of protein loss upon AA withdrawal, but with only a small contribution of TEX264 in degradation (Figure S6C,D), suggesting that these receptors may be degraded independently of TEX264. Of note, the average percent decrease of 310 ER proteins (luminal, high curvature, and ER-membrane proteins) after 10h of AA withdrawal was ~7.8% in WT 293T cells, and this value was reduced to ~3.3% in TEX264−/− cells, suggesting that TEX264 alone can be responsible for ~50% of the total ER-phagy flux during starvation in 293T cells (see Table S4).
Figure 6. A role for TEX264 in ER-phagy revealed by quantitative proteomics.

(A) Schematic for global proteome analysis of WT and TEX264−/− 293T cells in response to AA withdrawal (10h). Cell extracts were either used for immunoblotting or for TMT-MS3.
(B) Lysates from cells from panel A were immunoblotted with the indicated antibodies.
(C) Volcano plot of -Log10 (p-value) versus Log2(TEX264−/− vs WT) in 293T cells under untreated conditions (left panel) or under 10h AA withdrawal conditions (right panel). Proteins with Log2-Ratio < −0.5 or > 0.5 (p-value <0.05) are indicated as colored empty circles, and filled colored circles indicate statistically significant hits (Welch’s t-test (S0 = 1), corrected for multiple comparison by permutation-based FDR (1%)). ER marker proteins are in orange.
(D) Correlation plot of data in panel C.
(E) Correlation plots (data from panel C) for nine organelle categories (material and methods). Results (p-value) of a two-tailed Mann-Whitney t-test comparing Log2(−AA/UT) between TEX264WT and TEX264−/− cell line is indicated for each organelle. See also Figure S5, S6 and Table S3.
TEX264 regulates ER-phagic flux
The finding that deletion of TEX264 stabilized ER protein abundance in response to starvation led us to directly examine the extent to which the abundance of TEX264 determines ER-phagic flux. Because the relatively small reductions in ER protein abundance measured by mass spectrometry (~10% for many ER proteins, Table S4) is difficult to directly visualize by immunoblotting (Figure S7A), we turned to flux measurements with Keima. For this, we selected two classes of ER proteins as candidate flux reporters. First, we adapted the previous ER-phagy reporter protein RAMP4 to the Keima system. RAMP4 (7.4 kDa) is localized to both ER tubules and sheets and has been validated as an ER-phagy reporter upon starvation (Liang et al., 2018). Fusion of Keima to the RAMP4 N-terminus places Keima in the cytosol, and trafficking of ER to the lysosome leads to cleavage of the Keima-RAMP4 fusion (33.9 kDa) by resident proteases to generate “processed” Keima (25 kDa) (Figure 7A). We found that HCT116 cells lacking TEX264 displayed reduced “processed” Keima in response to AA withdrawal when compared with WT cells (Figure 7B,C, S7B). The impact on Keima processing was about 50% of that seen with in ATG5−/− cells performed in parallel (Figure 7B,C, S7B). Moreover, we stably expressed either TEX264-eGFP or TEX264F273A-eGFP in HCT116 cells, thereby increasing the abundance of total TEX264 by ~3-fold, and examined Keima-RAMP4 processing (Figure 7D,E, S7C). We found that WT TEX264, but not the LIR mutant, increased “processed” Keima in full media, indicating that higher levels of TEX264 can promote ER-phagic flux under basal conditions (Figure 7D,E, S7C). TEX264 overexpression also increased Keima processing under starvation conditions and this also required the LIR (Figure 7D,E, S7C). As we previously reported (An and Harper, 2018), the hydrolysis of N-acyl group in Keima chromophore during SDS-PAGE sample denaturation resulted in the loss of N-terminal amino acids in Keima (6.7 kDa), generating a band corresponding to 27.2 kDa (asterisk in Figure 7B,D) that is not related to “processed” Keima.
Figure 7. TEX264 status determines ER-phagic flux.

(A) Schematic of Keima-RAMP4 as a reporter for ER-phagy. See text for details.
(B, C) Cells lacking TEX264 display reduced ER-phagic flux. HCT116 WT or TEX264−/− cells expressing the Keima-RAMP4 ER-phagy reporter were either left untreated or subjected to starvation (EBSS, 20h) and cell extracts analyzed for Keima-RAMP4 and “processed” Keima using immunoblotting (panel B). Quantification (Odyssey) of immunoblot signals from duplicate experiments are shown in panel C. Data are represented as mean ± SD for duplicate experiments. Asterisk (red) indicates a band resulting from N-acyl group hydrolysis in Keima chromophore during denaturation, leading to a loss of N-terminus amino acids (6.7 kDa) from intact Keima-RAMP4 protein.
(D, E) Overexpression of TEX264 promotes ER-phagy. HCT116 cells (WT or ATG5−/−) expressing Keima-RAMP4 and either TEX264-eGFP or TEX264F273A-eGFP were left untreated or subjected to starvation (EBSS, 20h). Cell extracts were examined for Keima-RAMP4 and “processed” Keima by immunoblotting (panel D). Quantification (Odyssey) of immunoblot signals from triplicate experiments are shown in panel E. Data are represented as mean ± SD for triplicate experiments.
(F) Histogram showing the relative abundance of processed Keima derived from either Keima-RAMP4, RTN4-Keima or EPHX1-Keima in response to MTOR inhibition (16h) (SD, n=3).
(G) Model for TEX264 in the ER membrane. The structure of the GyrI-like domain was predicted based on Swiss-model based molecular modeling (see STAR METHODS).
(H) Working model for capture of TEX264-positive ER. The red structure is the isolation membrane (IM) and extends through incorporation of vesicles. Expansion of the IM then allows interaction of ATG8 proteins (primarily LC3A and B) on the phagophore with TEX264 on nearby ER tubules. The interaction of ATG8 proteins with TEX264 may be analogous to a “zipper” allowing the ER membrane to come into close contact with the inner phagophore membrane through a trans interaction. Precisely how the IM is cleaved from the ER and how ER tubules may be cleaved is unknown. See also Figure S7 and Table S4. p****< 0.0001, p***<0.001, p*<0.1
In a second approach, we selected 4 ER proteins whose abundance in 293T cells was decreased upon nutrient stress in a partially TEX264-dependent manner (RTN4, EPHX1, REEP4, and PTDSS) (Figure S7D, Table S4), and examined their mRNA levels together with three ER-phagy receptors in WT and TEX264−/− cells before and after amino acid withdrawal (Figure S7E). RT-qPCR results indicated negligible difference between the two TEX264−/− clones and WT cells both in nutrient rich and deficient conditions, thus ruling out the possibility that the TEX264 dependent decrease of these ER proteins upon starvation (Figure S7D) was caused by altered transcription. Next, we measured autophagic flux of RTN4 and EPHX1 individually using ER-localized Keima Fusion proteins (Figure S7F). When introduced to HCT116 TEX264−/− cells reconstituted with TEX264WT-GFP or TEX264F273A-GFP, both constructs localized to ER-like structures and had low basal flux as measured by “processed” Keima. This flux was increased upon nutrient stress with MTOR inhibition in a manner that was partially dependent upon the LIR in TEX264 (Figure S7F), consistent with a role for TEX264 in their turnover via autophagy.
Relationship of TEX264 with other ER-phagy receptors
We compared the trafficking of TEX264-eGFP with that of other reported ER-phagy receptors. Ectopically expressed FAM134B-mTurquoise substantially accumulated in TEX264-eGFP-positive puncta in response to nutrient stress in COS7 cells (Figure S7G). In contrast, the extent of mCherry-CCPG1 signal enrichment within TEX264-eGFP positive puncta in response to nutrient stress was significantly lower than that of FAM134B, while RTN3L remained reticular upon starvation (Figure S7G). We also examined whether the ability of TEX264 to accumulate in puncta required FAM134B or CCPG1, and whether accumulation of FAM134B puncta required TEX264. Depletion of FAM134B or CCPG1 by RNAi had no obvious effect on the ability of TEX264-eGFP to form puncta in response to EBSS in HCT116 cells (Figure S7H–J). Likewise, overexpression of FAM134B-mTurquoise still trafficked into puncta in TEX264−/− HCT116 cells to an extent similar to that seen in TEX264-expressing cells (Figure S7K). These data are consistent with distinct roles for individual ER-phagy receptors in response to nutrient stress.
DISCUSSION
The ER constitutes one of the most abundant and dynamic membranous compartments within cells (Hu and Rapoport, 2016). As such, multiple mechanisms ensure ER quality control, including via ER-phagy (Dikic, 2018; Smith and Wilkinson, 2017). Moreover, the ER serves as a site of initiation of autophagosomal membranes through the formation of initial isolation membrane (IM) structures (Hayashi-Nishino et al., 2009; Mercer et al., 2018). Here, we identify TEX264 as a previously unrecognized ER-phagy receptor. TEX264 is tethered to ER through its N-terminal transmembrane segment, placing its central GyrI-like domain and C-terminal LIR in the cytosol (Figure 7G). TEX264 is degraded through autophagy and this requires its C-terminal LIR, the ATG8 conjugation system, the ULK1-RB1CC1 kinase complex, and the VPS34 PI3P kinase complex. Flux of TEX264 through autophagy is greatly increased in response to nutrient stress, including AA withdrawal or MTOR inhibition. When expressed at equivalent levels, TEX264 associates most efficiently with the ATG8 proteins LC3A, LC3B and GABARAPL1, but the relative abundance of ATG8 proteins found associated with endogenous TEX264 likely will depend upon their levels of endogenous expression, which vary in distinct cell types.
How is TEX264 trafficked from the ER to autophagosomes? Through live-cell imaging, we found that TEX264-eGFP accumulates in LC3-positive puncta, which are often tethered near 3-way ER tubule junctions and merge with lysosomes in a process that can occur in ~15 min in nutrient-depleted COS7 cells (Figure 4 and Movie S1,S2). Previous studies have suggested that a subdomain of ER forms a cradle-like curved structure that physically connects with and is encircling the IM at an early step in autophagosome formation (Hayashi-Nishino et al., 2009; Mercer et al., 2018). This study could not distinguish between two distinct initiating structures: Model 1 in which the IM extends from the end of an ER tubule to form a cradle, and Model 2 in which the IM emerges from a structure reminiscent of a 3-way junction (Hayashi-Nishino et al., 2009). The IM is thought to be assembled, in part, through vesicular tubular clusters that localize near the IM and are marked by autophagy regulators (WIPI, VPS34, ULK1) and lipidated ATG8s, but the extent to which it contains ER-derived proteins is less clear. These prior studies, together with our live-cell imaging and APEX2 studies, suggest a working model shown in Figure 7H. In this model, the ATG8-containing IM cradles a tubular segment of TEX264-positive ER near a 3-way junction, allowing the formation of a synapse of two juxtaposed membranes with trans interaction between TEX264 and ATG8 proteins. Expansion of the IM would extend capture of ER possibly through a “zipper-like” process involving continued trans TEX264-ATG8 interactions, until poorly understood mechanisms lead to fission of relevant membranes and ultimately autophagosomal membrane closure. This model is consistent with our finding that TEX264-APEX2 staining in EM thin sections is adjacent to the inner membrane of autophagosome-like structures and appears to extend across a large portion of the inner membrane as viewed in cross-section (Figure 4, S4C). It is also possible that TEX264 can traffic into autophagosomes at structures distinct from 3-way junctions. Interestingly, our APEX2 and interaction proteomics experiments identified not only ATG8 proteins but also other autophagy regulators in proximity to TEX264, including cargo adaptors (SQSTM1 and TAX1BP1), the VPS34 complex, and WIPI2, which would be expected to also be present at the luminal side of the inner membrane (Mercer et al., 2018) (Figure 7H). Further studies are required to fully understand the topological mechanisms driving TEX264 incorporation into autophagosomes. We note, however, that our EM data in TEX264-APEX2 expressing cells suggests that a cis model of TEX264 incorporation into growing autophagosomes through lateral transfer within the same membrane is unlikely.
Quantitative proteomics of WT and TEX264−/− cells during AA withdrawal revealed that a large cohort of ER proteins are reduced in abundance by an average of 7.9%, and about half of this flux was TEX264-dependent (see Table S4). We validated a partial TEX264-dependence for turnover of the previously reporter pan ER-phagy reporter (Keima-RAMP4), as well RTN4-Keima and EPHX1-Keima, which were identified as candidate ER-phagy targets of TEX264 by proteomics (Figure 6). Taken together, our results suggest two broad conclusions. First, the extent of turnover of ER-proteins during nutrient stress can be quite distinct in terms of dynamic range. For example, while the abundance of TEX264 is reduced by ~50%, the reduction of most ER proteins is far lower (see Table S4). This could reflect distinct sub-ER localization for the various proteins being degraded, thereby affecting the extent of turnover for individual proteins. Alternatively, TEX264 may be actively “sorted” into domains that emerge from the ER and traffic to ATG8-positive autophagosomes, leading to a form of selectivity in turnover via autophagy. Second, the finding that TEX264-deletion doesn’t fully block ER protein turnover, and that some ER proteins are affected more severely than others upon TEX264 depletion (see Table S4), is consistent with some level of functional redundancy among ER-phagy receptors, as previously noted for FAM134B, CCPG1, and RTN3L (Fumagalli et al., 2016; Grumati et al., 2017; Khaminets et al., 2015; Smith et al., 2018). Consistent with this, we found that trafficking of TEX264 to autophagosomes doesn’t require FAM134B and vice versa, despite the fact that both proteins co-localize within the same puncta during nutrient stress (Figure S7G–K). This result is also consistent with the finding that mutation of the LIR in TEX264 blocks its accumulation into puncta, indicating that FAM134B isn’t sufficient for trafficking of TEX264 into autophagosomes. In contrast, CCPG1 is not appreciably enriched into, or required for the formation of, TEX264-containing puncta, and we did not detect specific puncta formation with RTN3L, suggesting that distinct mechanisms may underlie ER-phagic flux through individual receptors. Moreover, we found that while the abundance of two additional ER-phagy receptors RTN3L and SEC62 is reduced by 13% and 21% upon AA withdrawal in WT cells, there is only a small contribution of TEX264 to their turnover (abundance reduced by 9% and 14% upon AA withdrawal in TEX264−/− cells, respectively) (Figure S6C,D), consistent with the idea that they could have largely distinct turnover mechanisms. Future studies are required to understand precisely when and how individual ER-phagy receptors are used to control ER remodeling during nutrient stress and to elucidate the extent to which individual receptors control morphologically distinct components of the ER network.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, J. Wade Harper (wade_harper@hms.harvard.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293 (human embryonic kidney, fetus, ATCC CRL-1573, RRID: CVCL_0045), HCT116 (human colorectal carcinoma, male, ATCC CCL-247, RRID: CVCL_0291), HEK293T (human embryonic kidney, fetus, ATCC CCL-3216, RRID: CVCL_0063), and COS7 (Cercopithecus aethiops kidney, ATCC CRL-1651, RRID: CVCL_0224) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM), high glucose, pyruvate and supplemented with 10% fetal calf serum and maintained in a 5% CO2 incubator at 37°C.
Generation of TEX264-eGFP knock-in cell lines using CRISPR-Cas9 gene editing
gRNAs targeting C-terminus region of human TEX264 gene was designed using the CHOPCHOP website (http://chopchop.cbu.uib.no/). The guide sequence for TEX264 gene (5’-GGGGACTACCAAGTGGCTC-3’) was assembled into pX459 plasmid (Ran et al., 2013). Donor vector was constructed by assembling eGFP transgene with upstream and downstream homology arms (650 nucleotide each) into digested pSMART plasmid by Gibson assembly. HEK293, HEK293T, and HCT116 cells were transfected with donor and gRNA vectors (1 to 1 ratio) by Lipofectamine 3000 (Invitrogen). Ten days after the transfection, eGFP fluorescent positive cells were sorted into 96 well plates by flow-cytometry (MoFlo Astrios EQ, Beckman Coulter). Three weeks later, the expanded single-cell colonies were screened for the integration of the eGFP transgene by immunoblotting with α-TEX264, followed by genotyping.
Generation of gene knock-out cell lines using CRISPR-Cas9 gene editing
TEX264, ATG5, and ATG7 knock-out on 293T, 293, and HCT116 cell lines was carried out by plasmid-based transfection of Cas9/gRNA using pX459 plasmid as described (Ran et al., 2013). Initially, six gRNAs for ATG5 and ATG7, and three guide RNAs for TEX264 knock-out were designed using CHOPCHOP website. Puromycin selection was followed 24 hours after the transfection. 48 hours after the transfection, gene cleavage efficiency of each guide RNA was measured by Surveyor assay. The following gRNA was shown the best cutting efficiency among the tested guides: 5’- TGATAAGTGCCGATGTGCCG-3’ (exon4) for TEX264, 5’- GATCACAAGCAACTCTGGAT-3’ (exon5) for ATG5, and 5’- ATCCAAGGCACTACTAAAAG-3’ (exon2) for ATG7. Single cell was sorted into 96-well plate using limiting dilution method, and expanded clonal cells were screened by immunoblotting with α-TEX264, α-ATG7, and α-ATG5 antibodies. For the 293T cells used for proteomics in Figure 1, gRNA sequences targeting human ATG7 (5’-CTCTTGTAAATACCATCTGT-3’) or RB1CC1 (5’- TTTCTAACAGCTCTATTACG-3’) were used. Complementary oligonucleotides encoding each guide sequence were designed with BbsI-compatible overhangs, annealed and ligated into the BbsI site of pU6-(BbsI)_CBh-Cas9-T2A-BFP (Addgene plasmid #64323). 293T cells were transfected with gRNA-containing plasmid using lipofectamine 3000 (Invitrogen). Two days post-transfection, BFP-expressing cells were sorted into 96-well plates by fluorescent activated cell sorting (MoFlo XDP, Beckman Coulter) and clonally expanded for 20 days. Individual isolates were screened by immunoblotting with α-ATG7 (CST, 8558S), α-LC3A/B (CST, 12741S) and/or α-RB1CC1 (CST, 12436S) antibodies to verify knockout.
Cloning and generation of stable cell lines
Cloning:
Full-length clones for human TEX264, LAMP1, LC3A, LC3B, LC3C, GABARAP, GABARAPL1, GABARAPL2, RAMP4, RTN4, and EPHX1 were obtained and cloned as previously reported (An and Harper, 2018). Tyrosine 27 to alanine and phenylalanine 273 to alanine substitutions in TEX264 were created using standard site-directed mutagenesis. The destination pHAGE vectors containing N-terminal or C-terminal eGFP, Keima, mTurquoise, and mCherry tags were prepared by Gibson assembly. Using the Gateway cloning system (Thermo-Fisher Scientific), lentiviral based expression vectors containing C-terminal tandem eGFP tagged TEX264 and ATG8 proteins, N-terminal tandem mCherry tagged LC3A, LC3B, RTN3L, and CCPG1, N-terminal tandem Keima tagged RAMP4, C-terminal tandem Keima tagged TEX264, RTN4, and EPHX1, C-terminal tandem mTurquoise tagged FAM134B constructs were generated. In the case of TEX264-APEX2 constructs, the APEX2 coding sequence was introduced to pHAGE vector using GIBSON assembly.
Stable cell line generation:
Stable cell lines were generated based on previously reported method (An and Harper, 2018). Briefly, 293T cells were transfected with the corresponding lentiviral expressing vectors using Lipofectamine 3000 (Invitrogen). Virus-containing supernatant was harvested 48h post-transfection. HEK293, HEK293T, COS7, and HCT116 cell lines were infected with the virus to create stable cell lines. In the case of stable cell lines used for eGFP immunoprecipitation experiments, TEX264-eGFP and TEX264F273A-eGFP virus were introduced to TEX264 knock-out HCT116 cells, and eGFP positive cells were single cell sorted by flow-cytometry to generate clonal cell lines. 19 days after, the expanded single cell clones were screened using immunoblotting method to select clones with TEX264-eGFP expression levels similar to the control endogenous TEX264. Similarly, TEX264-APEX2 and TEX264F273A-APEX2 virus were introduced to TEX264 knock-out HCT116 cells and selected with puromycin (Invitrogen) for one week. Single cell was sorted into 96 well plate using limiting dilution method, and expanded clonal cells were screened by immunoblotting with anti-TEX264. TEX264-eGFP, TEX264-Keima, HA-Flag-eGFP, eGFP-ATG8 proteins, mCherry-LC3A, mCherry-LC3B, mCherry-CCPG1, mCherry-RTN3L, FAM134B-mTurquoise, RTN4-Keima, EPHX1-Keima, and Keima-RAMP4 expressing cell lines were first selected with puromycin for at least one week, then sorted by flow-cytometry (MoFlo Astrios EQ, Beckman Coulter) in order to equalize the eGFP and Keima expression levels.
Method Details
Amino acid free medium preparation (500ml)
DMEM powder (4.16g, US biological) and 1.85g sodium bicarbonate were dissolved in 400 ml H2O. To the solution, 1.75 g of glucose, 5 ml of 100x sodium pyruvate (final conc. = 1mM) and 50 ml of dialyzed fetal calf serum were added. 150 µl of 6N HCl was slowly added to bring the pH to 7.4. The final volume was adjusted to 500 ml. The media was filtered through 0.2 µm filter and kept at 4 °C.
Cell lysis and immunoblotting assay
Cells were cultured in the presence of the corresponding chemicals to ~50–60% confluency in 6-well plates or 10 cm dishes. After removing the media, the cells were washed with DPBS twice, then RIPA buffer containing mammalian protease inhibitor cocktail (Sigma) and 20 unit/ml Benzonase (Millipore) were added directly onto the cells. Cell lysates were collected by cell scrapers and sonicated on ice three times with level 5, followed by centrifugation (13000 rpm, 5 min). The concentration of the supernatant was measured by Bradford assay, and the whole cell lysate was denatured by the addition of LDS sample buffer supplemented with 100 mM DTT, followed by boiling at 75°C for 5 minutes. 20 µg of each lysate was loaded onto the 4–20% Tris-Glycine gel (Thermo Fisher Scientific) or 4–12% NuPAGE Bis-Tris gel (Thermo Fisher Scientific), followed by SDS-PAGE with Tris-Glycine SDS running buffer (Thermo Fisher Scientific) or MES SDS running buffer (Thermo Fisher Scientific), respectively. The proteins were electro-transferred to PVDF membranes (0.45 µm, Millipore), and then the total protein was stained by Revert total protein stain kit (LI-COR) or Ponceau staining (Thermo Fisher Scientific). The membrane was then blocked with 5% non-fat milk, incubated with the indicated primary antibodies (4°C, overnight), washed three times with TBST (total 30 min), and further incubated with fluorescent IRDye 680RD Goat anti-Mouse IgG H+L or IRDye 800CW Goat anti-Rabbit IgG H+L secondary antibody (1:20000) for 1 hour. After harsh wash with TBST for 30 min, near infrared signal was detected using OdysseyCLx imager and quantified using ImageStudioLite (LI-COR). For quantitative immunoblotting of Keima reporter cells, at least 70 µg of total lysate was loaded on to SDS-PAGE gel due to low expression level of Keima construct and small amount of processed Keima level. 4–20% Tris-Gly gel was used to resolve the proteins.
Flow-cytometry analysis
Flow-cytometry analysis was performed based on previously reported method (An and Harper, 2018). Briefly, cells were grown in the absence of nutrient or in the presence of Torin1 to 60% confluency in 6-well plates, then collected after trypsin treatment. After re-suspending the cells in a FACS buffer (1x DPBS, 1 mM EDTA, 1% FBS, 25 mM HEPES, final pH 7.3–7.5), the cells were analyzed by flow-cytometry (MoFlo Astrios EQ, Beckman Coulter). The data was processed by FlowJo software. The 561/488 nm ratio distribution graph was processed in Prism software after exporting the 488 and 561 nm ex/em intensity of individual cells (10000 cells) were exported.
Confocal microscopy
Live-cell confocal microscopy was performed based on previously reported method (An and Harper, 2018). Briefly, cells were plated onto 35 mm-glass bottom dish (No. 1.5, 14 mm glass diameter, MatTek) pre-treated with poly-L-lysine, then incubated in phenol-red free medium (FluoroBrite™DMEM, Thermo Fisher) containing 10% fetal bovine serum for 48 hours. Followed by the treatment with corresponding chemicals or EBSS, the cells were imaged using a Yokogawa CSU-X1 spinning disk confocal with Spectral Applied Research Aurora Borealis modification on a Nikon Ti motorized microscope equipped with a Nikon Plan Apo 60×/1.40 N.A or 100x/1.40 N.A objective lens. Pairs of images for ratiometric analysis of mKeima fluorescence were collected sequentially using 100 mW 442 nm and 100 mW 561 solid state lasers attenuated and controlled with an AOTF (Spectral Applied Research LMM-5), and emission collected with a 620/60 nm filter (Chroma Technologies). Wide-field fluorescence images of Hoeschst were collected using a Lumencor SOLA light source, 395/35nm excitation and 480/40nm emission filters (Chroma Technologies). Both confocal and widefield images were acquired with the same Hamamatsu ORCA-ER cooled CCD camera and MetaMorph software. For the analysis of HCT116, 293T, and 293 cells expressing endogenously tagged TEX264-eGFP, six z-series optical sections were collected with a step-size of 0.6 microns, using the Nikon Ti internal focus motor. Z-series are displayed as maximum z-projections, and gamma, brightness, and contrast were adjusted for each image equally using FiJi software. Without any cutting, GFP puncta number per cell in HCT116, 293T, and 293 cells expressing endogenously tagged TEX264-eGFP was counted unbiasedly using FiJi software. Five to six image frames per condition were analyzed without exclusion. Puncta size bigger than 0.7 µm with intensity higher than 1 (arbitrary unit) were automatically detected and manually assigned to each cell. For measurement of ER area/cell, we employed ER-tracker and image analysis as previously described (Smith et al., 2018) with modification. In particular, ER positive area per cell changes along the position of focal plane on z-axis; therefore, maximum z-projection image of six z-stacks encompassing the entire cell was used to measure ER positive area.
Time-lapse imaging analysis
COS7 cells stably expressing TEX264-eGFP and/or mCherry-LC3 were sorted by flow-cytometry to equalize the expression levels and plated on a 35 mm glass bottom dish 24 hours before the analysis. The cells were maintained in 37 °C with 5% CO2 condition using Okolab Stage Top Incubator through-out the imaging acquisition. Images were acquired at intervals of 15 or 30 seconds for 30 min, and multiple stage positions were analyzed using Metamorph software. Laser intensity and exposure time were optimized to prevent photobleaching during the course of imaging acquisition. For the detection of TEX264-eGFP puncta fusing with lysosome, 25 nM of LysoTracker Red DND-99 (Thermo Fisher, L-7528) was added to the TEX264-eGFP expressing cells 5 minutes before the live cell imaging. Size of TEX264-eGFP puncta in stable COS7 cells were measured by FiJi software. Note that the resolution limit for the fluorescent microscope used in this study is ~0.220 µm for 525 nm emission. An intensity cut-off (190) was chosen such that pixels with higher intensity than the background ER membrane resident TEX264-eGFP were only measured.
APEX2 DAB staining and electron microscopy
Cells were plated onto a 6-well plate with 20% confluency. 48 hours after, the cells were treated with warm EBSS (Sigma) and BafA1 (25 nM, Sigma) for 3 hours at 37 C. Media was then removed, and cells were washed with DPBS. 2% glutaraldehyde solution was added to the cells and kept at 4°C for 45 min. The fixation buffer was then removed, 2 ml of chilled sodium cacodylate buffer (100 mM, pH7.3, Sigma) was added to each well, incubated for 2 min, and the buffer was washed out. This was repeated for five times. Sodium cacodylate buffer (100 mM, pH7.3) containing 5 mM glycine was then added to quench un-reacted aldehyde, and incubated for 5 min. Following the removal of 5 mM glycine buffer, the cells were washed with sodium cacodylate buffer for 5 times. Sodium cacodylate buffer containing 3,3′-Diaminobenzidine tetrahydrochloride hydrate (0.5 mg/ml, 1.4 mM final concentration) and H2O2 (10 mM) was added to the cells and incubated on ice for 5 min. The buffer was then removed, and the cells were washed with sodium cacodylate buffer 5 times again. Following osmication and uranyl acetate staining, dehydration in alcohols and embedment in Taab 812 Resin (Marivac Ltd, Nova Scotia, Canada) were performed (Hung et al., 2017). Sections were cut with Leica ultracut microtome, and picked up on formvar/carbon-coated copper slot grids. The samples were imaged under the Phillips Tecnai BioTwin Spirit transmission electron microscope.
APEX2 Biotin tyramide IP – WB and TMT-MS3 Samples
APEX2 experiments were performed as described but with modifications (Hung et al., 2017). The indicated cells were plated onto a 15 cm dish to 30% confluency, 24 hours before the starvation. Amino acid deficient media containing 25 nM Bafilomycin A was added to the cells for starvation, while the control cells were treated with fresh rich DMEM media. 2.5 hours later, biotin tyramide dissolved in DMSO (500 mM stock in DMSO, 500 µM final conc.) was added to the cells and incubated for 30 min at 37 °C. Three plates were taken out from the incubator each time and treated with H2O2 (1 mM final conc.) for 1.5 min at room temperature. Media was immediately removed, and the cells were washed with cold DPBS containing 10 mM sodium ascorbate, 10 mM sodium azide, and 5 mM Trolox three times to reduce H2O2, followed by washing with DPBS twice. The plates were then kept on ice, and DPBS was completely removed. 800 µl RIPA (50 mM HEPES, 150 mM NaCl, pH7.6, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, one tablet of Phos-STOP, 1X mammalian protease inhibitor cocktail (Sigma)) containing 1 mM sodium ascorbate was directly added to the plate, and the lysate was gathered by a cell scraper. The cell lysate was then sonicated 3 times with power 5. Bradford assay was followed to measure the total protein concentration, and each lysate was adjusted to become 1.5 mg/ml using RIPA buffer. 50 µl of the lysate was taken and snap frozen for input. 200 µl of high capacity streptavidin agarose beads was washed with RIPA buffer twice, and distributed to 11 tubes (final bead volume was 17.5 µl per condition). 1 ml of each lysate (1.5 mg/ml, total 1.5 mg) was added to the beads and incubate at 4 °C overnight. Flow-through was collected, and the beads were washed with 1 ml RIPA X 2, 1 M KCl, 0.1 M Na2CO3 (pH 9), 2 M Urea with 1 M NaCl in 10 mM HEPES (pH 8), and RIPA X 2. Finally, the beads were washed with water for two times, then the water was completely removed. In parallel, input and flow-through were analyzed by western blotting to ensure the proper mTOR inhibition and autophagy induction, and efficient capture of biotinylated proteins by the streptavidin beads. Lastly, 50 µl of 1,1,1,3,3,3-Hexafluoro-2-propanol was added to the beads, incubated for 5 min with agitation at room temperature, and eluted through a hydrophobic filter. This was repeated once more, and the gathered eluate was evaporated on speed vac. For Western blotting analysis, the samples were resuspended in 40 µl 1x SDS buffer containing reducing agent and boiled at 75 °C for 3 min. For mass spectrometry, samples were resuspended in 100 µl of 6M Urea in 100 mM EPPS pH 8.5 buffer.
GFP-Trap IP – WB and TMT-MS3 Samples
The indicated cells were plated onto a 15 cm dish with ~30% confluency so that the final confluency becomes 50–60 %. 24 hours later, amino acid deprived DMEM containing 25 nM BafA1 was added to the cells for starvation, and incubated at 37 °C for 3 hours. Following the removal of media, cells were washed with DPBS three times for WB, and five times for MS. 800 µl of lysis buffer (50 mM HEPES, 150 mM NaCl, 0.1% NP-40, 10 mM glycerophosphate, 10 mM sodium pyrophosphate, 1x mammalian protease inhibitor cocktail, one tablet of Phos-STOP, pH 7.5) was directly added to the dish, then the cells were collected by a scraper and pipetted up and down for 15 times followed by centrifugation at 7000 rpm for 30 sec. The concentration of each supernatant was measured by Bradford assay, and adjusted to become 1 mg/ml. The lysate was then filtered through 0.4 µm filter, and 50 µl of each lysate was taken for input. 66 µl of GFP-Trap beads (bead volume only) was washed with lysis buffer twice to remove ethanol, and distributed into 11 × 1.5 ml Eppendorf tubes (6 µl beads were used per condition). 1 ml of the lysate was added to the beads, and incubated for 1.5 hours. The flow-through was collected by centrifuging the beads at 2.5 rcf 2 min, 4 °C (4725 rpm) and snap frozen. The beads were then washed with 1 ml of wash buffer 1 (50 mM HEPES, 150 mM NaCl, 0.1% NP-40, pH 7.5) and 1 ml of wash buffer 2 (50 mM HEPES, 150 mM NaCl, pH 7.5,) for twice. The beads were then incubated with 50 µl of 2% SDS (HEPES 50 mM, ~pH 7) at 60 °C for 5 min with agitation and the eluate was further processed for WB or TMT-MS analysis.
GFP-Trap and APEX2 TMT Proteomics analysis
For mass spectrometry, samples were subjected to reduction (5 mM tris (2-carboxyethyl)phosphine (TCEP) for 10 min at room temperature) and alkylation (25 mM chloroacetamide for 20 min at room temperature) followed by single-pot solid-phase-enhanced sample preparation (SP3) protein clean-up (Moggridge et al., 2018). Samples were briefly dried and resuspended in 30 µl digestion buffer (100 mM EPPS pH 8.5, 0.1% RapiGest) and digested for 2 h at 37°C with Lys-C (0.25 µg) and then a fur ther 6–8 h with trypsin (0.5 µg) at 37°C. Digests, separated from the beads were then labeled with TMT. A total of 2 µL of the 20 ng/µL stock of TMT reagent was added to the peptides along with acetonitrile to achieve a final acetonitrile concentration of approximately 30% (v/v). Following incubation at room temperature for 1 h, the reaction was quenched with hydroxylamine to a final concentration of 0.5% (v/v) for 15 min. The TMT-labeled samples were then pooled together at a 1:1 ratio. The sample were acidified with equal volume of 1% formic acid (FA) to a pH ~ 2, incubated for 15 min, vacuum centrifuged to near dryness and subjected to C18 solid-phase extraction (SPE) (10 mg SOLA HRP, Thermo). Sample was then fractionated according to manufacturer’s instructions using High pH reversed-phase peptide fractionation kit (Thermo Fisher Scientific) for a final 6 (APEX2 sample) or 4 (GFP-Trap sample) fractions and subjected to C18 StageTip desalting. Mass spectrometry data were collected using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) coupled to a Proxeon EASY-nLC1200 liquid chromatography (LC) pump (Thermo Fisher Scientific). Peptides were separated on a 100 µm inner diameter microcapillary column packed in house with ~35 cm of Accucore150 resin (2.6 µm, 150 Å, Thermo Fisher Scientific, San Jose, CA) with a gradient consisting of 5%–24% (0–90 min), 24–32% (95–110min) (ACN, 0.1% FA) over a total 120 min run at ~500 nL/min. For analysis, we loaded 1/2 of each fraction onto the column. Each analysis used the Multi-Notch MS3-based TMT method (McAlister et al., 2014), to reduce ion interference compared to MS2 quantification. The scan sequence began with an MS1 spectrum (Orbitrap analysis; resolution 120,000 at 200 Th; mass range 400−1400 m/z (GFP-Trap) or 400–1600 (APEX2); automatic gain control (AGC) target 1×106; maximum injection time 246 ms). Precursors for MS2 analysis were selected using a Top10 method. MS2 analysis consisted of collision-induced dissociation (quadrupole ion trap analysis; Turbo scan rate; AGC 2.0×104; isolation window 0.7 Th; normalized collision energy (NCE) 35; maximum injection time 90 ms (GFP-Trap) or 120 ms (APEX2). Monoisotopic peak assignment was used and previously interrogated precursors were excluded using a dynamic window (120 s ± 7 ppm). Following acquisition of each MS2 spectrum, a synchronous-precursor-selection (SPS) MS3 scan was collected on the top 10 most intense ions in the MS2 spectrum (McAlister et al., 2014). MS3 precursors were fragmented by high energy collision-induced dissociation (HCD) and analyzed using the Orbitrap (NCE 65; AGC 3×105; maximum injection time 150 ms, resolution was 50,000 at 200 Th).
Total proteomics analysis using TMT
Cells are plated into a 10 cm dish per condition. 18 hours later, control cells were ~ 40% confluent, and -AA cells were ~50–60% confluent. Amino acid deprived DMEM was added to the -AA cells and incubated for 10 hours. Cells were then washed with ice cold PBS five times. After the complete removal of DPBS, 800 µl of RIPA buffer (50 mM HEPES, 150 mM NaCl, pH7.6, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 10 mM glycerophosphte, 10 mM sodium pyrophosphate, mammalian protease inhibitor cocktail, one tablet of Phos-stop, pH 7.5) was added to the dishes. The lysate was then collected and sonicated three times with level 5. Bradford assay was performed to measure the protein concentration. The lysate was then adjusted to become 3 mg/ml concentration through-out the samples using RIPA buffer. 100 µg of each sample was taken, and reduced by the incubation in the presence of 5 mM TCEP at 55 °C for 10 min. The lysate was then cooled down to room temperature, and fresh chloroacetamide solution (final conc. 20 mM) was added and incubated at room temperature for 15 min, followed by chloroform/methanol precipitation.
Samples were resuspended in 100 mM EPPS, pH 8.5 containing 0.1% RapiGest and digested at 37°C for 2 h with LysC protease at a 20 0:1 protein-to-protease ratio. Trypsin was then added at a 100:1 protein-to-protease ratio and the reaction was incubated for 6 h at 37°C. Tandem mass tag labeling of each sample was performed by adding 10 µL of the 20 ng/µL stock of TMT reagent along with acetonitrile to achieve a final acetonitrile concentration of approximately 30% (v/v). Following incubation at room temperature for 1 h, the reaction was quenched with hydroxylamine to a final concentration of 0.5% (v/v) for 15 min. The TMT-labeled samples were pooled together at a 1:1 ratio. The sample was vacuum centrifuged to near dryness, and subjected to C18 solid-phase extraction (SPE) (50 mg, Sep-Pak, Waters).
Dried TMT-labeled sample was resuspended in 100 µl of 10 mM NH4HCO3 pH 8.0 and fractionated using BPRP HPLC (Paulo et al., 2016). Briefly, samples were offline fractionated over a 90 min run, into 96 fractions by high pH reverse-phase HPLC (Agilent LC1260) through an aeris peptide xb-c18 column (Phenomenex; 250 mm x 3.6 mm) with mobile phase A containing 5% acetonitrile and 10 mM NH4HCO3 in LC-MS grade H2O, and mobile phase B containing 90% acetonitrile and 10 mM NH4HCO3 in LC-MS grade H2O (both pH 8.0). The 96 resulting fractions were then pooled in a non-continuous manner into 24 fractions (as outlined in Supplemental Figure 5 of (Paulo et al., 2016)) and 12 fractions (even numbers) were used for subsequent mass spectrometry analysis. Fractions were vacuum centrifuged to near dryness. Each consolidated fraction was desalted via StageTip, dried again via vacuum centrifugation, and reconstituted in 5% acetonitrile, 1% formic acid for LC-MS/MS processing.
Mass spectrometry data were collected using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) coupled to a Proxeon EASY-nLC1200 liquid chromatography (LC) pump (Thermo Fisher Scientific). Peptides were separated on a 100 µm inner diameter microcapillary column packed in house with ~35 cm of Accucore150 resin (2.6 µm, 150 Å, Thermo Fisher Scientific, San Jose, CA) with a gradient consisting of 5%–21% (0–125 min), 21–28% (125–140min) (ACN, 0.1% FA) over a total 150 min run at ~500 nL/min. For analysis, we loaded 1/10 of each fraction onto the column. Each analysis used the Multi-Notch MS3-based TMT method (McAlister et al., 2014), to reduce ion interference compared to MS2 quantification. The scan sequence began with an MS1 spectrum (Orbitrap analysis; resolution 120,000 at 200 Th; mass range 400−1400 m/z; automatic gain control (AGC) target 5×105; maximum injection time 50 ms). Precursors for MS2 analysis were selected using a Top10 method. MS2 analysis consisted of collision-induced dissociation (quadrupole ion trap analysis; Turbo scan rate; AGC 2.0×104; isolation window 0.7 Th; normalized collision energy (NCE) 35; maximum injection time 90 ms). Monoisotopic peak assignment was used and previously interrogated precursors were excluded using a dynamic window (150 s ± 7 ppm). Following acquisition of each MS2 spectrum, a synchronous-precursor-selection (SPS) MS3 scan was collected on the top 10 most intense ions in the MS2 spectrum (McAlister et al., 2014). MS3 precursors were fragmented by high energy collision-induced dissociation (HCD) and analyzed using the Orbitrap (NCE 65; AGC 3×105; maximum injection time 150 ms, resolution was 50,000 at 200 Th).
Proteomics data analysis
Mass spectra were processed using a Sequest-based in-house software pipeline (Huttlin et al., 2010) (Figure 1; S1) or Sequest-HT using Proteome Discoverer (v2.3.0.420 - Thermo Fisher Scientific) (Figure 3, 5, 6, S6, and S7).
For Fig 1 and S1, spectra were converted to mzXML using a modified version of ReAdW.exe. Database searching included all entries from the human UniProt database. This database was concatenated with one composed of all protein sequences in the reversed order. Searches were performed using a 20 ppm precursor ion tolerance for total protein level analysis. The product ion tolerance was set to 0.9 Da. These wide mass tolerance windows were chosen to maximize sensitivity in conjunction with Sequest searches and linear discriminant analysis (Huttlin et al., 2010). TMT tags on lysine residues and peptide N termini (+229.163 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification. Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR) and PSM filtering was performed using a linear discriminant analysis, as described previously (Huttlin et al., 2010), while considering the following parameters: XCorr, ∆Cn, missed cleavages, peptide length, charge state, and precursor mass accuracy. For TMT-based reporter ion quantitation, we extracted the summed signal-to-noise (S:N) ratio for each TMT channel and found the closest matching centroid to the expected mass of the TMT reporter ion. For protein-level comparisons, PSMs were identified, quantified, and collapsed to a 1% peptide false discovery rate (FDR) and then collapsed further to a final protein-level FDR of 1%. Moreover, protein assembly was guided by principles of parsimony to produce the smallest set of proteins necessary to account for all observed peptides. Proteins were quantified by summing reporter ion counts across all matching PSMs using in-house software, as described previously (Huttlin et al., 2010). PSMs with poor quality, MS3 spectra with more than 9 TMT reporter ion channels missing, or isolation specificity less than 0.7, or with TMT reporter summed signal-to-noise ratio that were less than 150, or had no MS3 spectra were excluded from quantification (Paulo et al., 2016).
For Fig 3, 5, S5 and 6, the identification of proteins was performed using the SEQUEST-HT engine against the UniProt Human Reference Proteome (2018) using the following parameters (post-recalibration): a tolerance level of 10 ppm for MS1 and 0.6 Da for MS2 and false discovery rate of the Percolator decoy database search was set to 1%. Trypsin was used as the digestion enzyme, three missed cleavages were allowed, and the minimal peptide length was set to 7 amino acids. TMT tags on lysine residues and peptide N termini (+229.163 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification. For TMT-based reporter ion quantitation, we extracted (integration tolerance of 0.003 Da) the summed signal-to-noise ratio for each TMT channel and found the closest matching centroid to the expected mass of the TMT reporter ion. For protein-level comparisons, PSMs were identified, quantified, and collapsed to a 1% peptide false discovery rate (FDR) and then collapsed further to a final protein-level FDR of 1%. Moreover, protein assembly was guided by principles of parsimony to produce the smallest set of proteins necessary to account for all observed peptides. Proteins were quantified by summing reporter ion counts across all matching PSMs for unique peptides only (Fig 3, 5) or unique and razor peptides (Fig S5, 6). PSMs with poor quality, MS3 spectra with TMT reporter summed signal-to-noise ratio that were less than 10 per channel, isolation specificity ≤0.4, or had less than 70% of correctly selected SPS ions were excluded from quantification (Paulo et al., 2016).
Protein quantification values were exported for further analysis in Microsoft Excel and Perseus (Tyanova et al., 2016). Each reporter ion channel was summed across all quantified proteins and normalized assuming equal protein loading of all 10 samples. For Figure 5, normalization was done by normalizing to the total streptavidin signal.
Supplemental data Tables list all quantified proteins as well as associated TMT reporter ratio change to control channels used for quantitative analysis.
Annotations for bona fide organellar protein markers were assembled using the proteins which had scored with confidence “very high” or “high” from the HeLa dataset previously published Itzhak D.N. (Itzhak et al., 2016).
For Label free quantification (Fig 3F), mass spectra were processed using Protein Discoverer using the Minora algorithm (set to default parameters). The identification of proteins was performed using the SEQUEST-HT engine against the UniProt Human Reference Proteome (2018) using the following parameters: a tolerance level of 15 ppm for MS1 and 0.6 Da for MS2 post-recalibration and the false discovery rate of the Percolator decoy database search was set to 1%. Trypsin was used as the digestion enzyme, three missed cleavages were allowed, and the minimal peptide length was set to 7 amino acids. TMT tags on lysine residues and peptide N termini (+229.163 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification. Precursor abundance quantification was determined based on area. Proteins were quantified based only on unique peptides. MS1-based protein quantification of each ATG8 proteins was divided by the respective MS3-based total sum-signal to noise across all 11 channels. Each individual TMT channel sum s/n was then multiplied by the previously obtain MS1 signal /total MS3 signal ratio and the obtained value representing the extracted MS1 precursor abundance for each individual TMT channel where plotted in Prism.
Cell proliferation and cell viability assay
1×104 293T cells were plated onto 24-well plates 24h prior to MTOR inhibition or ER stress induction. Cells were collected 24 h post treatment and stained with Trypan Blue Stain (Thermo Fisher Scientific). Viable cells were then counted using Countess™ (Thermo Fisher Scientific).
RT-qPCR
mRNA was purified from ~1.0 × 106 cells using RNeasy RNA purification kit (Qiagen), and cDNA was produced using High Capacity cDNA Reverse Transcription kit (Applied Biosystems) according to the manufacturer protocol. qPCR reagent mixture was prepared by adding 10 ng of cDNA to TaqMan Fast Advanced Master mix (Thermo Fisher Scientific) and the corresponding TaqMan probes (FAM), which was then quantified by QuantStudio 7 Flex Real-Time PCR System (Thermo Fisher Scientific). GAPDH and 18S rRNA genes were used for data normalization.
siRNA transfection
siRNAs for FAM134B and CCPG1 (Dharmacon) were transfected into HCT116 TEX264-eGFP K/I cell-line using RNAiMAX reagent (Thermo Fisher Scientific). Total mRNA level was measured by RT-qPCR 52h post transfection, and the cells were collected for immunoblotting as well. In parallel, the cells were plated onto glass-bottom flasks, which were then subjected to EBSS and BafA (50 nM) treatment (4h), followed by live-cell imaging analysis.
Domain analysis
The TEX264 sequence was analyzed by Swiss-model (https://swissmodel.expasy.org/). This led to the identification of tertiary structure models based on multiple bacterial proteins containing a GyrI-like domain. The model based on Protein Database entry 1jyh (SbmC protein) was rendered in Pymol. Transmembrane domains in TEX264 were examined using http://www.cbs.dtu.dk/services/TMHMM/.
Quantification and Statistical Analysis
Statistics
All statistical data were calculated using GraphPad Prism 7 or Perseus. Comparisons of data were performed by two-way ANOVA with Tukey’s multiple comparisons test; p-values <0.01 were considered significant.
Reproducibility
All experiments were repeated at least three times unless otherwise indicated.
Supplementary Material
Supplemental Movie S1. Visualization of ER tubules marked by TEX264-eGFP (green) and autophagosomes marked by mCherry-LC3A in COS7 cells after EBSS treatment. Relevant to Figure 4.
Supplemental Movie S2. Visualization of ER tubules marked by TEX264-eGFP (green) and lysosomes marked by Lysotracker-Red in COS7 cells after EBSS treatment. Relevant to Figure 4.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| TEX264 | Sigma | HPA017739: RRID:AB_1857910 |
| ATG7 | Cell Signaling Technology | 8558S; RRID:AB_10831194 |
| Keima | MBL international | M126–3M; RRID:AB_843710 M182–3; RRID:AB_10794910 |
| LC3B | MBL international | M186–3; RRID:AB_10897859 |
| ATG5 | Cell Signaling Technology | 12994; RRID:AB_2630393 |
| BECN1 | Cell Signaling Technology | 3495; RRID:AB_1903911 |
| p70 S6K phospho-T389 | Cell Signaling Technology | 9234S; RRID:AB_2269803 |
| phospho-S6 ribosomal protein Ser253, 236 | Cell Signaling Technology | 4858 |
| GFP | Proteintech Group | 50430–2-AP; RRID:AB_11042881 |
| REEP5 | Proteintech Group | 14643–1-AP; RRID:AB_2178440 |
| Calnexin | Proteintech Group | 10427–2-AP; RRID:AB_2069033 |
| SEC62 | GeneTex | GTX129853 |
| Tubulin | Abcam | ab7291; RRID:AB_2241126 |
| SQSTM1 | Novus Biologicals | H00008878-M01; RRID:AB_548364 |
| CALR | Cell Signaling Technology | 12238S; RRID:AB_2688013 |
| CALCOCO2 | Proteintech Group | 12229–1-AP; RRID:AB_11182600 |
| RPS6 | Cell Signaling Technology | 2217; RRID:AB_331355 |
| 4EBP1 | Cell Signal Technology | 9644; RRID:AB_2097841 |
| ATG14 | Cell Signaling Technology | 5504, RRID:AB_10695397 |
| RPS3 | Cell Signaling Technology | 9538; RRID:AB_10622028 |
| FAM134B | Proteintech Group | 21537–1-AP |
| CHOP | Thermo Fisher Scientific | MA1–250; RRID:AB_2292611 |
| BIP | Cell Signaling Technology | 3177 |
| eIF-2a-pS52 | Thermo Fisher Scientific | 44–728G; RRID:AB_1500038 |
| CCPG1 | Proteintech Group | 13861–1-AP, RRID:AB_2074010 |
| IRDye 800CW Goat anti-Rabbit IgG H+L | LI-COR | 925–32211; RRID:AB_2651127 |
| IRDye 680 RD Goat anti-Mouse IgG H+L | LI-COR | 926–68070; RRID:AB_10956588 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Torin1 | Cell Signal Technology | 14379 |
| SAR405 | APExBio | A8883 |
| Bafilomycin A | Sigma | B1793 |
| Lysotracker Red DND-99 | Thermo Fisher Scientific | L7528 |
| ER-Tracker Red | Thermo Fisher Scientific | E34250 |
| EBSS | Sigma | E3024 |
| Biotin tyramide | Iris Biotech | LS-3500 |
| Hydrogen peroxide solution | Sigma | H1009 |
| Sodium cacodylate trihydrate | Sigma | C0250 |
| 3,3′-Diaminobenzidine tetrahydrochloride hydrate | Sigma | D5637 |
| Sodium ascorbate | VWR international | 95035–692 |
| Trolox | Cayman Chemical | 53188–07-1 |
| Poly-L-lysine solution | Sigma | P4832 |
| GFP-Trap_A | Chromotek | GTA-20 |
| Lipofectamine 3000 | Invitrogen | L3000008 |
| FluoroBrite DMEM | Thermo Fisher Scientific | A1896701 |
| Benzonase Nuclease HC | Millipore | 71205–3 |
| SURVEYOR Mutation Detection Kit | Integrated DNA Technologies | 706025 |
| REVERT™ Total Protein Stain kit | LI-COR | P/N926–11010 |
| CellLight™ ER-RFP | Thermo Fisher Scientific | C10591 |
| 1,1,1,3,3,3-Hexafluoro-2-propanol | Sigma | 52517 |
| Dulbecco’s MEM (DMEM), high glucose, pyruvate | Gibco / Invitrogen | 11995 |
| Dulbecco’s MEM (DMEM), Low Glucose, w/o Amino Acids | US Biological | D9800–13 |
| Pyruvic Acid powder | Sigma | |
| TCEP | Gold Biotechnology | TCEP2 |
| Puromycin | Gold Biotechnology | Gold Biotechnology |
| Formic Acid | Sigma-Aldrich | 94318 |
| DAPI | Thermo Fisher Scientific | D1306 |
| Protease inhibitor cocktail | Sigma-Aldrich | P8340 |
| PhosSTOP | Sigma-Aldrich | 4906845001 |
| Trypsin | Promega | V511C |
| Lys-C | Wako Chemicals | 129–02541 |
| Rapigest SF Surfactant | Glixx Laboratories | Cat#GLXC-07089 |
| EPPS | Sigma-Aldrich | Cat#E9502 |
| 2-Chloroacetamide | Sigma-Aldrich | C0267 |
| Tandem Mass Tags | Thermo Fisher Scientific | Cat#90406 |
| Empore™ SPE Disks C18 | 3M - Sigma-Aldrich | 66883-U |
| Urea | Sigma | Cat#U5378 |
| SeraMag Speed Beads | GE Healthcare | Cat#45152105050250 Cat#65152105050250 |
| High pH Reversed-Phase Peptide Fractionation Kit | Thermo Fisher Scientific | Cat#84868 |
| Bio-Rad Protein Assay Dye Reagent Concentrate | Bio-Rad | #5000006 |
| Pierce Quantitative Colorimetric Peptide Assay | Thermo Fisher Scientific | #23275 |
| Trypan Blue Stain | Thermo Fisher Scientific | T10282 |
| On-Target plus SMART pool, FAM134B | Dharmacon | L-016936–02-0005 |
| On-Target plus SMART pool, CCPG1 | Dharmacon | L-013998–00-0005 |
| TaqMan Fast Advanced Master Mix | Thermo Fisher Scientific | 4444556 |
| TaqMan GAPDH probe | Thermo Fisher Scientific | Hs02786624_g1 |
| TaqMan 18S rRNA probe | Thermo Fisher Scientific | Hs99999901_s1 |
| TaqMan CCPG1 probe | Thermo Fisher Scientific | Hs01043676_m1 |
| TaqMan EPHX1 probe | Thermo Fisher Scientific | Hs01116806_m1 |
| TaqMan REEP4 probe | Thermo Fisher Scientific | Hs00951335_g1 |
| TaqMan RTN4 probe | Thermo Fisher Scientific | Hs00199671_m1 |
| TaqMan RTN3 probe | Thermo Fisher Scientific | Hs01581961_g1 |
| TaqMan FAM134B probe | Thermo Fisher Scientific | Hs00375273_m1 |
| High Capacity cDNA Reverse Transcription Kit | Applied Biosystems | 4368814 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Human: HEK293 | ATCC | CRL-1573; RRID:CVCL_0045 |
| Human: HCT116 | ATCC | CCL-247; RRID:CVCL_0291 |
| Human: HEK293T | ATCC | CRL-3216; RRID:CVCL_0063 |
| Cercopithecus aethiops: COS-7 | ATCC | CRL-1651; RRID:CVCL_0224 |
| Oligonucleotides | ||
| Guide RNA sequence for TEX264 KO: TGATAAGTGCCGATGTGCCG | This paper | N/A |
| Guide RNA sequence for ATG7 KO: ATCCAAGGCACTACTAAAAG | This paper | N/A |
| Guide RNA sequence for ATG5 KO: GATCACAAGCAACTCTGGAT | An and Harper, 2018 | N/A |
| Guide RNA sequence for RB1CC1 KO: TTTCTAACAGCTCTATTACG | This paper | N/A |
| Guide RNA sequence for TEX264-eGFP K/I: TGATAAGTGCCGATGTGCCG | This paper | N/A |
| FW primer for genotyping of TEX264-eGFP K/I:TTCCAAGCTTGAAGTGGGGG | This paper | N/A |
| RV primer for genotyping of TEX264-eGFP K/I:CTCACCTGGGCACAGCTTTA | This paper | N/A |
| Recombinant DNA | ||
| pDONR-FAM134B | Khaminets et al., 2015 | N/A |
| pDONR-RTN3L | Grumati et al., 2017 | N/A |
| pDONR-CCPG1 | Smith et al., 2018 | N/A |
| pHAGE-TEX264-eGFP | This paper | N/A |
| pHAGE-TEX264F273A-eGFP | This paper | N/A |
| pHAGE-TEX264Y27A-eGFP | This paper | N/A |
| pHAGE-TEX264-APEX2 | This paper | N/A |
| pHAGE-TEX264F273A-APEX2 | This paper | N/A |
| pHAGE-mCherry-LC3B | This paper | N/A |
| pHAGE-mCherry-LC3A | This paper | N/A |
| pHAGE-TEX264-Keima | This paper | N/A |
| pHAGE-TEX264F273A-Keima | This paper | N/A |
| pHAGE-LAMP1-eGFP | An and Harper, 2018 | N/A |
| pHAGE-Keima-Ramp4 | This paper | N/A |
| pHAGE-RTN4-Keima | This paper | N/A |
| pHAGE-EPHX1-Keima | This paper | N/A |
| pHAGE-FAM134B-mTurquoise | This paper | N/A |
| pHAGE-mCherry-CCPG1 | This paper | N/A |
| pHAGE-mCherry-RTN3L | This paper | N/A |
| pHAGE-eGFP-LC3A | This paper | N/A |
| pHAGE-eGFP-LC3B | An and Harper, 2018 | N/A |
| pHAGE-eGFP-LC3C | This paper | N/A |
| pHAGE-eGFP-GABARAP | This paper | N/A |
| pHAGE-eGFP-GABARAPL1 | This paper | N/A |
| pHAGE-eGFP-GABARAPL2 | This paper | N/A |
| Software and Algorithms | ||
| Prism | GraphPad, v7 | https://www.graphpad.com/scientific-software/prism/ |
| Proteome Discoverer | Thermo Fisher Scientific, v2.3 | https://www.ThermoFisher.com/order/catalog/product/OPTON-30795 |
| In-house mass spectrometry data analysis software | Huttlin et al Cell. (2010) 143:1174–89. | N/A |
| SEQUEST | Eng et al., 1994 | N/A |
| Perseus | Tyanova et al., Nat Methods. (2016) 13:731–40. | http://www.perseus-framework.org |
| FlowJo™ | V10.5.2 | https://www.flowjo.com |
| ImageStudioLite | V 5.2.5 | https://www.licor.com/bio/products/software/image_studio_lite |
| FiJi | ImageJ V.2.0.0 | https://imagej.net/Fiji |
| Other | ||
| Orbitrap Fusion Lumos Mass Spectrometer | Thermo Fisher Scientific | Cat#IQLAAEGAAPFADBMBHQ |
| Easy-nLC 1200 | Thermo Fisher Scientific | LC140 |
| Odyssey CLx Imager | LI-COR bioscience | N/A |
| QuantStudio7 | Thermo Fisher Scientific | |
| Countess Automated Cell Counter | Thermo Fisher Scientific | |
HIGHLIGHTS.
Quantitative global proteomics of cells during nutrient stress
TEX264 identified as ER-associated selective autophagy receptor during starvation
TEX264 traffics into autophagosomes at ER tubule junctions
TEX264-deficient cells defective in ER-phagy during nutrient stress
ACKNOWLEDGMENTS
This work was supported by NIH (R37 NS083524, RO1 NS110395, and RO1 GM095567 to J.W.H., K01 DK098285 to J.A.P., R00 GM117218 to C.J.S., and R35 GM127136 to V.D.), and a generous gift from Ned Goodnow (J.W.H.). We thank Ivan Dikic for plasmids and advice, Simon Wilkinson for plasmids, Tom Rapoport for helpful discussions, and the Nikon Imaging Center at Harvard Medical School for imaging assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
J.W.H. is a consultant and founder of Rheostat Therapeutics and a consultant for X-Chem Inc.
Data and Software Availability
All data are available by request.
REFERENCES
- An H, and Harper JW (2018). Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat Cell Biol 20, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman V, and Aravind L (2004). The SHS2 module is a common structural theme in functionally diverse protein groups, like Rpb7p, FtsA, GyrI, and MTH1598/TM1083 superfamilies. Proteins 56, 795–807. [DOI] [PubMed] [Google Scholar]
- Anding AL, and Baehrecke EH (2017). Cleaning House: Selective Autophagy of Organelles. Dev Cell 41, 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikic I (2018). Open questions: why should we care about ER-phagy and ER remodelling? BMC Biol 16, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F, Noack J, Bergmann TJ, Cebollero E, Pisoni GB, Fasana E, Fregno I, Galli C, Loi M, Solda T, et al. (2016). Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol 18, 1173–1184. [DOI] [PubMed] [Google Scholar]
- Grumati P, Morozzi G, Holper S, Mari M, Harwardt MI, Yan R, Muller S, Reggiori F, Heilemann M, and Dikic I (2017). Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 6, 10.7554/eLife.25555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Ordureau A, and Heo JM (2018). Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol 19, 93–108. [DOI] [PubMed] [Google Scholar]
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, and Yamamoto A (2009). A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 11, 1433–1437. [DOI] [PubMed] [Google Scholar]
- Hu J, and Rapoport TA (2016). Fusion of the endoplasmic reticulum by membrane-bound GTPases. Semin Cell Dev Biol 60, 105–111. [DOI] [PubMed] [Google Scholar]
- Hung V, Lam SS, Udeshi ND, Svinkina T, Guzman G, Mootha VK, Carr SA, and Ting AY (2017). Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 6, 10.7554/eLife.24463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, and Young LN (2017). Mechanisms of Autophagy Initiation. Annu Rev Biochem 86,225–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhak DN, Tyanova S, Cox J, and Borner GH (2016). Global, quantitative and dynamic mapping of protein subcellular localization. Elife 5, 10.7554/eLife.16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama H, Kogure T, Mizushima N, Yoshimori T, and Miyawaki A (2011). A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 18, 1042–1052. [DOI] [PubMed] [Google Scholar]
- Khaminets A, Behl C, and Dikic I (2016). Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol 26, 6–16. [DOI] [PubMed] [Google Scholar]
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, et al. (2015). Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358. [DOI] [PubMed] [Google Scholar]
- Ktistakis NT, and Tooze SA (2016). Digesting the Expanding Mechanisms of Autophagy. Trends Cell Biol 26, 624–635. [DOI] [PubMed] [Google Scholar]
- Liang JR, Lingeman E, Ahmed S, and Corn JE (2018). Atlastins remodel the endoplasmic reticulum for selective autophagy. J Cell Biol 217, 3354–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Khor S, Hackett SR, Rabinowitz JD, Perlman DH, and White E (2014). Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol Cell 55, 916–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejlvang J, Olsvik H, Svenning S, Bruun JA, Abudu YP, Larsen KB, Brech A, Hansen TE, Brenne H, Hansen T, et al. (2018). Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J Cell Biol 217, 3640–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TJ, Gubas A, and Tooze SA (2018). A molecular perspective of mammalian autophagosome biogenesis. J Biol Chem 293, 5386–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, and Ohsumi Y (2011). The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27, 107–132. [DOI] [PubMed] [Google Scholar]
- Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, and Nakatogawa H (2015). Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 522, 359–362. [DOI] [PubMed] [Google Scholar]
- Moggridge S, Sorensen PH, Morin GB, and Hughes CS (2018). Extending the Compatibility of the SP3 Paramagnetic Bead Processing Approach for Proteomics. J Proteome Res 17, 1730–1740. [DOI] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, and Lazarou M (2016). Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol 215, 857–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, and Shimizu S (2009). Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461, 654–658. [DOI] [PubMed] [Google Scholar]
- Paulo JA, O’Connell JD, Everley RA, O’Brien J, Gygi MA, and Gygi SP (2016). Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J Proteomics 148, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontano Vaites L, Paulo JA, Huttlin EL, and Harper JW (2017). Systematic analysis of human cells lacking ATG8 proteins uncovers roles for GABARAPs and the CCZ1/MON1 regulator C18orf8/RMC1 in macro and selective autophagic flux. Mol Cell Biol 10.1128/MCB.00392-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronan B, Flamand O, Vescovi L, Dureuil C, Durand L, Fassy F, Bachelot MF, Lamberton A, Mathieu M, Bertrand T, et al. (2014). A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat Chem Biol 10, 1013–1019. [DOI] [PubMed] [Google Scholar]
- Smith M, and Wilkinson S (2017). ER homeostasis and autophagy. Essays Biochem 61, 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, von Kriegsheim A, Behrends C, and Wilkinson S (2018). CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev Cell 44, 217–232 e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Lin MG, Stowe TR, Chen S, Zhu M, Stearns T, Franco B, and Zhong Q (2013). Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 502, 254–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboyama K, Koyama-Honda I, Sakamaki Y, Koike M, Morishita H, and Mizushima N (2016). The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354, 1036–1041. [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Wirth M, Joachim J, and Tooze SA (2013). Autophagosome formation--the role of ULK1 and Beclin1-PI3KC3 complexes in setting the stage. Semin Cancer Biol 23, 301–309. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Movie S1. Visualization of ER tubules marked by TEX264-eGFP (green) and autophagosomes marked by mCherry-LC3A in COS7 cells after EBSS treatment. Relevant to Figure 4.
Supplemental Movie S2. Visualization of ER tubules marked by TEX264-eGFP (green) and lysosomes marked by Lysotracker-Red in COS7 cells after EBSS treatment. Relevant to Figure 4.