

Abstract
This chapter reviews genes and syndromes associated with predisposition to colorectal cancer (CRC), with an overview of gene variant classification. We include updates on the application of preventive and therapeutic measures, focusing on the use of non-steroidal anti-inflammatory drugs (NSAIDs) and immunotherapy. Germline pathogenic variants in genes conferring high or moderate risk to cancer are detected in 6–10% of all CRCs and 20% of those diagnosed before age 50. CRC syndromes can be subdivided into nonpolyposis and polyposis entities, the most common of which are Lynch syndrome and familial adenomatous polyposis. In addition to known and novel genes associated with highly penetrant CRC risk, identification of pathogenic germline variants in genes associated with moderate-penetrance cancer risk and/or hereditary cancer syndromes not traditionally linked to CRC may have an impact on genetic testing, counseling and surveillance. The use of multigene panels in genetic testing has exposed challenges in the classification of variants of uncertain significance. We provide an overview of the main classification systems and strategies for improving these. Finally, we highlight approaches for integrating chemoprevention in the care of individuals with genetic predisposition to CRC and use of targeted agents and immunotherapy for treatment of mismatch repair deficient and hypermutant tumors.
Keywords: Hereditary colorectal cancer, cancer predisposition, cancer syndromes, polyposis, cancer genes, variants of uncertain significance, VUS, chemoprevention, checkpoint inhibitors, immune-oncology
Introduction
Colorectal cancer (CRC) is the third most common cancer diagnosed in men and women. While there has been an overall decrease in CRC incidence and mortality among individuals age 50 and older, recent epidemiological studies demonstrate increasing incidence of CRC among young individuals which remains unexplained [1]. Genetic predisposition, due to pathogenic germline variants in genes associated with high cancer risk has been implicated in 2–8% of all CRCs, 6–10% when considering pathogenic mutations in known high- and moderate- penetrance genes [2–4] (1 in 5 of those diagnosed at age<50) [5–7]. For individuals with certain hereditary cancer syndromes lifetime risks for CRC may approach 50–80% in the absence of endoscopic and/or surgical intervention. In addition to family history, tumor histology and molecular phenotypes are instrumental not only for identifying individuals with genetic predisposition to CRC but also for guiding cancer treatment. The following review is an overview of the known CRC predisposing genetic conditions, the challenge of variant classification, and chemoprevention and treatment strategies in hereditary CRC syndromes.
Genetic Susceptibility to Colorectal Cancer: Polyposis and Non-Polyposis Syndromes
Genetic susceptibility to CRC appears to be more common than previously appreciated. Several recent studies have identified pathogenic germline variants in a broad spectrum of high and moderate penetrance cancer susceptibility genes in > 10% of individuals with advanced cancer diagnoses [8, 9] and the prevalence of 1 in 10 appears to also be true among individuals with CRC [2]. In a cohort of unselected CRC patients evaluated at a tertiary care cancer center, 105/1058 (9.9%) had pathogenic germline variants identified through next generation sequencing with a multigene panel, half of which were in cancer genes not previously associated with CRC risk [2]. Genetic susceptibility appears to be even more prevalent among young CRC patients, with several studies documenting prevalence of germline mutations of 16–33% among those diagnosed age<50 [5–7].
The hereditary CRC syndromes, characterized by dramatic increases in risk for colorectal neoplasia, are phenotypically divided into polyposis and nonpolyposis syndromes, based largely on the number and histology of colorectal polyps (Figure 1). Tumor molecular features characteristic of CRC-predisposing syndromes caused by altered DNA repair are shown in Table 1, and current colonoscopy surveillance recommendations for the well-known high penetrance CRC syndromes, in Table 2.
Figure 1.
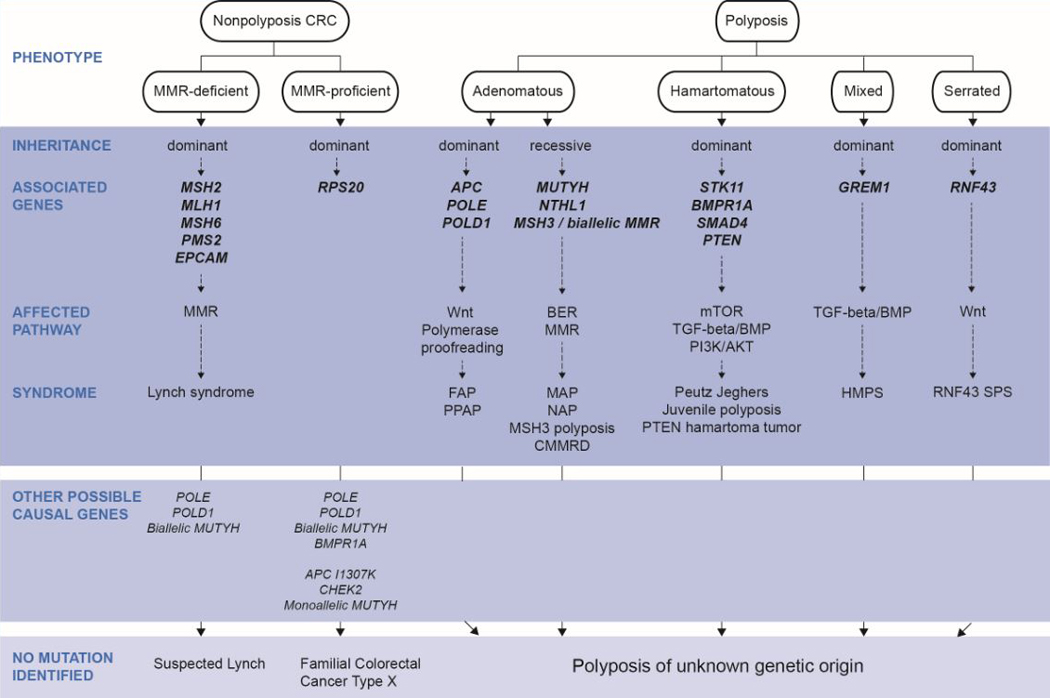
Phenotypic classification of nonpolyposis and polyposis CRC syndromes, mode of inheritance, causal genes and affected molecular pathways.
Note: Germline AXIN2 autosomal dominant mutations (Wnt pathway) may cause oligodontia-colorectal cancer syndrome characterized by severe permanent tooth agenesis and the presence CRC or precancerous colonic or gastric lesions of variable types (adenomas, hyperplastic polyps) [132–134]. Due to the still undefined CRC and polyposis phenotype, it has not been included in the figure.
Abbreviations: BER, base excision repair; CMMRD, constitutional mismatch repair deficiency; HMPS, hereditary mixed polyposis syndrome; MAP, MUTYH-associated polyposis; MMR, DNA mismatch repair; PPAP, polymerase proofreading-associated polyposis; SPS, serrated polyposis syndrome.
Table 1.
Molecular alterations detected in the tumors developed by carriers of germline mutations in DNA repair genes.
| Syndrome | Causal gene | Tumor molecular features |
COSMIC mutational signaturesb |
|---|---|---|---|
| Lynch syndrome | MSH2 | MSI | Single base substitution: SBS6, SBS15, SBS21, SBS26, SBS44 |
| MLH1 | IHC: loss of MMR protein | Doublet base substitution: DBS7, DBS10 | |
| MSH6 | hypermutated | Insertion and deletion: ID7, (ID1), (ID2) | |
| PMS2 | |||
| PPAP | POLEa | Ultramutated | Single base substitution: SBS10a, SBS10b, SBS14 (concurrent POLE mutation and MMR deficiency), SBS20 (concurrent POLD1 mutation and MMR deficiency) Doublet base substitution: DBS3 |
| POLD1a | ↑C:G>A:T (context TCT) | ||
| ↑C:G>T:A (context TCG) | |||
| MAP | Biallelic MUTYH | ↑G:C>T:A | Single base substitution: SBS36 |
| KRAS G12C | |||
| NTHL1-associated polyposis | Biallelic NTHL1 | ↑G:C>A:T | Single base substitution: SBS30 |
| MSH3-associated polyposis | Biallelic MSH3 | MSI of di- and tetranucleotides (EMAST) | - |
| CMMRD | Bilalellic MSH2, | MSI |
See Lynch syndrome |
| MLH1, MSH6, | IHC: loss of MMR protein | ||
| PMS2 | (tumor and normal tissues) | ||
Mutations affecting the proofreading (exonuclease) activity of the polymerases.
Alexandrov et al. 2018 [129].
Abbreviations: CMMRD, constitutional mismatch repair deficiency; EMAST, elevated microsatellite alterations at selected tetranucleotide repeats; IHC, immunohistochemistry; MAP, MUTYH-associated polyposis; MMR, DNA mismatch repair; MSI, microsatellite instability; PPAP, polymerase proofreading-associated polyposis.
Table 2.
Colonoscopy surveillance recommendations for individuals with germline pathogenic variants (high penetrance syndromes) [130].
| Syndrome (Gene) | Family history of CRC | Age at CRC screening initiation | Screening interval if no adenomas |
|---|---|---|---|
| No mutationa | No | 50 | 10 years |
| Yes (≥1 FDR) | 40b | 5–10 years | |
| FAP (APC) | N/A | 10–15 | 1 year, colectomy if polyps too numerous |
| Lynch syndrome (MLH1, MSH2, MSH6, PMS2) | N/A | 20–25 | 1–2 years until age 40, then every 1 year |
| MAP (MUTYH Biallelic) | N/A | 25–30 | 1–3 years depending on polyp burden |
| Juvenile polyposis (SMAD4, BMPR1A) | N/A | 15 | 1–3 years depending on polyp burden |
| Peutz-Jeghers (STK11) | N/A | 15 | 2–3 years depending on polyp burden |
| Li Fraumeni (TP53) | N/A | 20–25 | 3 years |
| Hereditary Breast | No | 50 | 10 years |
| Ovarian Cancer | Yes | 50 or per family history | 5 years |
| (BRCA1/BRCA2) | |||
Recommendations based in the guidelines from the National Comprehensive Cancer Network [71].
40 years old or 10 years earlier than the youngest-onset CRC in the family.
Abbreviations: CRC, colorectal cancer; FDR, first-degree relative; SDR, second-degree relative.
Polyposis syndromes
Familial adenomatous polyposis (FAP) is characterized by multiple (typically dozens to hundreds) colorectal adenomas, with potential for significant variability in clinical phenotype. FAP is associated with pathogenic germline variants in APC, a tumor suppressor instrumental in regulation of WNT signaling. While FAP exhibits autosomal dominant inheritance, approximately 30% of affected individuals have no family history and represent de novo mutations [10]. Phenotypes vary, with some individuals exhibiting classic polyposis (100s-1000s polyps) requiring surgical colectomy, while others may manifest more subtle presentations (20–100 polyps), often referred to as attenuated polyposis (or AFAP). Most individuals with FAP also develop neoplasia in the upper GI tract, including gastric fundic gland polyps and duodenal and ampullary adenomas..Adenocarcinomas of the duodenum and ampullanowadays represent the second leading cause of cancer death after CRC requiring ongoing endoscopic surveillance. Although gastric fundic gland polyps rarely exhibit neoplastic transformation, gastric adenocarcinomas have been reported. A rare germline point mutations in Exon 1B of APC have been identified in individuals with Gastric Adenocarcinoma and Proximal Polyposis Syndrome (GAPPS), conferring severe gastric polyposis and high risk for gastric cancer without colorectal polyposis [11]. Extraintestinal manifestations in FAP can include increased risk for papillary thyroid cancers (particularly the cribiform-morular variant). Desmoid tumors develop in some individuals, and mesenteric desmoid disease can be a source of significant morbidity and mortality. Although some studies have found associations between mutations in codons 543–713 and 1310–2011 and risk for desmoid disease [12], factors contributing to desmoid disease remain largely unknown.
MUTYH-associated polyposis (MAP) is an autosomal recessive syndrome associated with biallelic germline variants in the base excision repair gene MUTYH. Individuals with MAP can exhibit a wide range of phenotypes including classic and attenuated polyposis. Two common founder mutations (Y165C and G382D) have a carrier frequency of 1% in populations of European ancestry [13]. Monoallelic MUTYH variants have been found to be associated with a moderate (1.5–2 fold) increased risk for CRC, particularly among individuals with a first degree relative with CRC [14].
Polymerase proofreading-associated Polyposis (PPAP) is associated with germline pathogenic variants in the exonuclease (proofreading) domains of polymerases epsilon (POLE) and delta (POLD1) [15]. Individuals may present with autosomal dominant classic or attenuated polyposis, CRCs and other tumors that exhibit somatic hypermutation, usually with DNA mismatch repair proficient phenotypes.
Adenomatous polyposis syndromes have been recently updated with the addition of two rare autosomal recessive forms caused by biallelic mutations in NTHL1, a DNA glycosylase involved in base excision repair [16], and in MSH3, an MMR gene not associated with Lynch syndrome [17].
Hamartomatous polyposis syndromes, characterized by the presence of gastrointestinal hamartomatous polyps, are rare, having only one tenth the prevalence of adenomatous polyposis syndromes. Hamartomatous polyposis syndromes exhibit autosomal dominant patterns of inheritance, and include Peutz-Jeghers, Juvenile polyposis and PTEN-hamartoma tumor syndromes.
Peutz-Jeghers syndrome (PJS) is characterized by multiple hamartomatous polyps throughout the GI tract and increased risk for various cancers including gastrointestinal (gastric, colorectal, pancreatic), breast, lung, and sex cord tumors. Individuals with PJS may have prominent mucocutaneous pigmentation and bowel obstructions due to polyp intussusceptions. Germline pathogenic variants in STK11 are identified in 50–70% of individuals.
Juvenile polyposis syndrome (JPS) is characterized by multiple gastric and/or colonic hamartomas. Germline pathogenic variants in BMPR1A and SMAD4 are identified in 50–70% of affected individuals. JPS is associated with increased risks for gastric and colorectal cancers. Individuals with SMAD4 mutations are at risk for hereditary hemorrhagic telangiectasia (HHT).
PTEN-hamartoma tumor syndrome (PHTS) is associated with increased risk for breast, thyroid, endometrial, and renal cancers resulting from germline pathogenic variants in PTEN. The gastrointestinal phenotype of the PTEN-hamartoma tumor syndrome can include gastric and colorectal hamartomas, adenomas, serrated polyps, hyperplastic polyps, lipomas and ganglioneuromas. PTEN pathogenic variants confer variable clinical phenotypes, which include several conditions such as Cowden, Bannayan-Riley-Ruvalcaba and Proteus-like syndromes [18].
Mixed polyposis is characterized by the presence of multiple colorectal polyps of mixed histological type, including serrated lesions, conventional adenomas and hamartomas, and is associated with increased risk of colorectal carcinoma. While the genetic cause remains elusive in most cases, germline variants in and upstream of GREM1 have been identified in some affected individuals. A founder mutation consisting of a duplication of 40Kb upstream GREM1 has been identified in several kindreds of Ashkenazi Jewish ancestry [19], while a duplication of 16kb has been reported in a Swedish family affected with hereditary mixed polyposis [20].
Serrated polyposis, previously referred to as hyperplastic polyposis, is defined by the World Health Organization on the basis of any of the following criteria 1) ≥5 serrated polyps proximal to the sigmoid colon with at least 2 measuring >10 mm; 2) any number of serrated polyps in the proximal colon in an individual with a first-degree relative with serrated polyposis; or 3) >20 serrated polyps of any size [21]. While germline mutations in the tumor suppressor gene RNF43 have been identified in rare cases of serrated polyposis [22, 23], the low mutation frequency among affected individuals tempers enthusiasm for including RNF43 in multigene panels [24, 25]. Although germline mutations in GREM1 and MUTYH have been reported, genetic testing is usually uninformative.
Hereditary nonpolyposis colorectal cancer
Although the CRC syndromes associated with polyposis phenotypes are the most easily recognized, the vast majority of individuals affected by genetic predisposition to CRC do not exhibit multiple polyps. Syndromic non-polyposis CRC is subdivided on the basis of molecular tumor phenotype as DNA mismatch repair deficient (MMR-d) or proficient (MMR-p) (Figure 1).
Lynch syndrome (LS) [previously known as hereditary nonpolyposis colorectal cancer (HNPCC)] is the most common of the hereditary CRC syndromes. LS is associated with pathogenic germline variants or epimutations in DNA mismatch repair genes (MLH1, MSH2, MSH6, PMS2), which predispose to development of neoplasms with distinctive molecular phenotypes of MMR-d. MMR-d tumors exhibit high instability at specific DNA microsatellites (MSI-H) and loss of expression of the corresponding DNA mismatch repair protein by immunohistochemistry. Although CRC and endometrial cancer are the most prominent cancers in most affected families, risks for ovarian, gastric, small intestinal, urinary tract, brain, pancreatic, prostate and sebaceous neoplasms of the skin are also increased among mutation carriers. Lynch-associated colorectal neoplasms tend to develop at younger ages and progress more rapidly compared with sporadic CRCs, requiring specialized surveillance (Table 2). Although risk prediction models use personal and family history to assess an individual’s probability of carrying a germline MMR gene mutation (e.g. PREMM1,2,6 [26], MMRpro [27]), universal screening of CRC tumors for MMR-d remains the most effective strategy for identifying individuals affected with LS (Figure 2) [28, 29].
Figure 2.
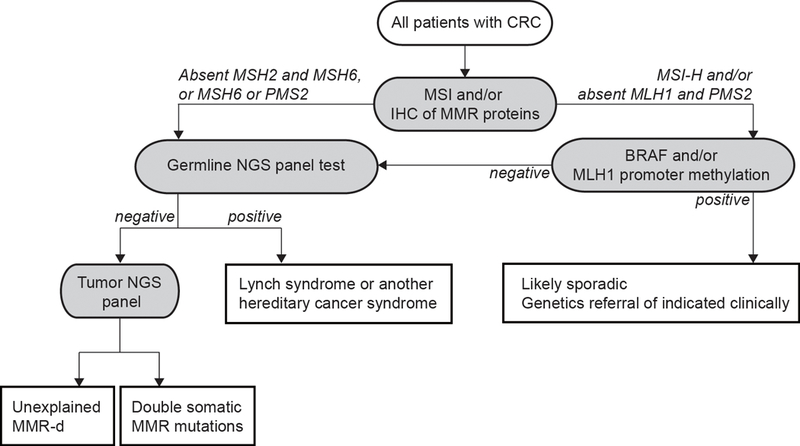
Strategy for universal tumor screening for Lynch syndrome in CRC patients (Adapted from Hampel et al. 2018 [29]). The different etiologies of MMR-d CRCs are: i) germline MMR gene mutation; ii) serrated pathway lesions (somatic BRAF mutation and/or MLH1 promoter hypermethylation); iii) double somatic MMR gene mutations; iv) somatic MMR gene mutation secondary to a POLE or POLD1 exonuclease mutation or to biallelic MUTYH mutations.
Abbreviations: CRC, colorectal cancer; IHC, immunohistochemistry; MMR, DNA mismatch repair; MMR-d, mismatch repair deficient; MSI, microsatellite instability; MSI-H, high level of microsatellite instability (microsatellite unstable); NGS, next generation sequencing.
MMR-proficient hereditary nonpolyposis colorectal cancer.
Half of CRC families meeting Amsterdam criteria (3 individuals with CRC over 2 generations, ≥1 diagnosed age<50) have MMR-p tumors, without identifiable germline mutations in the MMR genes. These families can be distinguished from LS in that the lifetime risk for CRC is lower (only 2-fold increased) and there is no increase in risks for extracolonic tumors [30]. Despite enormous efforts to identify new genes that could explain the apparently dominantly inherited forms of MMR-p nonpolyposis CRC, the only candidate gene that has shown consistent association with hereditary nonpolyposis CRC is RPS20 (ribosomal protein S20) [31, 32]. Although the scarce available data suggest high penetrance for RSP20 mutations and absence of extracolonic manifestations, data from additional mutation carriers are required to estimate risks and recommend surveillance measures. Many other putative familial CRC genes have been proposed, but most are extremely uncommon, and others may only moderately increase the risk to CRC, complicating the assessment of their contribution to predisposition to CRC [33–36].
Prevalence and penetrance
The prevalence of mutations in CRC-predisposing genes has traditionally been estimated from patients diagnosed with CRC (or endometrial cancer for LS). This approach reveals that LS accounts for about 3% of CRC [2, 37–40] and 2% of endometrial cancer cases [41–43]; additionally, FAP accounts for 0.3–0.5% of diagnosed colorectal tumors [2, 3]. When LS is ascertained from individuals with a personal/familial history of cancer, germline mutations in MSH2 and MLH1 consistently account for the majority of LS cases (60–87.5%), with a minority of cases carrying mutations in MSH6 and PMS2 [2, 5, 6, 37, 39, 44].
Recent epidemiologic data have shed more light on the true prevalence of LS in the general population, finding that LS is more common and less penetrant than traditionally estimated. Recently, Win et al. studied 5,744 CRC patients and 37,634 first-degree relatives, 2% of whom had been diagnosed with CRC, recruited through the Colon Cancer Family Registry [45] (http://coloncrf.org) and for whom germline genetic testing results were available [46]. They estimated that 0.36% (1 in 279) of the population carry pathogenic mutations in the MMR genes: 0.140% (1 in 714) in PMS2, 0.132% (1 in 758) in MSH6, 0.051% (1 in 1,946) in MLH1and 0.035% (1 in 2,841) in MSH2. Regarding MUTYH, 2.2% (1 in 45) would be monoallelic and 0.012% (1 in 8,073), biallelic carriers. Interestingly, pathogenic variants in PMS2 and MSH6 are the most prevalent in the general population, but least prevalent among LS cases ascertained based on their personal history of cancer, suggesting that the PMS2 and MSH6 mutations confer more modest risk of cancer when compared with MLH1 and MSH2 mutations [47–53]. Of note, the presence of founder mutations in specific populations may increase the prevalence of the syndrome and influence the relative proportion of mutations in each gene in those populations [54–58].
For other less common CRC syndromes, the estimated prevalence is very low, ranging from 1 in 10,000–31,250 for APC-associated adenomatous polyposis [59, 60], to 1 in 100,000–250,000 for hamartomatous polyposis syndromes [61–63]. The prevalences of more recently described syndromes, such as those caused by mutations in POLE and POLD1, NTHL1, RNF43 or MSH3, remain unknown.
Significant interpatient heterogeneity exists among patients with a priori the same CRC predisposing syndrome, posing challenges for diagnosis and clinical management. For years, lack of prospectively-obtained information has led current clinical guidelines to rely upon retrospective data from patient cohorts whose selection for molecular testing was biased (CRC risk estimates from retrospectively collected cohorts were reviewed by Lorans et al. [64]). The Prospective LS Database (PLSD) provides estimates of cancer risks in LS, both in individuals who have yet to develop a cancer and those who have survived a cancer (http://lscarisk.org/). According to these data, the relative cumulative incidence (relative risk) of cancer at age 75 is: 10–12% for CRC and 25–35% for endometrial cancer in MLH1 and MSH2 mutation carriers; 30% for endometrial cancer in MSH6 mutation carriers, and for PMS2, the increased cancer risks did not reach statistical significance when compared to population incidence [65]. More recently, the analysis of 284 families, including 4,878 first- and second-degree family members, 513 of whom were PMS2 mutation carriers, concluded that PMS2 mutation carriers are at small increased risk of CRC (cumulative risk at age 80: 12–13%) and endometrial cancer (cumulative risk at age 80 for female carriers: 13%) [66].
In addition to the gene-specific risks, cancer risks in hereditary CRC syndromes may also vary by the type of mutation, ethnicity or geographic location. There is heterogeneity even among family members sharing the same mutation, suggesting that other factors, such as environmental and polygenic factors may influence phenotypic expression [67].
Moderate-penetrance colorectal cancer gene mutations
The inclusion of moderate-penetrance cancer susceptibility genes in multigene panel testing poses challenges regarding the optimal management of carriers of pathogenic mutations in these genes. In fact, the associated clinical significance of these mutations remains unclear. In the case of CRC, the most prevalent mutations are APC p.I1307K, CHEK2 c.1100delC, CHEK2 p.I157T, and monoallelic MUTYH mutations [2] (Figure 1). Recently, Katona et al. [68] defined a counseling framework for these moderate-penetrance mutations based on the estimated CRC risk associated with each variant [69] and the estimated CRC risk for average-risk individuals [70 April 2017. #171]. Based on this analysis, colonoscopy screening initiation is recommended at age 45 (at age 50 for average-risk individuals [71]) for APC p.I1307K and CHEK2 mutation carriers, however, no earlier initiation of colonoscopy screening is recommended for monoallelic MUTYH mutation carriers, in line with current National Comprehensive Cancer Network recommendations [71]. Such recommendations apply to patients without family history of CRC; importantly, however, earlier and more frequent colonoscopy screening is recommended for individuals with a family history of CRC, even in the absence of gene-based findings [72].
Mutations in genes associated with hereditary cancer syndromes not traditionally linked to CRC
Both classical testing strategies and multigene panel tests in CRC cases have uncovered germline pathogenic variants in cancer susceptibility genes associated with syndromes that do not classically include CRC, such as hereditary breast and ovarian cancer syndrome. Studies with several hundred up to roughly 2,000 CRC cases and controls have yielded evidence for ATM, BRCA1, BRCA2, CDKN2A, PALB2, and TP53 as moderate-risk CRC susceptibility genes (Table 3). The most frequently mutated non-CRC hereditary genes identified in CRC patients are BRCA1 and BRCA2 (0.7–1.3% of CRC patients, regardless of selection criteria), followed by the moderate-penetrance gene ATM (0.7–0.9% of CRC patients, regardless of selection criteria). The debate whether pathogenic BRCA mutations, or mutations in any of the above mentioned genes, increase the risk of CRC is still ongoing. A recent meta-analysis based on 14 studies [73] estimated a 1.22-fold increased risk of CRC in BRCA mutation carriers, and this was attributable largely to a 1.48-fold greater risk in BRCA1 mutation but not in BRCA2 mutation carriers, regardless of age.
Table 3.
Characteristics and results of key published studies on multigene germline testing for CRC predisposition.
| Study | Tested patients | Country | Multigene panel | Hereditary CRC genes | Other cancer genesa |
|---|---|---|---|---|---|
| Non-selected CRC patients | |||||
| Yurgelun 2017 [2] | 1,058 CRC patients (clinic-based) | USA | Commercial 25-gene panelb |
High penetrance 3.1% MMR gene (Lynch sd.) 0.5% APC 0.3% biallelic MUTYH Moderate/Low penetrance 1.7% monoallelic MUTYH 1.3% APC*I1307K 0.2% CHEK2 |
High penetrance 1.0% BRCA1/2 0.2% PALB2 0.1% CDKN2A 0.1% TP53 Moderate/Low penetrance 0.9% ATM 0.3% BRIP1 0.2% NBN 0.1% BARD1 |
| AlDubayan 2018 [3] | 680 CRC patients (NHS, HPFS, CanSeq study) | USA | 14 CRC-risk genes and 40 DNA repair genes associated with (non-CRC) cancer phenotypes |
High penetrance 0.6% MMR gene (Lynch sd.) 0.3% APC 0% biallelic MUTYH Moderate/Low penetrance 1.62% monoallelic MUTYH 1.18% APC*I1307K 0.6% CHEK2 |
High penetrance 0.7% BRCA1/2 0.4% PALB2 0.3% TP53 Moderate/Low penetrance 0.7% ATM 0.3% BRIP1 0.1% BARD1 |
| DeRycke 2017 [4] | 548 CRC patients (Colon Cancer Family Registry) | Australasia USA Canada | 36-gene custom panelc (known or putative CRC genes) |
High penetrance 6% MMR gene (Lynch sd.) 0.9% APC 0.4% biallelic MUTYH Moderate/Low penetrance 0.4% CHEK2 |
High penetrance 0.2% TP53 0.5% FLCN |
| Non-selected young-onset CRC patients | |||||
| Pearlman 2017 [5] | 450 CRC patients age <50 | USA | Commercial 25-gene panelb |
High penetrance 8.4% MMR gene (Lynch sd.) 1.3% APC 0.9% biallelic MUTYH 0.2% SMAD4 Moderate/Low penetrance 1.6% monoallelic MUTYH 0.9% APC*I1307K 0.2% CHEK2 |
High penetrance 1.3% BRCA1/2 0.4% PALB2 0.2% CDKN2A Moderate/Low penetrance 0.9% ATM |
| DeRycke 2017 [4] | 333 CRC patients age ≤50 (MMR-proficient or unknown MMR status) | Australasia USA Canada | 36-gene custom panelc (known or putative CRC genes) |
High penetrance 4.8% MMR gene (Lynch sd.) 2.1% APC 1.5% biallelic MUTYH 0.3% SMAD4 0.3% BMPR1A Moderate/Low penetrance 0.3% CHEK2 |
High penetrance 0% TP53 |
| High-risk patients (familial CRC) | |||||
| Yurgelun 2015 [44] | 1,260 patients referred for Lynch sd. germline testing | USA | Commercial 25-gene panelb |
High penetrance 9.0% MMR gene (Lynch sd.) 0.4% APC 0.2% biallelic MUTYH 0.08% STK11 Moderate/Low penetrance 2.1% monoallelic MUTYH 0.4% CHEK2 |
High penetrance 1.2% BRCA1/2 0.08% PALB2 Moderate/Low penetrance 0.7% ATM 0.2% BRIP1 0.08% NBN 0.08% BARD1 0.08% RAD51C |
| Stoffel 2018 [6] | 430 CRC patients age <50 evaluated by a clinical genetics service | USA | Germline DNA sequencing (n=293) Commercial multigene panel (n=22)b 67-gene custom panel (n=117)d |
High penetrance 13.0% MMR gene (Lynch sd.) 2.3% APC 1.9% biallelic MUTYH 0.5% SMAD4 0.2% POLE Moderate/Low penetrance 0.5% CHEK2 |
High penetrance 0.5% TP53 0.2% BRCA1/2 |
| Hansen 2017 [131] | 274 patients (263 families) fulfilling the Amsterdam (n=262) or revised Bethesda (n=12) criteria with no pathogenic MMR mutations | Norway Australia | 122-gene custom paneld |
High penetrance 1.14% MMR gene (Lynch sd.)e 0.8% POLE 0.4% biallelic MUTYH 0.4% PTEN 0.4% AXIN2 (oligodontia-CRC sd.) 0% APC Moderate/Low penetrance 1.5% monoallelic MUTYH 0.4% CHEK2 |
High penetrance 1.1% BRCA1/2 Moderate/Low penetrance 0.8% ATM |
The causal role of these genes in CRC predisposition has not been unequivocally proven.
NTHL1, MSH3, POLE and POLD1 and RPS20 are not included in the panel.
NTHL1, POLE and POLD1 and RPS20 are not included in the panel.
NTHL1 and RPS20 are not included in the panel.
Previously identified MMR mutation carriers had been excluded from the analysis.
Whether these findings are the result of detecting the background population prevalence of such mutations or the result of pleiotropism, i.e., a germline variant manifests itself in a variety of clinical phenotypes, which would suggest that, for example, BRCA1/BRCA2 mutations increase the risk to CRC, is still a matter of debate. In order to clarify the contribution of non-CRC susceptibility genes to CRC predisposition, Dobbins et al. analyzed 114 hereditary cancer genes in approximately 850 unexplained early-onset/familial CRC and 1,609 controls. Globally, no statistically significant enrichment of pathogenic and likely pathogenic variants was detected between cases and controls (6.7% vs. 5.3%), not even for BRCA or TP53 mutations, thus arguing against the hypothesis supporting pleiotropism [74]. Recently, AlDubayan et al. evaluated the presence of germline mutations in 40 DNA repair genes linked to (non-CRC) inherited cancer predisposition in 591 unselected CRC patients from two prospective population-based studies and 89 clinic-based unselected CRC patients (total n=680) and compared the mutation frequency with that observed in 27,728 ancestry-matched cancer-free adults from the Exome Aggregation Consortium (ExAC). This study revealed significantly higher rates of ATM and PALB2 mutations in CRC patients than in cancer-free controls, results that were independently validated in 1,661 unselected CRC patients for both genes and in 1,459 early-onset (age<56) CRC patients only for ATM [3]. On the other hand, no differences were observed for BRCA1 and BRCA2 or other non-CRC DNA repair genes. The consequences of one or the other situation (background population mutation prevalence vs. pleiotropism) are different and highly relevant for the management of the families, therefore, requiring further research to provide definitive evidence.
Genetic testing for predisposition to CRC
The well-established, clinically-actionable susceptibility genes with quantified magnitude of risk form the core of current familial CRC and polyposis genetic testing (Table 3). In recent years, clinical genetic testing has transitioned from phenotype-driven single gene sequencing to multigene panel testing using targeted massively parallel sequencing. The criteria for identifying individuals most likely to benefit from genetic testing continue to evolve along with our understanding of the variability in disease penetrance and expressivity associated with germline alterations in cancer predisposition genes [71].
Overview on variants of uncertain significance
When patients are tested for germline susceptibility gene mutations, most outcomes fall into one of three categories: a pathogenic variant is found, a variant of uncertain significance (VUS) is found, or no reportable variant is found. When a pathogenic variant is found, patients can be counseled and managed on the basis of their personal and family cancer history plus gene-specific guidelines. “Cascade testing” of at-risk relatives can identify additional carriers. These newly identified carriers benefit because they can be offered earlier and intensified screening; this is particularly valuable for CRC syndromes because there is credible evidence that exposure to colonoscopy reduces incidence and mortality from CRC [75, 76]; put simply, cascade testing followed by intensified screening of carriers can add years to these individuals’ lives. Non-carriers benefit from knowing that their CRC risk is lower than that of carriers in their family, and may be spared intensified family history-based screening [77, 78].
Observation of a VUS presents a quandary, since it is not known where on a spectrum from pathogenic to benign any given VUS falls, carrier status does not stratify members of a family into those with higher or lower risk. Thus, detection and reporting of the VUS provides no medical management benefit to sentinel carriers or their relatives. Unfortunately physicians may misinterpret or miscommunicate a VUS test result, resulting in management of a patient with a VUS as if they carried a pathogenic variant, which is clearly incorrect [79, 80]. Moreover, there is a lack of tools for updating clinical oncologists and genetic counselors after a VUS has been reclassified [81].
Main categories of variants of uncertain significance
Most VUS fall into one of three categories: missense substitutions, splice junction variants, and in-frame insertion or deletions variants (in-frame indels), with missense substitutions being the most numerous. Because of the patterns biophysical similarity and dissimilarity between the 20 naturally occurring amino acids, a missense substitution can fall anywhere in a spectrum from innocuous to ablating protein function to creating new protein functions. Similarly, because splicing machinery has varying dependencies on the individual nucleotide positions within splice donor and splice acceptor consensus sequences, sequence variants within these regions may ablate, reduce, or even increase the efficiency of splicing at the affected intron-exon junction.. Thus, the key analytic problem is that the effects of VUS need not to be all-or-none. Whether variants are assayed one-by-one or en masse using high-throughput gene editing techniques, it is difficult to determine what proportion reduction of normal function from a damaged protein, or of productive transcript from a damaged allele, is required to confer clinically relevant increased risk of cancer [82–88].
A second problem is that VUS are individually very rare, but summed across the population, numerous. Indeed, in a study of the ExAC data, Kobayashi et al. found that most pathogenic variants with continental population allele frequencies above 0.01% are already well characterized [89]; this means that most VUS (at least for dominant CRC susceptibility genes) with allele frequencies of above 0.01% are actually neutral. Yet on reviewing the lists of MLH1 and MSH2 sequence variants recorded in the InSiGHT and gnomAD databases, we found 1,299 distinct VUS missense substitutions (Table 4). This is just the tip of the iceberg, as, based on the estimated per generation germline de novo mutation rate and the size of the human population [90, 91], the human gene pool actually includes multiple missense substitutions at most protein coding codons, and most of these are pedigree-specific.
Table 4.
MLH1 and MSH2 missense substitutions in the InSiGHT and gnomAD databases.
| Variant class and source | Missense substitution count | Average sequence analysis-based probability of pathogenicity |
|---|---|---|
| Class 4, 5 | 144 | 0.784 |
| InSiGHTonly | 135 | 0.790 |
| InSiGHT and gnomAD | 9 | 0.690 |
| Class 3 | 1,299 | 0.391 |
| InSiGHTonly | 408 | 0.579 |
| InSiGHT and gnomAD | 176 | 0.388 |
| gnomAD | 891 | 0.306 |
| Class 1, 2 | 45 | 0.260 |
| InSiGHTonly | 5 | 0.374 |
| InSiGHT and gnomAD | 40 | 0.246 |
Variant classification frameworks
In 2008, an International Agency for Research on Cancer (IARC) Working Group on VUS in cancer susceptibility genes created the five-tiered variant classification scheme shown in Table 5 [92]. This scheme was adopted by the International Society for Gastrointestinal Hereditary Tumors (InSiGHT), and with minor modification for general use, by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) [86, 93].
Table 5.
The IARC variant classification scheme (Modified from Plon et al. 2008 [92]).
| Category | Synonym | Definition |
|---|---|---|
| Pathogenic | Post_P> 0.99 | |
| Likely Pathogenic | 0.99 ≥ Post_P> 0.95 | |
| VUS | Unclassified variant (UV) | 0.95 ≥ Post_P ≥ 0.05 |
| Likely Not Pathogenic | Likely Benign | 0.05 >Post_P ≥0.001 |
| Not Pathogenic | Benign | Post_P< 0.001 |
There are two basic frameworks for variant classification: qualitative rules-based and quantitative Bayesian classifications. Since methods for evaluation of VUS in LS genes are better developed than for most other CRC susceptibility genes, this discussion will focus on evaluation of MMR gene VUS. The essence of rules-based classification is to set up a series of points of evidence, and then to define rules governing which combinations of evidence result in a specific categorical classification. Data used in the qualitative InSiGHT MMR gene variant classifier [86, 87] fall into two broad categories: i) patient observational data, such as details of the patient’s personal and cancer family history, segregation (or not) in pedigrees, or presence (or not) of MSI in tumors with the VUS; and ii) variant specific data such as sequence analysis evidence, functional assay results including mismatch repair proficiency and protein stability assays, mRNA splicing assays, and allele frequency in control populations. Rules that result in classification as Pathogenic can use stand-alone data such as the variant is a protein truncating variant in a coding exon other than the final exon, or combine several pieces of individually weaker data such as reduced activity in a functional assay plus co-segregation with CRC plus multiple tumors with MSI plus very low allele frequency in continental level populations. There are also corresponding rules for classification as Not Pathogenic. For a succinct summary, see Figure 1a of Thompson et al., 2014 [86]. Critically, if a variant is not associated with enough data to meet either of the Pathogenic or Not Pathogenic rules, then it will be classified as VUS.
Bayesian classification views the points of evidence as data. Each data type is calibrated so that it can be re-expressed as a prior probability of pathogenicity (Prior_P), odds in favor of pathogenicity (Odds_Path), or a likelihood ratio (LR) in favor of pathogenicity. The data from each individual sequence variant are then combined using Bayes’ rule to obtain a posterior probability of pathogenicity (Post_P). The Post_P is then interpreted through a quantitative classifier [92] to obtain the categorical classifications (Table 5).
The challenge with quantitative classification is to calibrate the data types – i.e., to convert from the units in which each type of data is naturally expressed to Odds_Path, LR, or Prior_P. For classification of MMR gene variants, data that have been calibrated include: a sequence analysis-based Prior; segregation in pedigrees; and degree of MSI in CRC tumors combined with somatic BRAF mutation status [86, 94, 95].. Because MSI plus BRAF status can be detected by somatic tumor mutation screening, this datum will become more widely available as tumor screening takes off [96].
Functional assays could make and important contribution to VUS classification [93, 97, 98]. Noting that failure of mismatch repair is thought to be the key molecular defect underlying LS, de Wind and colleagues developed an in vitro MMR Activity (CIMRA) assay [99–101], which has now been calibrated to convert % wild-type activity into CIMRA Odds_Path [102]. When the sequence analysis Prior_P and CIMRA assay provided concordant evidence in favor of pathogenicity, results met the IARC Likely Pathogenic criterion that >95% of variants should actually be pathogenic. On the other hand, when the sequence analysis Prior_P and CIMRA assay provided concordant evidence against pathogenicity, results fell short of the IARC Likely Not Pathogenic criterion that <5% of variants should be pathogenic [86, 87, 102]. In fact, this asymmetry is somewhat expected because a purely in vitro assay is intrinsically not able to detect functional defects such as loss of subcellular localization or reduced protein half-life.
In their 2017 update to the MMR gene variant classification system, Tricarico and colleagues argued that “variants attaining thresholds for assignment to clinically actionable classes … with limited contribution from clinical or laboratory evidence be considered of uncertain significance until further evidence is accrued” [87]. Whether integrated into LS variant classification through the rules-based or quantitative Bayesian approach, the systematic application of the computational Prior_P and CIMRA assay is likely to dramatically accelerate classification to Likely Pathogenic of missense substitutions observed in patients with CRC or other LS spectrum tumor. Further acceleration may be possible if the high-throughput gene editing techniques that Findlay et al. recently applied to BRCA1 sequence variants can be applied to MMR VUS [85].
Precision medicine in hereditary colorectal cancer syndromes
Precision Prevention
Among patients with FAP and other polyposis syndromes, prophylactic surgery continues to be the current gold standard for prevention of CRC. However, those patients that elect rectal-sparing surgeries continue to develop adenomas, thus retaining an excessive risk for rectal cancer. Also, duodenal cancer has become a major cause of mortality among the FAP population. Therefore, the development of chemopreventive agents is still an unmet need in the care of patients with polyposis syndromes. Initial studies focused in the efficacy of non-steroidal anti-inflammatory drugs (NSAIDs) and more specifically Sulindac, Aspirin and COX-2 inhibitors. Sulindac was the first NSAID to show an effect decreasing the number and size of polyps in a cohort of 22 patients compared with placebo (44%, P=0.014; and 35%, P<0.001, respectively) [103]. Subsequently, a follow-up randomized double-blind placebo-controlled study was launched for primary prevention. Unfortunately, this study was only able to accrue 41 FAP patients and therefore was underpowered, which led to no significant differences in the number of polyps between the two arms [104]. The field moved next to explore agents with specificity for inhibition of COX-2. Celecoxib demonstrated that treatment of pre-surgical patients with 2 different doses (100 and 400 mg daily) reduced the number and burden of polyps with excellent tolerance from the safety standpoint in this young population.[105, 106]. However, the translation of Celecoxib into the general population rendered an unacceptable cardiovascular toxicity profile (2.5% of subjects in the Celecoxib group and 1.9% in the placebo group) and the drug development was halted despite of a reduction on the occurrence of adenomas (RR 0.64; 95% CI, 0.56 to 0.75) [107, 108]. Based on the safety data developed in sporadic populations, the benefit of regular use of coxibs in terms of delaying the growth of polyps and delaying prophylactic surgery in patients with FAP needs to be weighed against the risk of toxic cardiovascular effects. Since the onset of polyps in patients with FAP begins occurring during the teenage years, the toxicity profile of coxibs in these patients with FAP may be essentially different from that in the general population. In fact, Lynch et al. demonstrated that Celecoxib at a dose of 16 mg/kg/day in children (10–14 years) with FAP is safe, and generated a significant reduction of the number of colorectal polyps [109]. Given the cardiovascular toxicity of coxibs, the focus of chemopreventive efforts in FAP turned to Aspirin and combinations of Sulindac with other agents. The Concerted Action Polyp Prevention (CAPP) group completed an international, multicenter, randomized, placebo-controlled trial (CAPP1 protocol) of Aspirin (600 mg daily) and/or resistant starch (30 mg daily) in young FAP patients [110]. After 17 months of treatment, the primary endpoint to observe a decrease in the polyp number in the rectum and sigmoid colon failed. Noteworthy, the diameter of the largest polyp detected by endoscopy at the end of intervention tended to be smaller in the Aspirin group (P=0.05). On the combination side, DMFO plus Sulindac is being explored as chemoprevention in FAP (NCT01483144) after the remarkable activity demonstrated in a phase III with 375 patients with history of resected adenomas for prevention of polyp recurrence [111]. Finally, chemoprevention of duodenal adenomas in FAP has made significant advances recently with the publications of the results of a clinical trial combining Sulindac and Erlotinib on preventing duodenal neoplasia [112]. This was a double-blind randomized placebo-controlled study including 92 FAP participants that were given 150 mg of Sulindac twice daily combined with 75 mg of Erlotinib once daily. The end point of the trial was met and a 71% reduction in duodenal polyp burden was observed between the treatment and placebo groups. This combination also rendered substantial modulation of the colorectal adenoma burden [113].
Aspirin has been the primary NSAID explored for chemoprevention in LS. In the CAPP2 study, a total of 861 LS patients were given 600 mg of Aspirin or placebo for up to 4 years [114–116]. Overall, 600 mg of Aspirin given over an average of 25 months was found to be effective in reducing CRC occurrence in LS patients. As a follow-up CAPP-3, which is a non-inferiority clinical trial, is now being conducted to study the long-term effect of Aspirin in 3000 LS patients at three different doses: 100, 300, or 600 mg per day [114]. We have recently completed a multi-center Phase Ib biomarker, placebo-controlled, trial of Naproxen, an NSAID with improved safety profile [117], in a total of 80 LS patients (NCT02052908). All participants underwent colonoscopy before and after the intervention as well as collection of blood, plasma, tissue, and urine for subsequent biomarker studies with mRNA-seq, miRNA-seq and determination of levels PGE2 in tissue, Naproxen in blood and plasma, and PGM in urine. The primary endpoint of this trial was safety and modulation of PGE2 levels in tissue. This study has completed accrual and the data is currently being analyzed [118].
Precision treatment: The role of immuno-oncology (IO) in hereditary CRC syndromes
IO has become a reality in the treatment of patients with hypermutant cancers in general and also in CRC displaying MSI. The activation of the immune system developed inn this type of tumors was noted several decades ago by pathologists that observed extensive involvement by tumor-infiltrating lymphocytes (TILs) located mainly at the invasive front [119]. In fact, the presence of TILs became a standard pathology criterion for the diagnosis of sporadic and hereditary MSI tumors [120].
Tumors with a germline MMR mutation must acquire a second somatic hit in the alternate allele of the same gene in order to become hypermutant. The inactivity of one of the heterodimers of the MMR complex (either the MutL or MutS complexes) leads to the accumulation of frameshift mutations that generate neoantigens [121–123]. Some of these neoantigens will be processed, presented by the HLA system (HLA-I and II), and recognized as foreign by T-cells. In fact, high levels of infiltration by activated CD8-positive cytotoxic T lymphocytes and activated Th1 cells with associated IFNγ production has been confirmed in detailed immune-pathologic studies. In order to counterbalance this active immune environment, multiple immune checkpoints such as PD-1, PD-L1, CTLA-4 and others are then activated by tumor cells, thus making them particularly susceptible to immune check-point blockade [124].
All of these data provided the biologic rationale for two phase II clinical trials assessing the activity of checkpoint inhibitors in MSI/Hypermutant tumors. The first trial demonstrated that Pembrolizumab, a humanized monoclonal anti-PD-1 antibody, given as single agent induced an immune-related objective response rate of 40% and an immune-related progression-free survival rate at 20 weeks of 78% in patients with metastatic MSI CRC [125]. This exceptional activity contrasted with almost negligible response observed among microsatellite stable (MSS)/non-hypermutant tumors. Of note, there were also no significant differences in the objective response rate between LS- and non-LS-associated tumors (46% versus 59%, respectively) [126]. The second trial tested Nivolumab, which is another IgG4 PD-1 blocking antibody, and also demonstrated activity as a single agent and in combination with Ipilimumab, a fully human immunoglobulin monoclonal anti-CTLA4 antibody, thus providing double checkpoint blockade. 31.1% of patients treated with single agent Nivolumab achieved an objective response rate with disease control for 12 weeks or longer in 51% [127]. The combination of both checkpoint inhibitors expanded further these results and 55% of treated patients achieved an objective response rate, and disease control rate for more than 12 weeks was present in 80% of the patients [128]. This remarkable antitumoral activity showed by checkpoint inhibitors led the FDA to approve the use of Pembrolizumab in May of 2017, then Nivolumab in July of 2017, and later the combination of Ipilimumab with Nivolumab in July 2018 for the treatment of stage IV hypermutant/MSI tumors after progression to standard chemotherapy. Therefore, these advances have placed hereditary CRC syndromes at the epicenter of precision medicine and immune-oncology in the last 2 years.
Summary
CRC remains one of the most prevalent cancers, but it is also preventable. Making the diagnosis of genetic predisposition to CRC provides opportunities for precision cancer treatment, early detection, as well as prevention of subsequent cancers in patients and their at-risk relatives. Implementation of routine screening of CRC tumors for DNA MMR deficiency has been shown to improve detection of Lynch Syndrome beyond family history criteria alone. As new strategies for surveillance and chemoprevention provide opportunities to reduce morbidity and mortality for individuals with Lynch Syndrome, FAP and other genetic diagnoses, it is increasingly important to implement effective strategies to improve identification and management of presymptomatic individuals at high risk for CRC.
Acknowledgements
We would like to thank Wendy Kohlmann and Bryony Thompson for their help in the variant interpretation sections. Laura Valle’s work is funded by the Spanish Ministry of Science, Innovation and Universities and co-funded by FEDER funds- a way to build Europe- [SAF2016–80888-R], CIBERONC [CB16/12/00234] and the Government of Catalonia [PERIS SLT002/16/0037 and 2017SGR1282] and Fundación Olga Torres. Eduardo Vilar’s work is supported by grants R21 CA208461 and R01 CA219463 (US National Institutes of Health/National Cancer Institute), The SWOG/Hope Foundation Impact Award, a gift from the Feinberg Family, and The University of Texas Anderson Cancer Center Core Support Grant P30 CA016672 (US National Institutes of Health/National Cancer Institute). Sean Tavtigian’s work is funded by US NIH NCI grants R01 CA164138 and R01 CA164944.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018; 68: 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Yurgelun MB, Kulke MH, Fuchs CS, et al. Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer. J Clin Oncol 2017; 35: 1086–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.AlDubayan SH, Giannakis M, Moore ND, et al. Inherited DNA-Repair Defects in Colorectal Cancer. Am J Hum Genet 2018; 102: 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeRycke MS, Gunawardena S, Balcom JR, et al. Targeted sequencing of 36 known or putative colorectal cancer susceptibility genes. Mol Genet Genomic Med 2017; 5: 553–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearlman R, Frankel WL, Swanson B, et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol 2017; 3: 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stoffel EM, Koeppe E, Everett J, et al. Germline Genetic Features of Young Individuals With Colorectal Cancer. Gastroenterology 2018; 154: 897–905 e891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mork ME, You YN, Ying J, et al. High Prevalence of Hereditary Cancer Syndromes in Adolescents and Young Adults With Colorectal Cancer. J Clin Oncol 2015; 33: 3544–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robinson DR, Wu YM, Lonigro RJ, et al. Integrative clinical genomics of metastatic cancer. Nature 2017; 548: 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mandelker D, Zhang L, Kemel Y, et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. Jama 2017; 318: 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valle L Genetic predisposition to colorectal cancer: where we stand and future perspectives. World J Gastroenterol 2014; 20: 9828–9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, Woods SL, Healey S, et al. Point Mutations in Exon 1B of APC Reveal Gastric Adenocarcinoma and Proximal Polyposis of the Stomach as a Familial Adenomatous Polyposis Variant. Am J Hum Genet 2016; 98: 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slowik V, Attard T, Dai H, et al. Desmoid tumors complicating Familial Adenomatous Polyposis: a meta-analysis mutation spectrum of affected individuals. BMC Gastroenterol 2015; 15: 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balaguer F, Castellvi-Bel S, Castells A, et al. Identification of MYH mutation carriers in colorectal cancer: a multicenter, case-control, population-based study. Clin Gastroenterol Hepatol 2007; 5: 379–387. [DOI] [PubMed] [Google Scholar]
- 14.Win AK, Dowty JG, Cleary SP, et al. Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology 2014; 146: 1208–1211 e1201–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 2013; 45: 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weren RD, Ligtenberg MJ, Kets CM, et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet 2015; 47: 668–671. [DOI] [PubMed] [Google Scholar]
- 17.Adam R, Spier I, Zhao B, et al. Exome Sequencing Identifies Biallelic MSH3 Germline Mutations as a Recessive Subtype of Colorectal Adenomatous Polyposis. Am J Hum Genet 2016; 99: 337–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gammon A, Jasperson K, Kohlmann W, et al. Hamartomatous polyposis syndromes. Best Pract Res Clin Gastroenterol 2009; 23: 219–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaeger E, Leedham S, Lewis A, et al. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat Genet 2012; 44: 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohlin A, Eiengard F, Lundstam U, et al. GREM1 and POLE variants in hereditary colorectal cancer syndromes. Genes Chromosomes Cancer 2016; 55: 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Snover DC, Ahnen DJ, Burt RW, et al. Serrated polyps of the colon and rectum and serrated polyposis In: Bosman FTCF, Hruban RH, et al. , ed. WHO classification of tumours of the digestive system. IARC: Lyon, France: 2010:160–165. [Google Scholar]
- 22.Gala MK, Mizukami Y, Le LP, et al. Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology 2014; 146: 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan HH, Lai JC, Ho SL, et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut 2016. [DOI] [PubMed] [Google Scholar]
- 24.Buchanan DD, Clendenning M, Zhuoer L, et al. Lack of evidence for germline RNF43 mutations in patients with serrated polyposis syndrome from a large multinational study. Gut 2017; 66: 1170–1172. [DOI] [PubMed] [Google Scholar]
- 25.Quintana I, Mejias-Luque R, Terradas M, et al. Evidence suggests that germline RNF43 mutations are a rare cause of serrated polyposis. Gut 2018. [DOI] [PubMed] [Google Scholar]
- 26.Kastrinos F, Steyerberg EW, Mercado R, et al. The PREMM(1,2,6) model predicts risk of MLH1, MSH2, and MSH6 germline mutations based on cancer history. Gastroenterology 2011; 140: 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen S, Wang W, Lee S, et al. Prediction of germline mutations and cancer risk in the Lynch syndrome. Jama 2006; 296: 1479–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med 2009; 11: 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hampel H, Pearlman R, Beightol M, et al. Assessment of Tumor Sequencing as a Replacement for Lynch Syndrome Screening and Current Molecular Tests for Patients With Colorectal Cancer. JAMA Oncol 2018; 4: 806–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. Jama 2005; 293: 1979–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nieminen TT, O’Donohue MF, Wu Y, et al. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology 2014; 147: 595–598 e595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Broderick P, Dobbins SE, Chubb D, et al. Validation of Recently Proposed Colorectal Cancer Susceptibility Gene Variants in an Analysis of Families and Patients-a Systematic Review. Gastroenterology 2017; 152: 75–77 e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valle L Recent Discoveries in the Genetics of Familial Colorectal Cancer and Polyposis. Clin Gastroenterol Hepatol 2017; 15: 809–819. [DOI] [PubMed] [Google Scholar]
- 34.Bellido F, Sowada N, Mur P, et al. Association Between Germline Mutations in BRF1, a Subunit of the RNA Polymerase III Transcription Complex, and Hereditary Colorectal Cancer. Gastroenterology 2018; 154: 181–194 e120. [DOI] [PubMed] [Google Scholar]
- 35.Franch-Exposito S, Esteban-Jurado C, Garre P, et al. Rare germline copy number variants in colorectal cancer predisposition characterized by exome sequencing analysis. J Genet Genomics 2018; 45: 41–45. [DOI] [PubMed] [Google Scholar]
- 36.Martin-Morales L, Feldman M, Vershinin Z, et al. SETD6 dominant negative mutation in familial colorectal cancer type X. Hum Mol Genet 2017; 26: 4481–4493. [DOI] [PubMed] [Google Scholar]
- 37.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005; 352: 1851–1860. [DOI] [PubMed] [Google Scholar]
- 38.Hampel H, Frankel WL, Martin E, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 2008; 26: 5783–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moreira L, Balaguer F, Lindor N, et al. Identification of Lynch syndrome among patients with colorectal cancer. Jama 2012; 308: 1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ward RL, Hicks S, Hawkins NJ. Population-based molecular screening for Lynch syndrome: implications for personalized medicine. J Clin Oncol 2013; 31: 2554–2562. [DOI] [PubMed] [Google Scholar]
- 41.Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res 2006; 66: 7810–7817. [DOI] [PubMed] [Google Scholar]
- 42.Hampel H, Panescu J, Lockman J, et al. Comment on: Screening for Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer) among Endometrial Cancer Patients. Cancer Res 2007; 67: 9603. [DOI] [PubMed] [Google Scholar]
- 43.Watkins JC, Yang EJ, Muto MG, et al. Universal Screening for Mismatch-Repair Deficiency in Endometrial Cancers to Identify Patients With Lynch Syndrome and Lynch-like Syndrome. Int J Gynecol Pathol 2017; 36: 115–127. [DOI] [PubMed] [Google Scholar]
- 44.Yurgelun MB, Allen B, Kaldate RR, et al. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology 2015; 149: 604–613 e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jenkins MA, Win AK, Lindor NM. The Colon Cancer Family Registry Cohort In: Valle L, Gruber SB, Capellá G, eds. Hereditary Colorectal Cancer Genetic Basis and Clinical Implications. 1st Edition ed. Springer International Publishing; 2018. [Google Scholar]
- 46.Win AK, Jenkins MA, Dowty JG, et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer Epidemiol Biomarkers Prev 2017; 26: 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Senter L, Clendenning M, Sotamaa K, et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 2008; 135: 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baglietto L, Lindor NM, Dowty JG, et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst 2010; 102: 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonadona V, Bonaiti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. Jama 2011; 305: 2304–2310. [DOI] [PubMed] [Google Scholar]
- 50.ten Broeke SW, Brohet RM, Tops CM, et al. Lynch syndrome caused by germline PMS2 mutations: delineating the cancer risk. J Clin Oncol 2015; 33: 319–325. [DOI] [PubMed] [Google Scholar]
- 51.Guindalini RS, Win AK, Gulden C, et al. Mutation spectrum and risk of colorectal cancer in African American families with Lynch syndrome. Gastroenterology 2015; 149: 1446–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goodenberger ML, Thomas BC, Riegert-Johnson D, et al. PMS2 monoallelic mutation carriers: the known unknown. Genet Med 2016; 18: 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ryan NAJ, Morris J, Green K, et al. Association of Mismatch Repair Mutation With Age at Cancer Onset in Lynch Syndrome: Implications for Stratified Surveillance Strategies. JAMA Oncol 2017; 3: 1702–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ponti G, Castellsague E, Ruini C, et al. Mismatch repair genes founder mutations and cancer susceptibility in Lynch syndrome. Clin Genet 2015; 87: 507–516. [DOI] [PubMed] [Google Scholar]
- 55.Lagerstedt-Robinson K, Rohlin A, Aravidis C, et al. Mismatch repair gene mutation spectrum in the Swedish Lynch syndrome population. Oncol Rep 2016; 36: 2823–2835. [DOI] [PubMed] [Google Scholar]
- 56.Goldberg Y, Barnes-Kedar I, Lerer I, et al. Genetic features of Lynch syndrome in the Israeli population. Clin Genet 2015; 87: 549–553. [DOI] [PubMed] [Google Scholar]
- 57.Haraldsdottir S, Rafnar T, Frankel WL, et al. Comprehensive population-wide analysis of Lynch syndrome in Iceland reveals founder mutations in MSH6 and PMS2. Nat Commun 2017; 8: 14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rossi BM, Palmero EI, Lopez-Kostner F, et al. A survey of the clinicopathological and molecular characteristics of patients with suspected Lynch syndrome in Latin America. BMC Cancer 2017; 17: 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bisgaard ML, Fenger K, Bulow S, et al. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat 1994; 3: 121–125. [DOI] [PubMed] [Google Scholar]
- 60.Bulow S Results of national registration of familial adenomatous polyposis. Gut 2003; 52: 742–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burt RW, Bishop DT, Lynch HT, et al. Risk and surveillance of individuals with heritable factors for colorectal cancer. WHO Collaborating Centre for the Prevention of Colorectal Cancer. Bull World Health Organ 1990; 68: 655–665. [PMC free article] [PubMed] [Google Scholar]
- 62.Nelen MR, Kremer H, Konings IB, et al. Novel PTEN mutations in patients with Cowden disease: absence of clear genotype-phenotype correlations. Eur J Hum Genet 1999; 7: 267–273. [DOI] [PubMed] [Google Scholar]
- 63.Allen BA, Terdiman JP. Hereditary polyposis syndromes and hereditary non-polyposis colorectal cancer. Best Pract Res Clin Gastroenterol 2003; 17: 237–258. [DOI] [PubMed] [Google Scholar]
- 64.Lorans M, Dow E, Macrae FA, et al. Update on Hereditary Colorectal Cancer: Improving the Clinical Utility of Multigene Panel Testing. Clin Colorectal Cancer 2018; 17: e293–e305. [DOI] [PubMed] [Google Scholar]
- 65.Moller P, Seppala TT, Bernstein I, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut 2018; 67: 1306–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ten Broeke SW, van der Klift HM, Tops CMJ, et al. Cancer Risks for PMS2-Associated Lynch Syndrome. J Clin Oncol 2018; 36: 2961–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Win AK, Scott RJ. Genetic and Environmental Modifiers of Cancer Risk in Lynch Syndrome In: Valle L, Gruber SB, Capellá G, eds. Hereditary Colorectal Cancer Genetic Basis and Clinical Implications. 1st Edition ed. Springer International Publishing; 2018. [Google Scholar]
- 68.Katona BW, Yurgelun MB, Garber JE, et al. A counseling framework for moderate-penetrance colorectal cancer susceptibility genes. Genet Med 2018. [DOI] [PubMed] [Google Scholar]
- 69.Ma X, Zhang B, Zheng W. Genetic variants associated with colorectal cancer risk: comprehensive research synopsis, meta-analysis, and epidemiological evidence. Gut 2014; 63: 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2014 Based on November 2016 SEER data submission, posted to the SEER web site, April 2017. [cited; Available from: https://seer.cancer.gov/csr/1975_2014/ [Google Scholar]
- 71.National Comprehensive Cancer Network. Genetic/Familial High-Risk Assessment: Colorectal (version 2.2017) and Colorectal Cancer Screening (version 2.2017). [cited 1 November 2017]; Available from: https://www.nccn.org/
- 72.Johns LE, Houlston RS. A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol 2001; 96: 2992–3003. [DOI] [PubMed] [Google Scholar]
- 73.Oh M, McBride A, Yun S, et al. BRCA1 and BRCA2 Gene Mutations and Colorectal Cancer Risk: Systematic Review and Meta-analysis. J Natl Cancer Inst 2018; 110: 1178–1189. [DOI] [PubMed] [Google Scholar]
- 74.Dobbins SE, Broderick P, Chubb D, et al. Undefined familial colorectal cancer and the role of pleiotropism in cancer susceptibility genes. Fam Cancer 2016; 15: 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Samadder N, Curtin K, Pappas L, et al. Risk of Incident Colorectal Cancer and Death After Colonoscopy: A Population-based Study in Utah. Clin Gastroenterol Hepatol 2016; 14: 279–286.e272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen C, Stock C, Hoffmeister M, et al. Public health impact of colonoscopy use on colorectal cancer mortality in Germany and the United States. Gastrointest Endosc 2018; 87: 213–221.e212. [DOI] [PubMed] [Google Scholar]
- 77.Girardi F, Barnes D, Barrowdale D, et al. Risks of breast or ovarian cancer in BRCA1 or BRCA2 predictive test negatives: findings from the EMBRACE study. Genet Med 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee A, Cunningham A, Tischkowitz M, et al. Incorporating truncating variants in PALB2, CHEK2, and ATM into the BOADICEA breast cancer risk model. Genet Med 2016; 18: 1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eccles B, Copson E, Maishman T, et al. Understanding of BRCA VUS genetic results by breast cancer specialists. BMC Cancer 2015; 15: 936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kurian A, Li Y, Hamilton A, et al. Gaps in Incorporating Germline Genetic Testing Into Treatment Decision-Making for Early-Stage Breast Cancer. J Clin Oncol 2017; 35: 2232–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Augusto B, Lake P, Scherr C, et al. From the laboratory to the clinic: sharing BRCA VUS reclassification tools with practicing genetics professionals. J Community Genet 2018; 9: 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vallee M, Di Sera T, Nix D, et al. Adding In Silico Assessment of Potential Splice Aberration to the Integrated Evaluation of BRCA Gene Unclassified Variants. Hum Mutat 2016; 37: 627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Whiley P, de la Hoya M, Thomassen M, et al. Comparison of mRNA splicing assay protocols across multiple laboratories: recommendations for best practice in standardized clinical testing. Clin Chem 2014; 60: 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tournier I, Vezain M, Martins A, et al. A large fraction of unclassified variants of the mismatch repair genes MLH1 and MSH2 is associated with splicing defects. Hum Mutat 2008; 29: 1412–1424. [DOI] [PubMed] [Google Scholar]
- 85.Findlay G, Daza R, Martin B, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thompson B, Spurdle A, Plazzer J, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet 2014; 46: 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tricarico R, Kasela M, Mareni C, et al. Assessment of the InSiGHT Interpretation Criteria for the Clinical Classification of 24 MLH1 and MSH2 Gene Variants. Hum Mutat 2017; 38: 64–77. [DOI] [PubMed] [Google Scholar]
- 88.Yeo G, Burge C. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 2004; 11: 377–394. [DOI] [PubMed] [Google Scholar]
- 89.Kobayashi Y, Yang S, Nykamp K, et al. Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation. Genome Med 2017; 9: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roach J, Glusman G, Smit A, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science 2010; 328: 636–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shendure J, Akey J. The origins, determinants, and consequences of human mutations. Science 2015; 349: 1478–1483. [DOI] [PubMed] [Google Scholar]
- 92.Plon S, Eccles D, Easton D, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 2008; 29: 1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Thompson B, Goldgar D, Paterson C, et al. A Multifactorial Likelihood Model for MMR Gene Variant Classification Incorporating Probabilities Based on Sequence Bioinformatics and Tumor Characteristics: A Report from the Colon Cancer Family Registry. Hum Mutat 2013; 34: 200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thompson B, Greenblatt M, Vallee M, et al. Calibration of multiple in silico tools for predicting pathogenicity of mismatch repair gene missense substitutions. Hum Mutat 2013; 34: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Middha S, Zhang L, Nafa K, et al. Reliable Pan-Cancer Microsatellite Instability Assessment by Using Targeted Next-Generation Sequencing Data. JCO Precis Oncol 2017; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Collins F BRCA1--lots of mutations, lots of dilemmas. N Engl J Med 1996; 334: 186–188. [DOI] [PubMed] [Google Scholar]
- 98.Rasmussen L, Heinen C, Royer-Pokora B, et al. Pathological assessment of mismatch repair gene variants in Lynch syndrome: past, present, and future. Hum Mutat 2012; 33: 1617–1625. [DOI] [PubMed] [Google Scholar]
- 99.Drost M, Zonneveld J, van Dijk L, et al. A cell-free assay for the functional analysis of variants of the mismatch repair protein MLH1. Hum Mutat 2010; 31: 247–253. [DOI] [PubMed] [Google Scholar]
- 100.Drost M, Zonneveld J, van Hees S, et al. A rapid and cell-free assay to test the activity of lynch syndrome-associated MSH2 and MSH6 missense variants. Hum Mutat 2012; 33: 488–494. [DOI] [PubMed] [Google Scholar]
- 101.Drost M, Koppejan H, de Wind N. Inactivation of DNA mismatch repair by variants of uncertain significance in the PMS2 gene. Hum Mutat 2013; 34: 1477–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Drost M, Tiersma Y, Thompson B, et al. An Integrated, Functional Assay-Based Procedure to Classify Mismatch Repair Gene Variants in Lynch Syndrome. Manuscript Submitted 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Giardiello FM, Hamilton SR, Krush AJ, et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med 1993; 328: 1313–1316. [DOI] [PubMed] [Google Scholar]
- 104.Giardiello FM, Yang VW, Hylind LM, et al. Primary chemoprevention of familial adenomatous polyposis with sulindac. N Engl J Med 2002; 346: 1054–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000; 342: 1946–1952. [DOI] [PubMed] [Google Scholar]
- 106.Phillips RK, Wallace MH, Lynch PM, et al. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut 2002; 50: 857–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Arber N, Eagle CJ, Spicak J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med 2006; 355: 885–895. [DOI] [PubMed] [Google Scholar]
- 108.Bertagnolli MM. Chemoprevention of colorectal cancer with cyclooxygenase-2 inhibitors: two steps forward, one step back. Lancet Oncol 2007; 8: 439–443. [DOI] [PubMed] [Google Scholar]
- 109.Lynch PM, Ayers GD, Hawk E, et al. The safety and efficacy of celecoxib in children with familial adenomatous polyposis. Am J Gastroenterol 2010; 105: 1437–1443. [DOI] [PubMed] [Google Scholar]
- 110.Burn J, Bishop DT, Chapman PD, et al. A randomized placebo-controlled prevention trial of aspirin and/or resistant starch in young people with familial adenomatous polyposis. Cancer Prev Res (Phila) 2011; 4: 655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Meyskens FL Jr., McLaren CE, Pelot D, et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev Res (Phila) 2008; 1: 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Samadder NJ, Neklason DW, Boucher KM, et al. Effect of Sulindac and Erlotinib vs Placebo on Duodenal Neoplasia in Familial Adenomatous Polyposis: A Randomized Clinical Trial. JAMA 2016; 315: 1266–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Samadder NJ, Kuwada SK, Boucher KM, et al. Association of Sulindac and Erlotinib vs Placebo With Colorectal Neoplasia in Familial Adenomatous Polyposis: Secondary Analysis of a Randomized Clinical Trial. JAMA Oncol 2018; 4: 671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Burn J, Mathers JC, Bishop DT. Chemoprevention in Lynch syndrome. Fam Cancer 2013; 12: 707–718. [DOI] [PubMed] [Google Scholar]
- 115.Burn J, Bishop DT, Mecklin JP, et al. Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N Engl J Med 2008; 359: 2567–2578. [DOI] [PubMed] [Google Scholar]
- 116.Burn J, Gerdes AM, Macrae F, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011; 378: 2081–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nissen SE, Yeomans ND, Solomon DH, et al. Cardiovascular Safety of Celecoxib, Naproxen, or Ibuprofen for Arthritis. N Engl J Med 2016; 375: 2519–2529. [DOI] [PubMed] [Google Scholar]
- 118.Reyes-Uribe L, Lin R, Stoffel EM, et al. Abstract CT065: A phase Ib biomarker trial of naproxen in patients at risk for DNA mismatch repair deficient colorectal cancer. Cancer Research 2018; 78: CT065. [Google Scholar]
- 119.Smyrk TC, Watson P, Kaul K, et al. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer 2001; 91: 2417–2422. [PubMed] [Google Scholar]
- 120.Greenson JK, Huang SC, Herron C, et al. Pathologic predictors of microsatellite instability in colorectal cancer. Am J Surg Pathol 2009; 33: 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol 2010; 7: 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xiao Y, Freeman GJ. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov 2015; 5: 16–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.The Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487: 330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Llosa NJ, Cruise M, Tam A, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov 2015; 5: 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015; 372: 2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017; 357: 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017; 18: 1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Overman MJ, Lonardi S, Wong KYM, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol 2018; 36: 773–779. [DOI] [PubMed] [Google Scholar]
- 129.Alexandrov L, Kim J, Haradhvala NJ, et al. The Repertoire of Mutational Signatures in Human Cancer. bioRxiv 322859 2018. [Google Scholar]
- 130.Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 2015; 110: 223–262; quiz 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hansen MF, Johansen J, Sylvander AE, et al. Use of multigene-panel identifies pathogenic variants in several CRC-predisposing genes in patients previously tested for Lynch Syndrome. Clin Genet 2017; 92: 405–414. [DOI] [PubMed] [Google Scholar]
- 132.Lammi L, Arte S, Somer M, et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet 2004; 74: 1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bergendal B, Klar J, Stecksen-Blicks C, et al. Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1, and PAX9 genes. Am J Med Genet A 2011; 155A: 1616–1622. [DOI] [PubMed] [Google Scholar]
- 134.Marvin ML, Mazzoni SM, Herron CM, et al. AXIN2-associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. Am J Med Genet A 2011; 155A: 898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]