

Abstract
Background
Cleidocranial dysplasia (CCD) is a rare autosomal dominant disorder mainly characterized by hypoplastic or absent clavicles, delayed closure of the fontanelles, multiple dental abnormalities, and short stature. Runt-related transcription factor 2 (RUNX2) gene variants can cause CCD, but are not identified in all CCD patients.
Methods
In this study, we detected genetic variants in seven unrelated children with CCD by targeted high-throughput DNA sequencing or Sanger sequencing.
Results
All patients carried a RUNX2 variant, totally including three novel pathogenic variants (c.722_725delTGTT, p.Leu241Serfs*8; c.231_232delTG, Ala78Glyfs*82; c.909C > G, p.Tyr303*), three reported pathogenic variants (c.577C > T, p.Arg193*; c.574G > A, p.Gly192Arg; c.673 C > T, p.Arg225Trp), one likely pathogenic variant (c.668G > T, p.Gly223Val). The analysis of the variant source showed that all variants were de novo except the two variants (c.909C > G, p.Tyr303*; c.668G > T, p.Gly223Val) inherited from the patient’s father and mother with CCD respectively. Further bioinformatics analysis indicated that these variants could influence the structure of RUNX2 protein by changing the number of H-bonds or amino acids. The experimental result showed that the Gly223Val mutation made RUNX2 protein unable to quantitatively accumulate in the nucleus.
Conclusions
The present study expands the pathogenic variant spectrum of RUNX2 gene, which will contribute to the diagnosis of CCD and better genetic counseling in the future.
Keywords: RUNX2, Pathogenic variant, Cleidocranial dysplasia, Targeted next-generation sequencing
Background
Cleidocranial dysplasia (CCD; OMIM #119600) is a rare autosomal dominant disorder mainly characterised by hypoplastic or absent clavicles, delayed closure of fontanelles, multiple dental abnormalities, and short stature [1–3]. Variants in runt-related transcription factor 2 (RUNX2) gene (OMIM *600211) can result in haploinsufficiency of the protein and have been related to CCD [1, 2]. The RUNX2 gene is located on chromosome 6p21.1 and encodes a transcription factor with a highly conserved Runt domain [4, 5]. The Runt domain is responsible for binding to a specific DNA motif (TGT/CGGT sequence) in the promoter region of its target genes and heterodimerization with CBFB (core-binding factor subunit beta) [6–8]. The former participates in regulating the transcription of multiple genes. The latter increases the DNA-binding affinity as well as protects and stabilizes RUNX2 against proteolytic degradation. The N-terminal side of the Runt domain links a Q/A region consisting of 23 consecutive glutamine residues followed by 17 alanine residues, which acts as a second transactivation domain [9]. The C-terminal side of the Runt domain links a PST (proline/serine/threonine)-rich region, which contains the phosphorylation sites and represents the third transactivation domain [9, 10]. The last five amino acids (VWRPY) of RUNX2 protein compose a conserved motif in all runt proteins, and functions as a transcriptional repression domain [9, 11].
RUNX2 is essential for osteoblastic differentiation and skeletal morphogenesis. In mouse models, the homozygous mutation of RUNX2 gene blocked both intramembranous and endochondral ossification and resulted in a complete lack of bone formation [12]. The heterozygous mutation (RUNX2+/−) caused a similar phenotype to that of human CCD [13]. To date, 184 publicly available mutations in RUNX2 gene have been deposited in the Human Gene Mutation Database (HGMD, www.hgmd.cf.ac.uk). Most of these mutations were missense and clustered in Runt domain. Additionally, nonsense mutations, insertions or deletions are also observed in the RUNX2 gene, which are predominant within the Q/A domain or the PST domain. Although many mutations in the RUNX2 gene have been identified in familial and sporadic cases, novel mutation is still reported recently, suggesting that mutational screening on RUNX2 gene is far from saturation [14–19].
In the present study, we conducted genetic evaluation for a cohort of seven Chinese children with CCD by targeted high-throughput DNA sequencing or Sanger sequencing, and found seven different variants in RUNX2 gene, including six pathogenic variants and one likely pathogenic variant. These results will contribute to the diagnosis of CCD and better genetic counseling in the future.
Material and methods
Genomic DNA extraction and genetic testing
A total of seven unrelated children with CCD ranging in age from 1 month to 12 years were enrolled for genetic evaluation (Table 1). Genomic DNA of probands and their family members was extracted from peripheral blood leukocytes using Lab-Aid Nucleic Acid Isolation Kit (Zeesan, China), according to the manufacturer’s instructions.
Table 1.
Genetic detection methods and basic characteristics of seven children with CCD
| Proband ID | Gender | Age | Family history | Genetic detection methods |
|---|---|---|---|---|
| Family_A_II1 | Male | 3Y | No | Inherited disease panel (Agilent) Hiseq4000(Illumina), Sanger sequencing |
| Family_B_II1 | Female | 1Y9M | No | Focused exome panel (Agilent) Hiseq2500(Illumina), Sanger sequencing |
| Family_C_II1 | Male | 9Y11 M | No | Sanger sequencing |
| Family_D_II1 | Male | 12Y | No | xGen Exome research panel v1.0 (IDT) HiSeq4000(Illumina), Sanger sequencing |
| Family_E_II1 | Female | 1 M | No | Sanger sequencing |
| Family_F_III1 | Male | 3Y | Father with CCD | xGen Exome research panel v1.0 (IDT) HiSeq4000(Illumina), Sanger sequencing |
| Family_G_III1 | Male | 6Y | Mother with CCD Uncle with CCD Grandmother with CCD | xGen Exome research panel v1.0 (IDT) HiSeq4000(Illumina), Sanger sequencing |
Y Year, M Month
Among these CCD patients, five patients were firstly detected by targeted high-throughput DNA sequencing, two patients directly by Sanger sequencing (Table 1). For targeted high-throughput DNA sequencing, the preparation of sequencing library was completed using Agilent Inherited Disease panel, Agilent Focused exome panel or xGen Exome research panel v1.0 (Integrated DNA Technologies, Coralville, Iowa). Sequencing was performed on the Illumina HiSeq 2500 or 4000 (Illumina, San Diego, CA), according to the manufacturer’s instructions. Burrows-Wheeler Aligner (BWA, version 0.7.10) was used to mapping reads to the human reference genome (GRCh37/hg19). Base calling, QC analysis and coverage analysis were performed with Picard tools-1.124 and GATK software. Variants were annotated using SnpEff version 4.2. Subsequently, the following variants were filtered out: (i) variants with > 1% frequency in the population variant databases including 1000 Genomes Project, Exome Variant Server (EVS) and Exome Aggregation Consortium (ExAC) or > 5% frequency in our inhouse database (based on 150 exome datasets), (ii) intergenic and 3′/5′ untranslated region variants, none splice-related intronic and synonymous variants.
For Sanger sequencing, all exons of the RUNX2 gene in these probands were amplified by PCR reaction. DNA sequence variants were identified by Mutation Surveyor V4.0.5 software with reference sequences (NG_008020.1).
Variant assessment
MutationTaster (http://www.mutationtaster.org), SIFT (http://sift.jcvi.org), and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) were used to assess pathogenic potential of the variants [20–22]. Combined with clinical manifestation and modes of inheritance, candidate variants were validated by Sanger sequencing for all family members, and classified according to standards and guidelines of the American College of Medical Genetics and Genomics (ACMG). For the putative pathogenic or likely pathogenic variants, SWISS-MODEL (https://swissmodel.expasy.org) and Swiss-PdbViewer 4.1 software (http://spdbv.vital-it.ch/) were used to analyze the effect of these variants on protein structure [23, 24].
Subcellular localization of the RUNX2 mutant protein
The cDNA of wide-type RUNX2 gene was synthesized by Sangon Biotech (Shanghai) Co., Ltd., and amplified by PCR. The forward primer was 5′-GACACAGATCTCGAGATGGCATCAAACAGCCTCTTCAGC-3′ and the reverse primer was 5′-GTGTCGTCGACTGATATGGTCGCCAAACAGATTCA-3′. The PCR fragment was subcloned into pEGFP-N1 vector with the XhoI and SaII restriction sites. The RUNX2 668G > T (Gly223Val) mutation was introduced into pEGFP-N1 vector with wide-type RUNX2 cDNA by site-directed mutagenesis. The mutant primers were 5′-GCCTTCTGGGTTCCCGAGGTACATCTACTGTAACTTT AAT-3′, and 5′-ATTAAAGTTACAGTAGATGTACCTCGGGAACCCAGAAGGC-3′. All recombinant vectors were fully sequenced to exclude any additional mutations. The empty vector acting as a negative control (NC) and pEGFP-N1 vectors bearing wild type (WT) and mutant (Mut) were transfected into U2OS cells by using lipofectamine 2000 (Invitrogen). The cells were visualized and photographed (magnification 10X and 40X) with a fluorescent microscope (Olympus IX73, Japan).
Results
Clinical features of CCD children
All children underwent a clinical evaluation and were diagnosed as CCD by an experienced pediatrician. The clinical features of these patients including two female and five male patients were summarized in Table 2. Besides the clavicle and skull dysplasia, short stature, scoliosis, enamel hypoplasia, delayed eruption of deciduous teeth, low nasal bridge, delayed mineralization of pubic bone, broad femoral head with short femoral neck, hypoplastic iliac wing, syringomyelia and special faces were also observed in CCD children. Furthermore, hypertelorism was observed in all CCD children, except Family_A_II1. Supernumerary teeth, retention cysts and long second metacarpal were observed in all CCD children, except Family_A_II1 and Family_B_II1.
Table 2.
Comparison of clinical features of CCD children with different RUNX2 gene variant
| Clinical synopsis | Family_A_II1 (c.577C > T) | Family_B_II1 (c.574G > A) | Family_C_II1 (c.673C > T) | Family_D_II1 (c.722_725delTGTT) | Family_E_II1 (c.231_232delTG) | Family_F_III1 (c.909C > G) | Family_G_III1 (c.668G > T) |
|---|---|---|---|---|---|---|---|
| GROWTH | |||||||
| Height | |||||||
| Short stature | √ | √ | √ | √ | √ | √ | √ |
| HEAD & NECK | |||||||
| Head | |||||||
| Delayed fontanelle closure | √ | √ | √ | √ | √ | √ | √ |
| Parietal bossing | √ | √ | √ | √ | √ | √ | √ |
| Anterior fontanelle open in adults | |||||||
| Face | |||||||
| Frontal bossing | √ | √ | √ | √ | √ | √ | √ |
| Metopic groove | √ | √ | √ | √ | √ | √ | √ |
| Midface hypoplasia | √ | √ | √ | √ | √ | √ | √ |
| Micrognathia | √ | √ | √ | √ | √ | √ | √ |
| Ears | |||||||
| Deafness | |||||||
| Eyes | |||||||
| Hypertelorism | √ | √ | √ | √ | √ | √ | |
| Nose | |||||||
| Low nasal bridge | √ | √ | √ | √ | √ | √ | √ |
| Mouth | |||||||
| Cleft palate | |||||||
| Narrow, high-arched palate | |||||||
| Teeth | |||||||
| Delayed eruption of deciduous teeth | √ | √ | √ | √ | √ | √ | √ |
| Delayed eruption of permanent teeth | |||||||
| Supernumerary teeth | √ | √ | √ | √ | √ | ||
| Retention cysts | √ | √ | √ | √ | √ | ||
| Enamel hypoplasia | √ | √ | √ | √ | √ | √ | √ |
| RESPIRATORY | |||||||
| Airways | |||||||
| Respiratory distress in early infancy | |||||||
| CHEST | |||||||
| External Features | |||||||
| Narrow thorax | √ | √ | √ | √ | √ | √ | √ |
| Abnormal facility in opposing the shoulders | √ | √ | √ | √ | √ | √ | √ |
| Ribs Sternum Clavicles & Scapulae | |||||||
| Small scapula | |||||||
| Hypoplastic clavicles | √ | √ | √ | √ | √ | √ | √ |
| Aplastic clavicles | |||||||
| Short ribs | |||||||
| Cervical ribs | |||||||
| SKELETAL | |||||||
| Osteosclerosis | |||||||
| Increased bone fragility | √ | √ | √ | √ | √ | √ | √ |
| Skull | |||||||
| Wormian bones | √ | √ | √ | √ | √ | √ | √ |
| Bossing of frontal bone | √ | √ | √ | √ | √ | √ | √ |
| Bossing of occipital bone | |||||||
| Bossing of parietal bone | √ | √ | √ | √ | √ | √ | √ |
| Calvarial thickening | |||||||
| Absent frontal sinuses | |||||||
| Absent paranasal sinuses | |||||||
| Hypoplastic frontal sinuses | |||||||
| Hypoplastic paranasal sinuses | |||||||
| Large foramen magnum | |||||||
| Spine | |||||||
| Spondylolysis | |||||||
| Spondylolisthesis | |||||||
| Scoliosis | √ | √ | √ | √ | √ | √ | √ |
| Kyphosis | |||||||
| Pelvis | |||||||
| Wide pubic symphysis | |||||||
| Delayed mineralization of pubic bone | √ | √ | √ | √ | √ | √ | √ |
| Broad femoral head with short femoral neck | √ | √ | √ | √ | √ | √ | √ |
| Coxa vara | |||||||
| Hypoplastic iliac wing | √ | √ | √ | √ | √ | √ | √ |
| Hands | |||||||
| Brachydactyly | |||||||
| Long second metacarpal | √ | √ | √ | √ | √ | ||
| Short middle phalanges of second and fifth fingers | |||||||
| Cone-shaped phalangeal epiphyses | |||||||
| NEUROLOGIC | |||||||
| Peripheral Nervous System | |||||||
| Syringomyelia | √ | √ | √ | √ | √ | √ | √ |
Genetic testing
All patients carried a RUNX2 variant, totally including four novel variants and three reported variants (Figs. 1, 2 and Table 3). Among the seven variants, there were two pathogenic missense variants (c.574G > A, p.Gly192Arg; c.673 C > T, p.Arg225Trp), one likely pathogenic missense variant (c.668G > T, p.Gly223Val), two pathogenic frameshift variants (c.722_725delTGTT, p.Leu241Serfs*8; c.231_232delTG, Ala78Glyfs*82), and two pathogenic stop-gain variants (c.577C > T, p.Arg193*; c.909C > G, p.Tyr303*). The analysis of the variant source showed that all variants were de novo except the two variants (c.909C > G, p.Tyr303*; c.668G > T, p.Gly223Val). The former variant was inherited from the patient’s father with CCD, who carried a de novo heterozygous RUNX2 variant (c.909C > G, p.Tyr303*). The latter variant was inherited from the patient’s mother with CCD, who carried a maternal inherited and heterozygous RUNX2 variant (c.668G > T, p.Gly223Val).
Fig. 1.

The pedigree of the family and Sanger sequence chromatograms of RUNX2 gene variants (The black arrow indicates the proband)
Fig. 2.

Relative positions of RUNX2 gene variants identified in seven children with CCD (Variants in the box indicates the reported variants)
Table 3.
Summarization of RUNX2 gene variants in seven children with CCD
| Proband ID | Variant location | Variant type | Variant source | Literature report | Bioinformatic prediction | ACMG classification | ||
|---|---|---|---|---|---|---|---|---|
| MutationTaster | SIFT | PolyPhen-2 | ||||||
| Family_A_II1 | NM_001024630.3: c.577C > T, p.Arg193* (Het) | Stopgain | De novo | Hum Mol Genet. 1999;8 (12):2311–6. | Disease causing | NA | NA | Pathogenic |
| Family_B_II1 | NM_001024630.3: c.574G > A, p.Gly192Arg (Het) | Missense | De novo | J Hum Genet. 2005;50 (12):679–83. | Disease causing | Damaging | Probably damaging | Pathogenic |
| Family_C_II1 | NM_001024630.3: c.673 C > T, p.Arg225Trp (Het) | Missense | De novo | Am J Hum Genet. 1999;65 (5):1268–78. | Disease causing | Damaging | Probably damaging | Pathogenic |
| Family_D_II1 | NM_001024630.3: c.722_725delTGTT, p.Leu241Serfs*8 (Het) | Frameshift | De novo | _ | Disease causing | NA | NA | Pathogenic |
| Family_E_II1 | NM_001024630.3: c.231_232delTG, Ala78Glyfs*82 (Het) | Frameshift | De novo | _ | Disease causing | NA | NA | Pathogenic |
| Family_F_III1 | NM_001024630.3: c.909C > G, p.Tyr303* (Het) | Stopgain | Paternal inheritance | _ | Disease causing | NA | NA | Pathogenic |
| Family_G_III1 | NM_001024630.3: c.668G > T, p.Gly223Val (Het) | Missense | Maternal inheritance | _ | Disease causing | Damaging | Probably damaging | Likely pathogenic |
NA Not available; * the stop codon
The effect of the RUNX2 variants on protein structure
Among these variants, there were three variants changing the number of H-bonds in RUNX2 protein, including two variants increasing H-bonds (c.574G > A, p.Gly192Arg; c.668G > T, p.Gly223Val) and one variant decreasing H-bonds (c.673 C > T, p.Arg225Trp). In addition, there were four variants (c.722_725delTGTT, p.Leu241Serfs*8; c.231_232delTG, Ala78Glyfs*82; c.577C > T, p.Arg193*; c.909C > G, p.Tyr303*) decreasing the number of amino acids in RUNX2 protein.
Subcellular localization of the RUNX2 mutant protein
To further explore the function of the missense mutation (c.668G > T, p.Gly223Val) not reported, the wide-type and mutant RUNX2 proteins binding green fluorescent protein (GFP) were constructed and transiently transfected into human osteosarcoma U2OS. The result showed that the Gly223Val mutation could affect the subcellular distribution of RUNX2 protein and made RUNX2 protein unable to quantitatively accumulate in the nucleus (Fig. 3).
Fig. 3.
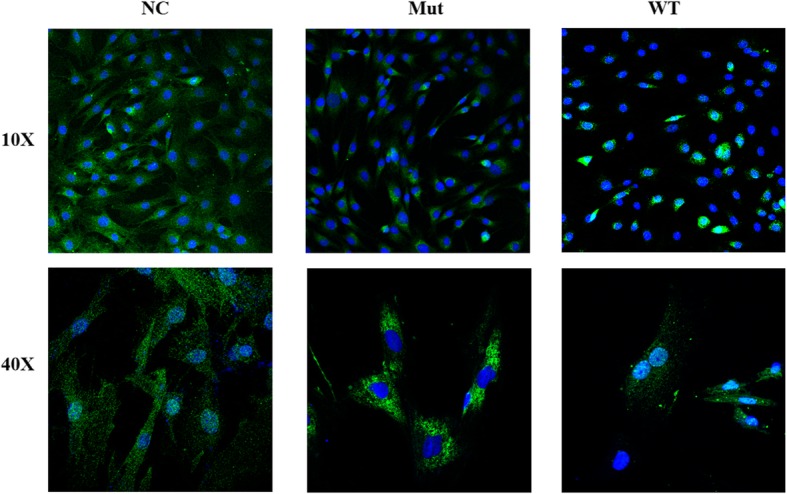
Subcellular localization of the RUNX2 mutant protein (NC, Mut, WT indicate control GFP, mutant Gly223Val RUNX2 and wild-type RUNX2, respectively. Magnification 10X and 40X)
Discussion
CCD is a skeletal dysplasia that represents a continuum of clinical findings ranging from classical CCD (dental abnormalities, hypoplastic or aplastic clavicles, and delayed closure of the cranial sutures) to mild CCD to isolated dental anomalies without other skeletal features. To date, no formal clinical diagnostic criteria for CCD have been established. Due to CCD inherited in an autosomal dominant manner, each child of an individual with CCD has a 50% chance of inheriting the pathogenic variant. If the pathogenic variant in the family is known, prenatal diagnosis for pregnancies at increased risk will be possible. Many kinds of molecular testing approaches, including single-gene testing, karyotype analysis and a multigene panel, can be currently used to detect the variants leading to CCD. For single-gene testing, sequence analysis of RUNX2 gene is performed first and followed by gene-targeted deletion/duplication analysis if no pathogenic variant is identified. For karyotype analysis, if RUNX2 testing is not diagnostic and strong suspicion persists in an individual with CCD features who also has multiple congenital anomalies and/or developmental delay, a karyotype analysis may be considered to evaluate complex chromosome rearrangements or translocations that involve RUNX2 locus but do not result in RUNX2 copy number changes [25, 26]. In addition, a multigene panel that includes RUNX2 and other genes of interest may also be considered.
In the present study, we utilized targeted high-throughput DNA sequencing or Sanger sequencing (single-gene testing) techniques to analyze genetic variants in seven CDD children, and found seven different variants in RUNX2 gene, including four novel variants (c.722_725delTGTT, p.Leu241Serfs*8; c.231_232delTG, Ala78Glyfs*82; c.909C > G, p.Tyr303*; c.668G > T, p.Gly223Val) and three reported variants (c.577C > T, p.Arg193*; c.574G > A, p.Gly192Arg; c.673 C > T, p.Arg225Trp) [27–29], which were all located in the transactivation region (Fig. 2). The bioinformatics analysis indicated that these variants were disease-causing, damaging and/or probably damaging variants. According to ACMG, six variants (c.574G > A, p.Gly192Arg; c.673 C > T, p.Arg225Trp; c.577C > T, p.Arg193*; c.722_725delTGTT, p.Leu241Serfs*8; c.231_232delTG, Ala78Glyfs*82; c.909C > G, p.Tyr303*) were classified as pathogenic variants, and one variant (c.668G > T, p.Gly223Val) as likely pathogenic variant. In addition, all variants were de novo except the following two variants: c.909C > G, p.Tyr303* and c.668G > T, p.Gly223Val. Thereinto the former variant (c.909C > G, p.Tyr303*) was inherited from the patient’s father, who is also a CCD patient carried a de novo heterozygous RUNX2 variant. The clinical features of the father included short stature and CCD, which were very similar to those of his 3-year-old son. The latter variant (c.668G > T, p.Gly223Val) was inherited from the patient’s mother with CCD, who carried a maternal inherited and heterozygous RUNX2 variant. Both of them also showed similar clinical phenotypes, such as short stature and CCD. By summarizing RUNX2 variants in HGMD and the current study, we found nine variant types, such as missense/nonsense, splicing, small deletions/insertions, gross insertions/duplications. Thereinto missense/nonsense variant was the most common variant type of RUNX2 gene (Table 4). A single amino acid (Gly) substitution at position 332 in RUNX2 protein was found not only in our lab (c.668G > T, p.Gly223Val), but also in Ott’ s study (c.667G > A, p.Gly223Arg) [1]. In addition, protein structure prediction showed that these variants could change the number of H-bonds or amino acids in RUNX2 protein (Fig. 4), suggesting that these variants played an important role in regulating the effective structure and function of RUNX2 protein. The experimental result showed that Gly223Val mutation, located in nuclear localization sequence (NLS) [29, 30], could affect the subcellular distribution of RUNX2 protein. The mutation made RUNX2 protein unable to quantitatively accumulate in the nucleus.
Table 4.
Summarization of RUNX2 gene variants in the HGMD and current study
| Variant type | Number of variants (%) | ||
|---|---|---|---|
| HGMD | The current study | Total | |
| Missense/nonsense | 77 (41.8%) | 5 (71.4%) | 82 (42.9%) |
| Splicing | 11 (6.0%) | – | 11 (5.8%) |
| Small deletions | 44 (23.9%) | 2 (28.6%) | 46 (24.1%) |
| Small insertions | 22 (12.0%) | – | 22 (11.5%) |
| Small indels | 2 (1.1%) | – | 2 (1.0%) |
| Gross deletions | 17 (9.2%) | – | 17 (8.9%) |
| Gross insertions/duplications | 5 (2.7%) | – | 5 (2.6%) |
| Complex rearrangements | 4 (2.2%) | – | 4 (2.1%) |
| Repeat variations | 2 (1.1%) | – | 2 (1.0%) |
Fig. 4.

The effect of RUNX2 gene variants on protein structure (The red arrow indicates the H-bonds)
In conclusion, the present study reveals some novel genetic causes of CDD, which not only expands the pathogenic variant spectrum of RUNX2 gene but also will contribute to the diagnosis of CCD and better genetic counseling in the future.
Acknowledgments
We thank the patients and their families for participating in our study.
Abbreviations
- ACMG
American College of Medical Genetics and Genomics
- CCD
Cleidocranial dysplasia
- EVS
Exome Variant Server
- ExAC
Exome Aggregation Consortium
- RUNX2
Runt-related transcription factor 2
Authors’ contributions
XG and KL analyzed and interpreted the patients’ data and were major contributors in writing the manuscript. All authors read and approved the final manuscript.
Funding
This work is supported by grants from the “Youth Research Project of the Shanghai Municipal Health and Family Planning Commission” (No. 20184Y0348, to GXR); the “National Natural Science Foundation of China” (No.81800780 to GXR; No.81670812 and No.81873671, to YYG); the “Jiaotong University Cross Biomedical Engineering” (No.YG2017MS72, to YYG); the “Shanghai Municipal Commission of Health and Family Planning” (No.201740192, to YYG); the “Shanghai Shen Kang Hospital Development Center new frontier technology joint project” (No.SHDC12017109, to YYG).
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
This study was carried out in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) and was certified by the Ethics Committee of Xinhua Hospital affiliated with the Shanghai Jiaotong University School of Medicine. Written informed consent was obtained from the patient’s parents.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xueren Gao and Kunxia Li contributed equally to this work.
Contributor Information
Xuefan Gu, Email: guxuefan@xinhuamed.com.cn.
Yongguo Yu, Email: yuyongguo@shsmu.edu.cn.
References
- 1.Ott CE, Leschik G, Trotier F, Brueton L, Brunner HG, Brussel W, et al. Deletions of the RUNX2 gene are present in about 10% of individuals with cleidocranial dysplasia. Hum Mutat. 2010;31(8):E1587–E1593. doi: 10.1002/humu.21298. [DOI] [PubMed] [Google Scholar]
- 2.Otto F, Kanegane H, Mundlos S. Mutations in the RUNX2 gene in patients with cleidocranial dysplasia. Hum Mutat. 2002;19(3):209–216. doi: 10.1002/humu.10043. [DOI] [PubMed] [Google Scholar]
- 3.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89(5):765–771. doi: 10.1016/S0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 4.Levanon D, Negreanu V, Bernstein Y, Bar-Am I, Avivi L, Groner Y. AML1, AML2, and AML3, the human members of the runt domain gene-family: cDNA structure, expression, and chromosomal localization. Genomics. 1994;23(2):425–432. doi: 10.1006/geno.1994.1519. [DOI] [PubMed] [Google Scholar]
- 5.Yoshida T, Kanegane H, Osato M, Yanagida M, Miyawaki T, Ito Y, et al. Functional analysis of RUNX2 mutations in Japanese patients with cleidocranial dysplasia demonstrates novel genotype-phenotype correlations. Am J Hum Genet. 2002;71(4):724–738. doi: 10.1086/342717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogawa E, Maruyama M, Kagoshima H, Inuzuka M, Lu J, Satake M, et al. PEBP2/PEA2 represents a family of transcription factors homologous to the products of the Drosophila runt gene and the human AML1 gene. Proc Natl Acad Sci U S A. 1993;90(14):6859–6863. doi: 10.1073/pnas.90.14.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kagoshima H, Shigesada K, Satake M, Ito Y, Miyoshi H, Ohki M, et al. The runt domain identifies a new family of heteromeric transcriptional regulators. Trends Genet. 1993;9(10):338–341. doi: 10.1016/0168-9525(93)90026-E. [DOI] [PubMed] [Google Scholar]
- 8.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89(5):747–754. doi: 10.1016/S0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 9.Thirunavukkarasu K, Mahajan M, McLarren KW, Stifani S, Karsenty G. Two domains unique to osteoblast-specific transcription factor Osf2/Cbfa1 contribute to its transactivation function and its inability to heterodimerize with Cbfbeta. Mol Cell Biol. 1998;18(7):4197–4208. doi: 10.1128/MCB.18.7.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pande S, Browne G, Padmanabhan S, Zaidi SK, Lian JB, van Wijnen AJ, et al. Oncogenic cooperation between PI3K/Akt signaling and transcription factor Runx2 promotes the invasive properties of metastatic breast cancer cells. J Cell Physiol. 2013;228(8):1784–1792. doi: 10.1002/jcp.24339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levanon D, Goldstein RE, Bernstein Y, Tang H, Goldenberg D, Stifani S, et al. Transcriptional repression by AML1 and LEF-1 is mediated by the TLE/Groucho corepressors. Proc Natl Acad Sci U S A. 1998;95(20):11590–11595. doi: 10.1073/pnas.95.20.11590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89(5):755–764. doi: 10.1016/S0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 13.Komori T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem Cell Biol. 2018;149(4):313–323. doi: 10.1007/s00418-018-1640-6. [DOI] [PubMed] [Google Scholar]
- 14.Peng YJ, Chen QY, Fu DJ, Liu ZM, Mao TT, Li J, et al. A novel gene mutation of Runx2 in cleidocranial dysplasia. J Huazhong Univ Sci Technolog Med Sci. 2017;37(5):772–776. doi: 10.1007/s11596-017-1803-z. [DOI] [PubMed] [Google Scholar]
- 15.Zhang T, Wu J, Zhao X, Hou F, Ma T, Wang H, et al. Whole-exome sequencing identification of a novel splicing mutation of RUNX2 in a Chinese family with cleidocranial dysplasia. Arch Oral Biol. 2019;100:49–56. doi: 10.1016/j.archoralbio.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Çamtosun E, Akıncı A, Demiral E, Tekedereli İ, Sığırcı A. A cleidocranial dysplasia case with a novel mutation and growth velocity gain with growth hormone treatment. J Clin Res Pediatr Endocrinol. 2018;11(3):301–5. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 17.Ma D, Wang X, Guo J, Zhang J, Cai T. Identification of a novel mutation of RUNX2 in a family with supernumerary teeth and craniofacial dysplasia by whole-exome sequencing: a case report and literature review. Medicine (Baltimore) 2018;97(32):e11328. doi: 10.1097/MD.0000000000011328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashmi JA, Almatrafi A, Latif M, Nasir A, Basit S. An 18 bps in-frame deletion mutation in RUNX2 gene is a population polymorphism rather than a pathogenic variant. Eur J Med Genet. 2019;62(2):124–128. doi: 10.1016/j.ejmg.2018.06.013. [DOI] [PubMed] [Google Scholar]
- 19.Zeng L, Wei J, Zhao N, Sun S, Wang Y, Feng H. A novel 18-bp in-frame deletion mutation in RUNX2 causes cleidocranial dysplasia. Arch Oral Biol. 2018;96:243–248. doi: 10.1016/j.archoralbio.2017.10.020. [DOI] [PubMed] [Google Scholar]
- 20.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 21.Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 25.Purandare SM, Mendoza-Londono R, Yatsenko SA, Napierala D, Scott DA, Sibai T, et al. De novo three-way chromosome translocation 46,XY,t (4;6;21)(p16;p21.1; q21) in a male with cleidocranial dysplasia. Am J Med Genet A. 2008;146A(4):453–458. doi: 10.1002/ajmg.a.31750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Northup JK, Matalon R, Lockhart LH, Hawkins JC, Velagaleti GV. A complex chromosome rearrangement, der (6) ins (6)(p21.1q25.3q27)inv (6)(p25.3q27), in a child with cleidocranial dysplasia. Eur J Med Genet. 2011;54(4):e394–e398. doi: 10.1016/j.ejmg.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 27.Zhou G, Chen Y, Zhou L, Thirunavukkarasu K, Hecht J, Chitayat D, et al. CBFA1 mutation analysis and functional correlation with phenotypic variability in cleidocranial dysplasia. Hum Mol Genet. 1999;8(12):2311–2316. doi: 10.1093/hmg/8.12.2311. [DOI] [PubMed] [Google Scholar]
- 28.Puppin C, Pellizzari L, Fabbro D, Fogolari F, Tell G, Tessa A, et al. Functional analysis of a novel RUNX2 missense mutation found in a family with cleidocranial dysplasia. J Hum Genet. 2005;50(12):679–683. doi: 10.1007/s10038-005-0311-3. [DOI] [PubMed] [Google Scholar]
- 29.Quack I, Vonderstrass B, Stock M, Aylsworth AS, Becker A, Brueton L, et al. Mutation analysis of core binding factor A1 in patients with cleidocranial dysplasia. Am J Hum Genet. 1999;65(5):1268–1278. doi: 10.1086/302622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanno T, Kanno Y, Chen LF, Ogawa E, Kim WY, Ito Y. Intrinsic transcriptional activation-inhibition domains of the polyomavirus enhancer binding protein 2/core binding factor alpha subunit revealed in the presence of the beta subunit. Mol Cell Biol. 1998;18(5):2444–2454. doi: 10.1128/MCB.18.5.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.