

Abstract
A primary challenge in type 2 diabetes (T2D) is the preservation of a functional population of β-cells, which play a central role in regulating blood glucose levels. Two congenital disorders, Bardet-Biedl syndrome (BBS) and Alström syndrome (ALMS), can serve as useful models to understand how β-cells are normally produced and regenerated. Both are characterized by obesity, loss of β-cells, and defects in primary cilia – the sensory center of cells. Primary cilia are cellular protrusions present in almost every vertebrate cell. This antenna-like organelle plays a crucial role in regulating several signaling pathways that direct proper development, proliferation, and homeostasis. Mutations in genes expressing ciliary proteins or proteins present at or near the base of the cilium lead to disorders, collectively called ciliopathies. BBS and Alström syndrome are such disorders. Though both BBS and Alström patients are obese, their childhood diabetes rates are vastly different, suggesting distinct pathogenesis underlying these two ciliopathies. Clinical studies suggest that BBS patients are protected against early onset diabetes by sustained or enhanced β-cell function. In contrast, Alström patients are more prone to develop diabetes. They have hyperinsulinemia, yet their β-cells fail to sense glucose and to regulate insulin secretion accordingly. These data suggest a potential role for primary cilia in maintaining a functional β-cell population and that defects in cilia or in ciliary proteins impair development and function of β-cells. Identifying the respective roles of primary cilia and ciliary proteins, such as BBS and ALMS1 may shed light on β-cell biology and uncover potentially novel targets for diabetes therapy.
Keywords: Primary cilia, Ciliopathy, Pancreas, β-cell, Type 2 diabetes, Bardet-Biedl syndrome, Alström syndrome
Introduction
Most nucleated vertebrate cells generate one immotile primary cilium [1]. This organelle acts as a signaling center, regulating a growing list of signal transduction pathways controlling developmental programs, cell proliferation, cell fate determination, and metabolic homeostasis [1]. Molecules involved in signal transduction such as ligands, channels, receptors, transcription factors, and ciliary structural proteins are specifically trafficked to primary cilium in a tightly regulated fashion. Any mutation perturbing the structure or operation of primary cilia can cause havoc on development and organogenesis. It also disrupts the normal functioning of differentiated cells [2]. The disorders of primary cilia and motile cilia are collectively referred to as ciliopathies.
Defects in primary cilia are also implicated in metabolic disorders, such as obesity and type 2 diabetes (T2D) [3,4]. Obesity is linked to T2D due to their association with insulin resistance, a risk factor of T2D. However, about 7.5 percent to 21 percent of T2D patients are lean [5-8], suggesting that diabetes in lean people is independent of obesity and may have other causes, such as anomalies in β-cells that produce insulin.
Successful production of β-cells, either by in vitro culture or by stimulation of in vivo regeneration, is uncommon, partially due to limited knowledge of the factors required for specification of these cells. Naturally congenital disorders with impaired β-cells, can serve as powerful models to unravel β-cell biology. In particular, disorders in which the onset of diabetes is directly linked to β-cell production and maintenance could be invaluable. Two ciliopathies Alström Syndrome and Bardet-Biedl syndrome represent such disorders. While both are characterized by highly penetrant obesity, they display vastly different childhood diabetes rates (75 percent and 2 to 6 percent respectively) [9-14]. This discrepancy raises the possibility that the incidence of diabetes in these two ciliopathies is independent of obesity and rather due to distinctions in the maintenance of β-cell population or function. Though the exact mechanism of how primary cilia maintain a functional population of β-cells have yet to be elucidated, the scientific community have started to acknowledge its significance at different aspects of β-cells. This review will focus on the current knowledge on roles of primary cilia and ciliopathy proteins in the production and maintenance of functional β-cell population.
Primary Cilium and Ciliopathies
Animals generate a variety of cilia and the synonymous flagella for a wide array of tasks. Defects in cilia underlie many congenital disorders in human, collectively referred to as ciliopathies. A primary cilium is immotile, supported by an axonemal structural scaffold consisting of nine microtubule doublets (Figure 1). The axoneme is enclosed by the ciliary membrane, that is physically connected to the plasma membrane. The protein composition in these two membranes differs substantially. Ciliary membrane proteins, such as receptors, channels and other molecules involved in the signaling cascade, are selectively transported into the ciliary compartment [15,16]. The axonemal microtubules are nucleated from the basal body, a modified centriole that contains nine triplet microtubules [17,18]. The ciliary pocket, an invagination of plasma membrane, is found at the base of each primary cilium [19]. Ciliary proteins assembled around the basal body are selectively transported into the cilium through the transition zone, which operates as a gate at the ciliary base [20-24]. The transition fibers that connect the distal basal body to the base of the ciliary membrane and the distinct Y-shaped structures between the membrane and axoneme form a diffusion barrier and, along with other proteins, they control the entry of ciliary proteins [25-30]. The movement between the ciliary base and the cilium is commonly called intraflagellar transport (IFT), a bi-directional trafficking mechanism carried out by IFT particles to transport cargoes into and out of the cilium. Anterograde motors, kinesins which are associated with IFT-B complexes are responsible for the tip-ward movement, whereas retrograde dynein motors associated with IFT-A complexes bring the cargo back to the cell body [31,32] (Figure 1).
Figure 1.
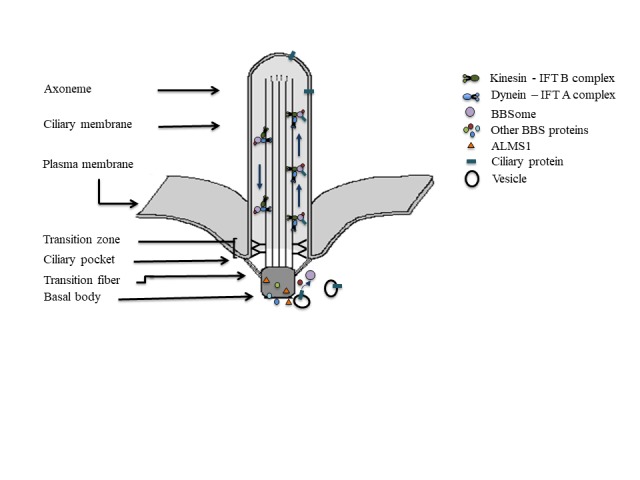
Primary cilium: Structural organization, protein trafficking by IFT and BBSome, and proposed functional localization of ALMS1 and BBS proteins.
Ciliopathies resulting from defective primary cilia are numerous, including Bardet-Biedl syndrome, Alström syndrome, polycystic kidney disease, nephronophthisis, Meckel-Gruber syndrome, Joubert syndrome, Senior-Loken syndrome, oral-facial-digital syndrome, Leber congenital amaurosis, and Early-onset severe retinal dystrophy, Jeune syndrome [33,34] (Table 1). Clinical overlap has been observed with mutations in the ciliopathy genes. The involvement of genetically distinct transcripts in multiple ciliopathies is a common feature as these proteins affect the same cellular structure: the primary cilium [33,34].
Table 1. Ciliopathies, causative genes, functional localizations of the proteins in primary cilia and the respective phenotypes of the syndromes.
| Ciliopathy | Genes | Ciliary localization | Phenotypes |
| Alström syndrome [58] | ALMS1 | Basal body | Truncal Obesity, T2D, Insulin resistance, hepatic dysfunction, hyperlipidemia, hypothyroidism, hypogonadism, short stature, wide feet, retinal degeneration, hearing loss, mental retardation |
| Bardet-Biedl syndrome [71,93] | BBS1-20 | Axoneme, Basal body | Obesity, T2D, hypertension, hyperlipidemia, polydactyly, male hypogonadism, retinal dystrophy, renal dysfunction, learning disabilities, cognitive impairment, fatty liver |
| Autosomal dominant polycystic kidney disease [94,95] | PKD1 | Axoneme | Renal cyst formation, loss of renal function, abnormalities in cardiovascular, portal, pancreatic and gastrointestinal systems |
| Nephronophthisis [96] | NPHP1-9 | Basal body Transition zone, Axoneme | Kidney cyst, tubulointerstitial nephropathy, retinal degeneration, liver fibrosis, cerebellar hypoplasia, situs inversus, and mental retardation |
| Meckel-Gruber syndrome [97] | MKS1, TMEM216, TMEM67, TMEM231 TMEM138, TMEM237, CEP290, RPGRIP1L, CC2D2A, NPHP3, TCTN2, B9D1 B9D2, EVC2, C5orf42, SEC8 | Basal body, Transition zone, Ciliary membrane | Lethal, cystic renal disease, central nervous system malformation, occipital encephalocele, polydactyly, hepatic fibrosis, polydactyly, situs inversus, skeletal defects |
| Joubert syndrome [98] | CEP290, CEP120, CEP41, INPP5E, ARL13B, CC2D2A, RPGRIP1L, TMEM216, TMEM67, TMEM237, TMEM231, TMEM138, NPHP1, AHI1, CXORF5, OFD1, TTC21B, KIF7, TCTN1, TCTN3 C5ORF42, ZNF423, CSPP1, ARMC9, FAM149B1 | Basal body, Transition zone | Cerebellum and midbrain abnormalities: molar tooth sign, hypotonia, psychomotor delay, irregular breathing pattern and oculomotor apraxia. developmental delay, truncal ataxia, speech apraxia, polydactyly, chorio-retinal colobomas, retinal degeneration, congenital hepatic fibrosis, fibrocystic kidney disease, cleft palate |
| Senior-Loken syndrome [99] | SDCCAG8, NPHP4, NPHP5, NPHP6, WDR19, TRAF3IP1 | Basal body Transition zone, Axoneme | renal nephronophthisis, retinal degeneration, retinitis pigmentosa |
| Oral-facial-digital syndrome (OFD) [100] | OFD1-15, two unclassified | Basal body Transition zone, Axoneme | abnormalities of the face, oral cavity and digits, pancreatic, renal, hepatic, ovarian cysts, cognitive defects |
| Leber congenital amaurosis and Early-onset severe retinal dystrophy [101] | GUCY2D, RPE65, SPATA7, AIPL1, LCA5, RPGRIP1, CRX, CRB1, NMNAT1, CEP290, IMPDH1, RD3, RDH12, LRAT, TULP1, KCNJ13, GDF6, OTX2, CABP4, CLUAP1, IQCB1, DTHD1, IFT140, ALMS1, PRPH2 | Basal body, Transition Zone, Axoneme | early infantile onset rod–cone dystrophies, retinal dystrophy |
| Jeune syndrome [102] | TCTEX1D2, DYNC2H1, WDR34, WDR60, IFT80, IFT172, IFT144, IFT139, IFT140, CEP120, CSPP1 | Basal body, Axoneme | multiple skeleto-muscular abnormalities, narrow thorax, shortened ribs, variable limb shortening, brachydactyly, polydactyly, renal dysfunction, hepatic dysfunction, retinal dystrophy |
As primary cilium is present in nearly every nucleated human cell transducing a wide array of stimuli [33], any defect in primary cilium-related genes could result in a spectrum of phenotypes ranging from retinal degeneration, skeletal defects, kidney disease, cognitive disorders, to more specific defects in cellular fate determination during tissue development [34]. The latter is being implicated in regulating energy metabolism, body weight maintenance and glucose homeostasis [3,4]. The review will focus on the role of primary cilium in maintaining glucose homeostasis by regulating a functional pancreatic β-cell mass.
Primary Cilium in Pancreas and β-cells
Pancreas, essential in maintaining glucose homeostasis, comprises exocrine and endocrine compartments. The endocrine pancreatic compartment, known as islets of Langerhans, contains distinct cell types producing different hormones: β-cells for insulin, α-cells for glucagon, δ-cells for somatostatin, ξ-cells for ghrelin, and pancreatic polypeptide cells for pancreatic polypeptides. Primary cilium is present in β, α, and δ-cells as well as in the ductal cells and centroacinar cells of exocrine pancreas [35]. As primary cilia regulate several signaling pathways, such as FGF, Sonic Hedgehog, Wnt, TGF-β, Notch, that are critical for proper pancreatic organogenesis and function, defects in this cellular organelle directly affect pancreas development [35].
Pancreatic β-cells play a central role in maintaining glucose homeostasis. Several studies identified multiple diabetes associated genes, that are involved in maintaining a functional β-cell population [36-41]. Loss of these cells is a primary cause of progression of diabetes. A significant loss of β-cell differentiation along with increased Shh signaling was observed when primary cilia are specifically ablated in β-cells [42]. The following sections will provide an overview of current knowledge, describing the roles of primary cilia and ciliary proteins in β-cells.
How do primary cilia affect insulin producing β-cells?
During pancreas development, progenitor cells expressing transcription factor NGN3 specify the fate of endocrine cells [43,44]. These NGN3 positive endocrine precursor cells and differentiated endocrine cells express another transcription factor RFX3, which is necessary for ciliogenesis [45,46]. Rfx3-/- mice possess fewer and shortened primary cilia, as well as fewer α, β, ξ-cells during perinatal stages [45,46]. Adult Rfx3-/- mice exhibit impaired glucose tolerance, consistent with their characteristic smaller islets, decreased insulin production and reduced glucose stimulated insulin secretion. Given its role in regulating critical developmental signaling, defects in cilia are probably causative to the observed abnormal β-cell development in embryonic (E15.5) Rfx3-/- mice [45].
Kinesin family member 3A (KIF3A) is another protein that is necessary for ciliogenesis as well as for intraflagellar transport. When suppressed, its deficiency leads to reduction in the number of primary cilia. This is also correlated with decreased proliferation of β-cells. This observation is consistent in cultured mouse insulinoma β-cells (Min6), dispersed primary mouse islet and human islet cells. [47]. Lack of primary cilia along with decreased proliferation, in Kif3a depleted β-cells, provide direct functional evidence for the involvement of cilia in β-cell proliferation. This phenomenon could be explained by the fact that the ciliary disassembly is important in proliferating cells. The assembly and disassembly of primary cilia and lifecycle of centrosomes are tightly linked to cell division [48-50]. The absence of primary cilia could prevent the β-cells from dividing, potentially by disrupting the communication between external signals that trigger β-cell proliferation and internal machineries that lead to cell division. Moreover, this gene is differentially expressed in non-diabetic and T2D human islets. A significantly higher expression is observed in non-diabetic human islets as well as in islets of obese non-diabetic mouse model when compared to the respective diabetic controls [47], further confirming its significance in maintaining functional β-cells.
The B6.V-Lepob/ob (B6-ob/ob) mouse develops severe obesity but is protected from diabetes, whereas the New Zealand Obese (NZO) mouse is obese and is a model for polygenic T2D. Interestingly, these obese mouse models are significantly different from each other in terms of number of primary cilia and expression of cilia-genes including Kif3a and genes involved in cell division in pancreatic islets. The diabetic NZO mouse islets are shown to have significantly less primary cilia and reduced expression of cilia-genes when compared to non-diabetic B6-ob/ob. The primary cilia in B6-ob/ob islets are able to disassemble in response to high carbohydrate diet and in turn can trigger β-cell proliferation, an important phenomenon that was not evident in NZO mice islets. The lack of primary cilia also correlated with increased apoptosis due to glucose toxicity in NZO mice [47]. An inability to fine tune insulin secretion in response to different carbohydrate concentrations is also observed in NZO mice, but not in B6-ob/ob mice. The observed insulin hypersecretion, even in low glucose conditions, in NZO mice is possibly due to the lack of proper regulation in glucose stimulated insulin secretion as a consequence of reduced primary cilia.
Other ciliary proteins that have been implicated in β-cell development are ALMS1 and BBS proteins. Mutations in these genes lead to autosomal recessive disorders — Alström syndrome and Bardet-Biedl syndromes respectively. They are also categorized as obesity ciliopathies due to highly penetrant childhood obesity [10,12,51-58]. However, they differ in susceptibilities to childhood onset T2D. About 75 percent of Alström patients develop T2D as early as by their first decade, while only 2 to 3 percent of BBS patients develop T2D in childhood. These discrepancies could be explained partly by severe insulin resistance observed in Alström patients [58], a risk factor that is not as apparent in BBS patients [59].
Alström syndrome is caused by loss-of-function mutations in the ALMS1 gene, which is enriched at the basal body of primary cilia [60] (Table 1). This syndrome is characterized by multi-organ dysfunctions, such as cardiomyopathy, retinal degeneration, fibrosis and renal dysfunction. In addition to that, Alström patients exhibit childhood obesity, severe insulin resistance, hyperinsulinemia, and early onset T2D [58]. Alms1 is highly expressed in adult and fetal pancreatic islets [61,62] and loss of Alms1 expression in mutant mice results in pancreatic hyperplasia, partial degranulation of β-cells and islet cysts [63,64]. Clinical reports suggest that the progression of diabetes in Alström patients is not due to further worsening of insulin resistance but as a result of failure in insulin secretion from β-cells [10]. In zebrafish, its absence during development leads to significant loss of β-cells [65], due to both decreased proliferation and increased apoptosis of β-cells specifically without any significant effect on progenitor cell population (Figure 2). These embryo sare also unable to regenerate β-cells after specific depletion [65]. An increased β-cell apoptosis correlates with the decrease in the expression of genes involved in RNA and protein processing in alms1-/- zebrafish model [66].
Figure 2.
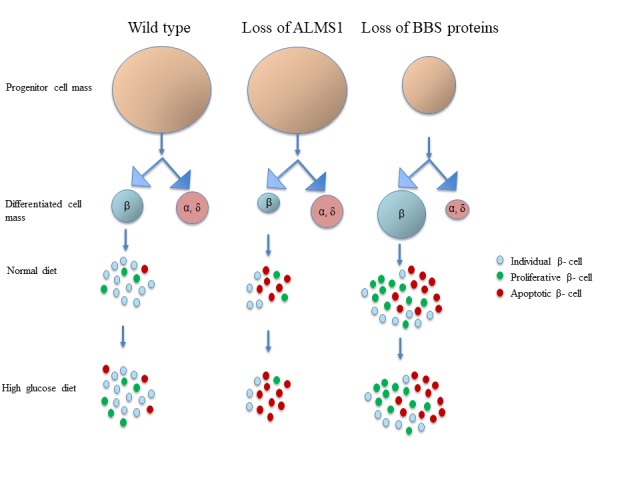
Alström and BBS genes regulate the pancreatic β-cell production. In wild-type pancreas, progenitor cells differentiate into endocrine cell types. β-cells proliferate in the presence of high glucose to meet the increased demand of insulin. Loss of ALMS1 does not have any effect on progenitor cell population but specifically decreases β-cell mass as a result of increased apoptosis and decreased proliferation. Elevated systemic glucose probably decreases β-cell mass due to further increase in the rate of apoptosis. ALMS1 depleted β-cells do not proliferate in high glucose condition. Loss of BBS genes leads to fewer pancreatic progenitors, that produce fewer endocrine α- and δ-cells. An increase in β-cell production is observed at the expense of other differentiated cell types. A compensatory increase in proliferation maintains the high number of β-cells, that are prone to apoptotic cell death in the absence of BBSs. The rate of proliferation and apoptosis remain unchanged and the elevated β-cell mass is maintained in high glucose environment in the absence on BBS proteins.
An important property of β-cells is to proliferate during increased demand [67]. However, zebrafish β-cells lacking Alms1 do not expand in response to prolonged exposure to glucose [65,66] (Figure 2). Cultured si-Alms1 β-cells (Min6) fail to alter Glut2 expression [66], a glucose transporter that also acts as glucose sensor in β-cells of rodents, and subsequent insulin secretion in high glucose condition [68], suggesting a potential involvement of Alms1 in glucose sensing. Consistent with hyperinsulinemia in patients bearing mutations in ALMS1 [13,58], alms1-/- zebrafish also exhibit hyperinsulinemia in the normal diet, but no further increase in total insulin after exposure to glucose. This observation is further confirmed by the insulin hypersecretion by Alms1-depleted Min6 cells at basal glucose concentration [66]. In zebrafish, alms1 regulates the expression of genes that are involved in cellular transport and secretion, including the expression of Ca2+ and K+ channels. A significantly increased expression of these genes in alms1-/- zebrafish explains the dysregulated insulin secretion in unstimulated conditions [66]. This ciliary protein probably regulates secretion by regulating the expression of genes involved in membrane potential regulation as well as in cAMP production and glucose sensing [66]. These data suggest a role for ALMS1 gene in both β-cell proliferation and function.
In contrast, BBS patients and mouse models of BBS display elevated insulin sensitivity, but normal glucose management [59,69]. BBS is an autosomal recessive disorder caused by mutations in one of 21 genes [70,71], out of which, eight BBS proteins form a complex, called BBSome. The BBSome is an integral component of basal body and plays crucial role in trafficking membrane-associated cargoes to primary cilium [72,73]. Mutations in these genes result in abnormal structure and functions of primary cilia. Defects in BBS genes have differential effects on insulin secretion. In Bbs4-/- mice, the absence of BBS4 leads to generation of β-cells that are inefficient in first phase insulin release, possibly due to deficient signaling through insulin receptor [74], but, downregulation of Bbs5, Bbs7, and Bbs9 lead to insulin hypersecretion in Min6 cells [69].
Interestingly, although β-cell proliferation increased, apoptotic β-cell numbers are also significantly higher both in basal and high glucose conditions in BBS models of zebrafish [65]. A decrease in endocrine progenitor cells along with differentiated α- and δ-cells also accompanied the increase in β-cell numbers [65] (Figure 2). These suggest that BBS patients possibly have higher β-cell mass to begin with, in the expense of other differentiated cell types and that the increased proliferation compensates for the increased apoptosis, at least for some time before patients start depleting their β-cell mass. This observation supports the low rate of diabetes susceptibility in BBS patients in the childhood [9,11,13,14].
These findings suggest that different ciliary proteins play different roles in the regulation of β-cells. ALMS1 is a basal body protein [60] (Figure 1) while the BBSome octamer (BBS 1, 2, 4, 5, 7, 8, 9, 18) functions as a cargo for anterograde and retrograde transport [75]. Other BBS proteins either form a chaperonin complex (BBS 6, 10, 12) to facilitate the BBSome formation [76,77] or function at the base of the cilium and in the basal body to recruit the BBSome to deliver cargo to ciliary compartment [78] (Figure 1). Different sub-ciliary locations of ALMS1 and BBS proteins as well as their specific functions in those locations allow the primary cilia to maintain a delicate balance in establishing a functional β-cell population.
β-cells are normally arranged as rosettes around capillaries in islets, where the primary cilia are generally found in the lateral surfaces of β-cells. Maintaining this organization of primary cilia, relative to blood vessels, is also an important factor in β-cells [79]. Defects in organization of primary cilia in β-cells, due to the absence of LKB1 protein, a tumor suppressor, impacts the size of β-cells and insulin secretion. Lkb1-/- mice have larger β-cells, where unlike wild type mice, primary cilia are not found in lateral surfaces of β-cells. Instead, they are positioned to the cell surface opposite to the blood vessels [79,80]. This change in cellular polarity potentially changes the cellular microenvironment and results in hyperactivation of mTOR pathway [81], leading to the generation of larger β-cells. Lkb1 deficient mice have an elevated blood glucose level after birth. However, the blood glucose level returns to normal in older pups. The adult mice tend to have faster clearance of glucose in glucose tolerance tests [82], a phenotype supported by insulin hypersecretion in those knockout mice [81]. Shifting the primary cilia away from the capillaries, where insulin secretion takes place, probably affects the intensity of how primary cilia sense and in turn regulate insulin secretion. Reduced sensing due to altered organization of primary cilia in β-cells could explain the observed insulin hypersecretion in Lkb1-/- mice.
As discussed in this review, maintaining the structural integrity and proper organization of primary cilia are important for maintaining a functional population of β-cells. Further investigation on the roles of primary cilia and associated proteins in the specification, proliferation, and functionality of β-cells, will identify the mechanisms contributing to their roles in modulation of signal transduction pathways during specific stages of β-cell specification.
Conclusion and Outlook
Recent studies have started to explore the role of primary cilia in β-cells. However, the direct association of insulin producing β-cells with ciliopathies are, so far, limited to only Bardet-Biedl and Alström syndromes, two ciliopathies that impart discrepant effects on β-cells. These disorders serve as useful models to identify novel factors involved in β-cell production and maintenance. Further investigations are needed to study previously unexplored roles of primary cilia, ciliary proteins and basal body proteins in the specification, maintenance, and regeneration of functional β-cells and to characterize the β-cell phenotype resulting from the loss of gene expression at different stages of development and to elucidate the mechanism by which disruption occurs. The opposing effects on β-cells from depletion of proteins localized to distinct structures central to the primary cilium, suggest the critical yet complicated roles of this organelle in β-cell biology. Therefore, these opposing effects of primary cilia offer a unique opportunity to elucidate the mechanism underlying β-cell production and survival.
Ciliopathy proteins are critical in the developmental program signaled by Sonic hedgehog (Shh), Wnt, and Notch [83-88]. These pathways are also key players at different stages of pancreatic development [89] and are targets in cellular reprogramming of induced pluripotent stem cells (iPS) into insulin producing β-cells [89-92]. Therefore, unraveling the roles of ciliary proteins including ALMS1, BBS proteins and primary cilium will shed critical insight on β-cell specification pathways in vivo and in vitro. These results will, in turn, identify novel potential targets, such as specific signaling pathways, specific molecules that regulate different aspects of β-cells through this evolutionary conserved organelle, for therapeutic intervention, and for the preservation and regeneration of β-cells.
Acknowledgments
I would like to thank Dr. Pinfen Yang (Marquette University) and Dr. Thomas Eddinger (Marquette University) for their critical inputs to help improve the manuscript.
Glossary
- T2D
Type 2 diabetes
- BBS
Bardet-Biedl syndrome
- ALMS
Alström syndrome
- IFT
Intraflagellar transport
- FGF
Fibroblast growth factor
- Wnt
Wingless/Integrated
- TGF-β
Transforming growth factor β
- NGN3
Neurogenin3
- RFX3
Regulatory Factor X3
- ALMS1
Alström syndrome protein 1
- iPS
induced pluripotent stem
References
- Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11(5):331–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satir P, Pedersen LB, Christensen ST. The primary cilium at a glance. J Cell Sci. 2010;123(Pt 4):499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Song J, Jung JH, Ko HW. Primary cilia in energy balance signaling and metabolic disorder. BMB Rep. 2015;48(12):647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song DK, Choi JH, Kim MS. Primary Cilia as a Signaling Platform for Control of Energy Metabolism. Diabetes Metab J. 2018;42(2):117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnethon MR, De Chavez PJ, Biggs ML, Lewis CE, Pankow JS, Bertoni AG, et al. Association of weight status with mortality in adults with incident diabetes. JAMA. 2012;308(6):581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman NJ, Miernik J, Philipson L, Fogelfeld L. Lean versus obese diabetes mellitus patients in the United States minority population. J Diabetes Complications. 2014;28(4):500–5. [DOI] [PubMed] [Google Scholar]
- Hartmann B, Lanzinger S, Bramlage P, Gross F, Danne T, Wagner S, et al. Lean diabetes in middle-aged adults: A joint analysis of the German DIVE and DPV registries. PLoS One. 2017;12(8):e0183235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobias DK, Pan A, Jackson CL, O’Reilly EJ, Ding EL, Willett WC, et al. Body-mass index and mortality among adults with incident type 2 diabetes. N Engl J Med. 2014;370(3):233–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437–46. [PMC free article] [PubMed] [Google Scholar]
- Bettini V, Maffei P, Pagano C, Romano S, Milan G, Favaretto F, et al. The progression from obesity to type 2 diabetes in Alstrom syndrome. Pediatr Diabetes. 2012;13(1):59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuillan PP, Ng D, Han JC, Sapp JC, Wetsch K, Spaulding E, et al. Patients with Bardet-Biedl Syndrome Have Hyperleptinemia Suggestive of Leptin Resistance. J Clin Endocrinol Metab. 2011;96(3):E528–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe E, Beales PL. Bardet-Biedl syndrome. Eur J Hum Genet. 2013;21(1):8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard D, Petrovsky N. Alstrom syndrome: insights into the pathogenesis of metabolic disorders. Nat Rev Endocrinol. 2011;7(2):77–88. [DOI] [PubMed] [Google Scholar]
- Grace C, Beales P, Summerbell C, Jebb SA, Wright A, Parker D, et al. Energy metabolism in Bardet-Biedl syndrome. Int J Obes. 2003;27(11):1319–24. [DOI] [PubMed] [Google Scholar]
- Christensen ST, Pedersen LB, Schneider L, Satir P. Sensory cilia and integration of signal transduction in human health and disease. Traffic. 2007;8(2):97–109. [DOI] [PubMed] [Google Scholar]
- Louvi A, Grove EA. Cilia in the CNS: the quiet organelle claims center stage. Neuron. 2011;69(6):1046–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho-Santos Z, Azimzadeh J, Pereira-Leal JB, Bettencourt-Dias M. Evolution: tracing the origins of centrioles, cilia, and flagella. J Cell Biol. 2011;194(2):165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisch C, Dupuis-Williams P. Ultrastructure of cilia and flagella - back to the future! Biol Cell. 2011;103(6):249–70. [DOI] [PubMed] [Google Scholar]
- Molla-Herman A, Ghossoub R, Blisnick T, Meunier A, Serres C, Silbermann F, et al. The ciliary pocket: an endocytic membrane domain at the base of primary and motile cilia. J Cell Sci. 2010;123(Pt 10):1785–95. [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43(8):776–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson EC, Dowdle WE, Ozanturk A, Garcia-Gonzalo FR, Li C, Halbritter J, et al. TMEM231, mutated in orofaciodigital and Meckel syndromes, organizes the ciliary transition zone. J Cell Biol. 2015;209(1):129–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Garcia G, 3rd, Van De Weghe JC, McGorty R, Pazour GJ, Doherty D, et al. Super-resolution microscopy reveals that disruption of ciliary transition-zone architecture causes Joubert syndrome. Nat Cell Biol. 2017;19(10):1178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegering A, Dildrop R, Kalfhues L, Spychala A, Kuschel S, Lier JM, et al. Cell type-specific regulation of ciliary transition zone assembly in vertebrates. EMBO J. 2018;37(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee LE, Garcia-Gonzalo FR, Bowie RV, Li C, Kennedy JK, Ashrafi K, et al. Conserved Genetic Interactions between Ciliopathy Complexes Cooperatively Support Ciliogenesis and Ciliary Signaling. PLoS Genet. 2015;11(11):e1005627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awata J, Takada S, Standley C, Lechtreck KF, Bellve KD, Pazour GJ, et al. NPHP4 controls ciliary trafficking of membrane proteins and large soluble proteins at the transition zone. J Cell Sci. 2014;127(Pt 21):4714–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Reiter JF. Scoring a backstage pass: mechanisms of ciliogenesis and ciliary access. J Cell Biol. 2012;197(6):697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Milenkovic L, Jin H, Scott MP, Nachury MV, Spiliotis ET, et al. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 2010;329(5990):436–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, Blacque OE, Leroux MR. The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep. 2012;13(7):608–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol. 2011;192(6):1023–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Reiter JF. Open Sesame: How Transition Fibers and the Transition Zone Control Ciliary Composition. Cold Spring Harb Perspect Biol. 2017;9(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen LB, Rosenbaum JL. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol. 2008;85:23–61. [DOI] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3(11):813–25. [DOI] [PubMed] [Google Scholar]
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364(16):1533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 2017;18(9):533–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodh S, O’Hare EA, Zaghloul NA. Primary cilia in pancreatic development and disease. Birth Defects Res C Embryo Today. 2014;102(2):139–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443(7110):453–7. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Kulkarni RN, Sarraf P, Ozcan U, Okada T, Hsu CH, et al. Targeted elimination of peroxisome proliferator-activated receptor gamma in beta cells leads to abnormalities in islet mass without compromising glucose homeostasis. Mol Cell Biol. 2003;23(20):7222–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa-Pineda B, Chowdhury K, Torres M, Oliver G, Gruss P. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature. 1997;386(6623):399–402. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Nammo T, Moriwaki M, Ihara A, Iizuka K, Yang Q, et al. Overexpression of dominant-negative mutant hepatocyte nuclear fctor-1 alpha in pancreatic beta-cells causes abnormal islet architecture with decreased expression of E-cadherin, reduced beta-cell proliferation, and diabetes. Diabetes. 2002;51(1):114–23. [DOI] [PubMed] [Google Scholar]
- Bonnefond A, Froguel P. Rare and common genetic events in type 2 diabetes: what should biologists know? Cell Metab. 2015;21(3):357–68. [DOI] [PubMed] [Google Scholar]
- Prasad RB, Groop L. Genetics of type 2 diabetes-pitfalls and possibilities. Genes (Basel). 2015;6(1):87–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landsman L, Parent A, Hebrok M. Elevated Hedgehog/Gli signaling causes beta-cell dedifferentiation in mice. Proc Natl Acad Sci USA. 2011;108(41):17010–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA. 2000;97(4):1607–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson KA, Dursun U, Jordan N, Gu G, Beermann F, Gradwohl G, et al. Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types. Dev Cell. 2007;12(3):457–65. [DOI] [PubMed] [Google Scholar]
- Ait-Lounis A, Baas D, Barras E, Benadiba C, Charollais A, Nlend Nlend R, et al. Novel function of the ciliogenic transcription factor RFX3 in development of the endocrine pancreas. Diabetes. 2007;56(4):950–9. [DOI] [PubMed] [Google Scholar]
- Bonnafe E, Touka M. The transcription factor RFX3 directs nodal cilium development and left-right asymmetry specification. Mol Cell Biol. 2004;24(10):4417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluth O, Stadion M, Gottmann P, Aga H, Jahnert M, Scherneck S, et al. Decreased Expression of Cilia Genes in Pancreatic Islets as a Risk Factor for Type 2 Diabetes in Mice and Humans. Cell Rep. 2019;26(11):3027–36.e3. [DOI] [PubMed] [Google Scholar]
- Irigoin F, Badano JL. Keeping the balance between proliferation and differentiation: the primary cilium. Curr Genomics. 2011;12(4):285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikova OV, Pugacheva EN, Golemis EA. Primary cilia and the cell cycle. Methods Cell Biol. 2009;94:137–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker RW, Pardee AB, Fujiwara K. Centriole ciliation is related to quiescence and DNA synthesis in 3T3 cells. Cell. 1979;17(3):527–35. [DOI] [PubMed] [Google Scholar]
- Adams M, Smith UM, Logan CV, Johnson CA. Recent advances in the molecular pathology, cell biology and genetics of ciliopathies. J Med Genet. 2008;45(5):257–67. [DOI] [PubMed] [Google Scholar]
- Badano JL, Mitsuma N, Beales PL, Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–48. [DOI] [PubMed] [Google Scholar]
- Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151c(4):281–95. [DOI] [PubMed] [Google Scholar]
- Cardenas-Rodriguez M, Badano JL. Ciliary biology: understanding the cellular and genetic basis of human ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151c(4):263–80. [DOI] [PubMed] [Google Scholar]
- Muller J, Stoetzel C, Vincent MC, Leitch CC, Laurier V, Danse JM, et al. Identification of 28 novel mutations in the Bardet-Biedl syndrome genes: the burden of private mutations in an extensively heterogeneous disease. Hum Genet. 2010;127(5):583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisse C, Reiter JF, Berbari NF. Cilia and Obesity. Cold Spring Harb Perspect Biol. 2017;9(7):a028217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36(6):437–46. [PMC free article] [PubMed] [Google Scholar]
- Marshall JD, Maffei P, Collin GB, Naggert JK. Alstrom Syndrome: Genetics and Clinical Overview. Curr Genomics. 2011;12(3):225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion V, Mockel A, De Melo C, Obringer C, Claussmann A, Simon A, et al. BBS-Induced Ciliary Defect Enhances Adipogenesis, Causing Paradoxical Higher-Insulin Sensitivity, Glucose Usage, and Decreased Inflammatory Response. Cell Metab. 2012;16(3):363–77. [DOI] [PubMed] [Google Scholar]
- Jagger D, Collin G, Kelly J, Towers E, Nevill G, Longo-Guess C, et al. Alstrom Syndrome protein ALMS1 localizes to basal bodies of cochlear hair cells and regulates cilium-dependent planar cell polarity. Hum Mol Genet. 2011;20(3):466–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearn T, Spalluto C, Phillips VJ, Renforth GL, Copin N, Hanley NA, et al. Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes. Diabetes. 2005;54(5):1581–7. [DOI] [PubMed] [Google Scholar]
- Li G, Vega R, Nelms K, Gekakis N, Goodnow C, McNamara P, et al. A role for Alstrom syndrome protein, Alms1, in kidney ciliogenesis and cellular quiescence. PLoS Genet. 2007;3(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsov T, Silva DG, O’Bryan MK, Sainsbury A, Lee NJ, Kennedy C, et al. Fat Aussie - A new Alstrom syndrome mouse showing a critical role for ALMS1 in obesity, diabetes, and spermatogenesis. Mol Endocrinol. 2006;20(7):1610–22. [DOI] [PubMed] [Google Scholar]
- Collin GB, Cyr E, Bronson R, Marshall JD, Gifford EJ, Hicks W, et al. Alms1-disrupted mice recapitulate human Alstrom syndrome. Hum Mol Genet. 2005;14(16):2323–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodh S, Hostelley TL, Leitch CC, O’Hare EA, Zaghloul NA. Differential effects on beta-cell mass by disruption of Bardet-Biedl syndrome or Alstrom syndrome genes. Hum Mol Genet. 2016;25(1):57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesmith JE, Hostelley TL, Leitch CC, Matern MS, Sethna S, McFarland R, et al. Genomic knockout of alms1 in zebrafish recapitulates Alström syndrome and provides insight into metabolic phenotypes. Human Mol Genet. 2019;28:13:2212-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison LA, Chen W. Nutrient excess stimulates beta-cell neogenesis in zebrafish. Diabetes. 2012;61(10):2517–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorens B, Sarkar HK, Kaback HR, Lodish HF. Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell. 1988;55(2):281–90. [DOI] [PubMed] [Google Scholar]
- Lee BH, Liu J, Wong D, Srinivasan S, Ashrafi K. Hyperactive Neuroendocrine Secretion Causes Size, Feeding, and Metabolic Defects of C. elegans Bardet-Biedl Syndrome Mutants. PLoS Biol. 2011;9(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heon E, Kim G, Qin S, Garrison JE, Tavares E, Vincent A, et al. Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum Mol Genet. 2016;25(11):2283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya S, Nampoothiri S, Sen P, Sripriya S. Bardet-Biedl syndrome: Genetics, molecular pathophysiology, and disease management. Indian J Ophthalmol. 2016;64(9):620–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klink BU, Zent E, Juneja P, Kuhlee A, Raunser S, Wittinghofer A. A recombinant BBSome core complex and how it interacts with ciliary cargo. eLife. 2017;•••:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129(6):1201–13. [DOI] [PubMed] [Google Scholar]
- Gerdes JM, Christou-Savina S, Xiong Y, Moede T, Moruzzi N, Karlsson-Edlund P, et al. Ciliary dysfunction impairs beta-cell insulin secretion and promotes development of type 2 diabetes in rodents. Nat Commun. 2014;•••:5. [DOI] [PubMed] [Google Scholar]
- Hernandez HV, Jenkins D. Advances in the understanding of the BBSome complex structure and function. Dovepress. 2015;6:191–201. [Google Scholar]
- Seo S, Baye LM, Schulz NP, Beck JS, Zhang Q, Slusarski DC, et al. BBS6, BBS10, and BBS12 form a complex with CCT/TRiC family chaperonins and mediate BBSome assembly. Proc Natl Acad Sci USA. 2010;107(4):1488–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Yu D, Seo S, Stone EM, Sheffield VC. Intrinsic protein-protein interaction-mediated and chaperonin-assisted sequential assembly of stable bardet-biedl syndrome protein complex, the BBSome. J Biol Chem. 2012;287(24):20625–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield JL, Lechtreck KF, Lorentzen E. Trafficking of ciliary membrane proteins by the intraflagellar transport/BBSome machinery. Essays Biochem. 2018;62(6):753–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiIorio P, Rittenhouse AR, Bortell R, Jurczyk A. Role of cilia in normal pancreas function and in diseased states. Birth Defects Res C Embryo Today. 2014;102(2):126–38. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Kishimoto T, Nemoto T, Kadowaki T, Kasai H. Fusion pore dynamics and insulin granule exocytosis in the pancreatic islet. Science. 2002;297(5585):1349–52. [DOI] [PubMed] [Google Scholar]
- Granot Z, Swisa A, Magenheim J, Stolovich-Rain M, Fujimoto W, Manduchi E, et al. LKB1 regulates pancreatic beta cell size, polarity, and function. Cell Metab. 2009;10(4):296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hezel AF, Gurumurthy S, Granot Z, Swisa A, Chu GC, Bailey G, et al. Pancreatic LKB1 deletion leads to acinar polarity defects and cystic neoplasms. Mol Cell Biol. 2008;28(7):2414–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezratty Ellen J, Stokes N, Chai S, Shah Alok S, Williams Scott E, Fuchs E. A Role for the Primary Cilium in Notch Signaling and Epidermal Differentiation during Skin. Dev Cell. 2011;145(7):1129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayeh MK, Yen HJ, Beck JS, Searby CC, Westfall TA, Griesbach H, et al. Genetic interaction between Bardet-Biedl syndrome genes and implications for limb patterning. Hum Mol Genet. 2008;17(13):1956–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin JL, Di Franco M, Eichers E, May-Simera H, Garcia M, Yan J, et al. Inhibition of neural crest migration underlies craniofacial dysmorphology and Hirschsprung’s disease in Bardet-Biedl syndrome. Proc Natl Acad Sci USA. 2008;105(18):6714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiens CJ, Tong Y, Esmail MA, Oh E, Gerdes JM, Wang J, et al. Bardet-Biedl Syndrome-associated Small GTPase ARL6 (BBS3) Functions at or near the Ciliary Gate and Modulates Wnt Signaling. J Biol Chem. 2010;285(21):16218–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaghloul NA, Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest. 2009;119(3):428–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulato E, Favaretto F, Veronese C, Campanaro S, Marshall JD, Romano S, et al. ALMS1-Deficient Fibroblasts Over-Express Extra-Cellular Matrix Components, Display Cell Cycle Delay and Are Resistant to Apoptosis. PLoS One. 2011;6(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver-Krasinski JM, Stoffers DA. On the origin of the β cell. Genes Dev. 2008;22(15):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Hebrok M. Stem Cells to Pancreatic beta-Cells: New Sources for Diabetes Cell Therapy. Endocr Rev. 2009;30(3):214–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoya M. Preparation of pancreatic beta-cells from human iPS cells with small molecules. Islets. 2012;4(3):249–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoya M, Kunisada Y, Kurisaki A, Asashima M. Induction of differentiation of undifferentiated cells into pancreatic beta-cells in vertebrates. Int J Dev Biol. 2012;56(5):313–23. [DOI] [PubMed] [Google Scholar]
- Novas R, Cardenas-Rodriguez M, Irigoin F, Badano JL. Bardet-Biedl syndrome: is it only cilia dysfunction? FEBS Lett. 2015;589(22):3479–91. [DOI] [PubMed] [Google Scholar]
- Cano DA, Murcia NS, Pazour GJ, Hebrok M. orpk mouse model of polycystic kidney disease reveals essential role of primary cilia in pancreatic tissue organization. Development. 2004;131(14):3457–67. [DOI] [PubMed] [Google Scholar]
- Kathem SH, Mohieldin AM, Nauli SM. The Roles of Primary cilia in Polycystic Kidney Disease. AIMS Mol Sci. 2014;1(1):27–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: disease mechanisms of a ciliopathy. J Am Soc Nephrol. 2009;20(1):23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker AR, Thomas R, Dawe HR. Meckel-Gruber syndrome and the role of primary cilia in kidney, skeleton, and central nervous system development. Organogenesis. 2014;10(1):96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming LR, Doherty DA, Parisi MA, Glass IA, Bryant J, Fischer R, et al. Prospective Evaluation of Kidney Disease in Joubert Syndrome. Clin J Am Soc Nephrol. 2017;12(12):1962–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur A, Dhir SK, Goyal G, Mittal N, Goyal RK. Senior Loken Syndrome. J Clin Diagn Res. 2016;10(11):Sd03–sd4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco B, Thauvin-Robinet C. Update on oral-facial-digital syndromes (OFDS). Cilia. 2016;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran N, Moore AT, Weleber RG, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. 2017;101(9):1147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidts M, Hou Y, Cortes CR, Mans DA, Huber C, Boldt K, et al. TCTEX1D2 mutations underlie Jeune asphyxiating thoracic dystrophy with impaired retrograde intraflagellar transport. Nat Commun. 2015;6:7074. [DOI] [PMC free article] [PubMed] [Google Scholar]