

Abstract
GPNMB is a glycoprotein observed upon tissue damage and inflammation and is associated with astrocytes, microglia, and macrophages. Gene variations in GPNMB are linked with Parkinson’s disease (PD) risk, and changes in protein levels of GPNMB have been found in lysosomal storage disorders, including Gaucher’s disease with glucocerebrosidase (GCase) deficiency. In the current study, GPNMB increases were seen in the substantia nigra (SN) of PD patients compared to age-matched controls. Such PD patients have a decrease in GCase activity and corresponding elevation of glycosphingolipids in the SN (Rocha et al., 2015a). Interestingly, transgenic mice modeling synucleinopathy did not show GPNMB elevations or altered GCase activity levels compared to wild-type mice. However, upon CBE-induced GCase lysosomal dysfunction with elevated glycosphingolipids in wild-type mice, there were similar changes in GPNMB levels in the brain as seen in PD patient brains. These results indicate that GPNMB levels do not depend on alpha-synuclein load per se but relate directly to the lipidopathy changes induced by CBE-mediated GCase inhibition. The experimental modeling of elevating glycolipids resulted in GPNMB elevations with glial activation in several brain regions in mice. This is the first demonstration of region-specific elevations of GPNMB protein in Parkinson’s disease. The presence of GPNMB in PD patient substantia nigra, the induction of GPNMB after experimental glycosphingolipid increases, but not with pure alpha-synucleinopathy, point towards the potential for primary lipid-induced degeneration in PD.
Keywords: Parkinson’s disease, Lysosome, GPNMB, Glucocerebrosidase, Lipidopathy, Post-mortem
Introduction
Recently a large genome-wide association study of Parkinson’s disease (PD) patients identified several novel genetic variants linked to the function of the autophagic-lysosomal pathway, with polymorphisms in the GPNMB gene found to associate with idiopathic PD (Kumaran and Cookson, 2015). Studies have identified transcriptional changes in GPNMB in rodent models of both PD and Alzheimer’s disease (Kanaan et al., 2015; Srinivasan et al., 2016), and a recent report based on data mining techniques identified elevations of GPNMB mRNA in the substantia nigra of PD patients (Neal et al., 2018). Furthermore, GPNMB protein levels are elevated and correlate with disease severity and progression in lysosomal storage disorders (Zigdon et al., 2015; Kramer et al., 2016; Murugesan et al., 2018).
The relationship between lysosomal storage disorders (LSDs) and Parkinson’s disease (PD) is becoming apparent through evidence of glycosphingolipid dysregulation and lysosomal dysfunction in the pathophysiology of PD. In fact, many of the genetic factors responsible for familial PD play a role in the autophagy-lysosomal pathway (Gan-Or et al., 2015). Haplo-insufficiency due to mutations in the lysosomal hydrolase glucocerebrosidase (GCase) is one of the largest genetic risk factors for the development of PD (Sidransky et al., 2009; Sidransky and Lopez, 2012). Studies have demonstrated that GCase activity is decreased in the substantia nigra (SN) of sporadic PD patients (Gegg et al., 2012; Rocha et al., 2015a) as well as in normal aging (Rocha et al., 2015a), and has been associated with driving alpha-synuclein pathology (Mazzulli et al., 2011; Murphy et al., 2014). Aging is the largest risk factor for PD and cellular changes observed in normal aging may be mirrored in neurodegenerative disorders, albeit at an accelerated rate (Collier et al., 2011, 2017). Lysosomal dysfunction plays an important role in driving the aging phenotype, and is implicated in many age-related disorders (Carmona-Gutierrez et al., 2016). The majority of LSDs typically manifest in early infancy or childhood (Nixon et al., 2008), which may reflect how severe and complete lysosomal dysfunction leads to early onset of disease in children, whereas progressive lysosomal dysfunction with minor lipid changes, as observed in PD, manifests as pathogenic only when compounded with aging.
The neuroinflammatory system also plays an important role in neurodegenerative diseases (Deleidi and Isacson, 2012; Dzamko et al., 2015). GPNMB has been widely studied in terms of its pathogenic role in driving tumor progression via its ability to dampen the inflammatory response around cancerous growth (Maric et al., 2013) and is upregulated in the CNS in response to inflammatory stimuli (Ripoll et al., 2007; Huang et al., 2012). In terms of neurodegeneration, GPNMB has been associated with modulating the neuroinflammatory response associated with neurodegenerative diseases, with potential neuroprotective consequences (Tanaka et al., 2012; Kanaan et al., 2015; Nagahara et al., 2015; Srinivasan et al., 2016; Neal et al., 2018).
Understanding the dynamics of how glycolipid changes can drive the neuroinflammatory and neurodegenerative phenotype will be crucial in driving the development of novel therapeutics for neurodegenerative diseases. In the current study, we tested whether PD-associated changes in GPNMB are observed at the protein level, and importantly, are uniquely elevated in the substantia nigra of post-mortem PD patient brains that also display an age-dependent decrease in GCase activity and corresponding elevation in glycosphingolipids or after severe experimental alpha-synucleinopathy. We also investigated how pharmacological inhibition of glucocerebrosidase and subsequent lysosomal dysfunction in mice recapitulates brain-region specific increases in GPNMB in regions that also display robust glial activation, and whether changes in GPNMB are associated with alpha-synuclein load. We find that changes in GPNMB directly reflect pathological changes in lysosomal pathway regulation and glycolipid homeostasis in the context of Parkinson’s disease, and are not influenced (and may be occurring prior to) by alpha-synucleinopathy.
Materials and Methods
Patients
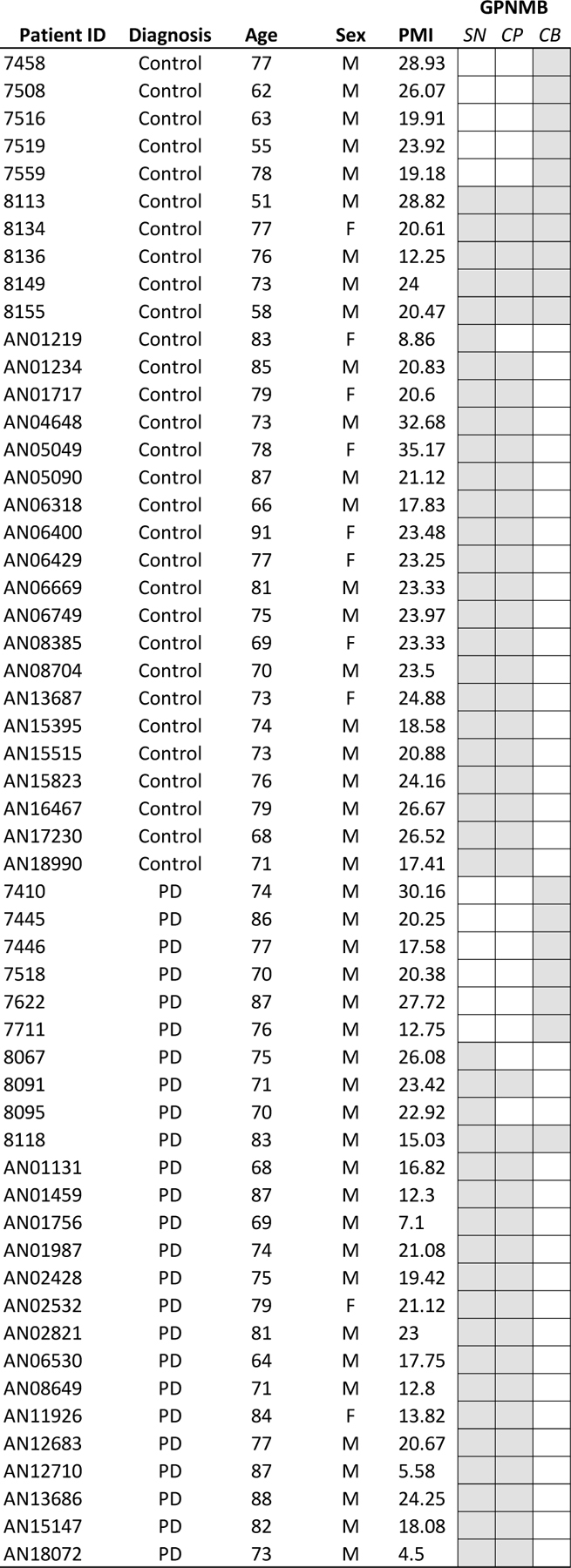
Frozen post-mortem brain tissue from neurologically unaffected patients (healthy control subjects, n=30) and pathologically-defined Parkinson’s disease patients (n=25, non-GBA mutant carriers) were obtained from the Harvard Brain Tissue Resource Center (HBTRC, McLean Hospital, Belmont, MA). The cohort of PD patient samples were previously sequenced to determine the absence of mutations in GBA1 (Rocha et al., 2015a). Tissue samples from the substantia nigra, caudate and putamen, and cerebellum were dissected (see Table 1 for patient information), and mechanically homogenized for 10 seconds in 400µL 10mM Tris buffer (KD-Medical, RGE-3340, pH 7.4) supplemented with protease inhibitors and 0.5mM EDTA (Thermo Fischer, 78430). Samples were subsequently centrifuged for 15 minutes at 4500xg (at 4°C) and the supernatant was transferred to a new tube and stored at −80°C until required. Total protein concentration in each supernatant sample was determined using the BCA protein quantification kit (Pierce, BCA protein assay kit, 23225).
Table 1:
Post-mortem brains from healthy subject controls and Parkinson’s disease patients used for GPNMB measurements. Patient’s age at death, sex, and post-mortem interval (PMI) are listed. Samples of substantia nigra (SN), caudate and putamen (CP), and cerebellum (CB) were dissected from fresh frozen brains, with grey boxes indicating which brain regions were obtained per patient.
 |
Paraffin-embedded tissue sections (6µm thick) containing substantia nigra from PD patients and age-matched healthy subjects were obtained from the NeuroBioBank at the HBTRC and processed for immunohistochemical analysis as outlined below.
Animals
Wild-type (WT) male mice (BDF1 strain, Charles River Laboratories), and mice modelling alpha-synucleinopathy (Thy1-aSYN; human WT alpha-synuclein under the promotion of the murine Thy1 promoter (Rockenstein et al., 2002; Chesselet et al., 2012)) were used in this experiment. Animals were housed (max 5 mice per cage) in standard conditions in a 12-h dark/12-h light cycle, with ad libitum access to food and water. Transgenic mice were maintained hemizygous by mating Thy1-aSYN females with BDF1 WT males, and transgene presence was determined via PCR analysis of genomic tail DNA using the following primers (ASO sense: 5’- GAC GGG TGT GAC AGC AGT AGC C −3’; ASO anti: 5’- GAT GAT GGC ATG CAG CAC TGG −3’, internal control THD 15: 5’- CAA ATG TTG CTT GTC TGG TG −3’; internal control THD 16: 5’- GTC AGT CGA GTG CAC AGT TT −3’). All animal procedures were performed in accordance with National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee (IACUC) at McLean Hospital, Harvard Medical School. Sample sizes for each experimental group are indicated below, and animals were randomly assigned to treatment and material harvest groups on day 1 of the experiment.
Pharmacological inhibition of lysosomal glucocerebrosidase
Mice aged 2–3 months, received a daily intraperitoneal (i.p.) injection of 100mg/kg conduritol-beta-epoxide (CBE; Millipore EMD, Cat nr. 234599) or vehicle (0.1% DMSO) for 28 days (between 9:30AM – 11AM every day). Animals were weighed daily to determine appropriate injection volume, and to monitor animals for treatment-induced weight loss. Early removal of mice based on loss of >30% of heaviest weight was set as euthanasia criteria, but no mice reached this cut-off point during the experiment. Mice were euthanized with sodium pentobarbital and subsequently transcardially perfused with 0.9% heparinized saline (for fresh dissection) or followed by 4% PFA (for fixed brain harvest) 24 hours after the final administration of DMSO or CBE.
Following perfusion with 0.9% heparinized saline, mouse brains (n=5–6 animals/treatment) were chopped into 750µm thick coronal sections using a McIlwain tissue chopper. The substantia nigra, hippocampus, striatum, a segment of the motor cortex and a portion of cerebellum were harvested, and flash frozen until required. Tissue was mechanically homogenized for 10 seconds in 100µL 10mM Tris buffer (KD-Medical, RGE-3340, pH 7.4) prepared in EmbryoMax Ultrapure water, supplemented with protease inhibitors and 0.5mM EDTA (Thermo Fischer, 78430). Samples were subsequently centrifuged for 15 minutes at 4500xg (at 4°C) and the supernatant was transferred to a new tube. Total protein concentration in each supernatant sample was determined using the BCA protein quantification kit (Pierce, BCA protein assay kit, 23225). Supernatants were stored at −80°C until required. Following perfusion with 4% PFA, mouse brains (n=5–6 animals/treatment) were isolated and post-fixed overnight. Following equilibration in 30% sucrose, free-floating serial coronal sections (40µm thick), stored in anti-freeze (30% glycerol [Sigma, G9012], 30% ethoxyethanol [Fischer, AC156020025] in PBS) at −30°C until required. Sections were processed for fluorescent immunostaining as outlined below.
Glucocerebrosidase activity and glycosphingolipid measurements
Glucocerebrosidase (GCase) activity was measured in mouse brain region homogenates diluted in GCase-activity sample diluent, as previously described (Rocha et al., 2015a; 2015b). Measurements of glycosphingolipids in mouse brain tissue were performed using a liquid chromatography / tandem mass spectrophotometry approach as described in (Rocha et al., 2015b; Hallett et al., 2018).
ELISA-based GPNMB measurement
For each sample, 25µg (human brain homogenates) or 20µg (mouse brain homogenates) total protein was loaded onto a sandwich-based enzyme-linked immunosorbent assay (ELISA) plate, coated with anti-GPNMB antibodies, and the ELISA was performed according to the manufacturer’s protocol (RayBiotech, Human Osteoactivin ELISA kit, ELH-Osteoactivin-1; or Mouse Osteoactivin ELISA kit, ELM-Osteoactivin-1). Absorbance values were read at 450nm SPECTRAmax plate reader (Molecular Devices), and GPNMB protein levels (pg/mL) from triplicate measurements were interpolated from a Human or Mouse Osteoactivin standard curve (as prepared according to kit manufacturer’s protocol).
Immunohistochemical staining
GPNMB DAB staining on human SN sections
Paraffin-embedded human SN sections from healthy subjects and Parkinson’s disease patients were de-paraffinized with xylene and rehydrated through a series of decreasing ethanol concentration solutions. Neuromelanin pigment was bleached by incubating slides in pre-warmed (55°C) 3% H2O2 in phosphate buffer (0.05M, pH 7.4) for 2 hours (Momose et al., 2011) so that this pigment would not interfere with the dark DAB-pigment generated by the immunostaining. Slides containing sections were subsequently washed in deionized H2O, followed by PBS, and then blocked with a 1-hour incubation at room temperature (RT) in 10% donkey serum (Jackson Immunoresearch, cat. nr. 017–000-121) in PBST (0.3% Triton X, Tx). The primary antibody (goat anti-human GPNMB polyclonal; 1:200, R&D systems, AF2550) was diluted in 10% donkey serum in PBS and applied to sections for an initial overnight incubation at RT, followed by 24 hours at 4°C. Slides were washed with PBS, and then incubated with a solution of biotinylated donkey anti-goat Ig (Biotin-SP AffiniPure IgG (H+L); 1:400, Jackson Immunoresearch, cat. nr. 705–065-003) in 10% donkey serum in PBS for 1hr at RT. Vectastain Elite ABC-peroxidase solution (Vector Laboratories, PK-6100) was then applied to the sections for 1hr at RT. The DAB Peroxidase Substrate kit with nickel chloride (Vector Laboratories, SK-4100) was used to produce a black precipitate identifying GPNMB-positive immunoreactivity. Slides were subsequently counterstained with 0.5% cresyl violet, differentiated with acetic acid, and then dehydrated with increasing concentrations of ethanol, cleared with xylene, and coverslipped with DPX (Electron Microscopy Sciences, #13512).
Fluorescent immunostaining for GPNMB and cell-specific markers on mouse brain sections
Briefly, free-floating sections were washed in PBS to remove anti-freeze, and then incubated in citrate-based antigen-retrieval solution (DAKO 1X, pH 6.1, S1699) for 20 minutes at 37°C. Following a couple of washes in PBS, sections were incubated with 10% donkey serum in PBST (0.3% Tx) for 1 hour at RT, before applying combinations of the following primary antibodies (diluted in 10% donkey serum in PBS) for overnight incubation at 4oC: goat anti-mouse GPNMB polyclonal (1:200, R&D systems, AF2330), rabbit anti-von Willebrand (1:1000, Sigma, F3520), mouse anti-NeuN (1:500, Millipore, MAB377), rabbit anti-TH (1:1000, Pel-Freez Biologicals, P40141), mouse anti-TH monoclonal (1:1000, Millipore, MAB318, clone LNC1), rabbit anti-GFAP (1:500, Abcam, ab7260), rabbit anti-Iba1 (1:200, Wako). The following day, fluorescently conjugated secondary antibodies were applied for 1 hour at RT in 10% donkey serum in PBS (all at 1:500, Invitrogen Alexa Fluor), followed by a 5-minute incubation with Hoechst (PromoKine, PK-CA707–40047). Sections were subsequently mounted onto Superfrost slides (Thermo Fischer) and coverslipped with Mowiol (Sigma, Cat nr. 81381) prior to imaging with the Keyence microscope system (40x objective, tiled images). High magnification images (63x objective) were obtained using the Leica TCS-SP8 Confocal microscope.
Statistical analysis
Data was tabulated in Microsoft Excel (2016), and statistical analysis was performed using GraphPad Prism v7.01. Outliers were identified using the GraphPad Prism-recommended ROUT method (Q set at 1%). One outlier was identified in the caudate-putamen cohort of healthy subjects and removed from the data set. All analyses were performed on the cleaned data set. A two-way ANOVA, with Sidak’s multiple comparison correction test when appropriate, was used to measure differences in GPNMB levels across brain regions between healthy subjects and sporadic PD patients, between mouse brain regions upon vehicle or CBE treatment, or between WT and Thy1-aSYN mouse brain regions. Tests reached significance when p<0.05.
Results
GPNMB levels are elevated in the substantia nigra of sporadic PD patients.
Neuropathological changes in the substantia nigra (SN) are a hallmark feature of Parkinson’s disease, therefore we asked whether GPNMB protein levels are altered in this region of the brain of sporadic PD patients. Quantitative enzyme-linked immunosorbent assay (ELISA) revealed that GPNMB protein levels in the substantia nigra of sporadic PD patients were highly elevated compared to healthy subjects (Figure 1A; two-way ANOVA, brain region F(1,97) = 26.64, p<0.0001; with Sidak’s multiple comparison test, p<0.0001). Interestingly, the levels of GPNMB in the cerebellum (CB) and caudate and putamen (CP) of sPD patients were not altered compared to healthy subjects, indicating that GPNMB is selectively elevated in the SN, and this is significantly influenced by disease state (Figure 1A; two-way ANOVA, interaction F(2,97) = 3.943, p=0.0226). Given that increasing age is a major risk factor for the development of PD (Collier et al., 2011), we sought to identify if GPNMB expression in the SN is associated with age in PD patients. Although not significant, in both healthy subject and PD patient SN samples, GPNMB displays a trend for increased expression with age (Supplementary Figure 1, panels C and G). Indeed, when GPNMB levels in the SN of healthy subject controls and sPD patients are grouped by decade of age at death, a significant increase in GPNMB is found in the SN of sPD patients who died in their 7th decade compared to age-matched healthy subjects (two-way ANOVA, disease status F(1,39) = 8.954, p=0.0048; with Sidak’s multiple comparison test, p=0.024, data not shown). GPNMB immunoreactivity was observed within the SN region of healthy and sPD patient brains, with higher levels observed in the SN of sPD patients (Figure 1B). Large, dense puncta were more common in the SN of sPD patients, while GPNMB-positive puncta in healthy subjects were generally smaller, and fainter, providing a visual confirmation of the ELISA-based elevations of GPNMB measured in the SN of sPD patients.
Figure 1: GPNMB is uniquely elevated in the substantia nigra of sporadic PD patients.

(A) Tissue homogenates from post-mortem cerebellum (CB) caudate and putamen (CP) and substantia nigra (SN) from healthy subject controls (n=31) and sporadic PD patients (n=25) were prepared. Levels of GPNMB in these brain regions were determined by ELISA. Results are means ±SEM, **** p <0.0001 (two-way ANOVA, with Sidak’s multiple comparison post-hoc test). (B) DAB-based immunostaining revealed that GPNMB-positive puncta exist throughout the human SN (white arrowheads) but are larger and more numerous in sporadic PD patients compared to healthy subjects. Additionally, some GPNMB-positive immunoreactivity produced a cell-like staining pattern (open arrowheads) in both healthy subjects and sporadic PD patients. Scale bar is 50µm.
To identify whether GPNMB levels are associated with disease severity and other clinicopathological measures, we obtained the neuropathologic reports of the sPD patient cohort. There was no correlation between age of onset or duration of disease with the GPNMB levels measured in the SN (Supplementary Figure 2). In this cohort of sPD patients, GPNMB levels in the SN did not correlate with pathological features such as neuronal loss (Supplementary Figure 3, panels A and E), or extra-neuronal pigment (Supplementary Figure 3, panels B and E). GPNMB levels in the SN were also not correlated with disease severity based on the Braak/Del Tredici score (Supplementary Figure 3, panels C and E) or the extent of neurofibrillary tangles observed with the Braak/Braak score (Supplementary Figure 3, panels D and E).
Brain region specific increase in GPNMB following complete pharmacological inhibition of glucocerebrosidase activity in WT mice.
Increased GPNMB levels following pharmacological inhibition of glucocerebrosidase (GCase) has previously been observed in whole brain homogenates from CBE-treated mice (Zigdon et al., 2015; Vardi et al., 2016). To identify whether brain region-specific changes in GPNMB, as observed in human PD brains, can occur upon pharmacological induction of lipidopathy, 2–3-month-old male WT mice were injected with CBE (via i.p. injection) for 28 days.
Pharmacological intervention with CBE was observed to completely inhibit GCase activity in all brain regions measured (Figure 2A; two-way ANOVA, treatment F(1,44)=372.4, p<0.0001; with Sidak’s multiple comparison test, p<0.0001). Interestingly, GPNMB levels were elevated in a brain-region specific manner in the motor cortex (MCtx), hippocampus (HP), and substantia nigra (SN) of CBE-treated WT mice compared to vehicle-treated WT mice (Figure 2B; two-way ANOVA, brain region F(4,484)=105.7, p<0.0001; with Sidak’s multiple comparison test, MCtx p<0.0001, HP p=0.0014, SN p<0.0001), and although GCase activity was significantly decreased in the striatum and cerebellum, there was no concurrent increase in GPNMB levels in these brain regions in CBE-injected mice compared to vehicle-treated mice (Figure 2B). It is important to note that same-paradigm pharmacological inhibition of GCase activity (Vardi et al., 2016) or transgenic abolishment of GCase activity in the GBA-D409V homozygote mouse (Sardi et al., 2011) leads to accumulation of GCase glycosphingolipid substrates in brain regions displaying diminished GCase activity. These findings indicate that specific brain-regions respond differently to pharmacologically-induced lysosomal stress, which may be influencing the differential vulnerability of neuronal populations to neurodegeneration as posited by the threshold theory (Engelender and Isacson, 2016).
Figure 2: GPNMB is elevated in mouse brain following complete pharmacological inhibition of glucocerebrosidase activity.

Tissue homogenates of freshly dissected striatum (STR), cerebellum (CB), motor cortex (MCtx), hippocampus (HP) and substantia nigra (SN) from mice receiving daily (for 28 days) i.p. injections of DMSO (0.1%, n=4–6) or CBE (100mg/kg, n=3–6) were prepared. (A) Glucocerebrosidase activity levels were determined using the artificial substrate 4-methylumbelliferyl-β-D-glucopyranoside (4-MU-Glc). GCase activity levels were significantly diminished in all brain regions in CBE-treated mice. (B) Levels of GPNMB across these brain regions were determined by ELISA, with significantly elevated GPNMB measured in the MCtx, HP, and SN of CBE-treated mice. GPNMB levels in the STR and CB were not affected by CBE-treatment. For panels A and B, results are means ±SEM, ** p <0.01, **** p <0.0001 (two-way ANOVA, with Sidak’s multiple comparison post-hoc test).
To determine the influence of alpha-synucleinopathy versus lipidopathy in driving brain-region specific elevations, GPNMB levels were measured in the brains of Thy1-aSYN mice. Mice modelling synucleinopathy (Chesselet et al., 2012; Hallett et al., 2012) do not display pathophysiological changes in GCase activity at a whole brain level (Figure 3A; unpaired Student’s T-test, two-tailed, not significant (ns)) or in a brain-region specific manner (Supplementary Figure 4). Substrate accumulation of glycosphingolipids (Figure 3B; two-way ANOVA, ns) is also not observed in Thy1-aSYN mice compared to age-matched WT mice. Importantly, Thy1-aSYN mice did not show differences in GPNMB levels across various brain regions, indicating that aSYN load per se is not a trigger for elevating GPNMB (Figure 3C; two-way ANOVA, ns). GPNMB immunofluorescence in the SN of Thy1-aSYN and WT mice visually corroborated the similar ELISA-based levels of GPNMB protein measured in Thy1-aSYN mice compared to WT mice (Supplementary Figure 5).
Figure 3: GPNMB levels in brain regions of mice modelling alpha-synucleinopathy are not different compared to age-matched WT mice.

Whole brain homogenates from age-matched wild-type (WT; n=6) or Thy1 alpha-synuclein overexpressing (Thy1-aSYN; n=6) mice display similar levels of (A) glucocerebrosidase (GCase) activity and (B) glycosphingolipids such as glucosylceramide (GlcCer) and glucosphingosine (GluSph). Results are mean ±SEM. (C) GPNMB levels in striatum (STR), motor cortex (MCtx), and substantia nigra (SN) motor cortex homogenates from age-matched WT (n=3–6) and Thy1-aSYN mice (n=6–9) were measured by ELISA and show that transgenic animals display similar GPNMB levels to WT mice in these brain regions. Results are mean normalized to corresponding WT brain region ±SEM. No significant differences were observed with statistical tests (Panel A: unpaired two-tailed Students T-test; Panels B and C: two-way ANOVA).
GPNMB expression has been observed in a variety of cells in response to inflammatory triggers (Ripoll et al., 2007; Huang et al., 2012), and is elevated in both peripheral and central glial cells in lysosomal storage disorders (Marques et al., 2016). To identify the potential cellular source leading to elevated GPNMB following GCase inhibition in the CBE model of lipidopathy, fluorescent immunostainings for GPNMB and various cell-specific markers (NeuN, vWF, GFAP, and Iba1) were performed on brain sections collected from CBE-treated and vehicle-treated WT mice. GPNMB immunoreactivity is elevated in the mouse ventral cortex, motor cortex, hippocampus and substantia nigra following CBE treatment (Figure 4A), visually corroborating ELISA-based measurements which show that GPNMB is significantly elevated in the motor cortex, hippocampus, and substantia nigra after GCase inhibition in mice (Figure 2B). GPNMB immunoreactivity is not observed in NeuN-positive cells, or with blood vessels (vWF) in cortical regions, hippocampus or substantia nigra (Figure 4A). Additionally, GPNMB does not colocalize with TH-positive neurons in the substantia nigra (Figure 4A, panels p - s). An increase in GPNMB-positive staining is found in regions which display elevated glial activation upon CBE-treatment (Figure 4A) and upon high magnification observation, GPNMB immunopositivity is found in and around some astrocytes (GFAP), and microglia (Iba1) (Figure 4B).
Figure 4: GPNMB is associated with glial cells in mouse brain following pharmacological inhibition of glucocerebrosidase activity.

(A) Fluorescent immunostaining in ventral cortex (a–e), motor cortex (f–j), and hippocampus (k–o) for GPNMB (green), DAPI (blue), and various cell-specific markers in red (NeuN, vWF, Iba1 and GFAP) illustrate that GPNMB immunoreactivity is elevated following CBE-treatment (b–e, g–j, l–o) compared to vehicle-treatment (a, f, k). A triple immunostaining was performed to visualize TH-positive cells (blue), GPNMB (green) and cell-specific markers (red): NeuN (p), vWF (q) GFAP (r), and Iba1 (s) in the substantia nigra (p–s), which also depicts that GPNMB immunoreactivity is increased upon CBE-treatment (p – s) compared to vehicle treatment (inset in p). (B) High magnification confocal microscopy images illustrate the variety of staining patterns obtained for GPNMB (green) in and around astrocytes (i; GFAP, red) or microglia (ii; Iba1, red). GPNMB immunoreactivity is found to be associated with GFAP-positive processes (closed arrow in top panel) or surrounded by Iba1-positive processes (open arrows in bottom panel). GPNMB-positive cell-like structures that are not GFAP-positive are also observed (open arrowheads). GFAP-positive astrocytes, or Iba1-positive microglia that are negative for GPNMB are also present (closed arrowheads). Scale bars in panel A = 100µm. Scale bars in panel B = 10µm.
Discussion
This investigation provides the first molecular evidence for GPNMB protein changes in human Parkinson’s disease brain. The experiments showed that GPNMB protein levels are specifically elevated in the substantia nigra (SN) of sporadic PD patient brains, and, using a pharmacological approach that models lipidopathy in mice, we identified that changes in GPNMB are not influenced by the enhanced presence of aSYN aggregates, but are associated with the lipid dysregulation that occurs in PD-relevant brain regions. We have previously shown that the SN of post-mortem sporadic PD patient brains mirrors the characteristic molecular pathology observed in Gaucher’s disease: the activity of the lysosomal hydrolase glucocerebrosidase (GCase) is diminished in the SN and this is coincides with an accumulation of glycosphingolipids (Rocha et al., 2015a). A meta-data analysis of several large genome-wide association studies identified that polymorphisms in the GPNMB gene represent one of the major risk factors for the development of idiopathic PD (Kumaran and Cookson, 2015), with elevated transcripts of GPNMB found in PD patient brain (Murthy et al., 2017; Neal et al., 2018), as well as altered DNA methylation patterns in the GPNMB gene (Plagnol et al., 2011). The functional implications with regards its role in PD have yet to be fully elucidated, but evidence exists linking GPNMB to disease progression and severity in Gaucher’s disease (Zigdon et al., 2015; Kramer et al., 2016; Vardi et al., 2016; Murugesan et al., 2018) and Niemann Pick’s C disease (Marques et al., 2016), lipid storage disorders caused by lysosomal dysfunction. Furthermore, GPNMB may facilitate lysosomal function in that it was found to be necessary for phagosome fusion to the lysosome, leading to efficient macroautophagy in the context of tissue repair (Li et al., 2010). Thus, GPNMB, in the context of lysosomal dysfunction in PD may be functioning to enhance protein degradation systems in response to cellular stressors such as elevated glycolipids.
GPNMB serves as a marker for lipidopathy in context of Parkinson’s disease.
Many lines of evidence have demonstrated a bidirectional link between decreased GCase activity and increased aSYN accumulation in the context of Parkinson’s disease (Blanz and Saftig, 2016), and this molecular pathology is mirrored in the brains of Gaucher disease patients, who also display aggregates of aSYN reminiscent of Lewy bodies (LB) (Wong et al., 2004). However, mice modelling alpha-synucleinopathy (Thy1-aSYN) (Chesselet et al., 2012) do not show altered GCase activity levels compared to WT mice (Richter et al., 2014; Rockenstein et al., 2016), and in these Thy1-aSYN mouse brains we observed comparable GPNMB levels as detected in WT mouse brains. This implies that the presence of elevated aSYN does not drive alterations in GPNMB levels, whereas direct induction of lipidopathy using CBE-mediated inhibition of GCase activity leads to GPNMB elevations that mirror those seen in the SN of PD patients. Given that glycolipid profiles are altered in the brains of PD patients (Rocha et al., 2015a) it is plausible that changes in lipids is one driver of the pathophysiological elevations of GPNMB observed in the SN of PD patient brains.
Various studies using human material and mouse models have demonstrated that GPNMB is directly correlated with lipidopathy severity in Gaucher’s disease (Zigdon et al., 2015; Kramer et al., 2016; Murugesan et al., 2018). In vivo evidence shows that GPNMB mRNA levels correlate with glucosylceramide (GlcCer) and glucosphingosine (GluSph) levels in the CBE-model of lipidopathy (Vardi et al., 2016), as well as CSF levels of GPNMB correlating with GluSph in human Gaucher disease patients (Murugesan et al., 2018). Direct evidence that manipulation of lipid accumulation itself (and subsequent lysosomal lipid pressure) and not just lysosomal dysfunction leads to GPNMB expression has been demonstrated in vitro. Using the macrophage RAW264.7 cell line, Marques and colleagues demonstrated that cholesterol accumulation after U18666A treatment, mimicking the Niemann Pick C disease phenotype, resulted in elevation of GPNMB (Marques et al., 2016). Furthermore, prevention of GSL production via glucosylceramide synthase inhibition in obese adipose tissue macrophages lowered U18666A-mediated increase in GSLs and GPNMB (Gabriel et al., 2014). Additionally, GPNMB upregulation had been observed following in vitro induction of lysosomal stress and lipid accumulation by treating RAW264.7 macrophages with the fatty acid palmitate (Gabriel et al., 2014). Stress on the lysosomal system seems to be the driver for elevating GPNMB levels since increasing the intra-lysosomal pH in obese adipose tissue macrophages leads to GPNMB induction, whereas thapsigargin-induced ER stress does not alter GPNMB levels (Gabriel et al., 2014). Thus, changes in GPNMB levels are directly associated with lysosomal dysfunction in a variety of well-defined models and disease scenarios, supporting the idea that the GPNMB changes observed in PD-relevant brain regions are driven by glycolipid changes and lysosomal dysfunction.
The functional consequences of dysregulated lipids in the context of neurodegeneration is important when considering the impact of future therapeutic interventions, since the appearance of many of the conventional pathological hallmarks of Parkinson’s disease are becoming directly associated with lipid changes. Elevation of reactive oxygen species and mitochondrial dysfunction in neurons leads to elevated lipid production in neurons and subsequent accumulation of lipid droplets, a lipid storage organelle, in glial cells (Liu et al., 2015). Appropriate lipid transfer between these cells via lipid transporters such as apolipoproteins, is essential for maintaining metabolic integrity of the neuron. Under conditions of oxidative stress, if lipids are unable to be transported correctly from neurons to glial for lipid droplet formation, this results in the accumulation of lipids within neurons and leads to neurodegeneration (Liu et al., 2017). Importantly, MPTP-induce elevations of reactive oxygen species in mice led to elevations of GPNMB in astroglial cells in the nigrostriatal system (Neal et al., 2018), and as such changes in GPNMB may reflect the glial response to ROS-induced lipid droplet transfer dysfunction from neurons, and ultimate lipid dysregulation within neurons.
Studies in Gaucher disease cells demonstrated that glycolipid accumulation severely altered membrane fluidity and lipid raft structures resulting in impaired endocytosis and immune signaling (Batta et al., 2018). Thus, alteration of lipid homeostasis within a neuron could also lead to dire consequences in terms of neuronal membrane function. In fact, the phospholipid content of the lipid bilayer was shown to directly impact the ability of aSYN to aggregate, and supports the concept that lipid dysregulation is a driving force behind pathological accumulation of aSYN aggregates (Pfefferkorn et al., 2012; Taguchi et al., 2017; Lv et al., 2018). Given that aSYN plays an important physiological role at the synapse (Burré et al., 2018), lipid-induced changes of the availability of aSYN could impact synaptic function, and thus neuronal integrity. Furthermore, the presence of lipids and membranous lipid-dense structures within LB aggregates has recently been described, and provides physical evidence implicating lipids and lipid membrane fragments as building blocks of LB pathology in human PD (Shahmoradian et al., 2017). Neurons experiencing (oxidative) stress and subsequent dysregulation of lipid bilayer homeostasis may try to contain the potentially damaging effects of dysregulated lipids and lipid-membranes, along with lipid-bound protein aggregates, by pooling these molecules into Lewy bodies, which may serve as a benefit for cellular health at first, but over time results in the loss of cellular integrity under continued stress. The current paper, and several emerging publications in this field (Shahmoradian et al., 2017; Taguchi et al., 2017; Kim et al., 2018), are pointing out the interactions that occur in the lipid-alpha synuclein biochemistry. Indeed, as we and others are finding, the LB may, after the re-examination with more current data, be considered a lipid-inclusion, with large amounts of aSYN attached as fibrils and other forms along with undigested organelles (Shahmoradian et al., 2017). These observations suggest a more coherent process, as far as pathophysiology is concerned, in which many mutations involved in the PD spectrum disorders precipitate dysfunctions that tend to, with age, lead to accumulation or aggregation of aSYN.
Differential response of vulnerable neurons to elevation of glycolipids.
Systemic inhibition of lysosomal glucocerebrosidase using conduritol-beta epoxide (CBE) has previously been shown to recapitulate specific PD-relevant pathologies, including accumulation aSYN aggregates, elevation of glycosphingolipids, and widespread neuroinflammation (Rocha et al., 2015b). We report here that systemic CBE-mediated GCase inhibition results in brain region-specific elevations of GPNMB, corroborating whole-brain measurements from other groups (Zigdon et al., 2015; Vardi et al., 2016). GPNMB is elevated in the substantia nigra of mice modelling lipidopathy, which is analogous to the SN-specific elevations of GPNMB observed in human PD patients that also display GCase deficiencies in the SN.
Neuronal populations throughout the brain are known to exhibit different vulnerability profiles in response to various stressors. Intra-nigral injection of the TLR3 agonist Poly(I:C), a synthetic dsRNA, which causes viral-like inflammation, and of the TLR4 agonist, LPS (inducing bacterial-like inflammation), induces a prominent upregulation of inflammatory cytokines and a long-lasting inflammatory reaction, and triggers neuronal changes observed in early neurodegeneration and predisposes midbrain dopamine neurons to be more vulnerable to subsequent oxidative stress (Koprich et al., 2008; Deleidi et al., 2010). Complex 1 inhibitors such as MPTP (Brownell et al., 1998) or rotenone (Sherer et al., 2003) also drive the specific degeneration of nigrostriatal dopaminergic neurons. On the other hand, induction of mitochondrial dysfunction via complex 2 inhibition in aged rats is specifically toxic to striatal neurons (Bossi et al., 1993). The threshold theory (Engelender and Isacson, 2016) encompasses the interplay between cell type, stressor level, region specificity, and temporal aspect to describe how a neuronal population would respond to a neurotoxic insult. However, the intensity of a stressor could also drive a differential response within the same neuronal population: in terms of pathophysiology, intense and rapid elevations of glycolipids, as found in lysosomal storage disorders, or following CBE-treatment, versus a gradual increase over time, as observed in PD brains, produce very different neuropathic responses and subsequent disease manifestations (Neudorfer et al., 1996; Platt, 2014; Aflaki et al., 2017).
GPNMB may function to modulate the inflammatory response in PD
The role of GPNMB in the context of PD remains obscure, but evidence that GPNMB has a role in inflammation may provide clues regarding the elevated GPNMB observed in PD-relevant brain regions. Inflammation plays an important role in the pathogenesis of PD (Deleidi and Isacson, 2012), and many of the genes implicated in the disease process have been shown to play a role in pathways involved in the regulation of immunity and inflammation (Dzamko et al., 2015). GPNMB is expressed in a variety of immune cells (macrophages and dendritic cells) (Ahn et al., 2002; Ripoll et al., 2007) and has been studied in many contexts, including cancer and inflammation (Huang et al., 2012; Maric et al., 2013). Activation of T-cells via interaction with antigen-presenting cells such macrophages and dendritic cells is an important step in mediating an active immune response. GPNMB was found to cause impairment of T-cell activation via interaction with the extracellular matrix protein syndecan-4 (Chung et al., 2007), and as such can act as a potent anti-inflammatory mediator. GPNMB can also exist as an extracellular protein through cleavage of the extracellular domain by ADAM10, and thus can be release as soluble protein into the extracellular matrix to have angiogenic properties (Rose et al., 2010) which could influence immune cell migration function. On the other hand, by interacting with syndecan-4 on tumor-reactive T-cells, GPNMB promotes melanoma growth (Tomihari et al., 2010). GPNMB has also been implicated in regulating microglial inflammation in response to LPS activation (Shi et al., 2014).
Mice modelling GD-relevant lipidopathy, either via pharmacological inhibition of GCase activity, or transgenic GBA1 knockout mouse, display similar neuroinflammatory changes in microglia and astrocytes (Farfel-Becker et al., 2011; Vardi et al., 2016). We observed that regions with high glial activation following pharmacological inhibition of GCase activity where those that displayed the greatest elevation of GPNMB. New evidence demonstrates that GPNMB can attenuate the astrocytic inflammatory response via interaction with CD44 in the MPTP mouse model of PD (Neal et al., 2018). Therefore, it is plausible that lipidopathy-driven elevations in GPNMB may be a cellular mechanism for attenuating neuroinflammation, in response to a variety of stress insults that directly (lysosomal dysfunction) or indirectly (ROS) affect lipid homeostasis.
Lipid changes leading to inflammation, with GPNMB reflecting cellular interactions.
We have previously demonstrated that pharmacological inhibition of GCase activity with subsequent lipid dysregulation leads to microglial activation (Rocha et al., 2015b). Lipid regulation and transport through tissue is also important for Alzheimer’s disease and several other neurodegenerative conditions. Interestingly, TREM2-mediated activation of APOE signaling can drive phagocytic microglia towards a chronic inflammatory phenotype, and this is dependent on TREM2-APOE transcriptional regulation of a number of targets, GPNMB included, in microglia (Krasemann et al., 2017). Furthermore, in the PS2APP mouse model of Alzheimer disease, transcript levels of GPNMB are elevated in the brain, and this elevation is driven by microglial-specific upregulation of the GPNMB transcript in aged PS2APP brains (Srinivasan et al., 2016). Thus, elevation of GPNMB potentially reflects neuronal-glial communication in diverse neurodegenerative disorders, in response to neuronal dysfunction induced by a variety of reasons be it lipid-induced, inflammatory, ischemia, or oxidative stress (Nakano et al., 2014; Murata et al., 2015; Vardi et al., 2016; Neal et al., 2018) and may serve to stabilize the microenvironment for the most optimal response to pathological cellular stressors.
Summary and conclusions.
Here we show region-specific elevation of GPNMB levels in Parkinson’s disease substantia nigra and in an experimentally-induced glycolipid disorder, but not in a severe model of alpha-synucleinopathy. An interesting interpretation of our data is that lipidopathy drives pathology in both human PD and the animal models and suggests that the alpha-synucleinopathy observed in PD pathology may be secondary to pathological lipid changes. Instead of the existence of a dichotomy for lipid- or alpha-synuclein pathology, our new data may support the idea of a convergence, and perhaps even a different sequence of events leading to alpha-synucleinopathy than previously considered. Furthermore, these findings illustrate that GPNMB may, as a biomarker, define cellular signals that, if used in concert with genetic or other biological risk factors (Schapira, 2013; Chahine et al., 2014; Kumaran and Cookson, 2015), can show ongoing early and late pathology relevant to Parkinson’s disease prevention and treatment.
Supplementary Material
Highlights.
GPNMB is uniquely upregulated in the SN of PD patients displaying lipidopathy changes.
Pharmacological induction of lipidopathy drives brain region-specific elevations of GPNMB in mice.
Mice modeling alpha-synucleinopathy do not display disease-relevant changes in GPNMB.
Acknowledgements
This research was supported by the Harold and Ronna Cooper Family, the Consolidated Anti-Aging Foundation, the Orchard Foundation, the Poul Hansen family, NIH/NINDSR01 NS092667, and by NIH/NIA R01 AG060195.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aflaki E, Westbroek W, Sidransky E. The Complicated Relationship between Gaucher Disease and Parkinsonism: Insights from a Rare Disease. Neuron 2017; 93: 737–746.Available from: http://www.ncbi.nlm.nih.gov/pubmed/28231462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn JH, Lee Y, Jeon C, Lee S-JJ, Lee B-HH, Choi KD, et al. Identification of the genes differentially expressed in human dendritic cell subsets by cDNA subtraction and microarray analysis. Blood 2002; 100: 1742–54.Available from: http://www.ncbi.nlm.nih.gov/pubmed/12176896 [PubMed] [Google Scholar]
- Batta G, Soltész L, Kovács T, Bozó T, Mészár Z, Kellermayer M, et al. Alterations in the properties of the cell membrane due to glycosphingolipid accumulation in a model of Gaucher disease. Sci. Rep 2018; 8: 157.Available from: http://www.ncbi.nlm.nih.gov/pubmed/29317695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanz J, Saftig P. Parkinson’s disease: acid-glucocerebrosidase activity and alpha-synuclein clearance. J. Neurochem 2016; 139: 198–215.Available from: http://www.ncbi.nlm.nih.gov/pubmed/26860955 [DOI] [PubMed] [Google Scholar]
- Bossi SR, Simpson JR, Isacson O. Age dependence of striatal neuronal death caused by mitochondrial dysfunction. Neuroreport 1993; 4: 73–76.Available from: http://www.ncbi.nlm.nih.gov/pubmed/8453041 [DOI] [PubMed] [Google Scholar]
- Brownell A-L, Jenkins BG, Elmaleh DR, Deacon TW, Spealman RD, Isacson O. Combined PET/MRS brain studies show dynamic and long-term physiological changes in a primate model of Parkinson disease. Nat. Med 1998; 4: 1308–1312.Available from: http://www.ncbi.nlm.nih.gov/pubmed/9809556 [DOI] [PubMed] [Google Scholar]
- Burré J, Sharma M, Südhof TC. Cell biology and pathophysiology of α-synuclein. Cold Spring Harb. Perspect. Med 2018; 8: a024091.Available from: http://www.ncbi.nlm.nih.gov/pubmed/28108534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Gutierrez D, Hughes AL, Madeo F, Ruckenstuhl C. The crucial impact of lysosomes in aging and longevity. Ageing Res. Rev 2016; 32: 2–12.Available from: http://linkinghub.elsevier.com/retrieve/pii/S1568163716300666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahine LM, Stern MB, Chen-Plotkin A. Blood-based biomarkers for Parkinson’s disease. Park. Relat. Disord 2014; 20: S99–103.Available from: http://www.ncbi.nlm.nih.gov/pubmed/24262199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesselet MF, Richter F, Zhu C, Magen I, Watson MB, Subramaniam SR. A Progressive Mouse Model of Parkinson’s Disease: The Thy1-aSyn (‘Line 61’) Mice. Neurotherapeutics 2012; 9: 297–314.Available from: http://www.ncbi.nlm.nih.gov/pubmed/22350713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J-S, Dougherty I, Cruz PD Jr., Ariizumi K. Syndecan-4 Mediates the Coinhibitory Function of DC-HIL on T Cell Activation. J Immunol Ref 2007; 179: 5778–5784.Available from: http://www.jimmunol.org/content/179/9/5778 [DOI] [PubMed] [Google Scholar]
- Collier TJ, Kanaan NM, Kordower JH. Ageing as a primary risk factor for Parkinson’s disease: evidence from studies of non-human primates. Nat. Rev. Neurosci 2011; 12: 359–366.Available from: http://www.nature.com/nrn/journal/v12/n6/full/nrn3039.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier TJ, Kanaan NM, Kordower JH. Aging and Parkinson’s disease: Different sides of the same coin? Mov. Disord 2017; 32: 983–990.Available from: http://www.ncbi.nlm.nih.gov/pubmed/28520211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleidi M, Hallett PJ, Koprich JB, Chung C-Y, Isacson O. The Toll-Like Receptor-3 Agonist Polyinosinic:Polycytidylic Acid Triggers Nigrostriatal Dopaminergic Degeneration. J. Neurosci 2010; 30: 16091–16101.Available from: http://www.ncbi.nlm.nih.gov/pubmed/21123556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleidi M, Isacson O. Viral and Inflammatory Triggers of Neurodegenerative Diseases. Sci. Transl. Med 2012; 4: 121ps3–121ps3.Available from: http://www.ncbi.nlm.nih.gov/pubmed/22344685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamko N, Geczy CL, Halliday GM. Inflammation is genetically implicated in Parkinson’s disease. Neuroscience 2015; 302: 89–102.Available from: http://www.ncbi.nlm.nih.gov/pubmed/25450953 [DOI] [PubMed] [Google Scholar]
- Engelender S, Isacson O. The Threshold Theory for Parkinson’s Disease. Trends Neurosci 2016: 896–912.Available from: http://www.ncbi.nlm.nih.gov/pubmed/27894611 [DOI] [PubMed] [Google Scholar]
- Farfel-Becker T, Vitner EB, Pressey SNR, Eilam R, Cooper JD, Futerman AH. Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease. Hum. Mol. Genet 2011; 20: 1375–1386.Available from: http://www.ncbi.nlm.nih.gov/pubmed/21252206 [DOI] [PubMed] [Google Scholar]
- Gabriel TL, Tol MJ, Ottenhof R, van Roomen C, Aten J, Claessen N, et al. Lysosomal Stress in Obese Adipose Tissue Macrophages Contributes to MITF-Dependent Gpnmb Induction. Diabetes 2014; 63: 3310–23.Available from: http://www.ncbi.nlm.nih.gov/pubmed/24789918 [DOI] [PubMed] [Google Scholar]
- Gan-Or Z, Dion PA, Rouleau GA. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015; 11: 1443–1457.Available from: http://www.ncbi.nlm.nih.gov/pubmed/26207393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Burke D, Heales SJR, Cooper JM, Hardy J, Wood NW, et al. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol 2012; 72: 455–63.Available from: http://www.ncbi.nlm.nih.gov/pubmed/23034917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett PJ, Huebecker M, Brekk OR, Moloney EB, Rocha EM, Priestman DA, et al. Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of mouse brain. Neurobiol. Aging 2018; 67: 189–200.Available from: http://linkinghub.elsevier.com/retrieve/pii/S0197458018301155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett PJ, McLean JR, Kartunen A, Langston JW, Isacson O. Alpha-synuclein overexpressing transgenic mice show internal organ pathology and autonomic deficits. Neurobiol. Dis 2012; 47: 258–267.Available from: http://www.ncbi.nlm.nih.gov/pubmed/22549133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JJ, Ma WJ, Yokoyama S. Expression and immunolocalization of Gpnmb, a glioma-associated glycoprotein, in normal and inflamed central nervous systems of adult rats. Brain Behav 2012; 2: 85–96.Available from: http://doi.wiley.com/10.1002/brb3.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan NM, Collier TJ, Cole-Strauss A, Grabinski T, Mattingly ZR, Winn ME, et al. The Longitudinal Transcriptomic Response of the Substantia Nigra to Intrastriatal 6-Hydroxydopamine Reveals Significant Upregulation of Regeneration-Associated Genes. PLoS One 2015; 10: e0127768.Available from: http://dx.plos.org/10.1371/journal.pone.0127768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Yun SP, Lee S, Umanah GE, Bandaru VVR, Yin X, et al. GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc. Natl. Acad. Sci 2018; 115: 201700465Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1700465115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprich JB, Reske-Nielsen C, Mithal P, Isacson O. Neuroinflammation mediated by IL-1β increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. J. Neuroinflammation 2008; 5: 8.Available from: http://www.ncbi.nlm.nih.gov/pubmed/18304357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer G, Wegdam W, Donker-Koopman W, Ottenhoff R, Gaspar P, Verhoek M, et al. Elevation of glycoprotein nonmetastatic melanoma protein B (gpNMB) in type 1 Gaucher disease patients and mouse models. FEBS Open Bio 2016; 6: 902–913.Available from: http://doi.wiley.com/10.1002/2211-5463.12078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017; 47: 566–581.e9.Available from: http://www.ncbi.nlm.nih.gov/pubmed/28930663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran R, Cookson MR. Pathways to Parkinsonism Redux: Convergent pathobiological mechanisms in genetics of Parkinson’s disease. Hum. Mol. Genet 2015; 24: R32–R44.Available from: http://www.ncbi.nlm.nih.gov/pubmed/26101198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Castano AP, Hudson TE, Nowlin BT, Lin S-L, Bonventre JV., et al. The melanoma-associated transmembrane glycoprotein Gpnmb controls trafficking of cellular debris for degradation and is essential for tissue repair. FASEB J 2010; 24: 4767–81.Available from: http://www.fasebj.org/cgi/doi/10.1096/fj.10-154757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, MacKenzie KR, Putluri N, Maletić-Savatić M, Bellen HJ. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab 2017; 26: 719–737.e6.Available from: http://linkinghub.elsevier.com/retrieve/pii/S155041311730551X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, et al. Glial Lipid Droplets and ROS Induced by Mitochondrial Defects Promote Neurodegeneration. Cell 2015; 160: 177–190.Available from: http://linkinghub.elsevier.com/retrieve/pii/S009286741401589X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Z, Hashemi M, Banerjee S, Zagorski K, Rochet C, Lyubchenko YL. Phospholipid membranes promote the early stage assembly of α-synuclein aggregates. bioRxiv 2018: 295782Available from: https://www.biorxiv.org/content/early/2018/04/05/295782 [Google Scholar]
- Maric G, Rose AAN, Annis MG, Siegel PM. Glycoprotein non-metastatic b (GPNMB): A metastatic mediator and emerging therapeutic target in cancer. Onco. Targets. Ther 2013; 6: 839–852.Available from: http://www.dovepress.com/glycoprotein-non-metastatic-b-gpnmb-a-metastatic-mediator-and-emerging-peer-reviewed-article-OTT [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques AR, Gabriel TL, Aten J, Van Roomen CPAA, Ottenhoff R, Claessen N, et al. Gpnmb is a potential marker for the visceral pathology in Niemann-Pick type c disease. PLoS One 2016; 11: e0147208.Available from: 10.1371/journal.pone.0147208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and a-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011; 146: 37–52.Available from: http://linkinghub.elsevier.com/retrieve/pii/S0092867411006015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momose M, Ota H, Hayama M. Re-evaluation of melanin bleaching using warm diluted hydrogen peroxide for histopathological analysis. Pathol. Int 2011; 61: 345–350.Available from: http://www.ncbi.nlm.nih.gov/pubmed/21615609 [DOI] [PubMed] [Google Scholar]
- Murata K, Yoshino Y, Tsuruma K, Moriguchi S, Oyagi A, Tanaka H, et al. The extracellular fragment of GPNMB (Glycoprotein nonmelanosoma protein B, osteoactivin) improves memory and increases hippocampal GluA1 levels in mice. J. Neurochem 2015; 132: 583–594.Available from: http://doi.wiley.com/10.1111/jnc.13010 [DOI] [PubMed] [Google Scholar]
- Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, et al. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014; 137: 834–848.Available from: http://www.ncbi.nlm.nih.gov/pubmed/24477431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy MN, Blauwendraat C, Guelfi S, Hardy J, Lewis PA, Trabzuni D. Increased brain expression of GPNMB is associated with genome wide significant risk for Parkinson’s disease on chromosome 7p15.3. Neurogenetics 2017; 18: 121–133.Available from: http://link.springer.com/10.1007/s10048-017-0514-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugesan V, Liu J, Yang R, Lin H, Lischuk A, Pastores G, et al. Validating glycoprotein non-metastatic melanoma B (gpNMB, osteoactivin), a new biomarker of Gaucher disease. Blood Cells, Mol. Dis 2018; 68: 47–53.Available from: http://www.ncbi.nlm.nih.gov/pubmed/28003098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara Y, Shimazawa M, Tanaka H, Ono Y, Noda Y, Ohuchi K, et al. Glycoprotein nonmetastatic melanoma protein B ameliorates skeletal muscle lesions in a SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neurosci. Res 2015; 93: 1552–1566.Available from: http://doi.wiley.com/10.1002/jnr.23619 [DOI] [PubMed] [Google Scholar]
- Nakano Y, Suzuki Y, Takagi T, Kitashoji A, Ono Y, Tsuruma K, et al. Glycoprotein nonmetastatic melanoma protein B (GPNMB) as a novel neuroprotective factor in cerebral ischemia–reperfusion injury. Neuroscience 2014; 277: 123–131.Available from: http://linkinghub.elsevier.com/retrieve/pii/S0306452214005508 [DOI] [PubMed] [Google Scholar]
- Neal ML, Boyle AM, Budge KM, Safadi FF, Richardson JR. The glycoprotein GPNMB attenuates astrocyte inflammatory responses through the CD44 receptor. J. Neuroinflammation 2018; 15: 73.Available from: https://jneuroinflammation.biomedcentral.com/articles/10.1186/s12974-018-1100-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, et al. Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 1996; 89: 691–694.Available from: http://www.ncbi.nlm.nih.gov/pubmed/8917744 [DOI] [PubMed] [Google Scholar]
- Nixon RA, Yang DS, Lee JH. Neurodegenerative lysosomal disorders: A continuum from development to late age. Autophagy 2008; 4: 590–599.Available from: https://www.tandfonline.com/doi/full/10.4161/auto.6259 [DOI] [PubMed] [Google Scholar]
- Pfefferkorn CM, Jiang Z, Lee JC. Biophysics of α-synuclein membrane interactions. Biochim. Biophys. Acta - Biomembr 2012; 1818: 162–171.Available from: http://www.ncbi.nlm.nih.gov/pubmed/21819966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagnol V, Nalls MA, Bras JM, Hernandez DG, Sharma M, Sheerin U-M, et al. A Two-Stage Meta-Analysis identifies several new loci for Parkinson’s Disease. PLoS Genet 2011; 7: e1002142.Available from: http://dx.plos.org/10.1371/journal.pgen.1002142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt FM. Sphingolipid lysosomal storage disorders. Nature 2014; 510: 68–75.Available from: http://www.nature.com/doifinder/10.1038/nature13476 [DOI] [PubMed] [Google Scholar]
- Richter F, Fleming SM, Watson M, Lemesre V, Pellegrino L, Ranes B, et al. A GCase chaperone improves motor function in a mouse model of synucleinopathy. Neurotherapeutics 2014; 11: 840–56.Available from: http://www.ncbi.nlm.nih.gov/pubmed/25037721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoll VM, Irvine KM, Ravasi T, Sweet MJ, Hume DA. Gpnmb Is Induced in Macrophages by IFN- and Lipopolysaccharide and Acts as a Feedback Regulator of Proinflammatory Responses. J. Immunol 2007; 178: 6557–6566.Available from: http://www.jimmunol.org/cgi/doi/10.4049/jimmunol.178.10.6557 [DOI] [PubMed] [Google Scholar]
- Rocha EM, Smith GA, Park E, Cao H, Brown E, Hallett P, et al. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol 2015; 2: 433–438.Available from: http://doi.wiley.com/10.1002/acn3.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EM, Smith GA, Park E, Cao H, Graham A-R, Brown E, et al. Sustained Systemic Glucocerebrosidase Inhibition Induces Brain α-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice. Antioxid. Redox Signal 2015; 23: 550–564.Available from: http://online.liebertpub.com/doi/10.1089/ars.2015.6307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Clarke J, Viel C, Panarello N, Treleaven CM, Kim C, et al. Glucocerebrosidase modulates cognitive and motor activities in murine models of Parkinson’s disease. Hum. Mol. Genet 2016; 25: 2645–2660.Available from: http://www.ncbi.nlm.nih.gov/pubmed/27126635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, et al. Differential neuropathological alterations in transgenic mice expressing α-synuclein from the platelet-derived growth factor and Thy-1 promoters. J. Neurosci. Res 2002; 68: 568–578.Available from: http://doi.wiley.com/10.1002/jnr.10231 [DOI] [PubMed] [Google Scholar]
- Rose AAN, Annis MG, Dong Z, Pepin F, Hallett M, Park M, et al. ADAM10 releases a soluble form of the GPNMB/Osteoactivin extracellular domain with angiogenic properties. PLoS One 2010; 5: e12093.Available from: http://www.ncbi.nlm.nih.gov/pubmed/20711474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. U. S. A 2011; 108: 12101–12106.Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.1108197108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AHV Recent developments in biomarkers in Parkinson disease. Curr. Opin. Neurol 2013; 26: 395–400.Available from: http://www.ncbi.nlm.nih.gov/pubmed/23823465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahmoradian SH, Genoud C, Graff-Meyer A, Hench J, Moors T, Schweighauser G, et al. Lewy pathology in Parkinson’s disease consists of a crowded organellar membranous medley. bioRxiv 2017: 137976Available from: https://www.biorxiv.org/content/early/2017/06/28/137976 [DOI] [PubMed] [Google Scholar]
- Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp. Neurol 2003; 179: 9–16.Available from: http://www.ncbi.nlm.nih.gov/pubmed/12504863 [DOI] [PubMed] [Google Scholar]
- Shi F, Duan S, Cui J, Yan X, Li H, Wang Y, et al. Induction of matrix metalloproteinase-3 (MMP-3) expression in the microglia by lipopolysaccharide (LPS) via upregulation of glycoprotein nonmetastatic melanoma B (GPNMB) expression. J. Mol. Neurosci 2014; 54: 234–242.Available from: http://link.springer.com/10.1007/s12031-014-0280-0 [DOI] [PubMed] [Google Scholar]
- Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol 2012; 11: 986–998.Available from: http://www.ncbi.nlm.nih.gov/pubmed/23079555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease. N. Engl. J. Med 2009; 361: 1651–1661.Available from: http://www.ncbi.nlm.nih.gov/pubmed/19846850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan K, Friedman BA, Larson JL, Lauffer BE, Goldstein LD, Appling LL, et al. Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun 2016; 7: 11295.Available from: http://www.nature.com/doifinder/10.1038/ncomms11295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, et al. Glucosylsphingosine promotes α-synuclein pathology in mutant GBA-associated Parkinson’s disease. J. Neurosci 2017; 37: 1525–17.Available from: http://www.ncbi.nlm.nih.gov/pubmed/28847804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Shimazawa M, Kimura M, Takata M, Tsuruma K, Yamada M, et al. The potential of GPNMB as novel neuroprotective factor in amyotrophic lateral sclerosis. Sci. Rep 2012; 2: 1–11.Available from: http://www.nature.com/articles/srep00573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomihari M, Chung JS, Akiyoshi H, Cruz PD, Ariizumi K. DC-HIL/glycoprotein Nmb promotes growth of melanoma in mice by inhibiting the activation of tumor-reactive T cells. Cancer Res 2010; 70: 5778–5787.Available from: http://www.ncbi.nlm.nih.gov/pubmed/20570888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardi A, Zigdon H, Meshcheriakova A, Klein AD, Yaacobi C, Eilam R, et al. Delineating pathological pathways in a chemically induced mouse model of Gaucher disease. J. Pathol 2016; 239: 496–509.Available from: http://www.ncbi.nlm.nih.gov/pubmed/27234572 [DOI] [PubMed] [Google Scholar]
- Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab 2004; 82: 192–207.Available from: http://www.ncbi.nlm.nih.gov/pubmed/15234332 [DOI] [PubMed] [Google Scholar]
- Zigdon H, Savidor A, Levin Y, Meshcheriakova A, Schiffmann R, Futerman AH. Identification of a biomarker in cerebrospinal fluid for neuronopathic forms of Gaucher disease. PLoS One 2015; 10: e0120194.Available from: http://dx.plos.org/10.1371/journal.pone.0120194 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.