

Abstract
Single-stranded oligonucleotides containing deoxyuridine are aptamers (SOMAmers) that can bind proteins with high specificity and affinity and slow dissociation rates. SOMAscan, an aptamer-based proteomic technology, allows measurement of more than 1,300 proteins simultaneously for the identification of new disease biomarkers. The aim of the present study was to identify new serum and plasma protein markers for diagnosis of acquired aplastic anemia (AA) and response to immunosuppressive therapies (IST). SOMAscan was used to screen 1,141 serum proteins in 28 AA patients before and after therapy and 1,317 plasma proteins in seven SAA patients treated with standard IST and a thrombopoietin receptor agonist. From our analysis, 19 serum and 28 plasma proteins were identified as possible candidate diagnostic and prognostic markers. A custom immunobead-based multiplex assay with five selected serum proteins (BMP-10, CCL17, DKK1, HGF, and SELL) was used for validation in a verification set (n = 65) of samples obtained before and after IST and in a blinded validation cohort at baseline (n = 16). After technical validation, four biomarkers were employed to predict diagnosis (accuracy, 88%) and long-term response to IST (accuracy, 79%). In conclusion, SOMAscan is a powerful tool for the identification of new biomarkers. We propose further larger studies to validate new candidate serum and plasma diagnostic and prognostic markers of AA. Published by Elsevier Inc. on behalf of ISEH – Society for Hematology and Stem Cells.
Acquired aplastic anemia (AA), a bone marrow (BM) failure syndrome characterized by pancytopenia and BM hypocellularity, is caused by hematopoietic stem and progenitor cell (HSPC) destruction by immune cells [1]. BM transplantation remains the first therapeutic choice for young patients with a matched sibling donor. Immunosuppressive therapy (IST), with or without the thrombopoietin (TPO) receptor agonist eltrombopag (EPAG), are considered the standard of care in older patients and a therapeutic option for younger patients without a matched sibling donor [1,2]. However, the exact mechanisms of action of IST and EPAG are still not well understood [3–5]. Hematologic improvement of blood counts after IST is one of the most supportive indirect evidence of the autoimmunity to HSPCs in BM failure [6]. Additional lines of indirect evidence for an immune pathophysiology include measurements of activated cytotoxic T cells that inhibit BM proliferation, circulating and exosomal microRNAs (miRNAs), and the presence of pro-inflammatory cytokines in the plasma [1,7–11].
Measurement of proteins on a large scale in biological samples, or proteomics, has improved slowly compared with other “omics” fields [12,13]. Electrophoresis, mass spectrometry, enzyme-linked immunosorbent assay (ELISA), and immunobead-based multiplex assays are the most utilized techniques to detect and quantify proteins, but large-scale studies often are not feasible due to the limited numbers of samples or analytes that can be studied simultaneously or because of technical limitations in the quantification of low-abundance proteins [12–15]. An aptamer-based multiplexed proteomic technology, the SOMAscan assay, was released in 2010 and currently between 1,300 and 5,000 human proteins can be detected simultaneously [15,16]. Aptamers are short DNA or RNA molecules that can bind proteins with low affinity. They are sensitive to nuclease degradation [15], but hydrophobic modifications at the 5-position of deoxyuridine nucleotides greatly increase DNase resistance. Modified single-stranded aptamers, or SOMAmers (slow off-rate modified aptamers), are tested against targeted proteins from large libraries of randomized sequences through a technique referred to as SELEX (selected evolution of ligands by exponential enrichment) [13–15]. Only aptamers with slow dissociation rates (> 30 min) are further selected in order to minimize nonspecific binding interactions. As a result, SOMAmers are highly specific for epitopes and residues on many human proteins [13,14]. SOMAmer-target protein complexes are captured by biotin-streptavidin beads and nonspecifically bound SOMAmers are removed with a polyanionic-containing buffer. SOMAmers are then released from their specific target protein complexes by denaturation, hybridized to complementary sequences on microarray, and quantified by fluorescence [13,15]. Using this platform, new biomarkers have been discovered in malignant and nonmalignant disorders such as mesothelioma and Alzheimer’s disease [17,18].
To broaden current knowledge of proteomics in BM failure, we used SOMAscan to screen serum and plasma proteins from AA patients before and after IST, allowing the identification of new biomarkers of AA and responsiveness to therapy. These proteins may also relate to the overall pathophysiology of BM failure.
Methods
Patients
Sera were collected from 109 AA patients after informed consent was obtained in accordance with the Declaration of Helsinki [19] and protocols approved by the National Heart, Lung, and Blood Institute Institutional Review Board of the National Institutes of Health (NIH) (www.clinicaltrials.gov identifiers: NCT00260689 and NCT01623167). All patients were diagnosed as severe AA (SAA) and hematologic response to IST was defined according to standard criteria [20,21]. Patients were divided in three cohorts: a discovery set (n = 28) for large-scale proteomics screening using SOMAscan; a verification set (n = 65) including 21 patients from the discovery cohort for technical validation of selected aptamers by Lumincx assay; and a validation cohort (n = 16) of SAA patients whose hematologic response to IST was not known at the time of the study. Specimens were collected at the time of diagnosis and after 6, 12, and/or 24 months of initiating IST. Plasma samples were collected in EDTA tubes from seven SAA patients (www.clinicaltrials.gov identifier: NCT01623167) and specimens were collected at the time of diagnosis and after 6 months of initiating IST and EPAG. Healthy controls were recruited from donors at the NIH Clinical Center Department of Transfusion Medicine. Clinical characteristics are summarized in Table 1. After centrifugation at 2000 RPM for 10 min, serum or plasma samples were collected and stored at −80°C until use.
Table 1.
Patient Characteristic
| Serum |
Plasma |
|||
|---|---|---|---|---|
| Discovery set (n = 28) | Verification set (n = 65) | Validation set (n = 16) | Discovery set (n = 7) | |
| Median age, years (range) | 30 (10–65) | 33 (2–75) | 47 (9–78) | 36 (7–65) |
| Sex (M/F) | 18/10 | 37/28 | 7/9 | 3/4 |
| Treatment | ||||
| ATG+CsA | 28 | 56 | ||
| EPAG+ATG+CsA | – | 9 | 16 | 7 |
| Clinical response | ||||
| NR | 13 | 31 | 3 | 3 |
| PR | 9 | 20 | 6 | 1 |
| CR | 6 | 14 | 3 | 3 |
| Relapse/unknown | – | – | 4 | |
| Baseline CBC | ||||
| Median ANC (cells/μL) | 392 (0–1, 430) | 485 (0–1, 881) | 1, 103 (0–4, 700) | 500 (20–1, 190) |
| Median ALC (cells/μL) | 1, 170 (290–2, 691) | 1358 (137–3, 243) | 1, 244 (370–2, 580) | 1, 300 (360–2620) |
| Median AMC (cells/μL) | 104 (0–250) | 123 (0–393) | 226 (0–1, 470) | 100 (10–240) |
| Median ARC (103 cells/μL) | 18.275 (2.3–45.8) | 27.809 (1–130) | 38.044 (6.6–105.7) | 36.8 (7.2–65.1) |
| Median Hb (g/dL) | 7.7 (5–11) | 8.6 (5.4–13.7) | 9.4 (7.2–13.2) | 8.3 (7.6–9.3) |
| Median platelet count (/μL) | 13, 750 (1, 000–78, 000) | 26, 497 (1,000–229, 000) | 57, 375 (12, 000–209, 000) | 33, 300 (17, 000–59, 000) |
| Post-treatment CBC | ||||
| Median ANC (cells/μL) | 1, 339 (30–3, 260) | 1, 262 (70–3, 260) | – | 1, 300 (320–2, 790) |
| Median ALC (cells/μL) | 1, 038 (260–1, 910) | 1, 120 (9–3, 162) | – | 1, 300 (570–2, 580) |
| Median AMC (cells/μL) | 302 (64–640) | 307 (10–1310) | – | 200 (50–560) |
| Median ARC (103 cells/μL) | 42 (1–97) | 49.55 (2.9–153) | – | 58.2 (9.8–143.6) |
| Median Hb (g/dL) | 10 (7–15) | 10.6 (7–16.1) | – | 10.4 (7.2–15) |
| Median platelet count (/μL) | 62, 536 (6, 000–231, 000) | 72, 989 (1, 000–346, 000) | – | 70, 400 (6, 000–181, 000) |
ATG = anti-thymocyte globulin; CsA = cyclosporine; EPAG = eltrombopag; CBC = complete blood count; ANC = absolute neutrophil count; ALC = absolute lymphocyte count; AMC = absolute monocyte count; ARC = absolute reticulocyte count; Hb = hemoglobin
SOMAscan assay
Large-scale proteomic analysis was performed on serum samples in our discovery set of 28 SAA patients: six complete response (CR), nine partial response (PR), and 13 nonresponders (NR) and on plasma samples from seven SAA patients (three CR, one PR, and three NR) before and after IST by SOMAscan (SomaLogic, Boulder, CO, USA) at the Trans-NIH Center for Human Immunology, Autoimmunity, and Inflammation (CHI), as described previously [22]. Specimens (50 μL) were diluted to three concentrations (0.005%, 1%, and 40%) to separate groups of high, medium, and low abundance proteins, respectively, and then combined with dilution-specific SOMAmers. Quality controls (QC) and calibrators provided by SomaLogic were run together with internal site QC samples. Data generated from these samples were used to assess interassay variability, as described previously [22].
Luminex validation
For independent validation of candidate serum biomarkers, a custom five-plex immunobead-bascd multiplex assay was designed based on commercial availability for measurement of bone morphogenetic protein 10 (BMP-10), C-C motif chemokine ligand 17 (CCL17), Dickkopf WNT signaling pathway inhibitor 1 (DKK1), hepatocyte growth factor (HGF), and L-selectin (SELL) in serum or plasma (R&D Systems, Minneapolis, MN). The assay was carried out following the manufacturer’s instructions, and a standard curve and one internal control were included in each plate to reduce interassay variability.
Statistics
Data were analyzed using SomaSuite version 1.0.3 (NEC Corporation, Minato, Tokyo, Japan) and web tools developed by the CHI (https://foocheung.shinyapps.io/adat_v02/ and https://foocheung.shinyapps.io/plotterII/) [23]. VENNY 2.1, an interactive tool for comparing lists with Venn diagrams, was used to find common or unique proteins between groups [24]. Unpaired or paired t tests for two group comparison, analysis of variance (ANOVA), or Kruskal–Wallis test for three-group comparison were performed with Tukey’s test for multiple comparisons and false discovery rate (FDR) for correction. Pearson correlation was performed using an online tool developed by CHI [25]. Specificity and sensitivity were calculated by receiver operating characteristic curves using the healthy control group as reference [26]. Logistic regression and generalized linear model analysis were performed to calculate the diagnostic and prognostic power of combined markers. By convention, p < 0.05 was considered statistically significant. Principal component analysis (PCA) and the t-distributcd stochastic neighbor embedding (t-SNE) algorithm for visualization of high-throughput data in two or three dimensions were carried out using RStudio software (version 0.99.896, RStudio Inc., Boston, MA, USA). Protein pathway analysis was performed employing open-source pathway databases [27,28].
Results
Serum proteomic signature of AA
SOMAscan data from the discovery set and a group of healthy controls (n = 14; M/F, 6/8; mean age, 32.3 years, range 21–62) were combined and normalized as described previously [22]. t-SNE of all proteins (listed in Supplementary Table E1, online only, available at www.exphem.org) was performed by dividing patients based on hematologic responses to IST at landmark time points (Figure 1A). Although no clear clusters were identified, AA patients, even after treatment, completely separated from healthy subjects and NR sera appeared different from CR and PR sera after treatment. When t-SNE was performed including blood counts, clearer separations were discerned (Figure 1B). Next, serum protein levels were compared between healthy controls and AA patients before or after IST by unpaired t test or compared between patients’ groups based on clinical response to therapy (Supplementary Table E2, online only, available at www.exphem.org). AA patients’ groups before and after IST were compared by unpaired t test and proteins higher in each respective group were used to build Venn diagrams (Figure 1C and Supplementary Table E3, online only, available at www.exphem.org). Proteins elevated in sera of CR patients before (n = 7) and after (n = 20) therapy compared with NR were selected as candidate biomarkers of responsiveness to IST. Proteins higher in NR before therapy (n = 15) and in PR before and after IST (n = 2) were selected as candidate biomarkers of nonresponsiveness to IST. Among 44 proteins in this group, Wilcoxon Mann–Whitney test with FDR correction was performed to remove proteins showing similar serum levels among groups after treatment and 19 proteins were selected for further investigations (WISP1, DDR2, FRZB, CNTN4, SELL, THBS1, PDGFA, NID2, HGF, BMP 10, TEC, CLEC7A, SGTA, TNFRSF4, PPIF, PRKCZ, CCL17, DKK4, and DKK1) (Figure 1D). Fifteen out of these 19 proteins were also different in the plasma of AA patients compared with healthy controls.
Figure 1.
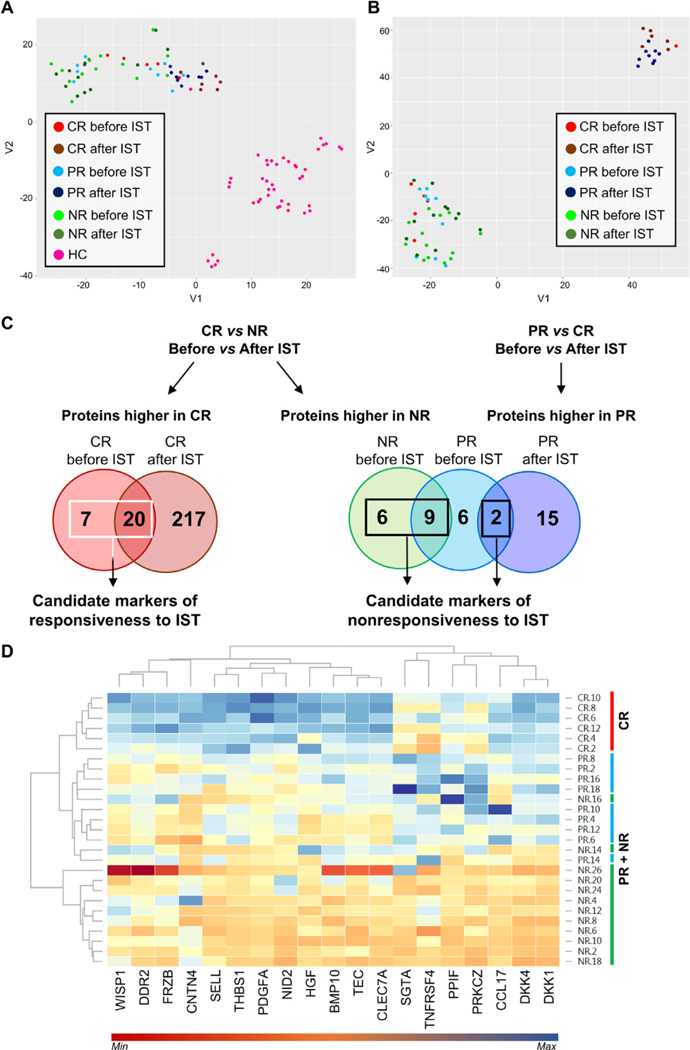
SOMAscan data analysis of serum samples from AA patients. t-SNE testing was carried out on AA patients at diagnosis and after IST and on healthy controls (HC) (A), also including blood counts (B). Patients were divided according to clinical response at 6 months or 1 year after treatment as CR, PR, or NR. (C) In order to find protein markers of responsiveness to therapy, unpaired t test was performed between CR and NR or PR before and after IST and proteins were grouped using Venn diagrams. Proteins higher in CR at diagnosis and after IST (n = 27) were chosen as candidate biomarkers of responsiveness to therapy, whereas proteins higher in NR and PR at baseline and after treatment (n =16) were selected as candidate biomarkers of nonresponsiveness to IST. (D) Protein levels after treatment of selected 19 candidate biomarkers are shown as a heat map with hierarchical clustering using a web-based tool. CR patients were grouped together and not mixed with PR or NR.
Because hematological improvements could lead to subsequent increases in serum proteins due to the appearance of adequate cells in the circulation, candidate protein markers were correlated to blood counts, such as hemoglobin level, platelets (PLT), absolute reticulocyte count (ARC), absolute neutrophil count (ANC), and absolute monocyte count (AMC), for each patient before and after therapy (Supplementary Figure E1, online only, available at www.exphem.org). Indeed, CCL17, DKK4, DKK1, PDGFA, and THBS1 were highly correlated with blood counts. Multiple correlations also were described for other proteins. Because transfusions could influence circulating protein levels, transfusion history was documented in our cohort of AA patients: 21 of them (two CR, seven PR, and 12 NR) had received transfusions before starting IST (mean time between last transfusion and starting drug administration, 144 days; range 1 day to 60 months). Protein pathway analysis using the Reactome Pathway Database revealed that proteins appeared related to the Wnt pathway, innate and adaptive immune responses, extracellular matrix or cell-to-cell interactions, and hematopoietic stem cell differentiation (Supplementary Table E4, online only, available at www.exphem.org). The STRING database was also employed for protein pathway analysis using proteins higher in HC compared with AA patients before IST or using the selected 19 markers (Supplementary Tables E5–E7, online only, available at www.exphem.org). Proteins were related to immune response, coagulation, regulation of apoptotic process, regulation of protein phosphorylation, cell adhesion, T-cell receptor, cytokine–cytokine receptor interaction, positive or negative response to cell surface receptor signaling, and Wnt, Ras, HIF-1, NF-κB, and Jak-STAT signaling cascades (Figure 2).
Figure 2.
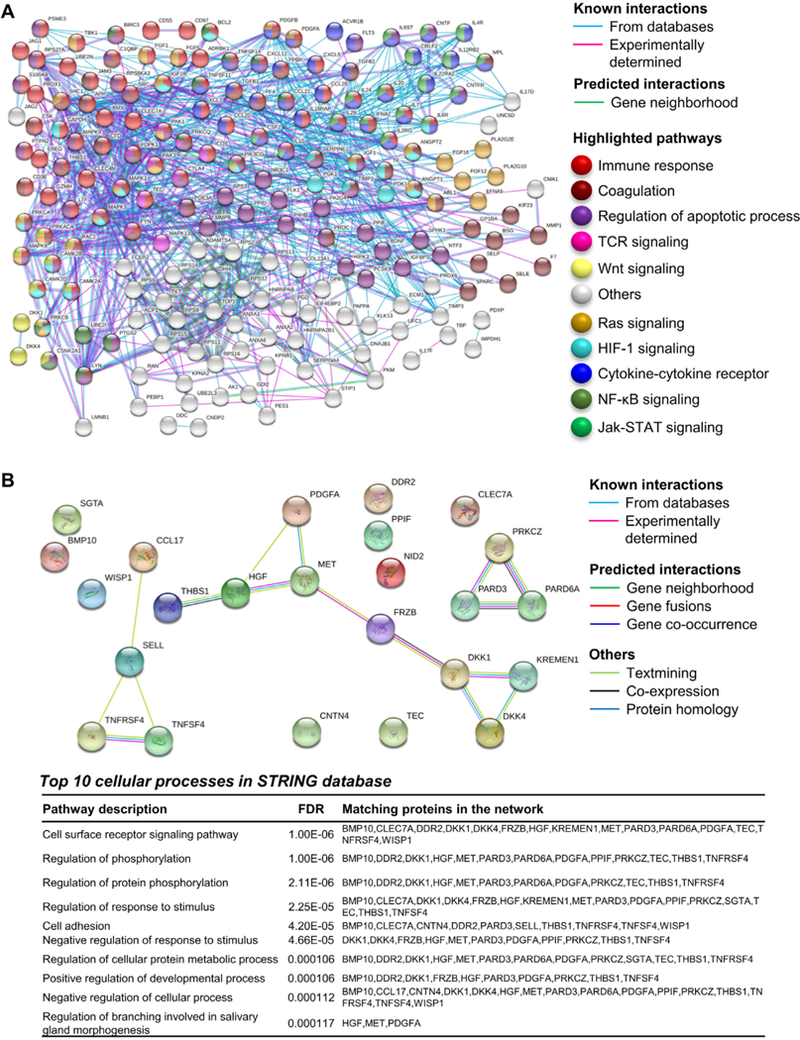
Protein pathway analysis using serum proteins present in healthy controls and/or AA patients. (A) STRING database was employed for protein pathway analysis using proteins higher in HC compared by unpaired t test with AA patients before IST. Known (from databases or experimentally determined) or predicted (based on gene neighborhood) interactions are shown. Proteins related to immune response, coagulation, regulation of apoptotic process, cytokine-cytokine receptor interaction, T-cell receptor (TCR), and Wnt, Ras, HIF-1, NF-κB and Jak-STAT signaling cascades are highlighted with different colors accordingly. (B) STRING database was employed for protein pathway analysis using the 19 selected serum candidate markers and the top 10 cellular processes are shown.
Validation of candidate serum markers
To assess generalizability of the preliminary SOMAscan findings, a five-plex immunobead-based multiplex assay was applied to a verification set of 65 SAA patients, with samples obtained before IST, at 6 months of treatment, and/or at 1 year after IST. A validation cohort of 16 patients at diagnosis and a group of age-and sex-matched healthy controls (n = 13; M/F, 7/6; mean age, 34.3 years, range 21–62) also was included. Among the 19 candidate serum markers, proteins linked to the Wnt pathway (DKK1 and BMP 10), innate and adaptive immune responses (CCL17 and SELL), and hematopoietic stem cell differentiation (HGF and DKK1) were selected for validation. Serum levels of DKK1, SELL, CCL17, and HGF showed significant correlations between the two techniques (all p < 0.01), whereas BMP-10 serum levels differed between the SOMAscan and Luminex assays (r = −0.075, p = 0.675). For this reason, BMP-10 was not included in further analyses. First, SAA patients from the verification cohort were compared with healthy controls, showing that all four selected proteins were significantly higher in healthy controls compared with patients (DKK1, SELL, and CCL17, all p < 0.0001; HGF, p = 0.037) (Figure 3A). All markers displayed a high specificity for AA (DKK1, area under the curve [AUC] = 0.74; SELL, AUC = 0.89; CCL17, AUC = 0.88; and HGF, AUC = 0.80) (Supplementary Figure E2A, online only, available at www.exphem.org). Subsequently, the diagnostic power of combined markers was assessed on verification (AUC = 0.974) (Supplementary Figure E2B, online only, available at www. exphem.org) and validation (AUC = 0.832) sets of SAA patients (Supplementary Figure E2C, online only, available at www.exphem.org).
Figure 3.
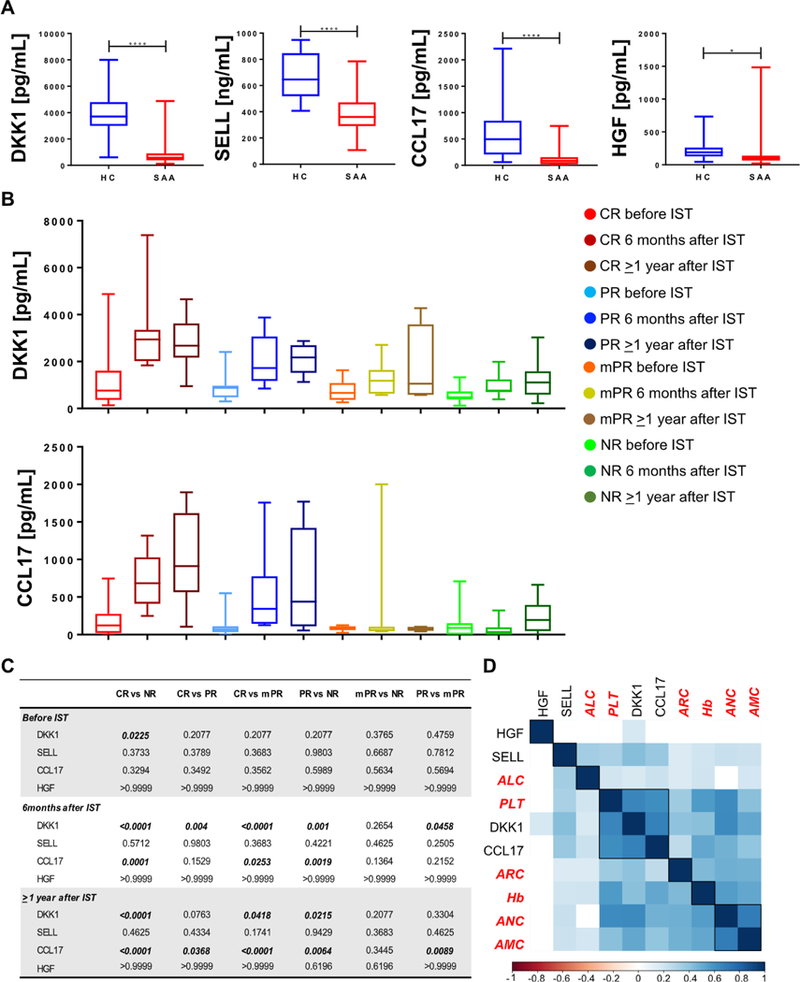
Diagnostic power of selected protein markers in the verification cohort. (A) Protein levels were compared between patients and a group of age- and sex-matched healthy controls (HC) by unpaired t test. Values are shown as minimum to maximum. (B) Protein levels were measured by Luminex and data for DKK1 and CCL17 are shown as minimum to maximum for each group of patients. (C) Protein levels were compared among groups and time points by one-way ANOVA. Values with p < 0.05 are bolded. (D) Correlation analysis between protein markers and blood counts was performed by Pearson analysis.
Next, SAA patients were divided based on clinical response at 6-month and/or 1-year time points and groups were compared by one-way ANOVA (Figures 3B and 3C and Supplementary Figure E3A, online only, available at www.exphem.org). In CR cases, DKK1 was significantly higher at baseline compared with NR patients and there were increased serum levels also at both the 6-month and 1-year time points compared with PR and NR sera. Similarly, CCL17 was higher in CR after IST compared with other groups. No significant differences were present for SELL and HGF. By comparing only CR with NR using unpaired t test, DKK1 was significantly increased in CR patients (p = 0.010), whereas SELL and CCL17 levels were only slightly higher than those in NR (p = 0.137 and p = 0.132). No differences were seen for HGF serum levels (p = 0.389). Pearson correlation analysis between selected protein markers and blood counts was performed as described above and multiple correlations were observed (Figure 3D and Supplementary Figure E3B, online only, available at www.exphem.org). Logistic regression and generalized linear model analysis were used to evaluate the prognostic power of combined markers. Data from patients at ≥ 1-year follow-up were used to generate a model and then functions were applied to patients from the verification and validation sets at baselines or at 6 months of therapy (Supplementary Figures E3C and E3D, online only, available at www.exphem.org). For patients at baseline, sensitivity to predict responsiveness to IST was low (48%), whereas specificity was high (82%); prediction at 6 months of therapy showed higher sensitivity and specificity (76% and 83%, respectively).
Plasma proteomic signature of AA
SOMAscan assay also was employed for screening plasma proteins in a small cohort of AA patients (n = 7) treated with IST and EPAG in order to identify common biomarkers with the serum signature and novel plasma proteins for diagnosis and disease progression. A group of healthy controls (n = 21; mean age, 57 years; range 37–62; M/F, 10/11) also was included. Heatmap (Figure 4A) and PCA were displayed using all 1,317 proteins to visualize a possible signature (Figure 4B). A proteomic profile of CR patients after IST was compared with those of CR and NR patients before IST. Unpaired t test with FDR correction (5%) was employed to compare a proteomic profile of healthy controls with that of AA patients; groups before or after IST were compared by unpaired t test without FDR correction because of the small number of subjects (CR, n = 3; NR, n = 3). In plasma, 600 proteins were different in healthy controls compared with AA and 35% of them were common to the serum proteomic profile. When plasma protein levels were compared between healthy controls and AA, 43 proteins were present in AA patients’ plasma and 43% of them (n = 27) were also present in the serum signature (Supplementary Figure E4 and Supplementary Table E8, online only, available at www.exphem.org). In AA patients, 28 proteins were present in CR or NR before IST and could potentially be used as diagnostic and/or prognostic biomarkers of AA (Figure 4C). After identification of groups of proteins as candidate signatures of responsiveness to IST, heatmaps were displayed using different combinations of those markers. Correlations with complete blood counts were also performed by Pearson analysis (Figures 4D and 4E). PCA was carried out using a group of 24 or 60 proteins present in CR or NR after therapy, respectively (Supplementary Figure E5, online only, available at www.exphem.org). In both cases, CR patients’ plasma proteins were different compared with those of NR patients. These two groups of different proteins were used for pathway analysis; except for complement and coagulation cascades, all pathways in NR cases were common to those found in CR cases (Supplementary Table E8, online only, available at www.exphem.org).
Figure 4.
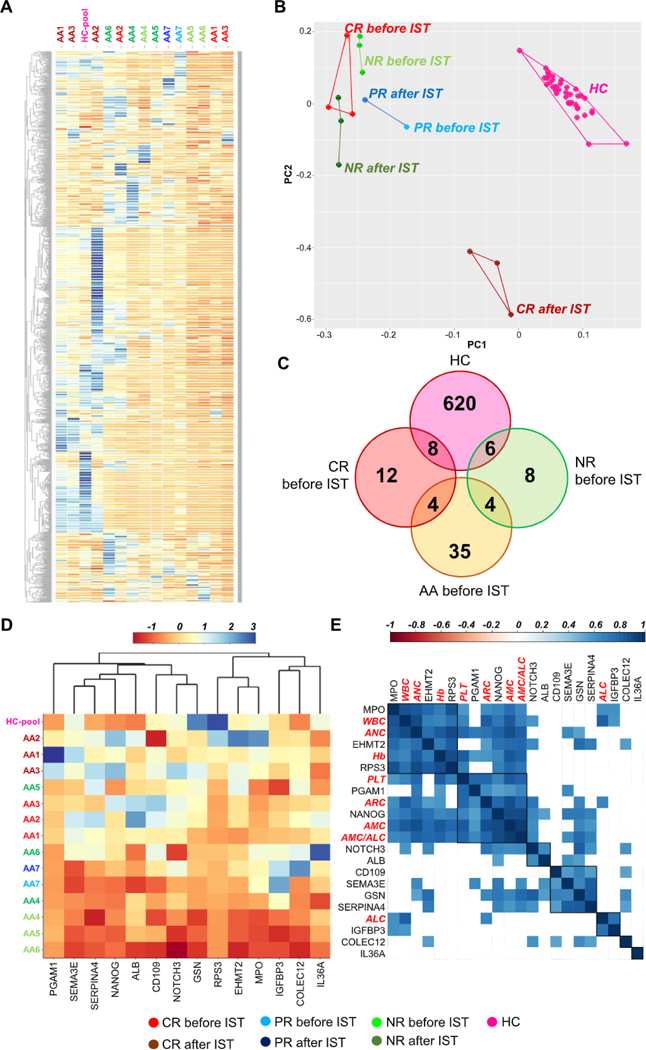
Analysis of SOMAscan data obtained from plasma samples of AA patients. (A) Patients were divided based on hematologic response in CR, PR, and NR and heat map with hierarchical clustering is shown using all 1, 317 proteins. Healthy controls (HC) were grouped together as a pool. (B) PCA was also performed using all 1, 317 proteins. (C) After unpaired t test between HC and patient groups, proteins present in each population before IST were grouped and are shown using Venn diagrams. (D) Plasma levels of 12 proteins present in CR patients before IST are displayed as heatmap. (E) Pearson correlations with blood counts were also performed (blood counts are highlighted in red). Correlations with p > 0.05 are shown as blank squares. WBC = white blood count; Hb = hemoglobin levels; AMC = absolute monocyte count; ALC = absolute lymphocyte count; ANC = absolute neutrophil count; ARC = absolute reticulocyte count; PLT = platelet count.
Large-scale proteomic profiles obtained from serum and plasma samples of CR patients after standard IST were compared. Because different types of body fluids were screened, we sought common or different proteins between serum and plasma in order to identify a protein signature independent from the type of body fluid used. More than 80 proteins were present in both serum of patients treated with standard IST and in plasma of CR treated with IST and EPAG. Common proteins were mostly linked to immune response and inflammation or to the extracellular/intracellular signaling pathways (Supplementary Figure E6 and Supplementary Table E9, online only, available at www.exphem.org). Protein pathway analysis was also performed with proteins present only in serum or plasma samples, identifying common pathways. Proteins in serum or plasma were grouped and PCA was performed accordingly (Figure 5). Using markers present in serum samples, NR treated with standard IST were different from CR with same treatment, whereas using proteins present in plasma samples, no clear clusters were visualized. Instead, for patients treated with IST and EPAG, NR and CR were different for both protein groups.
Figure 5.
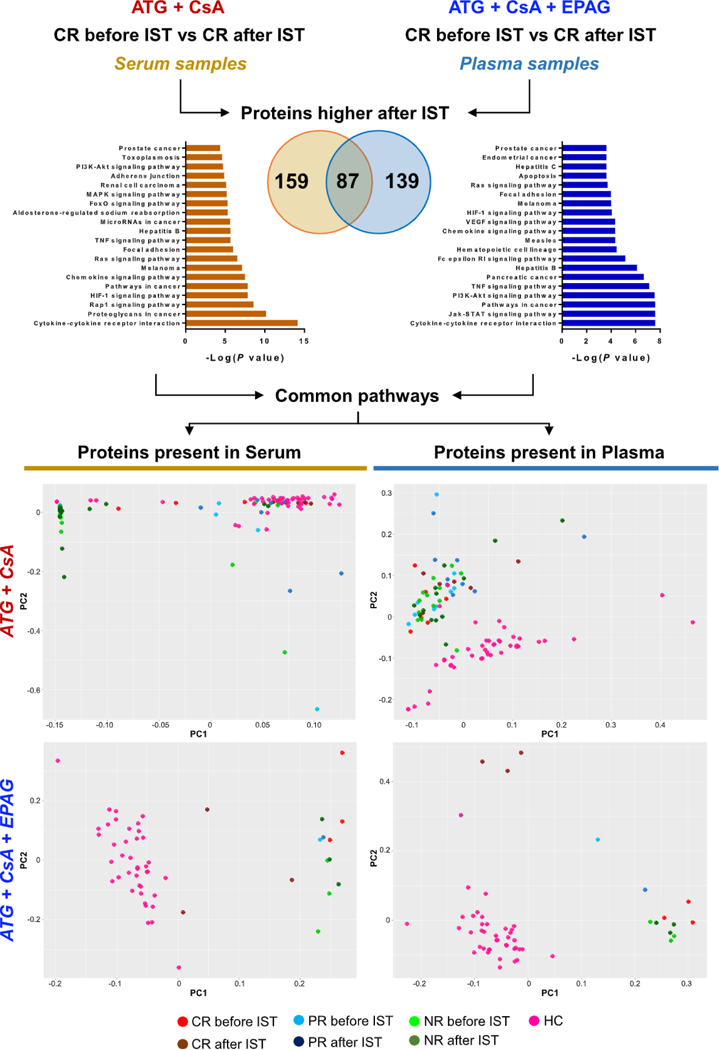
Plasma and serum proteomic signatures in patients after different ISTs. Unpaired t test was performed between CR before and after therapy using two SOMAscan datasets obtained from serum samples in patients treated with standard IST (anti-thymocyte globulin [ATGJ and cyclosporine A [CsA]) or from plasma samples in patients treated with standard IST and EPAG. Proteins higher in CR after therapy were grouped and interpolated using Venn diagrams. Using proteins present only in serum (159) or plasma (139), protein pathway analysis was performed and the top 20 pathways are shown (left and right for serum and plasma proteins, respectively). Multiple common pathways were identified and proteins from serum and plasma signatures were grouped. Using these two groups of proteins, PCA was performed in patients treated with standard IST (upper row) and in those who received standard IST and EPAG (bottom row). Patients were divided according to hematologic responses to therapy in CR, PR, and NR. HC ==healthy controls.
Validation of aptamer-based plasma proteomic profiling
As an indirect method to validate our large-scale plasma proteomic profiling, we investigated plasma levels of circulating cytokines already reported in AA [10]. Although our discovery cohort was small, we confirmed lower CXCL5, CCL5, CXCL11, and EGF plasma levels in our patients at diagnosis (all p < 0.0001) and, for higher granulocyte colony-stimulating factor and lower CD40 ligand, a similar trend with reported data was also present but did not reach statistical significance (p = 0.168 and p = 0.197, respectively) (Supplementary Figure E7, online only, available at www.exphem.org). In our cohort of treatment-naive A A patients who received IST and EPAG, TPO was higher in AA patients before treatment (p = 0.0008). After therapy, TPO decreased in CR plasma and there were no differences compared with healthy controls. Circulating c-MPL was also investigated and AA patients showed persistent lower levels in the plasma and serum (Supplementary Figure E7D, online only, available at www.exphem.org). Multiple linear regression analysis was also performed to assess correlation between disease severity and plasma/serum protein levels. Disease severity, defined as the combination of ANC, ARC, and PLT count [20], was assessed to serum proteins (DKK1, CCL17, SELL, and HGF) in both serum and plasma discovery sets at baseline (Supplementary Figure E8, online only, available at www.exphem.org). No correlations were described between disease severity and combined markers (p = 0.083 and p = 0.206 for serum and plasma signatures, respectively).
Discussion
The discovery of biomarkers has greatly enhanced clinical management of patients with neoplastic and non-neoplastic diseases [29,30]. A high-quality biomarker should be informative, sensitive, and specific for the disease, useful in guiding therapeutic strategy, and measurable in a time- and cost-effective manner [15]. Although mass spectrometry remains the standard, anti-body-based technologies are more sensitive and can detect low-abundance proteins, but multiplexing is limited [15]. The SOMAscan assay, an aptamer-based proteomic technology, is 20 times faster than mass spectrometry and applicable to much larger cohort screening [31–33]. Using this platform, thousands of proteins are measured simultaneously with sensitivity and specificity similar to high-quality ELISAs, and large-scale proteomics can be performed for the identification of new minimally invasive biomarkers in malignant and chronic diseases [12–18,33–35]. In this study, we performed for the first time a large-scale aptamer-based proteomic analysis of serum and plasma samples from patients with AA, and many proteins were associated with a clinical response to immunosuppressive therapies. Regardless of the body fluid screened, more than 600 proteins were proposed for further validation in larger cohorts as potential biomarkers of AA. In addition, 28 plasma and 19 serum proteins were identified as candidate predictors of responsiveness to IST at diagnosis. From the 19 serum protein signature, five candidate markers were selected for validation in a larger cohort of patients (based on availability for Luminex assay). We further examined four new serum candidate markers of AA (DKK1, SELL, CCL17, and HGF) in larger and independent cohorts, showing high specificity for diagnosis (AUC of 0.83 and accuracy of 88%) and long-term response to therapy (accuracy of 79%). Additional candidate markers could be included in this signature based on upcoming analytes available in Luminex. We also propose more than 250 proteins commonly present in both serum and plasma aptamer-based signatures as candidate biomarkers of AA for further validation in larger cohorts. Serum and plasma are not equivalent for analyte measurement (e.g., glucose) because anticoagulants and debris alter plasma composition, whereas cells and platelets release metabolites during clot formation in serum samples [36]. Serum is preferred for low-abundance proteins, whereas plasma shows better general reproducibility [36–38]. In our study, 35% of proteins were present in healthy controls in both serum and plasma and 43% in AA at diagnosis. This diversity in proteomics signature may reflect different composition of these body fluids; however, further studies are needed to better investigate the effects of matrices for biomarker measurement in clinical management of AA patients. Although our study confirms SOMAscan as a powerful and highly specific and sensitive discovery platform for large-scale screening of new biomarkers also in small cohorts [13,14], technical validation of our initial findings is required because SOMAmers recognize conformational epitopes [34].
In addition to technical issues related to data normalization [22], other challenges in assessing high-throughput data are the large amount of information and the need to select proteins linked to the disease itself and not to confounding phenomena, such as drug administration or surgical procedures [33]. In our study, an interactive user-friendly web-based tool was employed for SOMAscan data visualization by PCA or heatmap using pathways listed in the KEGG database and statistical analysis [23]. The t-SNE algorithm provided better visualization of the high-throughput data than PCA. Because of the possible influence of blood count improvements after IST, we rigorously sought confounding in the selection of candidate protein markers by correlations with blood counts using these web-based tools. In particular, DKK1 is expressed by several cell types, such as osteoblasts and osteocytes, but platelets are the major source of circulating DKK1. Circulating DKK1 levels are elevated in diseases in which bone is disrupted (e.g., multiple myeloma), contributing to tumor growth and bone destruction [39,40]. In our cohort, circulating DKK1 levels were decreased in AA patients at diagnosis and in NR after treatment compared with healthy controls and correlated with platelet counts. Decreased levels of platelet-related proteins might not only reflect the platelet count, but could also influence several biological functions because platelet-derived DKK1 is involved in macrophage and neutrophil activation and migration during acute inflammation [40,41]. In addition, anticoagulants used for serum collection can cause platelet activation and release of DKK1 [39]. In our cohort of AA patients, we found circulating DKK1 decreased in both serum (1.5-fold) and plasma (2.3-fold) compared with healthy controls.
Signatures of responsiveness to IST were extrapolated from our dataset using two independent statistical analyses: two-way ANOVA across all groups before and after therapy and unpaired t test with FDR correction (5%) between groups. Proteins that showed differences only after treatment were not included in further analyses because we believed that these changes might also reflect drug effects and not be predictive of clinical response from baseline. Pursuing this strategy, 19 serum and 28 plasma proteins were selected as candidate biomarkers of AA and disease progression. Here, we show that, in the current analyses, only one of several possible interpolations of the abundant SOMAscan data was pursued and we may have failed to identify other biomarkers because of our conservative approach and the small sample size of the discovery cohort.
Protein pathways can disclose known or predicted interactions between selected proteins from open-access databases. For the selected candidate markers, several enriched networks were found, such as Wnt pathway proteins in complete responders. Wnt signaling is important in early hematopoietic ontogeny and maintenance of self-renewing long-term hematopoietic stem cells (LT-HSCs) after stress [42–44]. Conversely, inhibition of Wnt by DKK1 impairs BM recovery after transplantation, reduces LT-HSCs, increases cell cycling, and promotes myeloid compartment expansion [43,45]. Regulation of Wnt is complex and multiple inhibitors and activators are involved [46]. From our signature proteins, four molecules were associated with the Wnt signaling pathway: three are negative regulators (DKK1, DKK4, and FRZB) [45], whereas WISP1, which is downstream of WNT1, is increased in many tumors and inflammatory diseases [47]. However, further in vitro studies are needed to elucidate the exact role of Wnt-related proteins in the pathophysiology of BM failure.
Some of our candidate markers, such as HGF, also are involved in HSC differentiation [48] and may sustain hematopoiesis together with DKKs during autologous immune attack. From our plasma signature, we confirmed that EPAG overcomes c-MPL blockade by binding the receptor with high affinity at a different site [3,4,49,50]. Circulating c-MPL was also measured and AA patients showed persistently lower levels in the plasma and serum. Decreased circulating c-MPL in responder patients with normal TPO levels might be due to c-MPL retention on the surface of megakaryocytes and young platelets [51]. However, multiple mechanisms are required to maintain a normal steady-state platelet count [51], and free c-MPL could be dispensable as a regulator of thrombopoiesis.
In conclusion, SOMAscan is a powerful tool for large-scale screening of new biomarkers and the availability of user-friendly software assists in handling high-throughput data in a reliable and reproducible manner. Our initial findings propose some novel candidate biomarkers for diagnosis and disease progression. Further validation in larger and more homogenous cohorts is required and should be supported by additional experiments. Due to the limited number of commercially available analytes for multiplexed assay, many other possible biomarkers were not examined in our validation analysis. The signature of responsiveness to IST in AA patients might be further improved by pursuing analysis and validation of deeper SOMAscan data.
Supplementary Material
Acknowledgments
The authors thank the Trans-NIH Center for Human Immunology, Autoimmunity, and Inflammation (National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD, USA). This work was supported by the Intramural Research Program of the NIH, the National Heart, Lung, and Blood Institute, and by the Trans-NIH Center for Human Immunology, Autoimmunity, and Inflammation.
VG, AB, XF, and NSY designed the study. VG, AB, XF, FC, JC, GF, SK, and ZW conducted the experiments, analyzed the data and interpreted the results. OR and DMT collected and analyzed clinical data. VG, AB, XF, SK, and NSY wrote the manuscript. All authors critically reviewed the manuscript content and agreed with the final submission of the manuscript.
Footnotes
Conflict of interest disclosure
The authors declare no competing financial interests.
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.exphem.2018.09.008.
References
- 1.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bacigalupo A How I treat acquired aplastic anemia. Blood. 2017;129:1428–1436. [DOI] [PubMed] [Google Scholar]
- 3.Cheng H, Cheruku PS, Alvarado L, et al. Interferon-γ perturbs key signaling pathways induced by thrombopoietin, but not eltrombopag, in human hematopoietic stem/progenitor cells. Blood. 2016;128:3870. [Google Scholar]
- 4.Alvarado LJ, Andreoni A, Huntsman HD, Cheng H, Knutson JR, Larochelle A. Heterodimerization of TPO and IFN7 impairs human hematopoietic stem/progenitor cell signaling and survival in chronic inflammation. Blood. 2017; 130:4.28684446 [Google Scholar]
- 5.Qu MM, Liu XN, Liu XG, et al. Cytokine changes in response to TPO receptor agonist treatment in primary immune thrombocytopenia. Cytokine. 2017;92:110–117. [DOI] [PubMed] [Google Scholar]
- 6.Young NS. Current concepts in the pathophysiology and treatment of aplastic anemia. Hematology Am Soc Hematol Educ Program. 2013;2013:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giudice V, Feng X, Lin Z, et al. Deep sequencing and flow cytometric characterization of expanded effector memory CD8 +CD57+ T cells frequently reveals T-cell receptor V(3 oligoclonality and CDR3 homology in acquired aplastic anemia. Haematologica. 2018;103:759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hosokawa K, Kajigaya S, Feng X, et al. A plasma microRNA signature as a biomarker for acquired aplastic anemia. Haematologica. 2017;102:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giudice V, Banaszak LG, Gutierrez-Rodrigues F, et al. Circulating exosomal microRNAs in acquired aplastic anemia and myelodysplastic syndromes. Haematologica. 2018;103:1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng X, Scheinberg P, Wu CO, et al. Cytokine signature profiles in acquired aplastic anemia and myelodysplastic syndromes. Haematologica. 2011;96:602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu W, Ge M, Lu S, et al. Anti-inflammatory effects of interleukin-35 in acquired aplastic anemia. Cytokine. 2015;76:409–416. [DOI] [PubMed] [Google Scholar]
- 12.Chandramouli K, Qian PY. Proteomics: challenges, techniques and possibilities to overcome biological sample complexity. Hum Genomics Proteomics. 2009;2009:239204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gold L, Walker JJ, Wilcox SK, Williams S. Advances in human proteomics at high scale with the SOMAscan proteomics platform. N Biotechnol. 2012;29:543–549. [DOI] [PubMed] [Google Scholar]
- 14.Gold L, Ayers D, Bertino J, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One. 2010;5:e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rohloff JC, Gelinas AD, Jarvis TC, et al. Nucleic acid ligands with protein-like side chains: modified aptamers and their use as diagnostic and therapeutic agents. Mol Ther Nucleic Acids. 2014;3:e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ostroff R, Foreman T, Keeney TR, Stratford S, Walker JJ, Zichi D. The stability of the circulating human proteome to variations in sample collection and handling procedures measured with an aptamer-based proteomics array. J Proteomics. 2010;73:649–666. [DOI] [PubMed] [Google Scholar]
- 17.Ostroff RM, Mehan MR, Stewart A, et al. Early detection of malignant pleural mesothelioma in asbestos-exposed individuals with a noninvasive proteomics-based surveillance tool. PLoS One. 2012;7:e46091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sattlecker M, Kiddle SJ, Newhouse S, et al. Alzheimer’s disease biomarker discovery using SOMAscan multiplexed protein technology. Alzheimers Dement. 2014;10:724–734. [DOI] [PubMed] [Google Scholar]
- 19.World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–2194. [DOI] [PubMed] [Google Scholar]
- 20.[No authors listed].Incidence of aplastic anemia: the relevance of diagnostic criteria. By the International Agranulocytosis and Aplastic Anemia Study. Blood. 1987;70:1718–1721. [PubMed] [Google Scholar]
- 21.Camitta BM, Thomas ED, Nathan DG, et al. Severe aplastic anemia: a prospective study of the effect of early marrow transplantation on acute mortality. Blood. 1976;48:63–70. [PubMed] [Google Scholar]
- 22.Candia J, Cheung F, Kotliarov Y, et al. Assessment of variability in the SOMAscan assay. Sci Rep. 2017;7:14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheung F, Fantoni G, Conner M, et al. Web tool for navigating and plotting SomaLogic ADAT files. J Open Res Softw. 2017;5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oliveros JC. Venny: An interactive tool for comparing lists with Venn’s diagrams 2007–2015. Available at: http://bioinfogp.cnb.csic.es/tools/venny/index.html.
- 25.Giudice V, Wu Z, Kajigaya S, et al. Circulating S100A8 and S100A9 protein levels in plasma of patients with acquired aplastic anemia and myelodysplastic syndromes. Cytokine. 2018. June 26. pii: SI043–4666(18)30274–6. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Florkowski CM. Sensitivity, specificity, receiver-operating characteristic (ROC) curves and likelihood ratios: communicating the performance of diagnostic tests. Clin Biochem Rev. 2008;29: S83–S87. [PMC free article] [PubMed] [Google Scholar]
- 27.Fabregat A, Jupe S, Matthews L, et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018;46:D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vargas AJ, Harris CC. Biomarker development in the precision medicine era: lung cancer as a case study. Nat Rev Cancer. 2016;16:525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gramolini A, Lau E, Liu PP. Identifying low-abundance biomarkers: aptamer-based proteomics potentially enables more sensitive detection in cardiovascular diseases. Circulation. 2016;134:286–289. [DOI] [PubMed] [Google Scholar]
- 31.De Groote MA, Sterling DG, Hraha T, et al. Discovery and validation of a six-marker serum protein signature for the diagnosis of active pulmonary tuberculosis. J Clin Microbiol. 2017;55: 3057–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gramolini AO, Peterman SM, Kislinger T. Mass spectrometry-based proteomics: a useful tool for biomarker discovery? Clin Pharmacol Ther. 2008;83:758–760. [DOI] [PubMed] [Google Scholar]
- 33.Ngo D, Sinha S, Shen D, et al. Aptamer-based proteomic profiling reveals novel candidate biomarkers and pathways in cardiovascular disease. Circulation. 2016;134:270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hathout Y, Brody E, Clemens PR, et al. Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2015;112:7153–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ganz P, Heidecker B, Hveem K, et al. Development and validation of a protein-based risk score for cardiovascular outcomes among patients with stable coronary heart disease. JAMA. 2016;315:2532–2541. [DOI] [PubMed] [Google Scholar]
- 36.Yu Z, Kastenmüller G, He Y, et al. Differences between human plasma and serum metabolite profiles. PLoS One. 2011;6: e21230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Bryant SE, Xiao G, Zhang F, et al. Validation of a serum screen for Alzheimer’s disease across assay platforms, species, and tissues. J Alzheimers Dis. 2014;42:1325–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Biancotto A, Feng X, Langweiler M, Young NS, McCoy JP. Effect of anticoagulants on multiplexed measurement of cytokine/chemokines in healthy subjects. Cytokine. 2012;60:438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voorzanger-Rousselot N, Goehrig D, Facón T, Clézardin P, Gar-nero P. Platelet is a major contributor to circulating levels of Dickkopf-1: clinical implications in patients with multiple myeloma. Br J Haematol. 2009;145:264–266. [DOI] [PubMed] [Google Scholar]
- 40.Behets GJ, Viaene L, Meijers B, et al. Circulating levels of scle-rostin but not DKK1 associate with laboratory parameters of CKD-MBD. PLoS One. 2017;12:e0176411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo Y, Mishra A, Howland E, et al. Platelet-derived Wnt antagonist Dickkopf-1 is implicated in ICAM-l/VCAM-1-mediated neutrophilic acute lung inflammation. Blood. 2015;126:2220–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lento W, Congdon K, Voermans C, Kritzik M, Reya T. Wnt signaling in normal and malignant hematopoiesis. Cold Spring Harb Perspect Biol. 2013;5:a008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fleming HE, Janzen V, Lo Celso C, et al. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell. 2008;2: 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Luis TC, Ichii M, Brugman MH, Kincade P, Staal FJ. Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia. 2012;26:414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Himburg HA, Doan PL, Quarmyne M, et al. Dickkopf-1 promotes hematopoietic regeneration via direct and niche-mediated mechanisms. Nat Med. 2017;23:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Function Niehrs C. and biological roles of the Dickkopf family of Wnt modulators. Oncogene. 2006;25:7469–7481. [DOI] [PubMed] [Google Scholar]
- 47.Chiang KC, Yeh CN, Chung LC, et al. WNT-1 inducible signaling pathway protein-1 enhances growth and tumorigenesis in human breast cancer. Sci Rep. 2015;5:8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grassinger J, Mueller G, Zaiss M, Kunz-Schughart LA, Andreesen R, Hennemann B. Differentiation of hematopoietic progenitor cells towards the myeloid and B-lymphoid lineage by hepatocyte growth factor (HGF) and thrombopoietin (TPO) together with early acting cytokines. Eur J Haematol. 2006;77:134–144. [DOI] [PubMed] [Google Scholar]
- 49.Pecquet C, Diaconu CC, Staerk J, et al. Thrombopoietin receptor down-modulation by JAK2 V617F: restoration of receptor levels by inhibitors of pathologic JAK2 signaling and of proteasomes. Blood. 2012;119:4625–4635. [DOI] [PubMed] [Google Scholar]
- 50.Zhao X, Feng X, Wu Z, et al. Persistent elevation of plasma thrombopoietin levels after treatment in severe aplastic anemia. Exp Hematol. 2018;58:39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grozovsky R, Giannini S, Falet H, Hoffmeister KM. Regulating billions of blood platelets: glycans and beyond. Blood. 2015;126:1877–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.